Multivalent helix mimetics for PPI-inhibition†
Received
28th September 2014
, Accepted 5th November 2014
First published on 5th November 2014
Abstract
The exploitation of multivalent ligands for the inhibition of protein–protein interactions has not yet been explored as a supramolecular design strategy. This is despite the fact that protein–protein interactions typically occur within the context of multi-protein complexes and frequently exploit avidity effects or co-operative binding interactions to achieve high affinity interactions. In this paper we describe preliminary studies on the use of a multivalent N-alkylated aromatic oligoamide helix mimetic for inhibition of p53/hDM2 and establish that protein dimerisation is promoted, rather than enhanced binding resulting from a higher effective concentration of the ligand.
Introduction
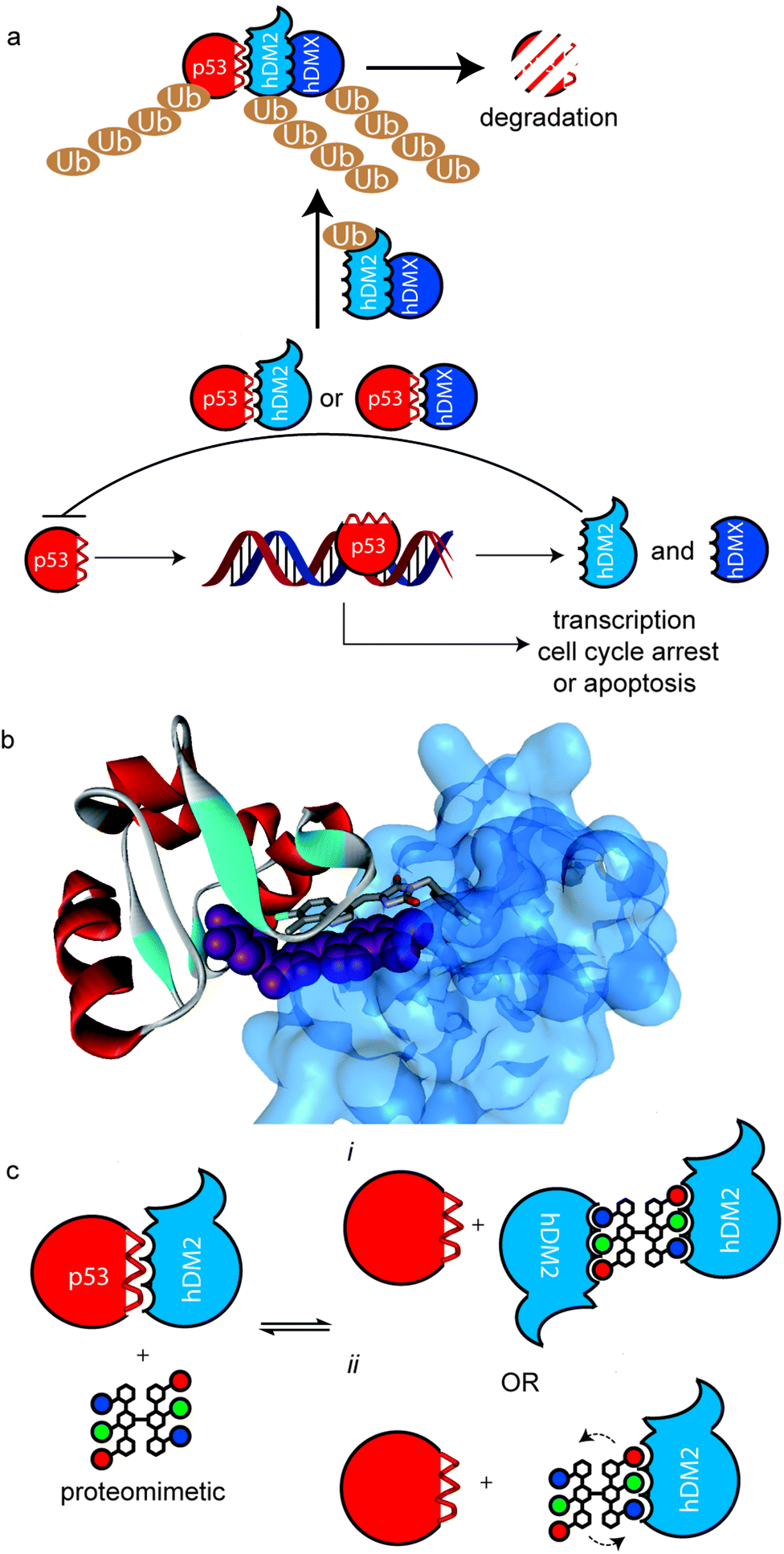
Protein–protein interactions (PPIs) regulate all essential biological processes and are frequently implicated in the development and progression of disease.1 As targets for molecular recognition, however, PPIs do not conform to conventional models used in drug design in that they are mediated by shallower surfaces with spatially distinct non-covalent contacts rather than the traditional lock and key model of biomolecular protein recognition.2,3 Therefore, PPIs represent an “ultimate challenge” in terms of complexity for supramolecular design.4 Several different approaches have been applied that exploit scaffolds that project recognition groups over a large area5–15 and/or target specific hot-spot residues on the surface.16,17 A particularly fertile avenue of investigation has concerned the development of secondary structure mimetics;18 α-helices are the most abundant secondary structure found in proteins and are frequently found at the interface of PPIs.19 α-Helix mimetics20–25 are designed to match the spatial arrangement of key binding residues from the helix involved in the interaction.18 We26–30 and others31–33 have developed several types of aromatic oligoamide helix mimetics34 with the binding groups attached to the 3O-,29,35 2O-36 and N-30,37 positions on the aromatic building blocks notably for the purposes of p53/hDM2 inhibition.28–30 This PPI is a key regulator of genomic stability and, as such, is of significant interest in the development of cancer treatments.38,39 p53 binds to hDM2 through its helical N-terminal transactivation domain. The interaction is mainly controlled by three key ‘hot-spot’ residues; Phe19, Trp23 and Leu26.40hDM2 and its partner hDMX act as negative regulators by blocking the transcriptional function of p53 and in the case of hDM2, through its ubiquitin ligase function which tags p53 for degradation by the proteasome (Fig. 1a).41 The process by which this occurs at the molecular level is complex, however what is clear is that ubiquitination is promoted by the formation of hDM2 homodimers and heterodimers (with hDMX), through their respective RING domains.42 Moreover, a recent study by the Roche group revealed that hDM2 and hDMX could be induced to form dimers, by a small molecule that spans the p53 binding site of two hDM2(X) monomers (Fig. 1b).43 This inspired the current study – we hypothesized that the use of multivalent44 helix mimetics might lead to co-operative binding properties either through the avidity resulting from chemically induced dimerisation of the target protein or the higher effective ligand concentration afforded by the proximity of additional covalently tethered copies of the protein-binding ligand (Fig. 1c).45 Multivalent ligands have been utilised for binding to a multitude of different biological targets including, toxins,46 heparin,47 DNA,48,49 β-tryptase50 and of particular note for foldamers, amyloid oligomers.51 Similarly, palindromic ligands have been utilised to inhibit amyloid assembly e.g. by stabilizing transthyretin tetramerization52,53 but not for inhibition of protein–protein interactions (PPIs). Of additional note, a number of dimeric helix mimetics have previously been reported.54,55 Herein we describe the synthesis of a dimeric N-alkylated aromatic oligoamide trimer and illustrate its ability to promote the inhibition of the p53/hDM2 interaction by dimerization/aggregation.
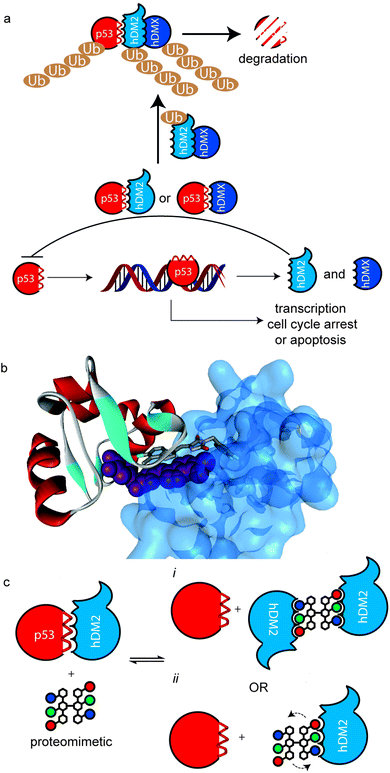 |
| | Fig. 1 Multivalent interactions of p53 and hDM2 (a) overview of p53 signalling pathway illustrating how hDM2(X) dimerisation enhances ubiquitination of p53. (b) X-ray structure of a small molecule inducer of hDM2 dimerisation (PDB ID: 3VBG). (c) Schematic depicting possible consequences of multivalent inhibition of p53/hDM2 (i) inhibition enhances hDM2 dimerization by additional intermolecular hDM2 contacts or (ii) proximity of additional ligand enhances affinity through more rapid association upon displacement of bound ligand. | |
Results and discussion
Synthesis of a divalent oligobenzamide
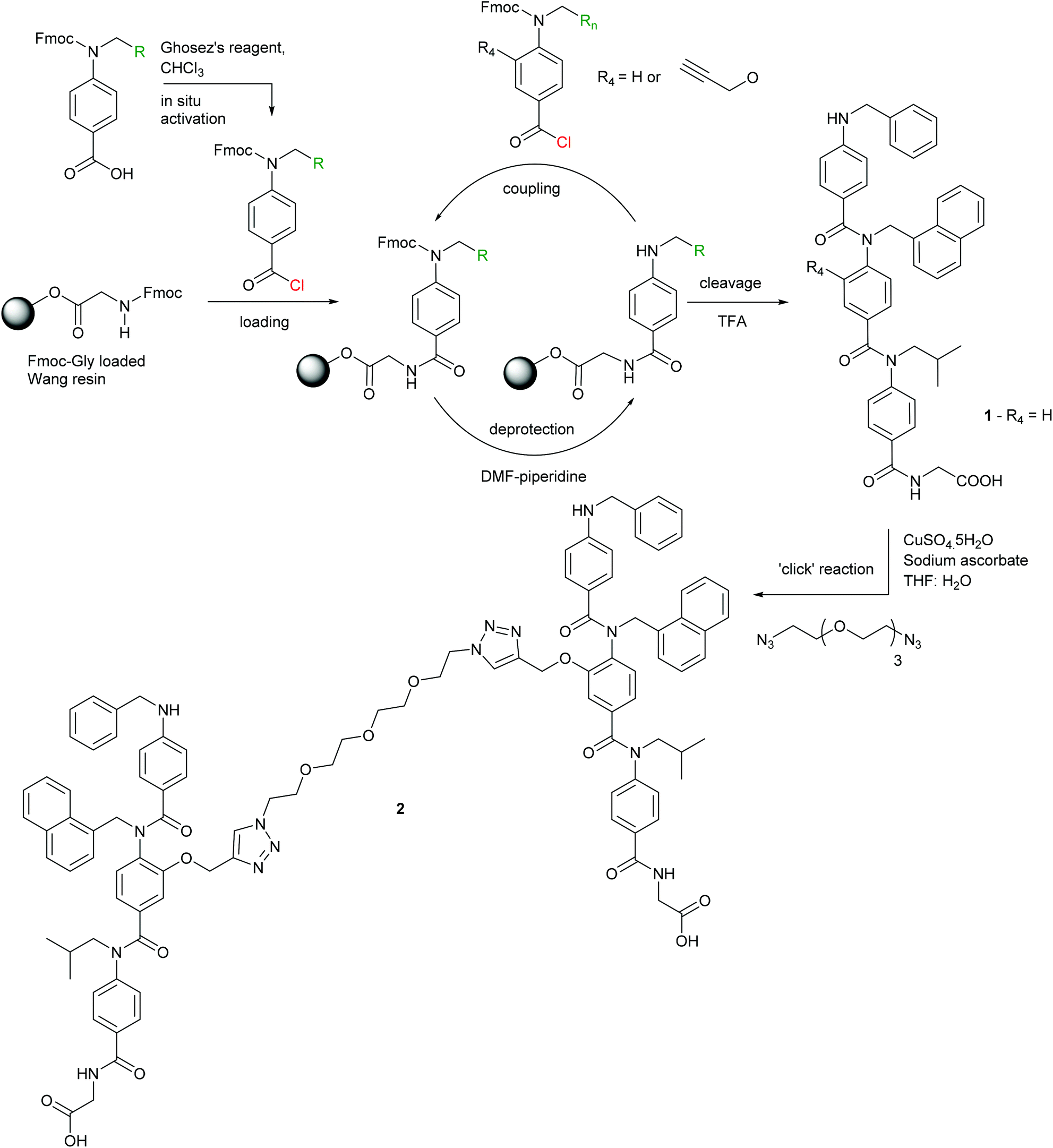
To obtain a divalent helix mimetic we employed the ‘click’ chemistry methodology for the synthesis of modified aromatic oligoamides recently reported by our group.56 A known N-alkylated inhibitor 1 of the p53/hDM2 interaction possessing phenyl, napthyl and isopropyl side chains to recapitulate the hot-spot residues of p53, was synthesised, functionalised with an alkyne, on solid-phase using an automated microwave peptide synthesiser. Once cleaved from the resin, the trimer was subjected to standard ‘click’ chemistry reaction conditions with a commercially available ethylene glycol di-azide (Scheme 1). After removal of the copper catalyst, the dimer 2 was isolated in good yield and purity. The equivalent monovalent trimer 1 was also synthesised using previously published methods.37
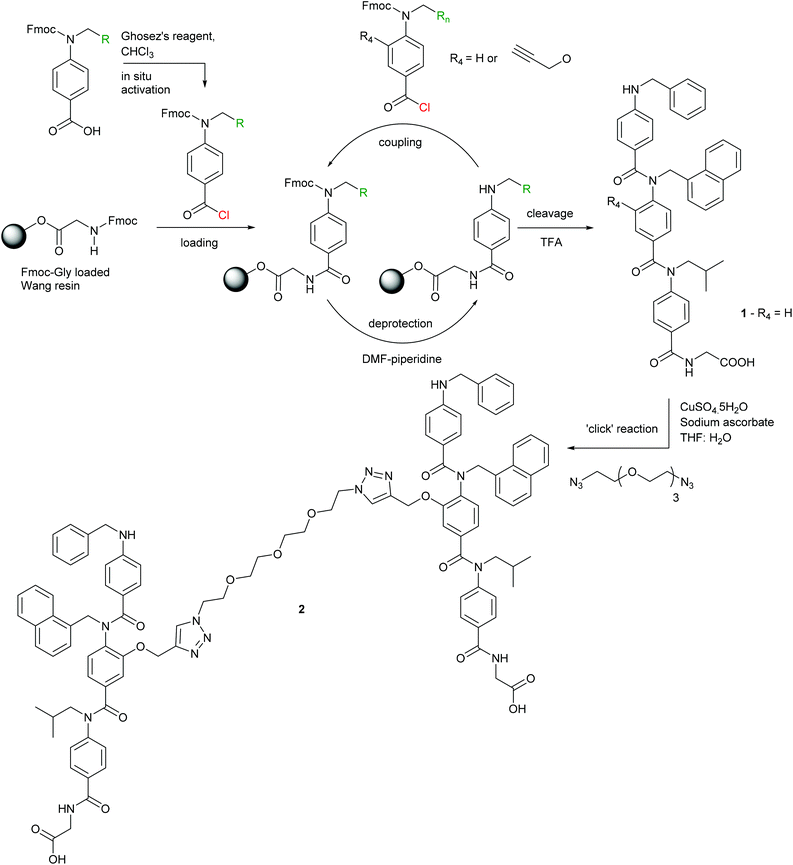 |
| | Scheme 1 Synthesis of helix mimetics 1 and 2. | |
Inhibition of p53/hDM2
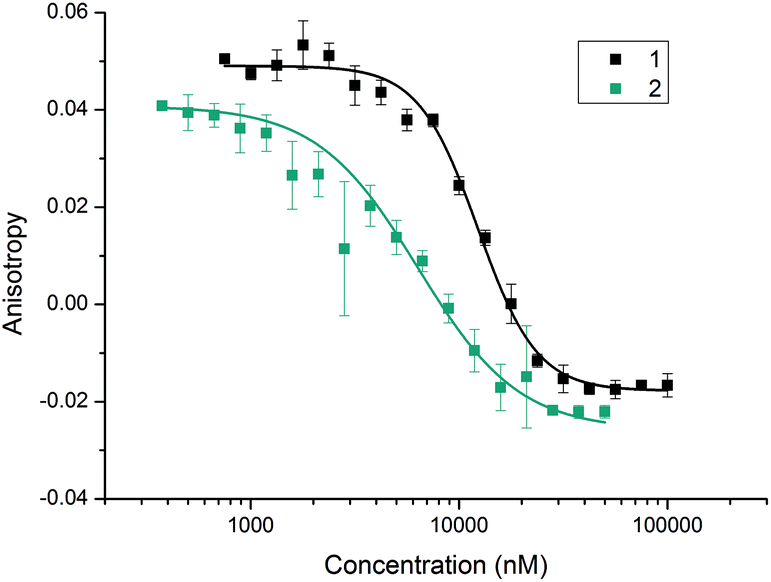
With the divalent mimetic in hand, we sought to determine if the increased valency would lead to an improvement in the inhibition of the p53/hDM2 interaction. We employed a fluorescence anisotropy competition assay whereby increasing concentrations of ligand 1 and 2 were used displace a fluorescein-labelled p53 peptide from the hDM2 binding cleft. The observed decrease in anisotropy was used to determine IC50 values (Fig. 2). Unfunctionalised monovalent trimer 1 was observed to act as a low μM inhibitor of the p53/hDM2 interaction (IC50 = 12.3 ± 0.4 μM). When compared against the divalent mimetic 2 a 2-fold improvement in inhibition was observed (IC50 = 6.3 ± 0.4 μM). However, as the effective concentration of binding groups is doubled this indicates an absence of positive cooperativity. The most likely cause of such an observation is that dimer 2 simultaneously interacts with two molecules of hDM2, but without the benefit of additional intermolecular interactions between the two proteins molecules.
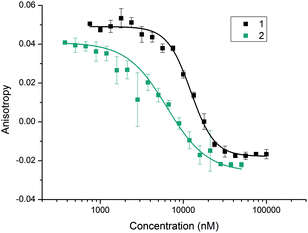 |
| | Fig. 2 Fluorescence anisotropy competition curves for the inhibition of the p53/hDM2 interaction by the monovalent trimer 1 (black) and the divalent trimer 2 (blue) in assay buffer (40 mM phosphate pH 7.5, 200 mM NaCl, 0.02 mg ml−1 BSA). hDM2 and FITC-p53 were added to a dilution series of the mimetics to give final concentrations of 154.2 and 54.5 nM, respectively. | |
Molecular modelling
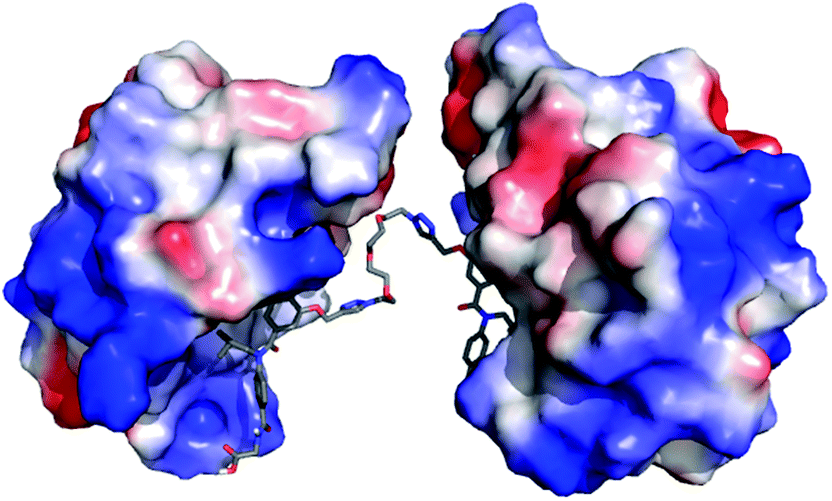
To assess if divalent inhibitor 2 was capable of simultaneous interaction with two copies of hDM2 we carried out a series of rudimentary modelling experiments. A series of conformers were generated from the structure of mimetic 2, from which an extended conformation was selected and each copy of the hDM2 binding pharmacophore was docked into the binding site of hDM2 taken from the crystal structure (Fig. 3). The resultant model indicated no steric clash between the two protein molecules, despite the ethylene glycol linker not adopting a fully extended conformation.
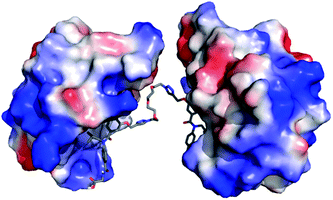 |
| | Fig. 3 Molecular modelling of compound 2; proposed binding mode of compound 2 in the p53 binding cleft of two hDM2 molecules (PDB ID: 1YCR). | |
Protein assembly
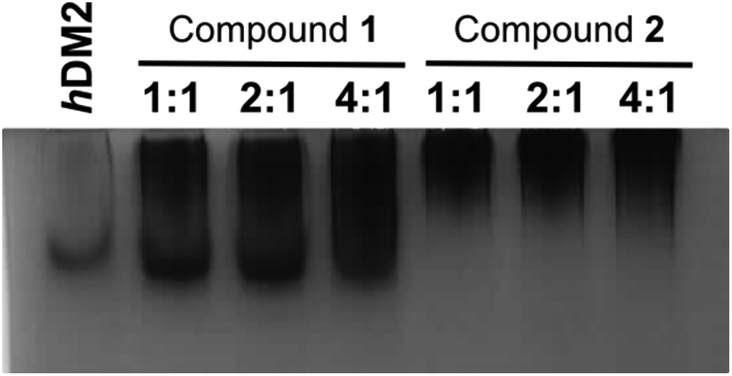
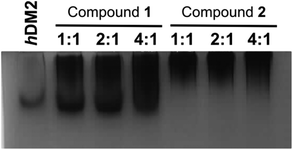
To obtain experimental evidence of protein assembly we ran a Native PAGE gel (Fig. 4) of hDM2 both alone (lane 1) and in the presence of either the monovalent ligand 1 (lanes 2–4) or divalent ligand 2 (lanes 5–7). The gel clearly indicates a difference in the mobility of the protein in the presence of the divalent trimer 2, consistent with an increase in size. In contrast no such change occurred for monovalent mimetic 1. We then sought to gain additional more quantitative evidence of protein aggregation. To this end, sedimentation velocity analytical centrifugation (svAUC) was performed on hDM2 both with and without DMSO and in the presence of compounds 1 and 2 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of protein: ligand (Fig. 5).
:4 ratio of protein: ligand (Fig. 5).
 |
| | Fig. 4
hDM2 analysed by native PAGE in the presence of increasing equivalent of both the monovalent ligand 1 (left) and divalent ligand 2 (right) run in 25 mM Tris buffer pH 8.6, 190 mM glycine. Bands were visualised with Coomassie staining. | |
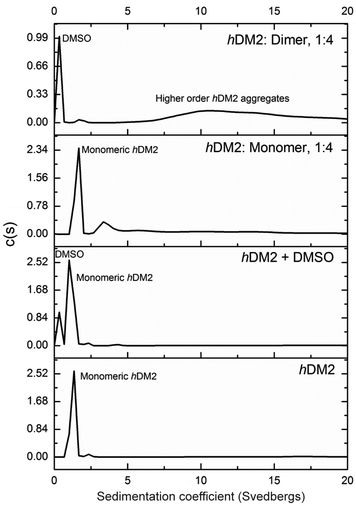 |
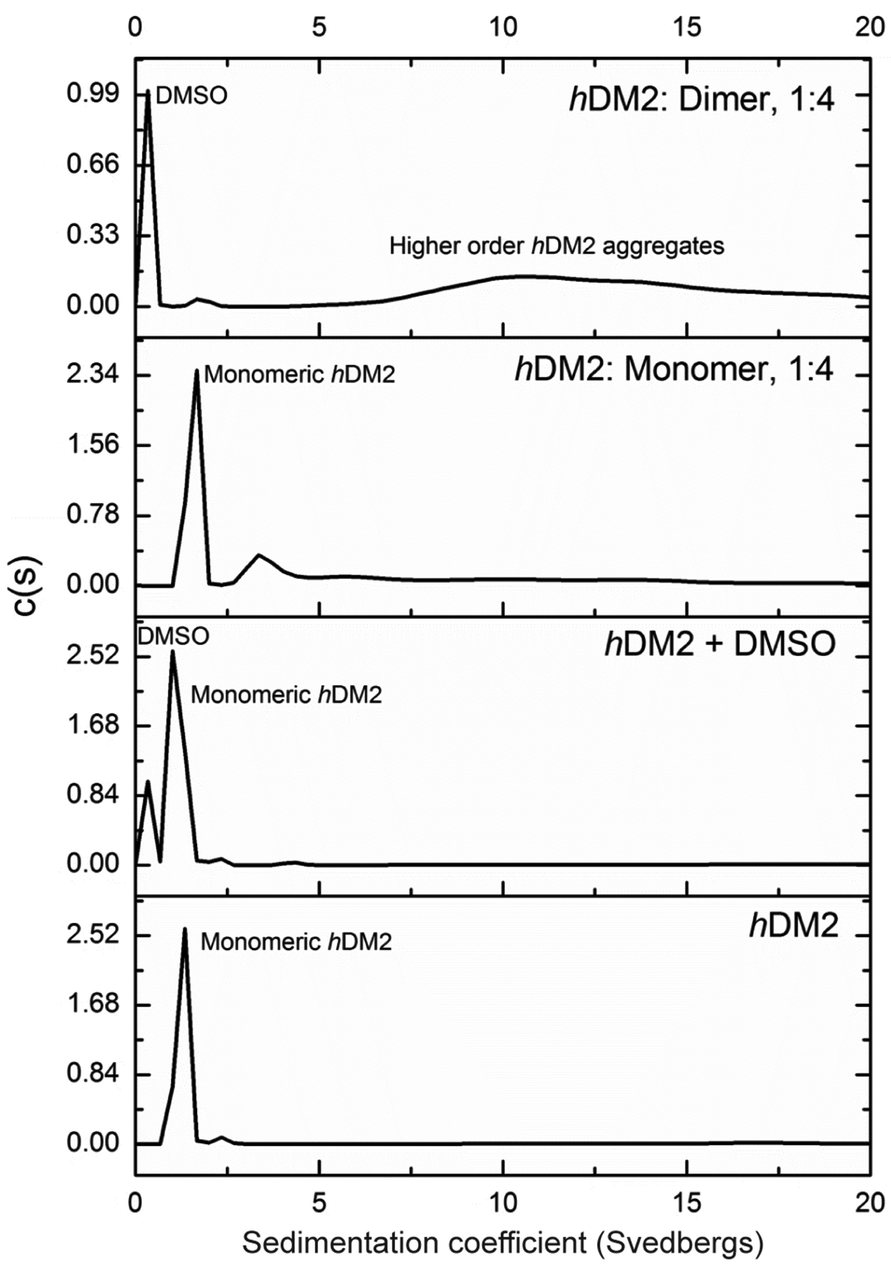
| | Fig. 5 Analytical ultracentrifugation analysis of hDM2 in the presence of monomer 1 and dimer 2 at a 1:4 concentration ratio. Samples were prepared in 40 mM phosphate buffer, 200 mM NaCl and 5% DMSO where appropriate. Samples were centrifuged at 48000 rpm and sedimentation coefficients calculated. | |
In the sample of hDM2 alone a clear peak was observed corresponding to the approximate molecular weight of the protein. Importantly, the peak was not perturbed by the presence of 5% DMSO, resulting from dilution of the ligand stock solution. When bound to mimetic 1 no significant changes in the overall size or shape of hDM2 were observed, a small peak at higher molecular weight also appears however, the majority of the species observed can be assigned as monomeric hDM2. The molecular weights were characterised by relating rate of sedimentation and diffusion coefficient to buffer density and the partial specific volume occupied by atoms in solution at a constant temperature, giving peaks as 14.5 kDa and 49.2 kDa for the major and minor peaks. The molecular weight for the major peak is in the region expected for hDM2 however the origin of the minor peak is unknown. In the sample containing mimetic 2, a complete loss of the peak for monomeric hDM2 was observed concomitant with the appearance of higher order aggregates. The peak is inconsistent with the mass corresponding to a protein dimer, rather it is the result of a much larger species. It is noteworthy that aggregation by multivalent ligands is not unprecedented57 and this unexpected result will be the focus of future investigations. It is plausible that the dimeric mimetic 2 induces protein dimerisation resulting in a ternary complex that is capable of further aggregation although the possibility that the mimetic is capable of interacting with additional binding sites on hDM2 cannot be discounted. Such a scenario, need not necessarily couple to the p53 tracer displacement occurring in the fluorescence anisotropy experiment.
Conclusions
A novel helix mimetic dimer 2 was prepared using ‘click’ chemistry methodology. This dimeric ligand 2 was found to be a potent inhibitor of the p53/hDM2 interaction being twice as potent as the monomeric analogue 1. When considered in terms of ligand copy number, 1 and 2 are similarly potent. Despite this, more detailed modelling and experimentation revealed the two compounds have differing modes of action. Whilst monomeric ligand 1 favours 1:1 stoichiometry with hDM2, compound 2 acts as a chemical inducer of dimerization/aggregation. This suggests that, with further refinement, such a strategy could be exploited for protein dimerisation assisted inhibition of protein–protein interactions and in synthetic biology58 for chemically induced dimerization59,60 and/or the controlled assembly of proteins.61,62
Experimental
General considerations
All chemicals and solvents were purchased and used without further purification. 1H, 13C and 2D NMR spectra were recorded with a Bruker DRX 500 MHz or DPX 300 MHz spectrometer. 1H NMR spectra are referenced to residual solvent and chemical shifts are given as parts per million downfield from TMS. Coupling constants are reported to the nearest 0.1 Hz. IR spectra were recorded with a Perkin-Elmer FTIR spectrometer and samples were analysed in the solid phase. Mass spectra (HRMS) were obtained with a Bruker maxis impact 3000 spectrometer using electrospray ionisation. LC-MS experiments were run on a Waters Micromass ZQ spectrometer. Analytical TLC was performed on 0.2 mm silica gel 60 F254 pre-coated aluminium sheets (Merck) and visualised by using UV irradiation. Flash chromatography was carried out on silica gel 60 (35–70 micron particles, FluoroChem). The convention used to assign the spectroscopic data and for naming compounds for this series of aromatic oligoamides has been described previously.37,56
Monomer syntheses and the synthesis of the monovalent trimer have been reported previously.
Expression of hDM2 and fluorescence anisotropy assays were performed as described previously.36,56
Synthesis of N-(N-(N-(benzyl-4-aminobenzoyl)-N-naphth-1-yl-4-aminobenzoyl)-N-isobutyl-4-aminobenzoyl)-glycine dimer
Glycine-loaded Wang resin (254 mg, 0.2 mmol) was swelled in anhydrous DMF (5 ml) 15 minutes prior to reaction. The monomers; benzyl (450 mg, 1.0 mmol), alkyne-1-naphthyl (554 mg, 1.0 mmol) and isobutyl (415 mg, 1.0 mmol) were each dissolved in anhydrous CHCl3 (10 ml) and pre-activated for coupling with Ghosez's reagent (630 μl, 20% in CHCl3, 0.96 mmol) for 1 hour at room temperature. The coupling reactions were carried out on a CEM Liberty™ microwave assisted automated peptide synthesiser. The trimer was then cleaved from the resin with TFA–DCM (1:1, 1 ml) and analysed by LC-MS to confirm formation of the desired trimer. The trimer (25.7 mg, 0.033 mmol) was dissolved in THF–H2O (1:1, 10 ml) and 1,11-diazido-3,6-9-trioxaundecance (3.47 μl, 0.017 mmol) was added followed by CuSO4·5H2O (0.42 mg, 1.6 × 10−3 mmol) and sodium ascorbate (0.43 mg, 3.3 × 10−3 mmol). The reaction mixture was stirred overnight at room temperature. The solvent was then removed and the residue taken up in DMSO (5 ml) and any copper precipitate removed by centrifugation. The DMSO was then removed under high vacuum affording the desired compound as a sticky orange solid (13.2 mg, 7.36 × 10−3 mmol, 43%). Rf = 0.3 (3:2, DCM–MeOH); 1H NMR (500 MHz, DMSO-d6) δ: 8.14–8.12 (m, 2–3°-T, 2H), 7.91–7.69 (br m, ArCH, 5H), 7.47–7.45 (m, ArCH, 4H), 7.27–7.05 (m, ArCH, 23H), 6.92–6.90 (m, 1-H2, 4H), 6.42–6.28 (m, 2-H5, 2-H6, 1-H3, 8H), 4.86–4.84 (s, 2-Hα, 4H), 4.55–4.53 (s, 1-NH, 2H), 4.26–4.24 (s, 4-Hα, 4H), 3.84–3.83 (s, 1-Hα, 4H), 3.64 (s, 3-Hα, 4H), 3.55–3.41(m, 2–3°-Hβ, 2–3°-Hγ, 2–3°-Hθ, 2–3°-Hι, 8H), 3.23 (s, 2–3°-Hδ, 2–3°-Hε, 2–3°-Hζ, 2–3°-Hη, 8H), 1.72–1.69 (m, 3-Hβ, 2H), 0.84 (s, 3-Hγ, 12H); HRMS: Calcd [M + H]+ (C104H105N14O15) m/z = 1789.787835, Found [M + H]+m/z = 1789.785716; νmax (cm−1): 3319s (O–H), 2916s (C–H), 1635s (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1601s (CO), 1504s, 1418m, 1385m, 1338w, 1284s, 1182m, 1130s, 1017s.
O), 1601s (CO), 1504s, 1418m, 1385m, 1338w, 1284s, 1182m, 1130s, 1017s.
Molecular modelling
The protein structure for hDM2 (PDB ID: 1YCR) was prepared using the protein preparation wizard within Maestro (Schrodinger) and the docking grid prepared by selecting the binding groove of the p53 helix using Glide (Schrodinger).
The monovalent inhibitor was docked using Glide (Schrodinger) and these docked structures superimposed onto a low energy structure of the divalent inhibitor to provide a model of protein dimerization.
Native PAGE
Protein samples were separated on a 10% acrylamide native PAGE gel in 25 mM Tris, 190 mM glycine buffer pH 8.6, gel was run at 25 mA. Bands were visualised with Coomassie staining. Samples were diluted from 10 mM DMSO stocks into 50 mM Tris buffer pH 8.0, 200 mM NaCl, 0.5 mM DTT.
Sedimentation velocity analytical ultracentrifugation
Samples (0.32 ml) were placed in a 1.2 cm pathlength 2-sector meniscus-matching epon centrepiece cell constructed with sapphire windows and centrifuged at 48000 rpm in an An50-Ti rotor in an Optima XL-I analytical ultracentrifuge at 20.0 °C. Changes in concentration of the solute were detected by interference optics, with a total of 500 scans being taken over approximately 8.3 hours. Buffer densities and viscosities were calculated by Sednterp version 1.09, omitting the presence of 5% DMSO.63 Radial interference profiles were fitted using the program Sedfit version 12.1b using a continuous distribution c(S) Lamm equation model.64
Acknowledgements
This work was supported by the European Research Council [ERC-StG-240324].
Notes and references
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Chem. Rev., 2008, 108, 1225–1244 CrossRef CAS PubMed.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- A. J. Wilson, Chem. Soc. Rev., 2009, 38, 3289–3300 RSC.
- P. B. Crowley, P. Ganji and H. Ibrahim, ChemBioChem, 2008, 9, 1029–1033 CrossRef CAS PubMed.
- A. Ojida, M.-a. Inoue, Y. Mito-oka, H. Tsutsumi, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 2052–2058 CrossRef CAS PubMed.
- Y. Mito-oka, S. Tukiji, T. Hiraoka, N. Kasagi, S. Shinkai and I. Hamachi, Tetrahedron Lett., 2001, 42, 7059–7062 CrossRef CAS.
- H. Bayraktar, P. S. Ghosh, V. M. Rotello and M. J. Knapp, Chem. Commun., 2006, 1390–1392 RSC.
- J. Ohkanda, R. Satoh and N. Kato, Chem. Commun., 2009, 6949–6951 RSC.
- J. Muldoon, A. E. Ashcroft and A. J. Wilson, Chem. – Eur. J., 2010, 16, 100–103 CrossRef CAS PubMed.
- A. J. Wilson, J. Hong, S. Fletcher and A. D. Hamilton, Org. Biomol. Chem., 2007, 5, 276–285 CAS.
- L. K. Tsou, C.-H. Chen, G. E. Dutschman, Y.-C. Cheng and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2012, 22, 3358–3361 CrossRef CAS PubMed.
- B. A. Rosenzweig, N. T. Ross, M. J. Adler and A. D. Hamilton, J. Am. Chem. Soc., 2010, 132, 6749–6754 CrossRef CAS PubMed.
- Y. Cheng, L. K. Tsou, J. Cai, T. Aya, G. E. Dutschman, E. A. Gullen, S. P. Grill, A. P.-C. Chen, B. D. Lindenbach, A. D. Hamilton and Y.-c. Cheng, Antimicrob. Agents Chemother., 2010, 54, 197–206 CrossRef CAS PubMed.
- D. Margulies, Y. Opatowsky, S. Fletcher, I. Saraogi, L. K. Tsou, S. Saha, I. Lax, J. Schlessinger and A. D. Hamilton, ChemBioChem, 2009, 10, 1955–1958 CrossRef CAS PubMed.
- D. Bier, R. Rose, K. Bravo-Rodriguez, M. Bartel, J. M. Ramirez-Anguita, S. Dutt, C. Wilch, F.-G. Klärner, E. Sanchez-Garcia, T. Schrader and C. Ottmann, Nat. Chem., 2013, 5, 234–239 CrossRef CAS PubMed.
- R. i. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- B. B. Lao, K. Drew, D. A. Guarracino, T. F. Brewer, D. W. Heindel, R. Bonneau and P. S. Arora, J. Am. Chem. Soc., 2014, 136, 7877–7888 CrossRef CAS PubMed.
- S. Kushal, B. B. Lao, L. K. Henchey, R. Dubey, H. Mesallati, N. J. Traaseth, B. Z. Olenyuk and P. S. Arora, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7531–7536 CrossRef PubMed.
- P. Ravindranathan, T.-K. Lee, L. Yang, M. M. Centenera, L. Butler, W. D. Tilley, J.-T. Hsieh, J.-M. Ahn and G. V. Raj, Nat. Commun., 2013, 4, 1923 CrossRef PubMed.
- W. E. Martucci, J. M. Rodriguez, M. A. Vargo, M. Marr, A. D. Hamilton and K. S. Anderson, MedChemComm, 2013, 4, 1247–1256 RSC.
- A. Kazi, J. Sun, K. Doi, S.-S. Sung, Y. Takahashi, H. Yin, J. M. Rodriguez, J. Becerril, N. Berndt, A. D. Hamilton, H.-G. Wang and S. d. M. Sebti, J. Biol. Chem., 2011, 286, 9382–9392 CrossRef CAS PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736–739 CrossRef CAS PubMed.
- G. M. Burslem, H. F. Kyle, A. L. Breeze, T. A. Edwards, A. Nelson, S. L. Warriner and A. J. Wilson, ChemBioChem, 2014, 15, 1083–1087 CrossRef CAS PubMed.
- P. Prabhakaran, A. Barnard, N. S. Murphy, C. A. Kilner, T. A. Edwards and A. J. Wilson, Eur. J. Org. Chem., 2013, 3504–3512 CrossRef CAS.
- V. Azzarito, P. Prabhakaran, A. I. Bartlett, N. S. Murphy, M. J. Hardie, C. A. Kilner, T. A. Edwards, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2012, 10, 6469–6472 CAS.
- J. P. Plante, T. Burnley, B. Malkova, M. E. Webb, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Commun., 2009, 5091–5093 RSC.
- F. Campbell, J. P. Plante, T. A. Edwards, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2010, 8, 2344–2351 CAS.
- A. Shaginian, L. Whitby, S. Hong, I. Hwang, B. Farooqi, M. Searcey, J. Chen, P. Vogt and D. Boger, J. Am. Chem. Soc., 2009, 131, 5564–5572 CrossRef CAS PubMed.
- T.-K. Lee and J.-M. Ahn, ACS Comb. Sci., 2010, 13, 107–111 CrossRef PubMed.
- J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535–539 CrossRef CAS PubMed.
- G. M. Burslem and A. J. Wilson, Synlett, 2014, 324–335 CAS.
- N. S. Murphy, P. Prabhakaran, V. Azzarito, J. P. Plante, M. J. Hardie, C. A. Kilner, S. L. Warriner and A. J. Wilson, Chem. – Eur. J., 2013, 19, 5546–5550 CrossRef CAS PubMed.
- V. Azzarito, P. Prabhakaran, A. I. Bartlett, N. S. Murphy, M. J. Hardie, C. A. Kilner, T. A. Edwards, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2012, 10, 6469–6472 CAS.
- K. Long, T. A. Edwards and A. J. Wilson, Bioorg. Med. Chem., 2013, 21, 4034–4040 CrossRef CAS PubMed.
- K. K. Hoe, C. S. Verma and D. P. Lane, Nat. Rev. Drug Discovery, 2014, 13, 217–236 CrossRef PubMed.
- K. Khoury, G. M. Popowicz, T. A. Holak and A. Domling, MedChemComm, 2011, 2, 246–260 RSC.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS.
- L. K. Linares, A. Hengstermann, A. Ciechanover, S. Müller and M. Scheffner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12009–12014 CrossRef CAS PubMed.
- K. Linke, P. D. Mace, C. A. Smith, D. L. Vaux, J. Silke and C. L. Day, Cell Death Differ., 2008, 15, 841–848 CrossRef CAS PubMed.
- B. Graves, T. Thompson, M. Xia, C. Janson, C. Lukacs, D. Deo, P. Di Lello, D. Fry, C. Garvie, K.-S. Huang, L. Gao, C. Tovar, A. Lovey, J. Wanner and L. T. Vassilev, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11788–11793 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 51, 6572–6581 CrossRef CAS PubMed.
- E. Fan, Z. Zhang, W. E. Minke, Z. Hou, C. L. M. J. Verlinde and W. G. J. Hol, J. Am. Chem. Soc., 2000, 2663–2664 CrossRef CAS.
- A. C. Rodrigo, A. Barnard, J. Cooper and D. K. Smith, Angew. Chem., Int. Ed., 2011, 50, 4675–4679 CrossRef CAS PubMed.
- P. Posocco, S. Pricl, S. Jones, A. Barnard and D. K. Smith, Chem. Sci., 2010, 1, 393–404 RSC.
- A. Barnard, P. Posocco, S. Pricl, M. Calderon, R. Haag, M. E. Hwang, V. W. T. Shum, D. W. Pack and D. K. Smith, J. Am. Chem. Soc., 2011, 133, 20288–20300 CrossRef CAS PubMed.
- P. R. Wich and C. Schmuck, Angew. Chem., Int. Ed., 2010, 49, 4113–4116 CrossRef CAS PubMed.
- L. Fülöp, I. M. Mándity, G. Juhász, V. Szegedi, A. Hetényi, E. Wéber, Z. Bozsó, D. Simon, M. Benkő, Z. Király and T. A. Martinek, PLoS One, 2012, 7, e39485 Search PubMed.
- N. S. Green, S. K. Palaninathan, J. C. Sacchettini and J. W. Kelly, J. Am. Chem. Soc., 2003, 125, 13404–13414 CrossRef CAS PubMed.
- S. E. Kolstoe, P. P. Mangione, V. Bellotti, G. W. Taylor, G. A. Tennent, S. Deroo, A. J. Morrison, A. J. A. Cobb, A. Coyne, M. G. McCammon, T. D. Warner, J. Mitchell, R. Gill, M. D. Smith, S. V. Ley, C. V. Robinson, S. P. Wood and M. B. Pepys, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20483–20488 CrossRef CAS PubMed.
- M. K. P. Jayatunga, S. Thompson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2014, 24, 717–724 CrossRef CAS PubMed.
- O. V. Kulikov, S. Thompson, H. Xu, C. D. Incarvito, R. T. W. Scott, I. Saraogi, L. Nevola and A. D. Hamilton, Eur. J. Org. Chem., 2013, 3433–3445 CrossRef CAS.
- A. Barnard, K. Long, D. J. Yeo, J. A. Miles, V. Azzarito, G. M. Burslem, P. Prabhakaran, T. A. Edwards and A. J. Wilson, Org. Biomol. Chem., 2014, 12, 6794–6799 CAS.
- C. Sisu, A. J. Baron, H. M. Branderhorst, S. D. Connell, C. A. G. M. Weijers, R. de Vries, E. D. Hayes, A. V. Pukin, M. Gilbert, R. J. Pieters, H. Zuilhof, G. M. Visser and W. B. Turnbull, ChemBioChem, 2009, 10, 329–337 CrossRef CAS PubMed.
- E. H. C. Bromley, K. Channon, E. Moutevelis and D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50 CrossRef CAS PubMed.
- M. Skwarczynska, M. Molzan and C. Ottmann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E377–E386 CrossRef CAS PubMed.
- E. M. Hobert and A. Schepartz, J. Am. Chem. Soc., 2012, 134, 3976–3978 CrossRef CAS PubMed.
- E. N. Salgado, X. I. Ambroggio, J. D. Brodin, R. A. Lewis, B. Kuhlman and F. A. Tezcan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1827–1832 CrossRef CAS PubMed.
- K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 3818–3821 CrossRef CAS PubMed.
-
H. Durchschlag, in Thermodynamic data for biochemistry and biotechnology, ed. H.-J. Hinz, Springer-Verlag, Berlin, Editon edn, 1986, pp. 45–182 Search PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compound 2. See DOI: 10.1039/c4ob02066a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence