
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpeciation of nanoscale objects by nanoparticle imprinted matrices†
Maria
Hitrik
a,
Yamit
Pisman
a,
Gunther
Wittstock
 *b and
Daniel
Mandler
*a
*b and
Daniel
Mandler
*a
aInstitute of Chemistry, the Hebrew University of Jerusalem, Jerusalem 9190401, Israel. E-mail: daniel.mandler@mail.huji.ac.il
bInstitute of Chemistry, Center of Interface Science, Faculty of Mathematics and Natural Sciences, Carl von Ossietzky University of Oldenburg, Oldenburg, D-26111, Germany
First published on 22nd February 2016
Abstract
The toxicity of nanoparticles is not only a function of the constituting material but depends largely on their size, shape and stabilizing shell. Hence, the speciation of nanoscale objects, namely, their detection and separation based on the different species, similarly to heavy metals, is of outmost importance. Here we demonstrate the speciation of gold nanoparticles (AuNPs) and their electrochemical detection using the concept of “nanoparticles imprinted matrices” (NAIM). Negatively charged AuNPs are adsorbed as templates on a conducting surface previously modified with polyethylenimine (PEI). The selective matrix is formed by the adsorption of either oleic acid (OA) or poly(acrylic acid) (PAA) on the non-occupied areas. The AuNPs are removed by electrooxidation to form complementary voids. These voids are able to recognize the AuNPs selectively based on their size. Furthermore, the selectivity could be improved by adsorbing an additional layer of 1-hexadecylamine, which deepened the voids. Interestingly, silver nanoparticles (AgNPs) were also recognized if their size matched those of the template AuNPs. The steps in assembling the NAIMs and the reuptake of the nanoparticles were characterized carefully. The prospects for the analytical use of NAIMs, which are simple, of small dimension, cost-efficient and portable, are in the sensing and separation of nanoobjects.
Introduction
Nanoparticles (NPs) are continuously introduced into new applications in different areas such as medicine,1–10 photonics,11–15 catalysis16–21 and others because of their unique size-dependent chemical and physical properties. The small size of NPs, which accounts for their attractive functional characteristics, also causes changes in the toxicological behaviour as compared to the corresponding bulk materials.2,22–31 Nanotoxicology, i.e. the study of the toxicity of nanomaterials, is an emerging field of research which receives increasing attention due to rising exposure of humans and the environment to artificial nanoobjects. Recent studies have clearly shown that the nanotoxicity of NPs depends on the size, shape, charge and chemical nature of the stabilizing shell and charge.25,31 These factors often affect the toxicity more than the toxicological properties of the bulk material. Even inert bulk materials, such as gold or TiO2, may become harmful as nanomaterials.26 For instance, Tsoli et al.32 demonstrated that 1.4 nm gold NPs (AuNPs) penetrated cells and nuclear membranes and could irreversibly coordinate to natural DNA. Pernodet et al.33 showed that citrate stabilized AuNPs of 14 nm diameter can easily penetrate the cell membrane and accumulate in vacuoles and decrease cell proliferation and motility. These examples show that there are completely new, often unknown modes by which NPs interfere with biological tissues (cells) and these interactions cannot be deduced from the known behaviour of solid bulk materials or dissolved ions, but uptake tests with NPs of different sizes, shells or shapes are required. This does not only limit the in vivo exploitation of AuNPs in medicine and related fields but also requires an extensive, rigorous and expensive investigation of the long- and short-term impacts of different NPs on living species before they can be brought into application.Interestingly, this situation is reminiscent of the speciation analysis of heavy metals, i.e. the toxicity as well as other physical and chemical properties of NPs depend largely on the specific species and not only on the elemental composition of the NP core. Hence, we believe that speciation of NPs based on their size, structure and stabilizing shell, coupled with the study of NP interactions with different materials, can contribute a new in vitro tool to the list of nanotoxicology tools for the fast and reliable detection of nanoobjects based on their structural parameters, which can be easily applied in laboratory and field settings.
Most of the classic methods for NP detection and characterization being used today (SEM, AFM, TEM, fluorescence etc.) are time-consuming and/or require bulky and expensive instrumentation with environmental control (vacuum, temperature, etc.). In addition, most of these techniques require analyte labelling or sample pretreatments (such as solvent pre-evaporation and others) that can affect the size and surface composition of the studied population of NPs. However, there are very few new methods for the detection and differentiation of NPs by their size, shape and shell that are based on optical methods, such as scattered light on-chip spectroscopy34 or Raman microlaser sensors.35,36 Nanoparticle voltammetry is an electrochemical sizing method based on individual collisions and conversions of NPs with microelectrodes.37–41
However, electrochemical NP detection should be carried out in electrolyte solutions, where NPs tend to aggregate, which might cause dramatic changes in the size distribution and shell structure of the studied NPs.
Recently, we have demonstrated a new approach coined ‘nanoparticles imprinted matrices’ (NAIMs) that enables size or shell differentiation of AuNPs from aqueous solutions.42,43 AuNPs were chosen as an example of nanoobjects because of their multidisciplinary applications,2,18,44,45 in particular in medical diagnostics46 and drug delivery.47 The NAIMs approach extends the well-known concept of molecularly imprinted polymers (MIPs)48–55 to nano-analytes. In MIPs the molecular analyte-template is imprinted in a polymeric matrix. After release, it leaves behind a cavity with the dimensions of the analyte molecule and functional groups in the walls of the matrix that favour interactions with just the analyte molecule. Those specific voids can be used for a selective reuptake of the analyte. MIPs have been used as sensing layers,56–59 catalysts60–62 and as chromatographic phases.63
Here, a new and simplified methodology is presented for NPs imprinting compared to the original assembly of the matrix by the Langmuir–Blodgett technique.42 The new method, based exclusively on self-assembly, is schematically shown in Scheme 1.
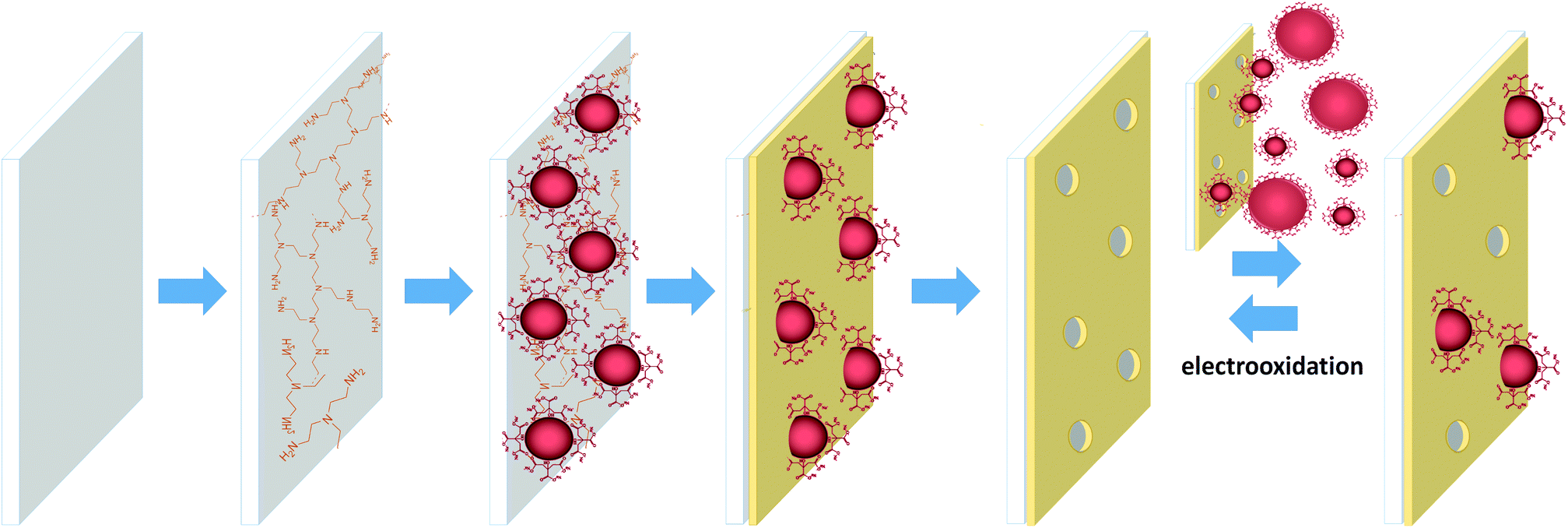 | ||
| Scheme 1 Schematic representation of the step-by-step formation of the NAIM system: 1 – modification of ITO with PEI, 2 – adsorption of AuNPs, 3 – adsorption of an organic layer in the non-occupied areas to form the matrix, 4 – removal of the AuNPs by electrooxidation, and 5 – reversible reuptake of template AuNPs that can be removed by electrooxidation. | ||
Specifically, negatively charged citrate-stabilized AuNPs were adsorbed on polyethylenimine (PEI) modified indium tin oxide (ITO), used as the solid electrode-substrate. The ITO areas, which were not occupied by the AuNPs, were coated with either oleic acid (OA) or poly(acrylic acid) (PAA) via self-assembly, forming the matrix. The NPs were dissolved by electrochemical oxidation leaving behind voids in the organic layer. The voids were complementary to the shape of the template AuNPs and showed remarkable size selectivity when exposed to AuNPs of different diameters. The selectivity was further improved by thickening the PAA template by self-assembly of an additional 1-hexadecylamine layer. The reuptake of silver NPs (AgNPs) with a similar shape, size and shell to the template AuNPs showed that the voids recognized different core NPs by their surface and geometric characteristics. Hence, this novel approach clearly shows promise towards the selective detection of NPs as well as the separation of them based on their structural properties.
Experimental
Materials
One side conductive ITO plates (7 × 50 × 0.7 mm3, surface conductivity Rs = 15–25 Ω per square) were purchased from Delta Technologies. Chloroauric acid hydrate (HAuCl4·H2O, 99.99% trace metals basis), polyethylenimine (PEI) aqueous solution (50% w/v, Mw = 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), oleic acid (OA, ≥99%), poly(acrylic acid) – PAA (analytical standard, Mw = 130000 g mol−1), K4[Fe(CN)6] – potassium hexacyanoferrate(II) (≥99.95%), tannic acid and 1-hexadecylamine (99%) were purchased from Sigma-Aldrich and used as received. Potassium chloride (Gadot, 99%), ethanol (absolute, J.T. Baker), acetone (for liquid chromatography, LiChrosolv) and trisodium citrate (BDH Chemicals, 99%) were also used without further purification. Ultrapure water (EasyPure UV, Barnstead 18.3 MΩ cm) was used for all the experiments. Solid AgNO3 (GR-grade) was purchased from Merck. Si wafers (n-type/As, resistivity <0.005 Ω cm, thickness (525 ± 20) μm) were obtained from Silicon Materials Inc., Belarus.
000 g mol−1), oleic acid (OA, ≥99%), poly(acrylic acid) – PAA (analytical standard, Mw = 130000 g mol−1), K4[Fe(CN)6] – potassium hexacyanoferrate(II) (≥99.95%), tannic acid and 1-hexadecylamine (99%) were purchased from Sigma-Aldrich and used as received. Potassium chloride (Gadot, 99%), ethanol (absolute, J.T. Baker), acetone (for liquid chromatography, LiChrosolv) and trisodium citrate (BDH Chemicals, 99%) were also used without further purification. Ultrapure water (EasyPure UV, Barnstead 18.3 MΩ cm) was used for all the experiments. Solid AgNO3 (GR-grade) was purchased from Merck. Si wafers (n-type/As, resistivity <0.005 Ω cm, thickness (525 ± 20) μm) were obtained from Silicon Materials Inc., Belarus.
Synthesis of AuNPs
Citrate stabilized spherical AuNPs (10 ± 2 nm and 40 ± 2 nm) were synthesized according to Bastus et al.64 based on the kinetic control of the NP growth. The final NP solution was cooled to room temperature and diluted by 2.5 (from 150 ml to 375 ml) with deionized water. This resulted in the formation of 10 nm diameter AuNPs. In order to obtain larger AuNPs of 40 nm diameter, the boiling purple solution was cooled to 90 °C without dilution followed by 14 additions of HAuCl4 and citrate every 30 min. These additions caused only the existing particles to grow without the formation of new AuNPs.64 The particles were examined by XHR-SEM. The final solution after addition of citrate was diluted to 375 ml (for details see ESI S-1.1†).Synthesis of AgNPs
Citrate stabilized spherical AgNPs were synthesized by the procedure of Bastus.65 The obtained AgNPs had a diameter of (10 ± 3) nm (for details see ESI S-1.2†).Instrumentations
See ESI S-1.3.†Procedures
ITO and Si surfaces were washed carefully prior to film adsorption by successive sonications for 10 min each in acetone, ethanol and deionized water. The ITO plates were placed in glass petri dishes filled with PEI (0.72 mg ml−1) with the conductive side facing up. After 2 h, the ITO plates were washed thoroughly with water and placed vertically into small (4 ml) vials filled with AuNP solution for 1 h in order to reduce the adsorption of aggregates. This was followed by immersion into an aqueous solution of OA (0.24 mg ml−1) or PAA (0.067 mg ml−1) for 1 h. The surfaces were washed carefully with deionized water and dried. The AuNPs were electrooxidized using linear sweep voltammetry (LSV) between 0.5 and 1.2 V at a scan rate of 0.05 V s−1. The surface after oxidation of the template NPs constitutes the NAIM. The reuptake of AuNPs was accomplished by immersing these (oxidized) surfaces into AuNP or AgNP solutions for 1 h followed by careful rinsing. The template thickening experiment was performed using electrooxidized NAIM ITO/PEI/AuNPs/PAA that was immersed into 1-hexadecylamine water solution (0.067 mg ml−1) for 2 h. The surfaces were washed with water.Results and discussion
NAIM formation and characterization
The first step in the formation of NAIM was the adsorption of a positively charged polymer, PEI, for enhancing the adsorption of the negatively charged, citrate stabilized AuNPs (Scheme 1) in neutral pH. Fig. S-2† compares the SEM image (S-2a to c†) and LSV (S-2d†) of AuNPs adsorbed on bare and PEI modified ITO plates at different PEI concentrations. Evidently, the surface density of the adsorbed NPs increased dramatically due to PEI. Based on these experiments, we employed a solution of 0.72 mg ml−1 PEI, which yielded a sufficient AuNP density without aggregation.The adsorption of the AuNPs is clearly time dependent as shown in Fig. S-3.† Specifically, SEM (S-3a–S-3d†) images of ITO identically treated with PEI show an increase of AuNP density with immersion time in the AuNP solution for 5, 30, 60 and 120 min. Interestingly, the density of AuNPs increased almost linearly with time from (100 ± 12) to (7700 ± 27) NPs μm−2 (Fig. S-3e†). This implies that the repulsion between NPs in solution is overcompensated by the strong surface–NP interaction. Based on these results, we decided to adsorb the AuNPs for 60 min, which is a compromise between sufficient density of the AuNPs and prevention of NP aggregation on the surface.
The next step involved self-assembled filling of the non-occupied area with the filler, namely, OA or PAA. Due to the small film thickness relative to the ITO roughness, it was impossible to monitor their adsorption with SEM or AFM. Therefore, the step-by-step adsorption of PEI, AuNPs and the fillers was visualized with AFM on a Si wafer (Fig. 1). While the clean Si surface showed a roughness of (0.31 ± 0.05) nm (Fig. 1a), a slightly but distinctly increased roughness of (0.52 ± 0.05) nm was measured after the adsorption and careful removal of PEI excess (Fig. 1b). Subsequent adsorption of AuNPs further increased the roughness to (5.6 ± 0.1) nm and individual AuNPs could be seen in Fig. 1c. The addition of a covering layer of either OA (Fig. 1d) or PAA (not shown) affected the topography of the surface. The obtained roughness values were (3.6 ± 0.1) nm and (4.0 ± 0.1) for OA and PAA, respectively. The decreased roughness was attributed to the adsorption of the fillers onto the PEI, in between the AuNPs. Moreover, rough estimations of the thicknesses of the template fillers are ca. 4 and 3 nm for the OA and PAA layers, respectively.
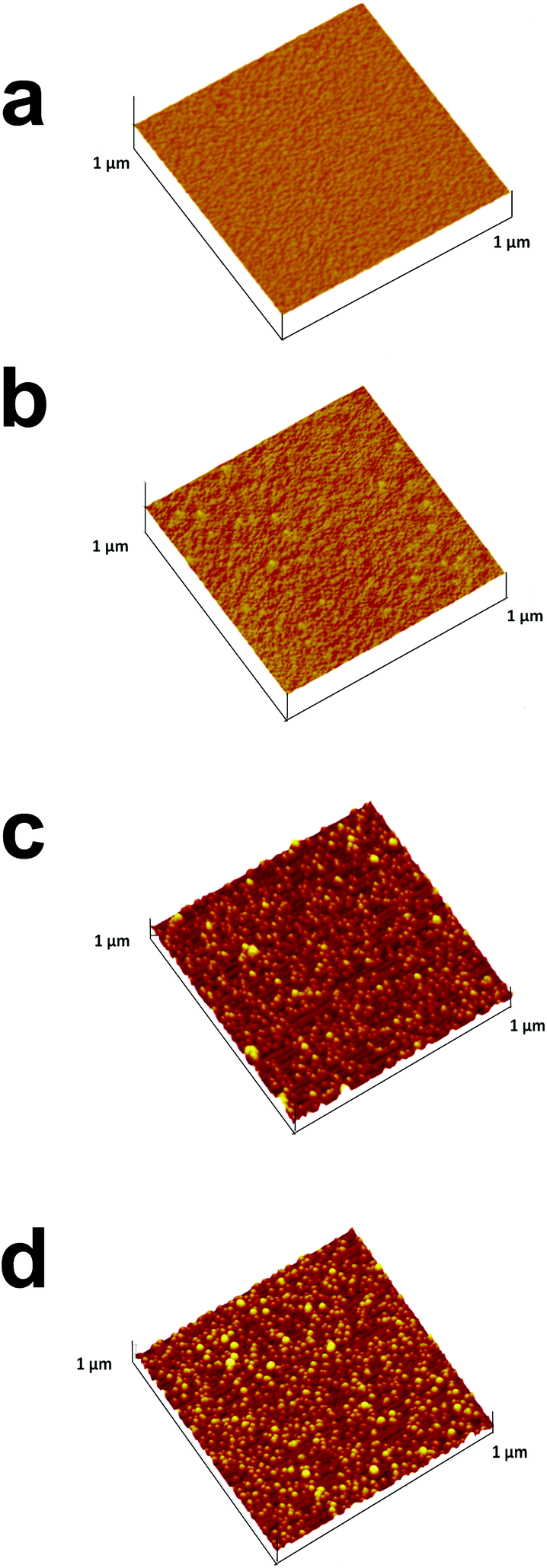 | ||
| Fig. 1 AFM images of sequentially assembled NAIM systems on Si wafers: (a) bare Si, (b) Si after treatment with PEI (1 h), (c) followed by AuNP adsorption for 2 h, and (d) after adsorbing OA for 1 h. Z-Axes max = 25 nm. | ||
Further evidence for the sequential assembly of the NAIM system was obtained by contact angle measurements (Table 1). The contact angle of ITO slightly increased after adsorbing PEI and was unaffected by the adsorption of the AuNPs. The significant change occurred upon adsorption of OA or PAA. While the former increased the contact angle to 87° ± 7° due to the hydrophobic nature of its tails, the latter formed a more hydrophilic surface, 62° ± 7°, probably due to the high charge density.
| ITO | ITO/PEI | ITO/PEI/AuNPs | ITO/PEI/AuNPs/ | ||
|---|---|---|---|---|---|
| PAA | OA | ||||
| WCA | 70° ± 6° | 77° ± 6° | 76° ± 4° | 62° ± 7° | 87° ± 7° |
The formation stages of the NAIM could be followed nicely by CV as shown in Fig. 2. The CV of [Fe(CN)6]4− was measured on bare ITO, ITO coated PEI, after adsorption of AuNPs and following the self-assembly of OA or PAA. As a result of PEI adsorption (curve 2), the faradaic current increased relative to the bare ITO (curve 1). This can be explained by the attractive forces between the positively charged PEI layer and the negatively charged redox species, i.e. [Fe(CN)6]4−. Hence, [Fe(CN)6]4− could accumulate in or on the PEI film leading to enhanced peak currents. Interestingly, the current further increased upon adsorption of the AuNPs (curve 3). This is not trivial and cannot be attributed to increasing the surface area as the diffusion layer is significantly thicker (a few micrometers) than the surface roughness due to AuNPs. Such behavior has been reported previously.66 We believe that the increase in the current was due to the enhanced rate of electron transfer on AuNPs as compared to ITO although this cannot be seen in decreasing the difference between the anodic and cathodic peak potentials. Nevertheless, it is expected to affect the faradaic current.
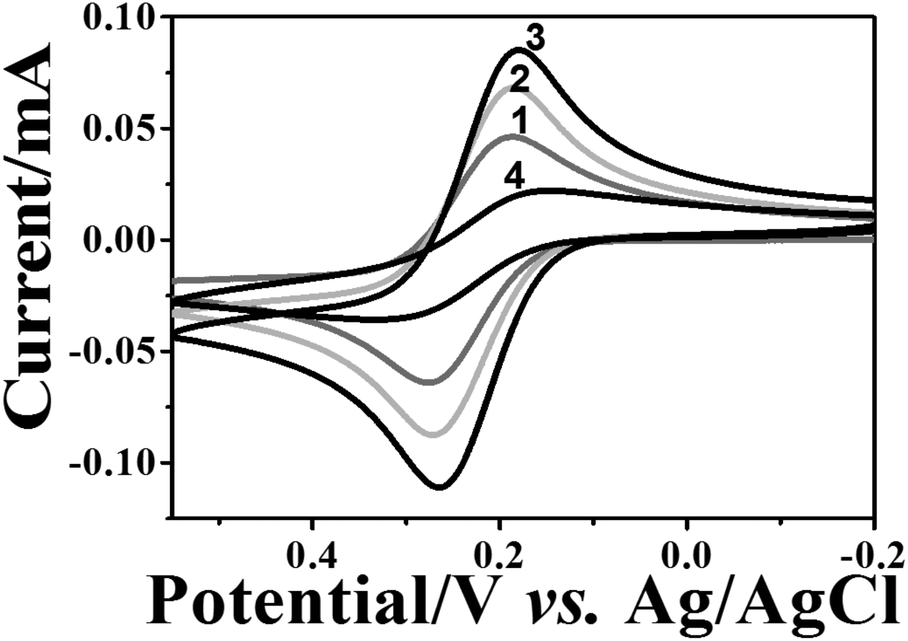 | ||
| Fig. 2 Electrochemical characterization of the step-by-step film assembly. CV recorded in 1 mM [Fe(CN)6]4− and 0.1 M KCl at (1) bare ITO; (2) after treatment with PEI; (3) after adsorption of AuNPs; and (4) after treatment with OA. The scan rate was 0.05 V s−1. | ||
Introduction of the filler layer of OA (curve 4) or PAA (not shown) for matrix formation decreased the faradaic current. In the case of OA, the current decline can be attributed to the insulating nature of OA that causes the decrease of the active electrode area. It is important to note that ITO covered with PEI and OA or PAA only, i.e. without AuNPs, resulted in a full blockage of the electrode towards electron transfer to [Fe(CN)6]4− (Fig. S-4a,† curves 2 and 3). However, for the PAA filler, a reversible CV was obtained when a negatively charged redox probe was replaced with a positively charged one – [Ru(NH3)6]3+ (Fig. S-4b†), demonstrating the non-insulating nature of the film. Therefore, the decrease in the redox current for [Fe(CN)6]4− in the case of the PAA filler must have been caused by the electrostatic repulsion between the negatively charged redox probe and the PAA layer. These results strongly suggest that the redox current of [Fe(CN)6]4− at ITO/PEI/AuNPs/OA or ITO/PEI/AuNPs/PAA passed through the uncovered AuNPs.
Electrochemical removal of AuNPs from NAIMs
As the next step, AuNPs were removed from the ITO/PEI/AuNPs/(PAA or OA) by successive LSV scans between 0.5 V and 1.3 V (Fig. S-5†). The peak area and peak height decreased in subsequent scans until almost complete removal of AuNPs. The oxidation potential of the metallic NPs depends on their size, i.e. the smaller the particle the less positive is the oxidation potential.67,68 For example, the oxidation peak of AuNPs with 10 nm diameter was at ca. 1.0 V or less, while that of AuNPs with 40 nm diameter was at 1.1–1.2 V (Fig. S-6†). Furthermore, the removal of larger AuNPs required more scans.Further support for the oxidation and removal of the AuNPs from the matrix was obtained by AFM. Fig. 3 shows an AFM image of ITO/PEI/AuNPs/PAA before (a) and after (b) electrooxidation of the AuNPs. The change of topography can be seen clearly. Prior to the oxidation of the NPs, the roughness of the matrix was higher and the topography possessed many “bumps” that disappeared upon oxidation of the AuNPs. OA NAIMs gave similar results.
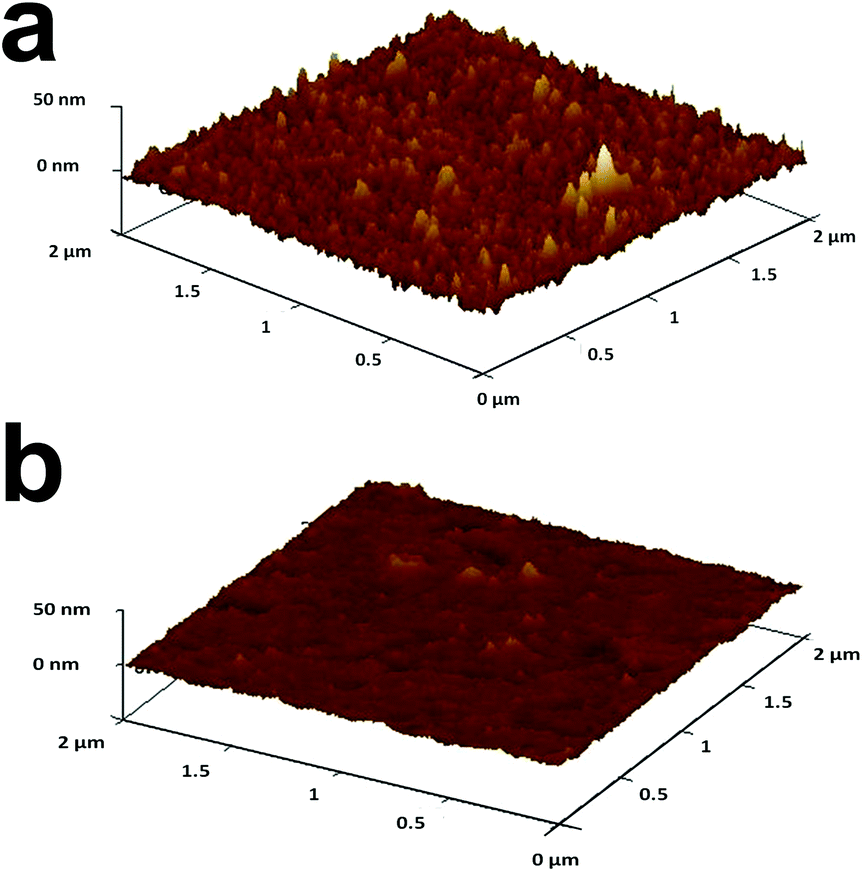 | ||
| Fig. 3 AFM images of topography changes of ITO/PEI/AuNPs/PAA before (a) and after (b) electrooxidation of AuNPs. LSV between 0.6 V and 1.3 V at 0.05 V s−1. X- and Y-axes max = 2 μm, Z-axes max = 50 nm. | ||
AuNPs reuptake experiments
At this stage, the NAIMs were ready for the most important challenge of the NAIM concept – the reuptake of the analyte template AuNPs. This was monitored by voltammetry as shown in Fig. 4. While Fig. 4a shows the CVs of the redox probe [Fe(CN)6]4− measured with ITO electrodes covered with the matrix after different preparation steps, Fig. 4b shows the LSVs of the AuNP oxidations. CV 1 and 2 (Fig. 4a) were recorded before and after oxidation of the original imprinted AuNPs, respectively. It is evident that the matrix became blocking towards [Fe(CN)6]4− as a result of the oxidative removal of AuNPs. We believe that this can be explained again by assuming that electron transfer to and from [Fe(CN)6]4− occurred at the AuNPs. This requires that the particles remain uncovered after the adsorption of the filler, in agreement with our results in Fig. 1 and 2. Moreover, the formation of areas not coated by the filler, i.e., voids, occurred due to the removal of the AuNPs. The voids contributed an almost undetectable faradaic current, implying that the area of the voids was negligible and the distance between the voids was substantially larger than their diameter. The difference between the charge transfer through the layers with and without AuNPs was drastic. We believe that this difference resulted from both faster electron transfer through the AuNPs and the geometry change of the conductive areas (see Scheme S-7†).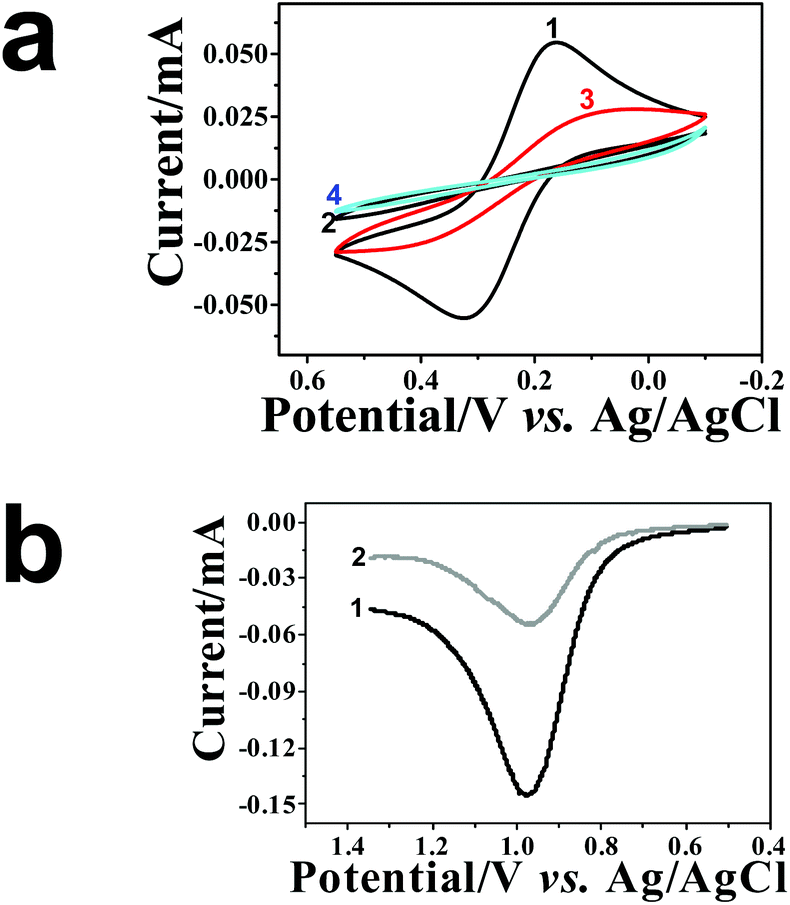 | ||
| Fig. 4 Electrochemical characterization of ITO/PEI/AuNPs/PAA: (a) CV of the ITO electrode covered with a PAA matrix: (1) before and (2) after AuNP oxidation; (3) after AuNPs reuptake and (4) after the second oxidation. Experiments were carried out in 1 mM [Fe(CN)6]4− at 0.1 M KCl. (b) LSV of (1) template AuNP oxidation and (2) oxidation of reuptaken AuNPs in 0.1 M KCl at a scan rate of 0.05 V s−1. | ||
