
Mild Ti-mediated transformation of t-butyl thio-ethers into thio-acetates†
Thomas C.
Pijper‡
a,
Jort
Robertus‡
b,
Wesley R.
Browne
ab and
Ben L.
Feringa
*ab
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 Groningen, The Netherlands
First published on 6th November 2014
Abstract
We report a straightforward method for the rapid conversion of thio-ethers to thio-acetates using TiCl4, in good to excellent yields. The reaction conditions tolerate a variety of functional groups, including halide, nitro, ether, thiophene and acetylene functionalities. A catalytic variant of this reaction is also described.
Introduction
Numerous nanoscale materials and devices1 are based on self-assembled monolayers (SAMs) of thiols on gold substrates.2 Thiols have proven to be versatile anchoring groups3 for immobilising functional units for photoswitching,4 molecular electronics,5 control of surface wettability,6 cell adhesion,7 to name but a few examples. Although thiol chemistry is often used in SAM formation,3 the introduction of a thiol group in a compound frequently presents synthetic challenges8 because the R–S–H group can be deprotonated, is nucleophilic, and is prone to oxidation.2 Hence, protected thiols such as thio-acetates are frequently used. Indeed, acetyl and trityl protected thiols can be deprotected readily in situ during self-assembly on gold surfaces.2 In particular acetyl protected thiol substituted arenes are convenient in their use as these can typically be cleaved in situ without requiring an exogenous base to form stable monolayers equivalent to those formed starting from free thiols.9–11However, the thio-acetate group is often unstable under aqueous reaction conditions, while the thio-trityl group is unstable under various other reaction conditions, such as those employed in Suzuki–Miyaura cross-coupling reactions.12 These drawbacks can be overcome through a method developed by Stuhr–Hansen in which the thiol is initially protected by a t-butyl protecting group and later exchanged for the desired acetyl protecting group by treatment with BBr3.13 The t-butyl thio-ether is beneficial as it is typically stable under both acidic14 and basic conditions.15 Furthermore, t-butyl thio-ethers can be synthesised with relative ease, either from the free thiol using t-butyl chloride or t-butanol, or from halides (R-X) using t-butyl thiol. Once the synthetic steps incompatible with the thio-acetate have been performed, exchange of the protecting groups can be achieved by deprotection of the t-butyl thio-ether by BBr3 followed by quenching with acetyl chloride at room temperature.13
Although S-t-butyl to S-acetyl exchange procedures have been reported (using BBr3,13 but also Br2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 or AlCl316), several important functionalities do not tolerate these conditions, examples being vinyl, TBDMSO, acetylene, aldehyde, and nitro functionalities.8,13 This prompted us to identify more versatile Lewis acids, with which the exchange reaction can be performed under mild conditions while tolerating a wider variety of functionalities. One such candidate is TiCl4, as there have been several examples of TiCl4/n-Bu4NI-mediated deprotection of ethers (R–O–R).17 Furthermore, TiCl4 has been used in the deprotection of silyl ether protected alcohols. The results of Tanabe and co-workers, who successfully deprotected aryl and aliphatic TBDMS-ethers in excellent yields (91–99%) using TiCl4-Lewis base (AcOEt, CH3NO2) complexes, are particularly encouraging.18 Finally, TiCl4 was used with great efficacy as a deprotection reagent in the hydrolysis of t-butyl esters in β-lactam chemistry, whereas the use of AlCl3, BF3, and FeCl3 resulted in degradation of the starting material or poor yields.19
8 or AlCl316), several important functionalities do not tolerate these conditions, examples being vinyl, TBDMSO, acetylene, aldehyde, and nitro functionalities.8,13 This prompted us to identify more versatile Lewis acids, with which the exchange reaction can be performed under mild conditions while tolerating a wider variety of functionalities. One such candidate is TiCl4, as there have been several examples of TiCl4/n-Bu4NI-mediated deprotection of ethers (R–O–R).17 Furthermore, TiCl4 has been used in the deprotection of silyl ether protected alcohols. The results of Tanabe and co-workers, who successfully deprotected aryl and aliphatic TBDMS-ethers in excellent yields (91–99%) using TiCl4-Lewis base (AcOEt, CH3NO2) complexes, are particularly encouraging.18 Finally, TiCl4 was used with great efficacy as a deprotection reagent in the hydrolysis of t-butyl esters in β-lactam chemistry, whereas the use of AlCl3, BF3, and FeCl3 resulted in degradation of the starting material or poor yields.19
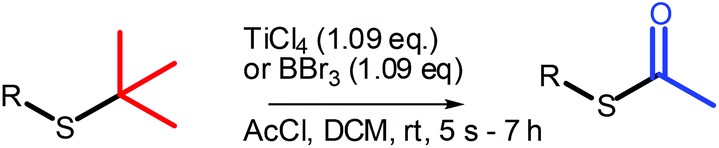
Herein, we present a robust method for the conversion of t-butyl thio-ethers to thio-acetates using TiCl4 instead of BBr3. We have found that TiCl4 is tolerant towards a wider variety of functional groups and performs consistently better than BBr3, providing the desired thio-acetates in high yields and in shorter reaction times (Scheme 1).
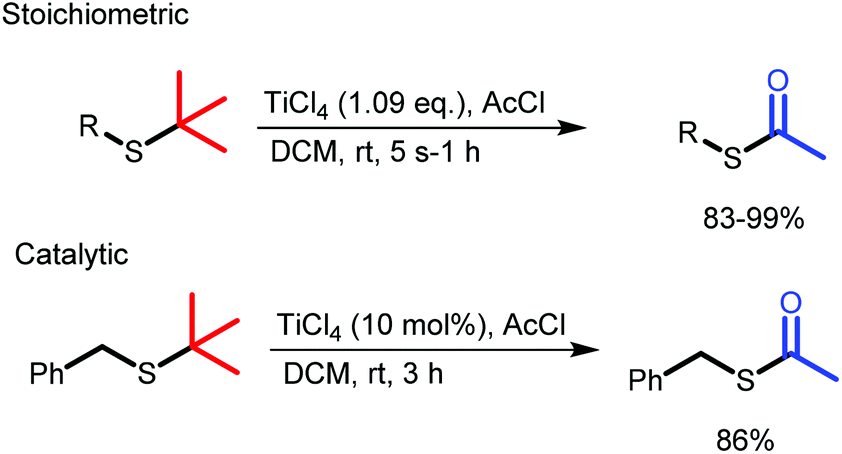 | ||
| Scheme 1 Rapid and clean conversion of thiotertbutyl ethers to acetyl protected thiols with stoichiometric and catalytic TiCl4. | ||
Results and discussion
12 substrates were examined to explore the utility of TiCl4 for the conversion of thio-ethers (a) to thio-acetates (b) (Table 1). t-Butyl thio-ethers 1–6a were converted to the corresponding thio-acetates 1–6b in good to excellent isolated yields using TiCl4 or BBr3. However, whereas the reactions using BBr3 were complete after 2.5 to 7 h, the use of TiCl4 allowed for substantially shorter reaction times, in several cases providing the thio-acetate within a few seconds (2a, 4a, and 6a as well as 8a and 10a). In addition, improved yields were observed for several substrates when TiCl4 was used (1a, 3a, and 5a). Conversion of 7a–10a using BBr3 was found to result in decomposition only. In sharp contrast, 7a and 8a were converted to their corresponding thio-acetates in high yield when TiCl4 was used.|
|
|||||
|---|---|---|---|---|---|
| Substrate | Reaction time BBr3 | Isolated yield | Reaction time TiCl4 | Isolated yield | |
a Forms multiple products.
b Full conversion to an unidentified product with an Rf = 0.19 on SiO2.
c The Friedel–Crafts acylation product p-CH3(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)C6H4SCH3 was isolated in 94% yield.22 Dec. = decomposition of the starting material. O)C6H4SCH3 was isolated in 94% yield.22 Dec. = decomposition of the starting material.
|
|||||
| 1a |
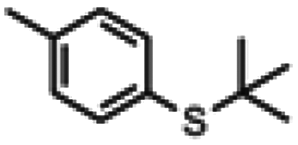
|
5 h | 81% | <1 h | 93% |
| 2a |

|
6 h | 96% | 5 s | 94% |
| 3a |

|
5 h | 76% | <1 h | 88% |
| 4a |

|
7 h | >99% | 5 s | >99% |
| 5a |
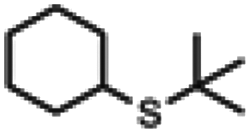
|
3 h | 92% | 1 h | >99% |
| 6a |
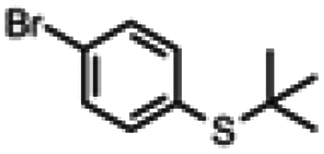
|
4 h | 65% | 5 s | 89% |
| 7a |

|
— | Dec. | <1 h | 88% |
| 8a |
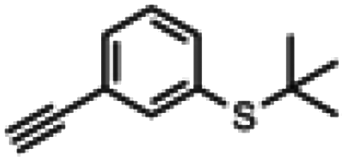
|
— | —a | 5 s | 83% |
| 9a |

|
— | Dec. | — | Dec. |
| 10a |

|
7 h | Dec. | 5 s | |
| 11a |
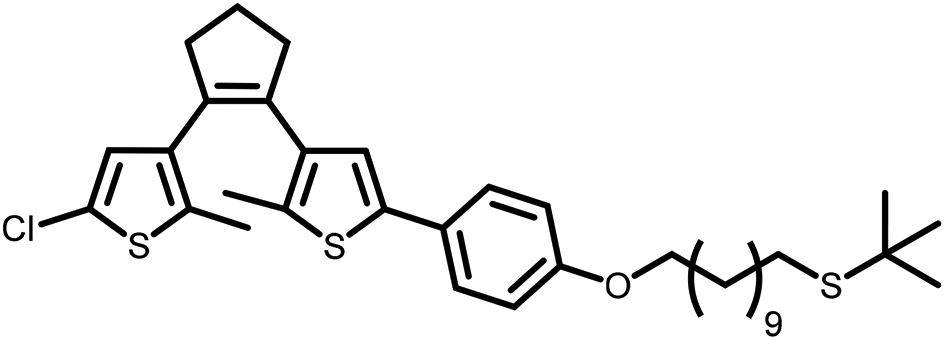
|
n/a | n/a | 2 h | 87% |
| 12a |
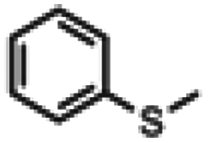
|
7 h | No conv. | 3 h | |
Aldehyde and pyrid-2-yl functionalized thioethers were examined to explore functional group limitations. Treatment of aldehyde 9a under these reaction conditions still resulted in decomposition. Analysis of the product revealed that with BBr3 the t-butyl group was cleaved while with TiCl4 the t-butyl group remained intact. In neither case, however, was thio-acetate 9b obtained. For thio-ether 10a, the TiCl4-mediated reaction did not provide the desired product either with full conversion to an unidentified compound instead.20,21 It was also attempted to use the above reaction conditions for the conversion of a methyl thio-ether group to the corresponding thio-acetate group. The methyl thio-ether group of 12a was found to be stable to both BBr3 and TiCl4. However, treatment of 12a with TiCl4 for 3 h resulted in the aromatic Friedel–Crafts acylation product.
The stability of the OTBDMS protecting group, alkenes and THP ethers under reaction conditions was examined (Scheme 2). p-Br-phenyl TBDMS ether was found to be stable under reaction conditions (see ESI, Fig. S2†), however, both the THP and alkene of 2-(dec-9-en-1-yloxy)tetrahydro-2H-pyran were found to react. It should be noted, however, that more complex structures such as 11a are stable under reaction conditions (vide infra).
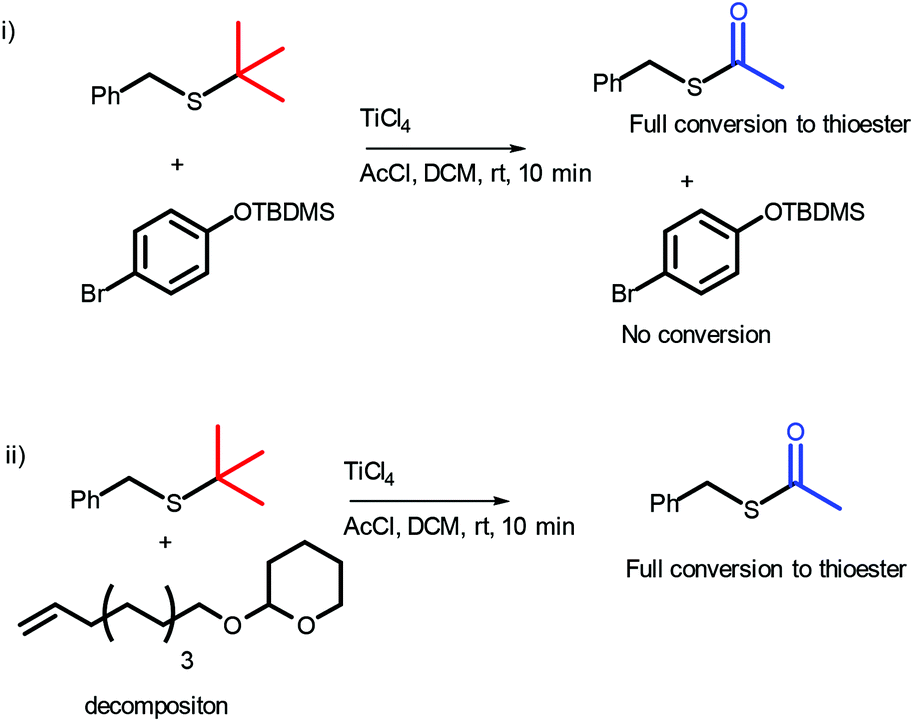 | ||
| Scheme 2 Stability of (i) the OTBDMS protecting group and (ii) terminal alkenes and THP ethers under reaction conditions with 1.09 eq. of TiCl4. | ||
A possible mechanism by which the reaction may proceed is that addition of TiCl4 to acetyl chloride results in the formation of the acylium ion (Scheme 3),23 as is supported by the Friedel–Crafts acylation product obtained with compound 12a. The acylium ion undergoes nucleophilic attack from the sulphur of the thio-ether. Expulsion of the 2-methylpropan-2-ylium carbocation subsequently results in product formation. Conversion of the anionic Ti species is achieved by dissociation of a chloride and the subsequent capture of the chloride ion by the carbocation resulting in the formation of 2-chloro-2-methylpropane and TiCl4, thus completing the catalytic cycle for the exchange of the t-Bu thio-ether to the thio-acetate. In d2-dichloromethane, the formation of iso-2-chloro-2-methylpropane was observed, whereas the formation of isobutene was not, which supports the proposed pathway (see ESI†).
 | ||
| Scheme 3 Proposed cycle for the conversion of thio-ethers to thio-acetates using TiCl4 as a catalyst. | ||
The reaction mechanism proposed furthermore implies that TiCl4 might be used catalytically. