
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An experimental and theoretical study into the facile, homogenous (N-heterocyclic carbene)2-Pd(0) catalyzed diboration of internal and terminal alkynes†
Melvyn B.
Ansell
a,
Vitor H.
Menezes da Silva
b,
Gabriel
Heerdt
 bc,
Ataualpa A. C.
Braga
*b,
John
Spencer
*a and
Oscar
Navarro
*a
bc,
Ataualpa A. C.
Braga
*b,
John
Spencer
*a and
Oscar
Navarro
*a
aDepartment of Chemistry, University of Sussex, Brighton, BN1 9QJ, UK. E-mail: j.spencer@sussex.ac.uk; o.navarro@sussex.ac.uk
bDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Avenida Professor Lineu Prestes, 748, São Paulo, 05508-000, Brazil. E-mail: ataualpa@iq.usp.br
cInstituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970, Campinas-SP, Brazil
First published on 19th August 2016
Abstract
Pd(ITMe)2(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh) acts as a highly reactive pre-catalyst in the unprecedented homogenous catalyzed diboration of terminal and internal alkynes, yielding a number of novel and known syn-1,2-diborylalkenes in a 100% stereoselective manner. DFT calculations suggest that a similar reaction pathway to that proposed for platinum phosphine analogues is followed, and that destabilization of key intermediates by the NHCs is vital to the overall success for the palladium-catalyzed B–B addition to alkynes.
CPh) acts as a highly reactive pre-catalyst in the unprecedented homogenous catalyzed diboration of terminal and internal alkynes, yielding a number of novel and known syn-1,2-diborylalkenes in a 100% stereoselective manner. DFT calculations suggest that a similar reaction pathway to that proposed for platinum phosphine analogues is followed, and that destabilization of key intermediates by the NHCs is vital to the overall success for the palladium-catalyzed B–B addition to alkynes.
Introduction
The transition metal catalysed π-insertion of homo and hetero element–element (E–E′) bonds into alkynes provides the most atom economical route for the stereoselective synthesis of tri- and tetrasubstituted alkenes.1 Among the various E–E′ reagents used in such transformations, B–B bonds (diborons) in particular have attracted substantial interest.2 The resulting 1,2-diboryl alkenes, due to their participation in Suzuki–Miyaura cross-coupling,3 are recognized as important building blocks in, for example, the synthesis of pharmaceuticals,4 chirotopical devices5 and optically/electronically active polymeric materials.6 A number of transition metals have been utilized in both homogenous and heterogenous catalytic addition of B–B bonds (diboration) to alkynes including cobalt,7 copper,8 iridium,9 rhodium,9 iron,10 platinum,11 and palladium.12 To date, platinum is by far the most effective and widely studied;13 this is attributed to the facile cleavage of the B–B bond and the lability of the corresponding bis(boryl)platinum complexes.14 As a result, even easily handled and often air stable tetraalkoxy- and tetraaryloxydiboron reagents can be utilized, regardless of their relatively high B–B bond strength.15 However, despite the extensive studies, a number of general limitations remain: the use of elevated temperatures, high catalyst loadings and long reaction times. Only two examples of palladium catalysed alkyne diborations have been described in the literature, both by Braunschweig and co-workers and involving the heterogenous catalysed diboration of alkynes using [2]borametalloarenophanes.12 The reactions required 6 mol% of Pd/C and proceeded over a period of 5–16 days at temperatures of 95–100 °C (Scheme 1). The dearth of reported palladium examples is attributed to the energetics of the B–B oxidative addition. The process is endothermic with a very low reverse activation barrier16 and therefore kinetically and thermodynamically unfavourable.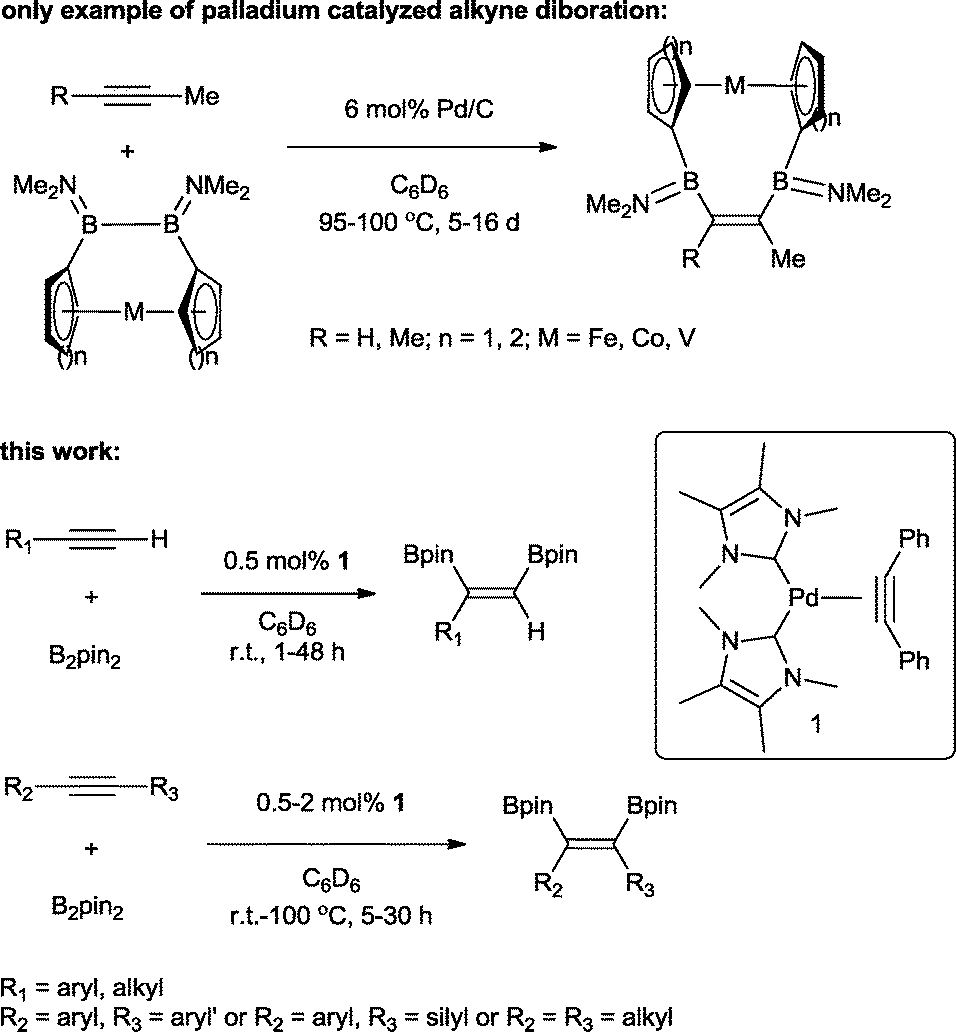 | ||
| Scheme 1 Palladium catalyzed diboration of alkynes. | ||
We recently reported the synthesis of the N-heterocyclic carbene bearing17 complex Pd(ITMe)2(PhCCPh) (ITMe = 1,3,4,5-tetramethylimidazol-2-ylidene) (1) and its high catalytic reactivity in bis-silylation18 and silaboration of internal and terminal alkynes.19 This prompted us to investigate its potential in the diboration of alkynes. Herein, we report the use of 1 in the unprecedented palladium catalysed diboration of sterically demanding internal and terminal alkynes, employing low catalytic loadings and mild reaction temperatures. In addition, a thorough density functional theory (DFT) study was conducted in order to establish a likely mechanistic pathway explaining this reactivity.
Results and discussion
The reaction parameters were optimized using diphenylacetylene and commercially available bis(pinacalato)diboron (B2pin2) as the model substrates, with C6D6 as solvent in order to monitor the progression by 1H NMR. To our delight, 100% stereoselective conversion to (Z)-1,2-diphenyl-1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethane (2) was observed using 0.5 mol% of 1 at room temperature in 21 h. Unfortunately, initial work-up procedures proved troublesome, with the use of either silica and alumina columns resulting in very low isolated yields presumably due to reactivity with, or strong binding to the stationary phase. Kugelrohr distillation is an alternative methodology reported in the literature,20 but is generally applicable to small quantities of material and therefore unviable as a scalable procedure. In our case, the more noticeable impurity was unreacted B2pin2. To remove it, the crude dry material was stirred in deionized H2O at room temperature over 24 h.21 Subsequent filtration and drying resulted in the clean isolation of 2 in a 99% yield. While there are several protocols in the literature for the synthesis of 2, the highest yield was reported by Jin and co-workers22 who obtained a comparable yield to ours using 2 mol% of nanoporous gold at 100 °C over 12 h. To test the potential of this protocol for scaling-up, the synthesis of 2 was also carried out in non-deuterated benzene and toluene on a larger practical scale, resulting in comparable isolated yields (Table 1). The potential of 1 to catalyse this reaction using other diboron reagents was also investigated, but unfortunately neither bis(catecolato)diboron nor bis(neopentylglycolato)diboron afforded any of the desired product.|
|
||
|---|---|---|
| B2pin2: 1–1.5 equiv. (see ESI for details). a Scale-up synthesis of 2: 0.8 mmol of substrate, benzene, r. t., 24 h (92%); 0.95 mmol of substrate, toluene, r. t., 24 h (95%). b Conversion of starting alkyne to 15. | ||
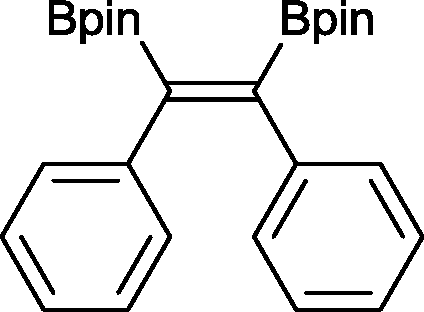 2 21 h 99%a
2 21 h 99%a |
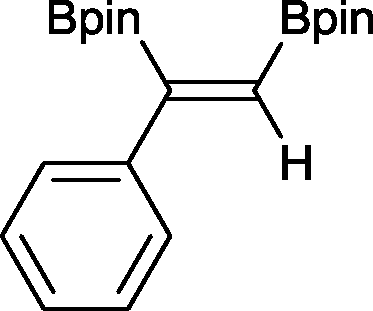 3 3 h, 91%
3 3 h, 91% |
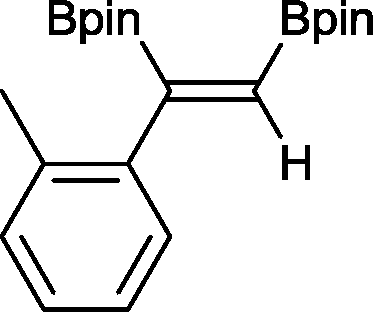 4 2 h, 79%
4 2 h, 79% |
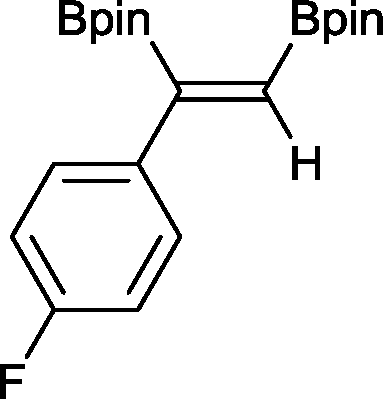 5 9 h, 95%
5 9 h, 95% |
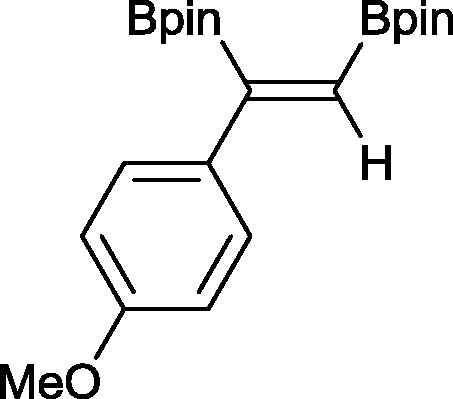 6 4 h, 88%
6 4 h, 88% |
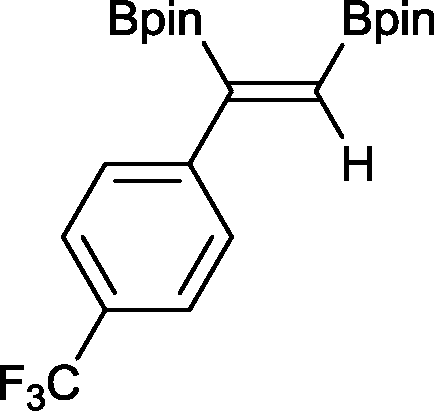 7 13 h, 94%
7 13 h, 94% |
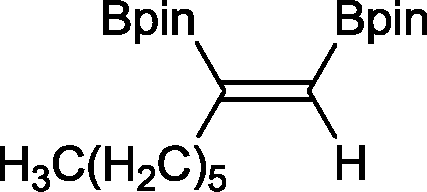 8 33 h, 87%
8 33 h, 87% |
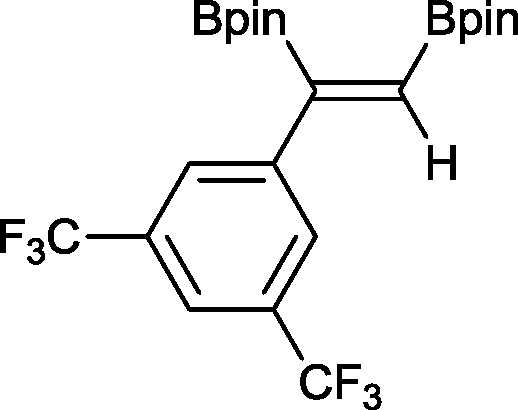 9 48 h, 79%
9 48 h, 79% |
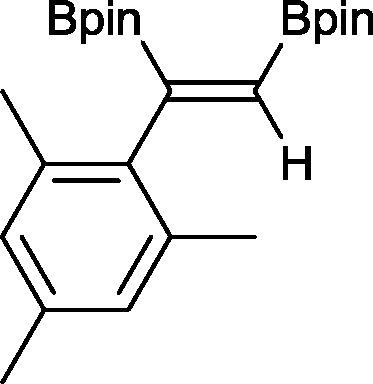 10 1 h, 94%
10 1 h, 94% |
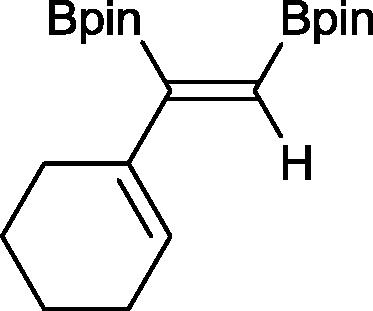 11 24 h, 89%
11 24 h, 89% |
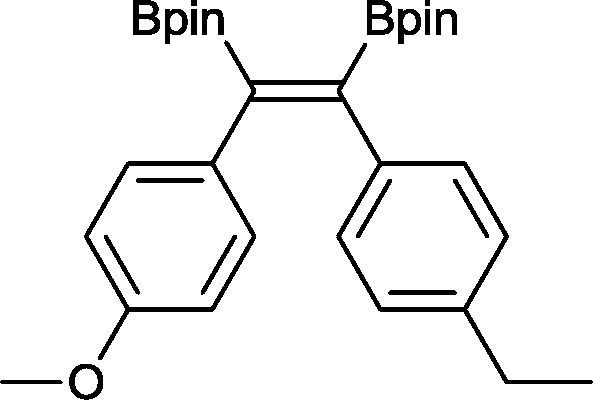 12 50 °C, 5 h, 85%
12 50 °C, 5 h, 85% |
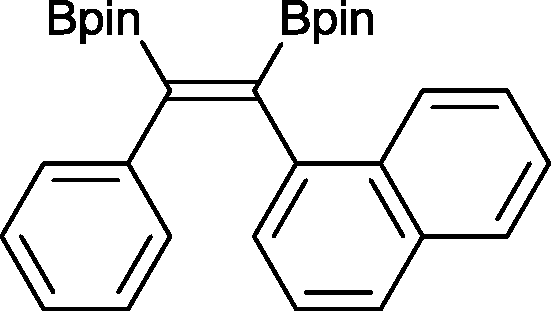 13 50 °C, 24 h, 94%
13 50 °C, 24 h, 94% |
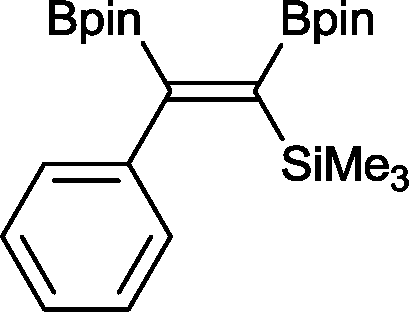 14 2 mol%, 100 °C, 30 h, 85%
14 2 mol%, 100 °C, 30 h, 85% |
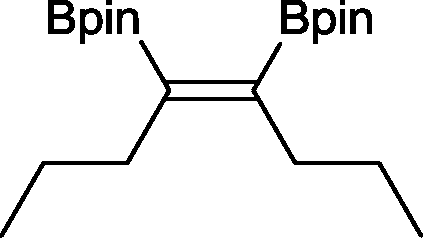 15 23 h, 39%b
15 23 h, 39%b |
|
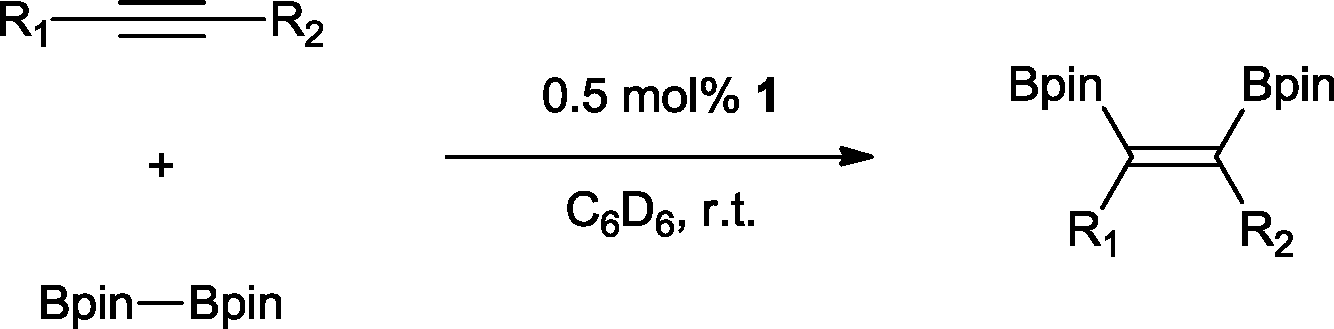
With this information in hand, a series of sterically and electronically demanding alkynes were reacted with B2pin2 (Table 1). The diboration of alkyl and aryl terminal alkynes proceeded using 0.5 mol% of 1 at room temperature over 1–48 h with 100% stereoselectivity. A wide range of functionalities on the aryl moiety was tolerated including fluoro, trifluoromethyl, methoxy and alkyl groups in the ortho, meta and para positions. Compounds 3, 4, 5 and 6 were synthesized using lower catalyst loadings, milder temperatures and in higher or comparable yields to the highest yielding protocol in the literature (2 mol% nanoporous gold, 100 °C),22 and 5 and 6 were synthesized with comparatively higher stereoselectivities. Low reaction temperatures have been reported for the synthesis of these compounds using both homo- and heterogenous platinum complexes, although at the expense of lower yields and in many cases higher catalyst loadings.13,23,24 Compound 7 was synthesized in a higher yield than the highest yielding protocol in the literature (0.2 mol% Pt/TiO2, 70 °C, 16 h).13 The highest yielding synthesis for compound 8 was reported by Miyaura and co-workers (94% yield)13 using 3 mol% Pt(CO)2(PPh3)2 at 80 °C in DMF over 24 h.
The novel compounds 9, 10 and 11 were synthesized with 100% syn-stereoselectivity as established by NOESY NMR spectroscopy. In the case of 11 chemoselectivity is achieved since the olefin remains unreacted. Unsymmetrical internal alkynes also reacted well under these conditions, albeit at higher- but still mild-temperatures (50 °C). The novel compounds 12 and 13 were synthesized with 100% syn-stereoselectivities. The diboration of 1-phenyl-2-trimethylsilane, resulting in the formation of 14, required an increased catalyst loading of 2 mol% and a much higher temperature (100 °C). The best procedure for the synthesis of 14 was detailed by Nishihara, obtaining a comparable yield using 5 mol% of Pt(PPh3)4 at 80 °C.13 Finally, the diboration of 4-octyne resulted in a maximum conversion to 15 of 39%. Even lower conversions and the formation of palladium black were observed when we carried out the reaction at higher temperatures. We presume that the electron-rich nature of the alkyne results in a low binding affinity to the very electron-rich, active catalyst and therefore discourages diboration.
We decided then to investigate the reasons behind this unprecedented activity. The accepted experimental and theoretical mechanism for platinum group transition metal catalysed diboration of alkynes involves: (i) oxidative addition of the B–B bond to a M(0)L2 centre forming L2M(II)(B)2, (ii) dissociation of an L ligand (a phosphine) and coordination of the alkyne in its place, (iii) insertion of the alkyne into the M–B bond, (iv) isomerization of the resulting complex, followed by re-coordination of the L ligand, and (v) stereoselective reductive elimination.25 This mechanism is general and applies to other E–E′ bond addition to alkynes.26 We recently proposed that the use of NHCs as a ligand set results in a different mechanism, in which both NHCs remained coordinated throughout. This alternative pathway was used as an explanation for the observed increase in reactivity of 1 compared to their phosphine and isocyanide analogues in alkyne bis-silylations18 and silaborations.19 To gain further insight into the mechanism and role of 1 in the diboration of alkynes, computational studies were carried out on the optimized model substrates (see ESI†). Additionally, a simultaneous study of Pd(0)(PMe3)2(PhCCPh) (1-PMe3) was performed to establish a direct comparison with the NHC ligand set.
Initially, the geometry of 1 was optimized at M06-L/BSI level of theory and compared to X-ray diffraction data.18 The optimized Pd–alkyne bond lengths Pd–C1 and Pd–C6 are longer, around 0.01 Å, than the results obtained by X-ray data. Both bond angles C6–C1–C5 and C1–C6–C25 are equal to 147.1°, in excellent agreement with the experimental values of 147.5(3)° and 146.03(3)°, respectively. A comparison between 1 and 1-PMe3 at the same level of theory was also undertaken (Fig. 1). The optimized bond lengths and angles of 1 (C9–C7 = 3.11 Å and C9–Pd–C7 = 94.9°) are similar 1-PMe3 (P1–P2 = 3.74 Å and P1–Pd–P2 = 105.8°). Furthermore, in 1-PMe3 a more accentuated out-of-plane distortion of the square planar geometry around the Pd centre, caused by the phosphine ligands, was observed. While the dihedral C1–C6–C7–C9 angle in 1 is 1.5°, in 1-PMe3 the dihedral C1–C6–P2–P1 angle is 7.5°.
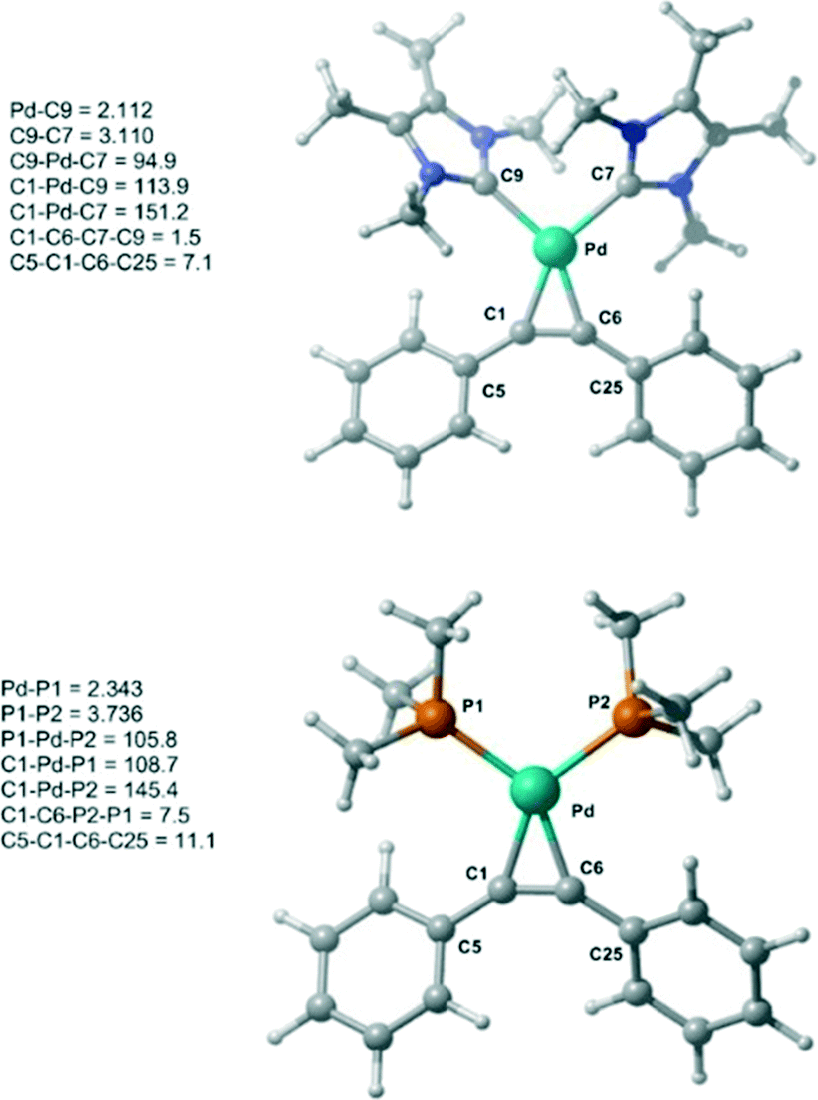 | ||
| Fig. 1 Optimized structures of 1 and 1-PMe3 at M06-L/BSI level of theory with selected bond lengths (Å) and angles (°). | ||
The oxidative addition of bis(pinacolato)diboron to 1 begins with the dissociation of the alkyne from the η2-complex, resulting in the formation of the 14 electron complex I1 (Fig. 2). This dissociation is favourable at 6.4 and 3.5 kcal mol−1 for the NHC and PMe3 complexes, respectively. The reaction continues through the incorporation of bis(pinacolato)diboron in the coordination sphere of I1, achieving the intermediate I2. The transition state TSA01 represents the step where the B–B bond is cleaved with concomitant formation of two σ Pd–B bonds. This process has a free energy activation barrier relative to the separated reactants at ΔG‡ = 9.7 kcal mol−1 for the NHC and 11.1 kcal mol−1 for the PMe3 bis-ligand complexes. These reaction barrier heights suggest that the oxidative addition for the phosphines is kinetically less favoured than the NHC ligands. Bis(boryl)palladium(II) complex (I3) is the product of the oxidative reaction step for both systems, and, energetically, is 8.7 kcal mol−1 and 6.5 kcal mol−1 above the reactants with NHC and PMe3 ligands, respectively. Alternatively, the oxidative addition step could proceed through the mono-ligand complexes. Scheme 2 depicts two possibilities (pathway A and pathway B) related to dissociative reaction routes. Dissociation of alkynes from 1 to form the 14-electron complex I1 is an exergonic process for the NHC (−6.4 kcal mol−1) and the PMe3 (−3.5 kcal mol−1) ligands (pathway A). However, in pathway B, the dissociation of ligand (L) from 1 to form the mono-ligand alkyne palladium(0) complex 1_alkyne is an endergonic process at 5.9 kcal mol−1 for L = NHC and 8.9 kcal mol−1 for L = PMe3. The thermodynamic driving force for the dissociation of L is greater going through pathway A over pathway B. Furthermore, the breaking of the second Pd–L bond to form the reactive mono-ligand Pd(0)–L (I4) is easier for L = PMe3, than for L = NHC systems. Indeed, the Pd–NHC bond typically is stronger than the Pd–phosphine bond. The bis-ligand complex is energetically preferred for both NHC and phosphine systems, in the oxidative addition of bis(pinacolato)diboron to the Pd centre (see ESI† for more details on the free energy profile of reaction pathways).
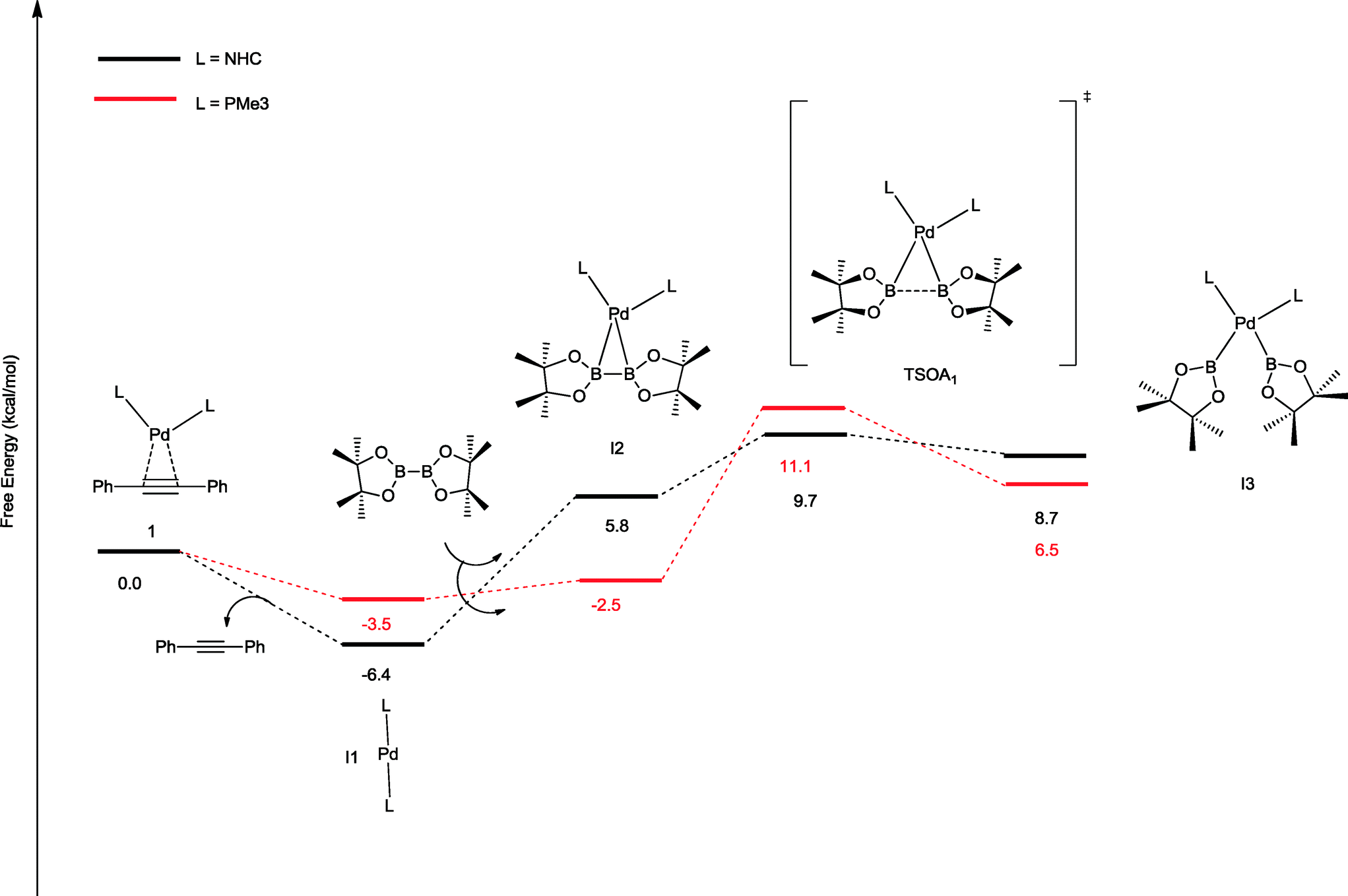 | ||
| Fig. 2 Free energy profile (in kcal mol−1) for the oxidative addition pathways with bis-ligand complexes. | ||
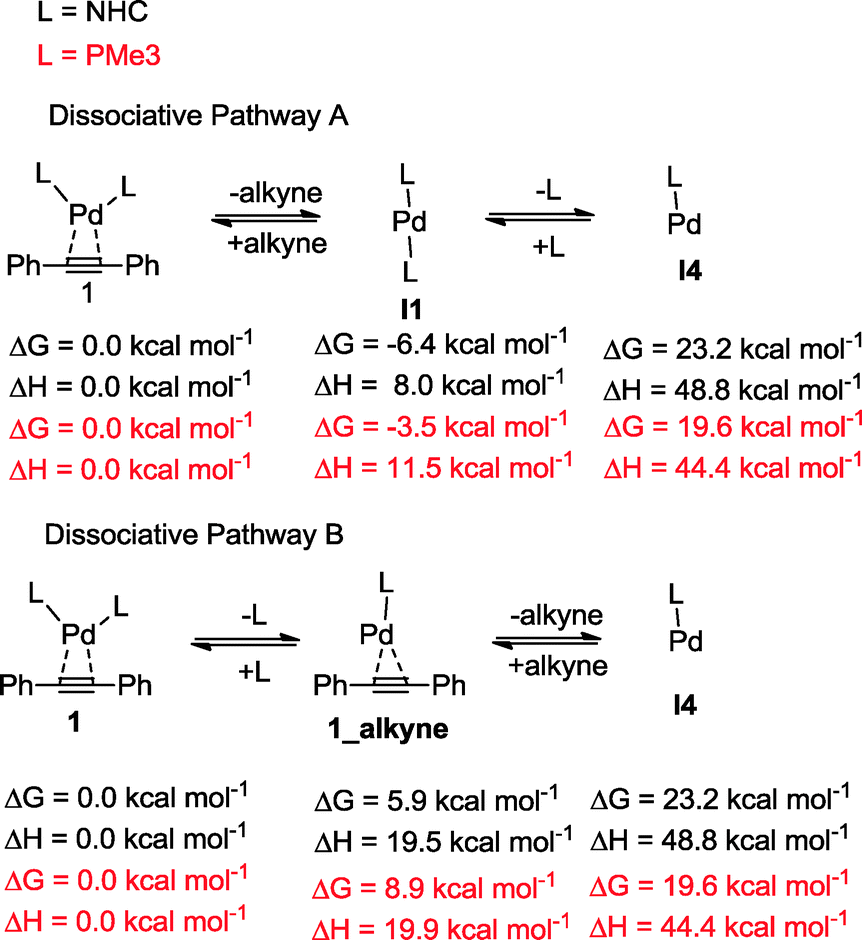 | ||
| Scheme 2 Dissociation pathways for the activation of catalyst. | ||
The next step is the alkyne insertion on the metal centre. The first proposal is that the insertion of alkyne occurs directly to the bis(boryl)palladium(II) complex I3. The product of the alkyne insertion can be obtained through the penta-coordinate transition state TSIA1 (Fig. 3 – see ESI† for further details and Scheme 3 for the proposed mechanism).
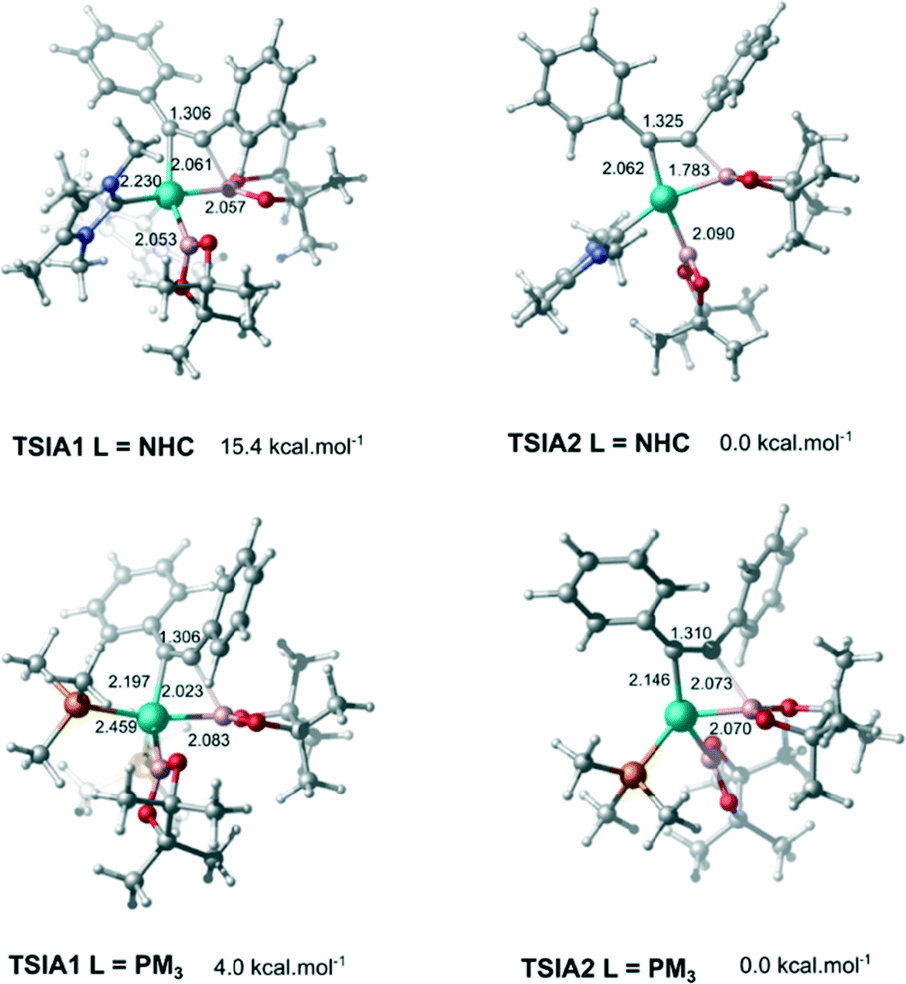 | ||
| Fig. 3 Associative (TSIA1) and dissociative (TSIA2) transition states for the alkyne insertion, respectively. Distances for selected bonds are given in angstrom units (Å). Relative free energies are given at 298.15 K. | ||
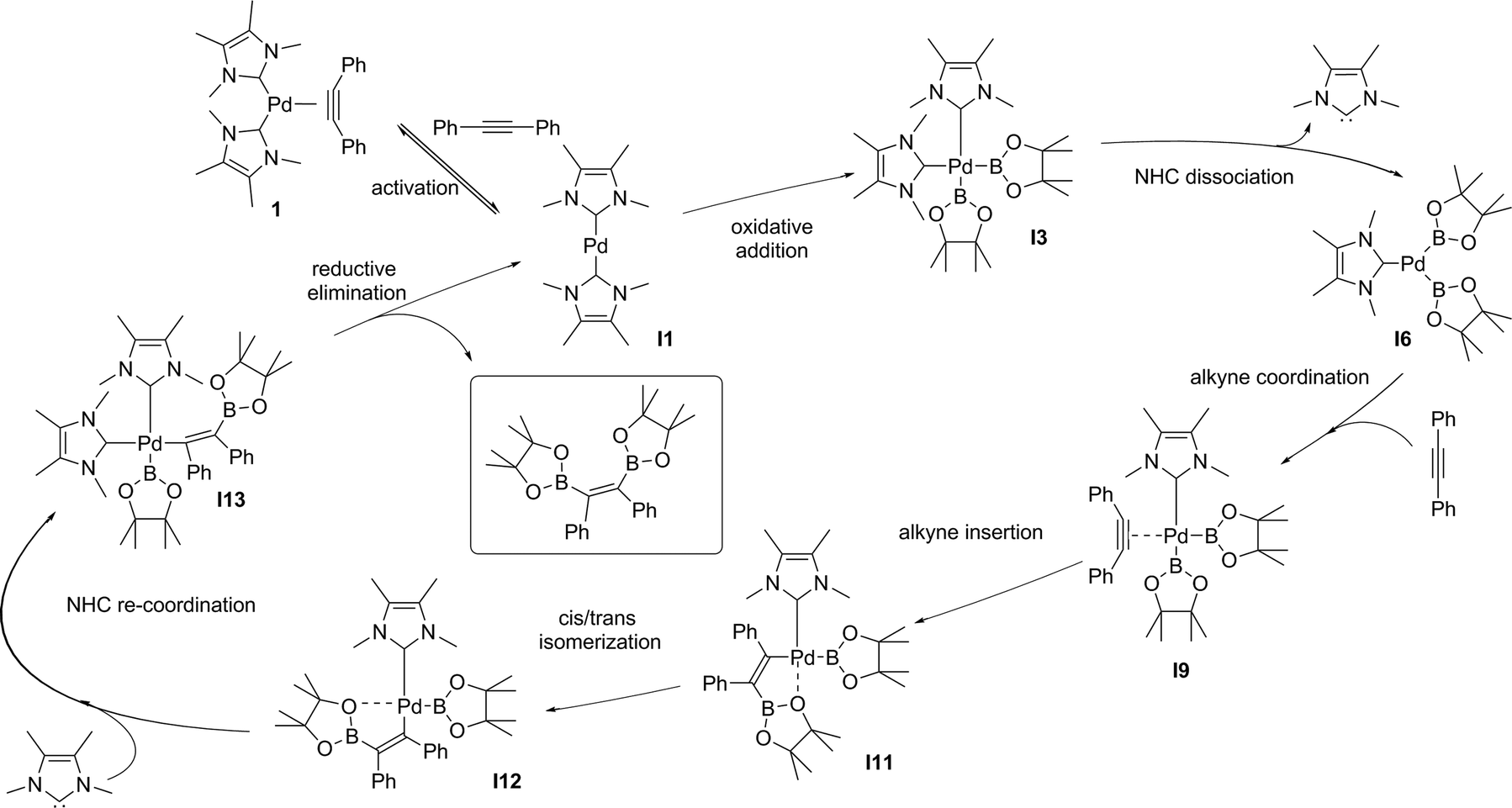 | ||
| Scheme 3 Proposed mechanism for the (NHC)2Pd(0) catalysed diboration of alkynes. | ||
Another possibility for the insertion of the alkyne proceeds via a dissociate pathway (TSIA2). Kinetic results on Pt(0)-catalyzed diboration reactions with phosphine ligands suggested that the alkyne insertion occurs from the three-coordinate species in the oxidative addition.27 Following this, it is a reasonable assumption that the dissociation of one L takes place from complex I3 to generate the mono(boryl)palladium(II) complex I6 (Scheme 3).The transition state TSIA2 is associated to the migratory insertion of the alkyne with one ligand attached on the metal centre. The alkyne triple bond and Pd–B bonds are broken forming a new C–B.
The dissociative pathway (TSIA2) has a lower relative reaction free energy barrier than on the associative pathway (TSIA1) at ΔΔG‡ = 4.0 kcal mol−1 for L = PMe3 and at ΔΔG‡ = 15.4 kcal mol−1 for L = NHC system. Based on these results, the reductive elimination should occur by the cis-complex I12 from the insertion of the alkyne via the dissociative mechanism for both ligands (further details in ESI†). The re-coordination of other ligand is reasonable, since the 16-electron configuration is achieved forming the palladium(II) complex I13 with a square planar geometry (Scheme 3).
In order to obtain the anti-adduct is necessary to proceed with a consecutive isomerization processes involving the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the C–Bpin moiety in the allyl ligand after the alkyne insertion is accomplished. Cui and co-workers suggested these isomerization pathways are energetically prohibitive because of the rigidity of the CC bond.25 Therefore, the substrate controls the stereoselectivity in the Pt(0)-catalyzed diboration reaction towards the syn-1,2-diborated product. Analogously, this mechanism for selectively could be expanded for the present reaction, since the same substrate was used (alkyne). In this case, if these isomerization processes take place very quickly, the selectivity would be defined solely by the relative energies of the transition states associated with the reductive elimination steps. Fig. 4 shows the optimized geometry of the transition states TSRE1 and TSRE2 reacted with the syn and anti-adducts, respectively. For the NHC complex, the relative free energy activation of ΔΔG‡ = 7.4 kcal mol−1, favouring the transitions state TSRE1, is in perfect agreement with the product detected experimentally.
C bond and the C–Bpin moiety in the allyl ligand after the alkyne insertion is accomplished. Cui and co-workers suggested these isomerization pathways are energetically prohibitive because of the rigidity of the CC bond.25 Therefore, the substrate controls the stereoselectivity in the Pt(0)-catalyzed diboration reaction towards the syn-1,2-diborated product. Analogously, this mechanism for selectively could be expanded for the present reaction, since the same substrate was used (alkyne). In this case, if these isomerization processes take place very quickly, the selectivity would be defined solely by the relative energies of the transition states associated with the reductive elimination steps. Fig. 4 shows the optimized geometry of the transition states TSRE1 and TSRE2 reacted with the syn and anti-adducts, respectively. For the NHC complex, the relative free energy activation of ΔΔG‡ = 7.4 kcal mol−1, favouring the transitions state TSRE1, is in perfect agreement with the product detected experimentally.
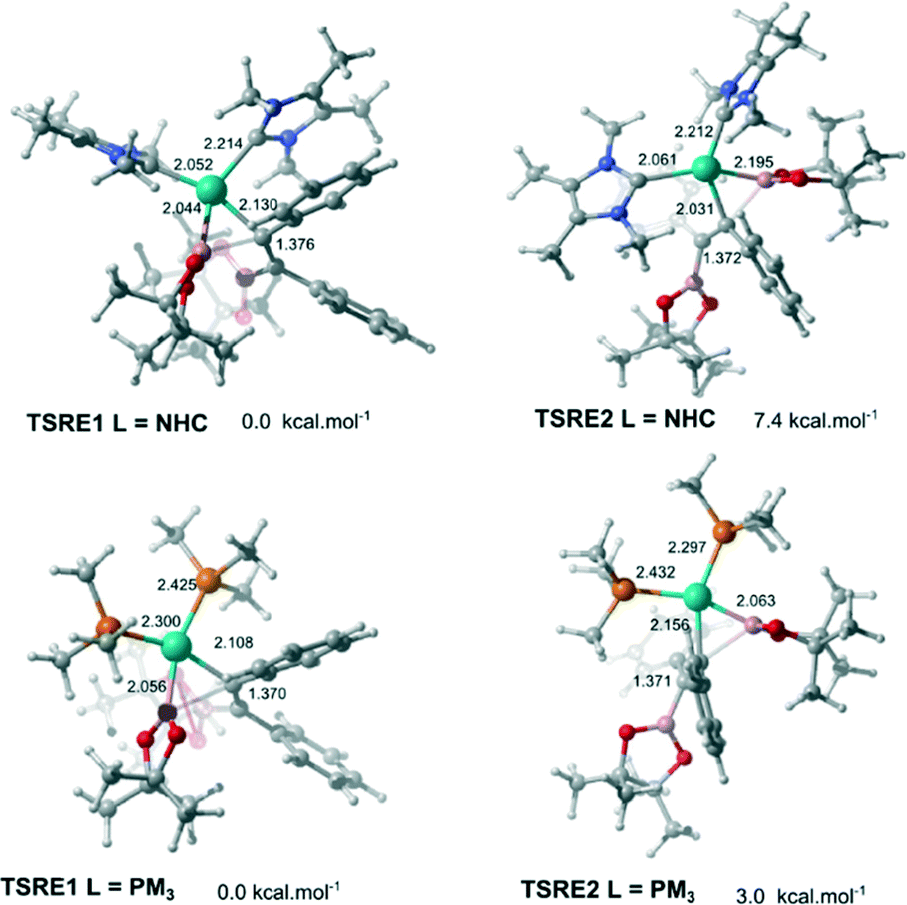 | ||
| Fig. 4 Calculated transition states for the reductive elimination step associated with syn- and anti-1,2-diborated adduct, respectively. Distances for selected bonds are given in angstrom units (Å). Relative free energies are given at 298.15 K. | ||
DFT calculation suggest the Pd(0)-catalysed alkyne diboration supported by NHC ligands proceeds through the same mechanism as the phosphine ligands (the free energy profile of the overall catalytic cycle is presented in the ESI†). This mechanism (Scheme 3) can therefore be summarized as: (i) the activation of the catalyst by alkyne dissociation from 1, (ii) oxidative addition of the B–B to Pd(0), (iii) ligand dissociation from bis(boryl)palladium(II) complex I3, (iv) insertion of the alkyne into a Pd–B bond via migratory insertion, (v) cis–trans isomerization involving the C–Bpin and the allyl ligands, and (vi) reduction of Pd(II) to Pd(0) with the elimination of the syn-1,2-diborylated product.
Cui and co-workers proposed that a reversible oxidative addition step is the reason of the null reactivity of a Pd(0)L2 catalyst (L = phosphine) in alkyne diboration reactions.16 The oxidative addition step was predicted to have an activation barrier of 8.6 kcal mol−1. However, the B–B oxidative addition to Pd(0) was characterized as an endothermic process with a reverse barrier of only 0.1 kcal mol−1. The cause of this low reverse barrier is attributed to the promotion energy from d10Pd(0)L2 with linear geometry (singlet – ground state) to d9s1 Pd(0)L2 with bent geometry (triplet – excited state). The energy between these two electronic configurations is larger for Pd(0)L2 than for Pt(0)L2 with phosphines. Sasaki and co-workers,27 studying the activity of Pd(0)L2 and Pt(0)L2 catalyst (L = phosphine) in the C–H activation of methane by oxidative addition, reported the destabilization of the M(0)L2 complexes as an important factor in smoother oxidative additions. Chelating phosphines were used to destabilize the M(0)L2 complexes by bringing the reactants closer in order to promote the oxidative addition transition state. (NHC)–Pd(0) catalysts were also investigated in the activation of methane by oxidative addition,28 and considered better candidates as catalysts than the analogous phosphine-based Pd(0) complexes. Based on these results, we propose that the considerably increased reactivity of NHC-bearing complex 1 in the alkyne diboration is a consequence of the oxidative addition step; more specifically, on the destabilization of the (diboron)Pd(0)L2 complex I2 by the NHC ligands resulting in a lower activation free energy for the oxidative addition (3.9 kcal mol−1) compared to PMe3 (13.3 kcal mol−1).
Conclusions
We have shown that complex 1 acts as highly active catalyst in the diboration of sterically and electronically demanding alkynes. For terminal alkynes, low catalyst loadings and temperatures were used for the 100% stereoselective synthesis of syn-1,2-diborylalkenes. Internal alkynes can react using this protocol, albeit requiring elevated temperatures. This represents the first example of homogenous palladium catalysed diboration of alkynes. DFT calculations, based on M06 suite density functionals, were performed to understand the activity of the NHC-bearing catalyst 1. The results suggest that 1 proceeds through the same mechanistic pathway as the corresponding phosphine analogues. The dissociation of one NHC is crucial part of the mechanism, unaccounted for in our proposed pathways for the other E–E′ bond additions to alkynes.18,19 Despite their strong coordination to metal centres, it has been previously shown that the reversible dissociation of an NHC from an oxidative addition products is a mechanistic possibility.29 The DFT study also showed that the destabilization of the (diboron)Pd(0)L2 adduct by the NHCs was key to the successful oxidative addition of the B–B bond. Further investigations into the scope and limitation of 1 in the B–B bond additions to other unsaturated organic substrates are currently ongoing in our laboratories.Acknowledgements
The authors wish to thank Dr. Iain Day and Dr. Alaa Abdul-Sada (University of Sussex) for helpful discussions. M. B. A. is funded as an EPSRC Standard Research Student (DTG) under grant no. EP/L505109/1. V. H. M. S (grants #2013/04813-6 and #2015/11840-5) and A. A. C. B (grant #2015/01491-3) are thankful to Fundação de Amparo à Pesquisa do Estado de São Paulo for financial support, and to the LCCA-USP for the computational support.Notes and references
- I. Beletskaya and C. Moberg, Chem. Rev., 1999, 99, 3435 CrossRef CAS PubMed.
- T. B. Marder and N. C. Marder, Top. Catal., 1998, 5, 63 CrossRef CAS; J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993 CrossRef; T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 4, 271 CrossRef PubMed; I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef PubMed; T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2000, 611, 392 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC; I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528 CrossRef PubMed.
- M. W. Carson, M. W. Giese and M. J. Goghan, Org. Lett., 2008, 10, 2701 CrossRef CAS PubMed; Q.-X. Lin and T.-L. Ho, Tetrahedron, 2013, 69, 2996 CrossRef; M. W. Carson, J. G. Luz, C. Suen, C. Montrose, R. Zink, X. Ruan, C. Cheng, H. Cole, M. D. Adrian, D. T. Kohlman, T. Mabry, N. Snyder, B. Condon, M. Maletic, D. Clawson, A. Pustilnik and M. J. Coghlan, J. Med. Chem., 2014, 57, 849 CrossRef PubMed.
- K. Yavari, S. Moussa, B. Ben Hassine, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2012, 51, 6748 CrossRef CAS PubMed.
- J. Yang, M. Chem, J. Ma, W. Huang, H. Zhu, Y. Huang and W. Wang, J. Mater. Chem. C, 2015, 3, 10074 RSC.
- C. J. Adams, R. A. Baber, A. S. Batsanov, G. Bramham, J. P. H. Charmont, M. F. Haddow, J. A. K. Howard, W. H. Lam, Z. Lin, T. B. Marder, N. C. Norman and A. G. Orpen, Dalton Trans., 2006, 1370 RSC.
- H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2013, 51, 235 CrossRef PubMed.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS PubMed.
- N. Nakagawa, T. Hatakeyama and N. Nakamura, Chem. – Eur. J., 2015, 21, 4257 CrossRef CAS PubMed.
- T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018 CrossRef CAS.
- H. Braunschweig, T. Kupfer, M. Lutz, K. Radacki, F. Seeler and R. Sigritz, Angew. Chem., Int. Ed., 2006, 45, 8048 CrossRef CAS PubMed; H. Braunschweig, M. Kaupp, C. J. Adams, T. Kupfer, K. Radacki and S. Schinzel, J. Am. Chem. Soc., 2008, 130, 11367 Search PubMed.
- K. M. Anderson, M. J. G. Lesley, N. C. Norman, A. G. Orpen and J. Starbuck, New J. Chem., 1999, 23, 1053 RSC; T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713 CrossRef CAS; R. L. Thomas, F. E. S. Souza and T. B. Marder, J. Chem. Soc., Dalton Trans., 2001, 1650 RSC; A. Grirrane, A. Corma and H. Garcia, Chem. – Eur. J., 2011, 17, 2467 CrossRef PubMed; F. Alonso, Y. Moglie, L. Pastor-Pérez and A. Sepúlveda-Escribano, ChemCatChem, 2014, 6, 857 CrossRef; J. Jiao, K. Hyodo, H. Hu, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 285 CrossRef PubMed.
- C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1995, 117, 4403 CrossRef CAS; G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J. Scott, W. Clegg and N. C. Norman, Organometallics, 1996, 14, 5137 CrossRef.
- A. Finch, P. J. Gardner and A. F. Webb, J. Chem. Thermodyn., 1972, 4, 497 CrossRef.
- Q. Cui, D. G. Musaev and K. Morokuma, Organometallics, 1998, 17, 742 CrossRef CAS.
- N-heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014 Search PubMed; N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Heidelberg, 1st edn, 2011 Search PubMed; N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Heidelberg, 1st edn, 2006 Search PubMed.
- M. B. Ansell, D. E. Roberts, F. G. N. Cloke, O. Navarro and J. Spencer, Angew. Chem., Int. Ed., 2015, 54, 5578 CrossRef CAS PubMed.
- M. B. Ansell, J. Spencer and O. Navarro, ACS Catal., 2016, 6, 2192 CrossRef CAS.
- J. Ramírez and E. Fernández, Synthesis, 2005, 10, 1698 Search PubMed.
- G.-Q. Li, S. Kiyomura, Y. Yamamoto and M. Miyaura, Chem. Lett., 2011, 40, 702 CrossRef CAS.
- Q. Chen, J. Zhao, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2013, 15, 5766 CrossRef CAS PubMed.
- J. B. Morgan and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 15338 CrossRef CAS PubMed.
- J. Ramírez and E. Fernández, Tetrahedron Lett., 2007, 48, 3841 CrossRef.
- C. N. Iverson and M. R. Smith III, Organometallics, 1996, 15, 5155 CrossRef CAS; Q. Cui, D. G. Musaev and K. Morokuma, Organometallics, 1997, 16, 1355 CrossRef.
- F. Ozawa, J. Organomet. Chem., 2000, 611, 332 CrossRef CAS; M. Tanabe and K. Osakada, Chem. – Eur. J., 2004, 10, 416 CrossRef PubMed; S. Sakaki, M. Ogawa and Y. Musashi, J. Organomet. Chem., 1997, 535, 25 CrossRef; A. Bottoni, A. P. Higueruelo and G. P. Miscione, J. Am. Chem. Soc., 2002, 124, 5506 CrossRef PubMed; S. Sakaki, S. Kai and M. Sugiomoto, Organometallics, 1999, 18, 4825 CrossRef; S. Sakaki, B. Biswas, Y. Musashi and M. Sugiomoto, J. Organomet. Chem., 2000, 611, 288 CrossRef.
- S. Sasaki, B. Biswas and M. Sugimoto, J. Chem. Soc., Dalton Trans., 1997, 803 RSC.
- T. R. Cundari and B. M. Prince, J. Organomet. Chem., 2011, 696, 3982 CrossRef CAS.
- A. K. de K. Lewis, S. Caddick, F. G. N. Cloke, N. C. Billingham, P. B. Hitchcock and J. Leonard, J. Am. Chem. Soc., 2003, 125, 10066 CrossRef PubMed; A. K. de K. Lewis, S. Caddick, O. Esposito, F. G. N. Cloke and P. B. Hitchcock, Dalton Trans., 2009, 7094 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy01266c |
| This journal is © The Royal Society of Chemistry 2016 |