The electrochemical activity of the voids depended on their size and their average spacing. This can be seen in the CVs obtained with ITO/PEI/(40 nm AuNPs)/OA (Fig. S-8†). Higher currents were obtained before and after oxidative removal of the 40 nm AuNPs compared to the corresponding layers with AuNPs of 10 nm diameter. The 40 nm AuNPs were less embedded into the PAA or OA thin filler layer. Therefore, a larger uncovered area was available for electron transfer to/from the redox probe. After electrooxidation of the 40 nm AuNPs, voids of larger area were formed which exposed more of the conductive “bottom” surface, when compared to the voids formed by AuNPs with 10 nm diameter. In addition, incomplete dissolution of the larger AuNPs may additionally provide surfaces with a faster electron transfer rate to the dissolved redox probe. Similar results were obtained with PAA as a filler.
Next, we studied the reuptake of the AuNPs by the voids of the NAIMs. The latter were immersed into the solution of AuNPs with the same diameter as that used for imprinting for 1 h. Fig. 4b shows the LSV of the oxidation of the imprinted AuNPs (curve 1) and for the oxidation of the AuNPs that were taken up by the voids (curve 2). Evidently, the oxidation wave after reuptake was smaller than the original wave of the template removal, indicating that fewer AuNPs were taken up by the voids during the same immersion conditions as those used for the initial immobilization of the template AuNPs on the PEI layer. It is known that the area under the LSV wave is proportional to the number of oxidized AuNPs provided that the AuNPs have the same size and are completely oxidized (see the explanation of the calculation in S9†). For ITO/PEI/AuNPs/PAA the ratio between the number of AuNPs reuptaken into the matrix and the originally adsorbed AuNPs was (55 ± 5)%, for the OA filler this ratio was (51 ± 6)%. In addition, the CV of [Fe(CN)6]4− recorded after AuNPs reuptake (Fig. 4a, curve 3) exhibited a higher current than the same electrode after template oxidation but before the reuptake (curve 2). However, the current after reuptake did not reach the values recorded before template oxidation (curve 1) in agreement with the LSV in Fig. 4b. In the field of molecular-imprinted polymers it is common to characterize the uptake of a non-imprinted film. In NAIM systems adsorption of NPs to the matrix material outside the void (termed here non-specific adsorption) may not necessarily lead to a detection of those NPs by electrochemical oxidation of the NPs because the matrix may prevent electronic communication between NPs and the ITO substrate. However, the non-specific adsorption outside the voids would decrease the concentration of NPs in the solution and lower the reuptake efficiency. Compared to our first NAIM system based on the Langmuir–Blodgett layer of neutral polyaniline,42 we aimed at decreasing the non-specific adsorption by employing a filler compound carrying the same charge as the citrate-capped AuNPs. Indeed, gold oxidation waves were not detected for ITO coated with only PEI/PAA or PEI/OA (Fig. S-10†). These results show that the adsorption of AuNPs occurred only in the imprinted voids. The fact that the voltammetric wave for the oxidation of the AuNPs was smaller than that during the release of the template AuNPs indicated that only a fraction of the voids were filled during the reuptake time. The tests were performed as separate tests for 10 nm and 40 nm AuNPs under strictly controlled reuptake conditions in order to avoid convolution with the faster mass transport for the smaller NPs.
Following the oxidation of the AuNPs reuptaken, the NAIMs returned to blocking behavior with respect to [Fe(CN)6]4− oxidation (Fig. 4a, curve 4). To summarize, AuNPs were electrochemically removed and then successfully reuptaken into the voids within the matrix previously assembled around the templates. The voids remained reproducibly active for reuptake for at least two successive experiments.
Fig. 5a shows a typical SEM image acquired after oxidizing the template AuNPs from ITO/PEI/AuNPs/PAA. The disappearance of the NPs is clearly noticed. Distinct AuNPs were seen following the reuptake process (Fig. 5b). It is evident that the density of the reuptaken AuNPs was significantly smaller than that of the template originally adsorbed (Fig. S-3c†). SEM images of blank experiments demonstrated that no or almost no particles were adsorbed when ITO covered with PEI/OA or PEI/PAA layers only was immersed into the AuNP solution for 1 h (Fig. S-11a and b†).
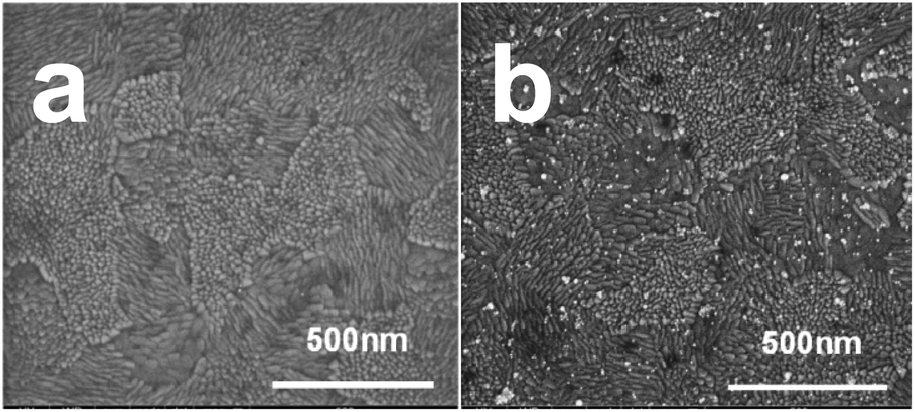 | ||
| Fig. 5 Morphology changes of the oxidized template upon particle reuptake. SEM images of ITO/PEI/AuNPs/PAA after oxidation (a) and after reuptake of the original AuNPs (b). | ||
Interestingly, the removal and reuptake of the AuNPs could also be followed by measuring the water contact angles. Table 2 summarizes the values for ITO/PEI/AuNPs/PAA and ITO/PEI/AuNPs/OA before and after AuNP removal as well as after AuNPs reuptake. The contact angle decreased upon removal of the AuNPs for the hydrophilic matrix PAA, whereas it increased slightly for the hydrophobic matrix OA. This is in agreement with the hydrophilic ligand shell of the AuNPs, which is more hydrophilic than the OA. However, based on contact angle results, AuNPs are more hydrophobic than the PAA layer. The contact angles changed again because of reuptaking the AuNPs, but they did not attain their original values, presumably due to the partial filling of the voids.
| Matrix | Before AuNP oxidation | After AuNP oxidation | After AuNPs reuptake |
|---|---|---|---|
| ITO/PEI/AuNPs/PAA | 62° ± 7° | 36° ± 8° | 57° ± 7° |
| ITO/PEI/AuNPs/OA | 87° ± 7° | 93° ± 5° | 90° ± 7° |
NAIMs selectivity towards NP size
In the next stage, we examined the size-selectivity of the imprinted matrices towards NP of different size and with identical shell. After removal of the 10 nm AuNPs from the matrices, the PAA and OA based films were immersed for 1 h into a solution containing bigger AuNPs of ca. 40 nm diameter. We were careful to apply exactly the same reuptake conditions AuNP concentration,64 time and temperature, as in the previous experiments. Fig. 6 shows the LSV of the PAA based NAIMs on ITO after reuptake of AuNPs with 40 and 10 nm diameters. Clearly, the resultant oxidation peak areas from 40 nm AuNPs were larger than those from the original 10 nm AuNPs. In order to compare the number of AuNPs that were reuptaken, the charge obtained from the oxidation wave must be normalized with respect to the size of the NPs. ESI S-9† details the considerations. To relate the number of 40 nm and 10 nm AuNPs, the charge for the oxidation of 40 nm AuNPs was divided by 64 (volume ratio between AuNPs of 10 nm and 40 nm diameters). The inset of Fig. 6 is shown here for illustration purposes only in order to visualize the difference between the peak areas when the normalization factor was taken into account. After this consideration, it became clear that much fewer AuNPs with 40 nm diameter were reuptaken as compared to 10 nm AuNPs. OA based NAIMs gave similar results (not shown). This was supported by SEM images shown in Fig. S-12.† It is evident that the number of small particles that were reuptaken by the 10 nm template voids is remarkably greater than that of larger particles.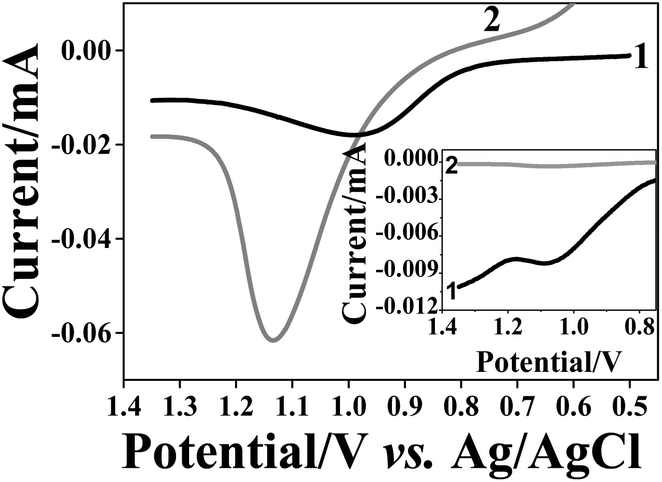 | ||
| Fig. 6 LSV recorded in 0.1 M KCl for NAIMs formed by ITO/PEI/10 nm AuNP/PAA after reuptake of 10 nm (curves 1) and 40 nm (curves 2) AuNPs for 1 h. The inset compares the curves after the currents were normalized to the particle volumes: for 10 nm AuNPs (curve 1) the current was divided by 1 and for 40 nm AuNPs (curve 2) the current was divided by 64. | ||
The quantitative analysis of the selectivity of the NAIMs was based on the normalized charges of the oxidation peaks of the reuptaken AuNPs. First, we considered the reuptake ratio, i.e., the normalized charge in the LSV oxidation wave of the reuptaken NPs (10 or 40 nm diameters) divided by the charge for oxidizing the 10 nm template AuNPs. This ratio was 50–55% for the 10 nm AuNPs while it was only 1.5–3% for the 40 nm AuNPs (Table 3). This clearly demonstrates the high preference of voids imprinted by 10 nm AuNPs to reuptake the NPs with matching size, i.e. the 10 nm AuNPs. Dividing the reuptake ratio for 10 nm AuNPs by that of the 40 nm AuNPs allowed estimating the selectivity factor associated with the reuptake process. The selectivity factors for PAA and OA based matrices (imprinted by 10 nm AuNPs) were 14–35 and 17–142, respectively (Table 3).
| Matrix | Reuptake ratio for 10 nm AuNPs [%] | Reuptake ratio for 40 nm AuNPs [%] | Selectivity factor |
|---|---|---|---|
| PAA | 55 ± 5 | 2.6 ± 0.9 | 14–35 |
| OA | 51 ± 6 | 1.5 ± 1.1 | 17–142 |
| PAA/1-hexadecylamine | 60 ± 8 | 0.6 ± 0.5 | 53–608 |
PAA layer thickening
In spite of the fact that the selectivity of the 10 nm imprinted NAIMs was at least 14 times higher towards the AuNPs used during imprinting compared to 40 nm AuNPs, we wished to increase it further. We attributed the observed limited reuptake of the larger AuNPs to the insufficient thickness of the PAA and OA layers around the voids. It resulted in shallow voids that enabled also the larger particles to partially penetrate some of the voids (S-13, Scheme S-3†). We hypothesized that thickening the matrix layer would improve the selectivity.Hence, we decided to form an additional layer of 1-hexadecylamine, a surfactant with a positively charged hydrophilic head group, on the negatively charged PAA at neutral pH. Therefore, after electrooxidation of the 10 nm template AuNPs, the ITO/PEI/AuNPs/PAA assembly was immersed into aqueous solution of 0.067 mg ml−1 1-hexadecylamine for 2 h. We expected that 1-hexadecylamine will self-assemble on top of the PAA layer avoiding the adsorption into the voids, due to electrostatic repulsion from positively charged PEI. The AuNPs were removed prior to this step because their negatively charged shell would likely lead to adsorption of 1-hexadecylamine, too. After adsorption of 1-hexadecylamine the contact angle of the ITO/PEI/(oxidized AuNPs)/PAA/1-hexadecylamine increased from (36 ± 8)° to (71 ± 3)° as expected for a self-assembled layer of surfactant molecules attached via the hydrophilic head group to the surface.
Subsequently, we studied the reuptake ability of this three-layered assembly for AuNPs with 10 and 40 nm diameters. Fig. 7 shows the AuNP oxidation waves obtained after reuptake. The peak areas (before normalization) of the 40 nm and 10 nm reuptaken AuNPs became similar in size indicating a dramatic decrease of the number of 40 nm AuNPs reuptaken as compared with the PAA matrix (Fig. 6). The reuptake ratio for the 10 nm diameter AuNPs was (60 ± 8)% while that of the 40 nm AuNPs was only (0.6 ± 0.5)%. Hence, the selectivity factor increased to 53–608 (Table 3). Therefore, it can be concluded that the matrix thickness and the void topology are important parameters that significantly influence the selectivity of the NAIM system.
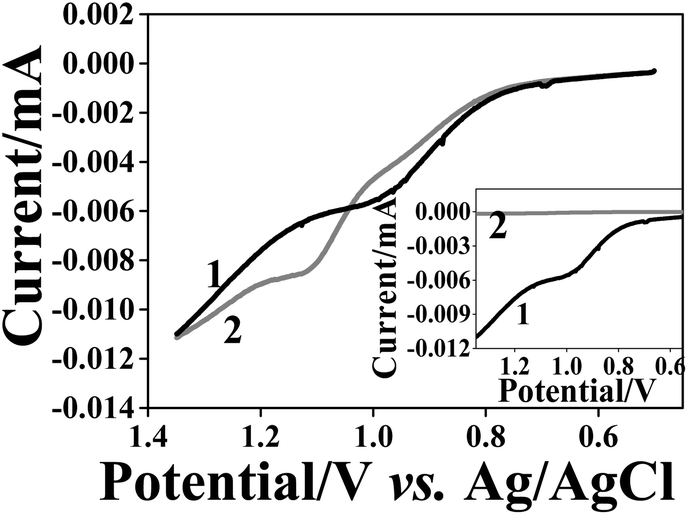 | ||
| Fig. 7 LSV recorded in 0.1 M KCl for ITO/PEI/10 nm AuNPs/PAA/1-hexadecylamine NAIM after reuptake of 10 nm (curves 1) and 40 nm (curves 2) AuNPs for 1 h. The inset compares the curves after the currents were normalized to the particle volumes. The currents were divided by 1 (curve 1) and 64 (curve 2) for the 10 and 40 nm AuNPs, respectively. | ||
Confirmation of the role of the NP shell
The reuptake abilities of the NAIMs were also tested for spherical silver NPs (AgNPs) with 10–13 nm diameter that were stabilized by the same citrate ligand shell as the AuNPs citrate stabilized. This test serves to prove the role of the ligand shell in the reuptake process of the NPs to the voids in the NAIM. NAIMs obtained after oxidative removal of AuNPs from ITO/PEI/AuNPs/OA or ITO/PEI/AuNPs/PAA were immersed into an aqueous solution containing a 50/50 (v/v) mixture of the original AuNP solution and freshly prepared AgNP solution. After the reuptake for 1 h, the NAIM surfaces were subjected to oxidation by LSV (Fig. 8). This system allowed the electrochemical observation of clearly separated oxidation waves for both AgNPs and AuNPs, indicating the successful reuptake of the particles from both types. The electrochemical signals not only prove the presence of the NPs at the surface, but must result from NPs located in the imprinted cavities because non-specifically adsorbed particles would not be in electrical contact with the ITO substrate. Therefore, we conclude that the NAIMs were able to detect NPs based on their size or shell rather than on the material they were made of.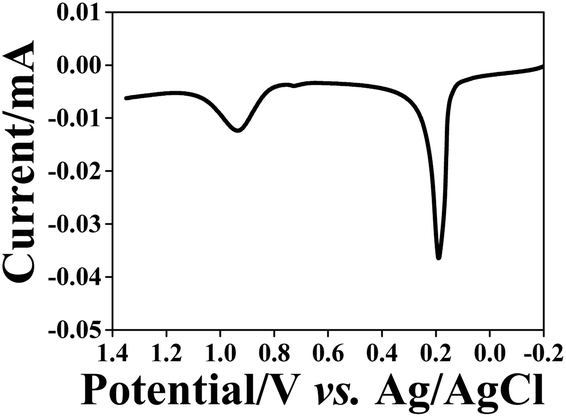 | ||
| Fig. 8 Oxidation of AgNPs and AuNPs reuptaken (1 h) from a NP mixture by the ITO/PEI/PAA NAIMs after oxidative removal of the 10 nm template AuNPs. LSV was recorded in 0.1 M KCl. | ||
Tests were also conducted with AuNP with a core diameter of 10 nm stabilized by a hydrophobic octadecylmercaptan shell and a positively charged hydrophilic chitosan polymer layer. In both cases, no adsorption of NP was detected. However, by exchanging the ligand shell also the overall particle size did change (13–14 nm for octadecylmercaptan and 60 nm for chitosan-stabilized AuNPs according to dynamic light scattering). Therefore, it is not necessarily the nature of the shell alone that excluded those particles from detection. This experiment highlights the difficulties of an isolated consideration of the core size, overall size, the nature of the ligand and shape with respect to uptake by NAIMs but most likely also for their biological effects.
Conclusions
Nanoparticle imprinted matrices (NAIMs) were prepared based entirely on self-assembly processes and layer-by-layer approaches. The process is based on forming voids within organic thin films by first adsorbing NPs of a narrow size-distribution followed by covering the free, non-occupied substrate surface by amphiphilic molecules serving as the filler. OA and the polymer PAA are suitable examples of filler molecules. The AuNPs were removed by anodic dissolution leaving behind nano-voids exhibiting high selectivity for the reuptake of NPs of the same size as the template NPs. The thickness of the organic matrix plays an important role in the recognition of the particles. Thickening the PAA matrix by adsorbing an additional long chain aliphatic amine deepened the voids and increased substantially the selectivity. Furthermore, we demonstrated that NPs made of different materials, such as silver and gold, could be reuptaken provided that their size and shell match those of the template NPs. The approach for NAIMs reuptake is simple, easy and inexpensive and can be developed further to obtain small and fast sensors for NPs based on their structural characteristics, namely, the size, shape and chemical nature of the shell – the most important parameters for the toxicity of NPs. In addition, this is the first step towards speciation of NPs. This will be accomplished by exploiting and improving the recognition of the NP shell by the matrix material of the NAIM. The NAIM concept allows designing the interaction of NAIMs with the NPs via supramolecular chemistry. We believe that NAIMs, which are simple, small, cost-efficient and portable, can further be exploited for sensing and separation of nanoobjects.Acknowledgements
The research was financially supported by the German–Israeli Foundation (Research Grant No. 1074-49.10/2009). The Harvey M. Krueger Family Centre for Nanoscience and Nanotechnology of the Hebrew University is acknowledged. M. Hitrik is indebted to the Wertheimer family for their support.References
- M. E. Akerman, W. C. W. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617–12621 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–U1140 CrossRef CAS PubMed.
- J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef CAS PubMed.
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef CAS PubMed.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS PubMed.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685–1706 CrossRef CAS.
- B. Luk'yanchuk, N. I. Zheludev, S. A. Maier, N. J. Halas, P. Nordlander, H. Giessen and C. T. Chong, Nat. Mater., 2010, 9, 707–715 CrossRef PubMed.
- S. A. Maier and H. A. Atwater, J. Appl. Phys., 2005, 98, 011101 CrossRef.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- L. Braydich-Stolle, S. Hussain, J. J. Schlager and M. C. Hofmann, Toxicol. Sci., 2005, 88, 412–419 CrossRef CAS PubMed.
- T. J. Brunner, P. Wick, P. Manser, P. Spohn, R. N. Grass, L. K. Limbach, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2006, 40, 4374–4381 CrossRef CAS PubMed.
- K. Donaldson, R. Aitken, L. Tran, V. Stone, R. Duffin, G. Forrest and A. Alexander, Toxicol. Sci., 2006, 92, 5–22 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- M. Heinlaan, A. Ivask, I. Blinova, H.-C. Dubourguier and A. Kahru, Chemosphere, 2008, 71, 1308–1316 CrossRef CAS PubMed.
- S. M. Hussain, K. L. Hess, J. M. Gearhart, K. T. Geiss and J. J. Schlager, Toxicol. In Vitro, 2005, 19, 975–983 CrossRef CAS PubMed.
- C. W. Lam, J. T. James, R. McCluskey, S. Arepalli and R. L. Hunter, Crit. Rev. Toxicol., 2006, 36, 189–217 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959–8964 CrossRef CAS PubMed.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS PubMed.
- M. Tsoli, H. Kuhn, W. Brandau, H. Esche and G. Schmid, Small, 2005, 1, 841–844 CrossRef CAS PubMed.
- N. Pernodet, X. H. Fang, Y. Sun, A. Bakhtina, A. Ramakrishnan, J. Sokolov, A. Ulman and M. Rafailovich, Small, 2006, 2, 766–773 CrossRef CAS PubMed.
- Q. Wei, H. Qi, W. Luo, D. Tseng, S. J. Ki, Z. Wan, Z. Gorocs, L. A. Bentolila, T.-T. Wu, R. Sun and A. Ozcan, ACS Nano, 2013, 7, 9147–9155 CrossRef CAS PubMed.
- B.-B. Li, W. R. Clements, X.-C. Yu, K. Shi, Q. Gong and Y.-F. Xiao, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14657–14662 CrossRef CAS PubMed.
- S. K. Oezdemir, J. Zhu, X. Yang, B. Peng, H. Yilmaz, L. He, F. Monifi, S. H. Huang, G. L. Long and L. Yang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3836–E3844 CrossRef PubMed.
- E. J. E. Stuart, K. Tschulik, C. Batchelor-McAuley and R. G. Compton, ACS Nano, 2014, 8, 7648–7654 CrossRef CAS PubMed.
- R. Dasari, K. Tai, D. A. Robinson and K. J. Stevenson, ACS Nano, 2014, 8, 4539–4546 CrossRef CAS PubMed.
- S. J. Kwon and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 7102–7108 CrossRef CAS PubMed.
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- X. Xiao, F.-R. F. Fan, J. Zhou and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16669–16677 CrossRef CAS PubMed.
- S. Kraus-Ophir, J. Witt, G. Wittstock and D. Mandler, Angew. Chem., Int. Ed., 2014, 53, 294–298 CrossRef CAS PubMed.
- N. Bruchiel-Spanier and D. Mandler, ChemElectroChem, 2015, 2, 795–802 CrossRef CAS.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- I. H. El-Sayed, X. H. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O'Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS PubMed.
- A. Moradian and K. Mosbach, J. Mol. Recognit., 1989, 2, 167–169 CrossRef CAS PubMed.
- D. J. O'Shannessy, L. I. Andersson and K. Mosbach, J. Mol. Recognit., 1989, 2, 1–5 CrossRef PubMed.
- B. Sellergren, M. Lepisto and K. Mosbach, J. Am. Chem. Soc., 1988, 110, 5853–5860 CrossRef CAS.
- G. Vlatakis, L. I. Andersson, R. Muller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS PubMed.
- G. Wulff, ACS Symp. Ser., 1986, 308, 186–230 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed., 1995, 34, 1812–1832 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1–27 CrossRef CAS PubMed.
- T. R. Ling, Y. Z. Syu, Y. C. Tasi, T. C. Chou and C. C. Liu, Biosens. Bioelectron., 2005, 21, 901–907 CrossRef CAS PubMed.
- N. M. Maier and W. Lindner, Anal. Bioanal. Chem., 2007, 389, 377–397 CrossRef CAS PubMed.
- A. Bossi, F. Bonini, A. P. F. Turner and S. A. Piletsky, Biosens. Bioelectron., 2007, 22, 1131–1137 CrossRef CAS PubMed.
- T. Takeuchi and J. Haginaka, J. Chromatogr., B: Biomed. Appl., 1999, 728, 1–20 CrossRef CAS.
- Z. Y. Cheng, L. W. Zhang and Y. Z. Li, Chem. – Eur. J., 2004, 10, 3555–3561 CrossRef CAS PubMed.
- R. Say, M. Erdem, A. Ersoz, H. Turk and A. Denizli, Appl. Catal., A, 2005, 286, 221–225 CrossRef CAS.
- A. Visnjevski, R. Schomacker, E. Yilmaz and O. Bruggemann, Catal. Commun., 2005, 6, 601–606 CrossRef CAS.
- H.-F. Wang, Y.-Z. Zhu, J.-P. Lin and X.-P. Yan, Electrophoresis, 2008, 29, 952–959 CrossRef CAS PubMed.
- N. G. Bastus, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef CAS PubMed.
- N. G. Bastus, F. Merkoci, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef CAS.
- A. T. B. Silva, A. G. Coelho, L. C. d. S. Lopes, M. V. A. Martins, F. N. Crespilho, A. Merkoci and W. C. da Silva, J. Braz. Chem. Soc., 2013, 24, 1237–1245 CrossRef CAS.
- K. Z. Brainina, L. G. Galperin, E. V. Vikulova, N. Y. Stozhko, A. M. Murzakaev, O. R. Timoshenkova and Y. A. Kotov, J. Solid State Electrochem., 2011, 15, 1049–1056 CrossRef CAS.
- O. S. Ivanova and F. P. Zamborini, J. Am. Chem. Soc., 2010, 132, 70–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: S1 – instrumentation, S2 – immobilization of AuNPs, S3 – time dependent immobilization, S4 – CVs at matrix-coated substrates, S5 – CVs at AuNP-loaded matrices, S6 – peak potentials for the oxidation of AuNPs of different sizes, S7 – schematics for the change of conductive area of the matrices, S8 – probe CVs before and after AuNPs oxidation, S9 – calculation of adsorbed and reuptaken AuNPs, S10 – CVs of AuNPs adsorbed on non-imprinted matrices, S11 – SEM images of AuNPs adsorbed on non-imprinted matrices, S12 – SEM images after reuptake of AuNPs, S13 – schematic of the effect of thickening the matrix. See DOI: 10.1039/c6nr01106c |
| This journal is © The Royal Society of Chemistry 2016 |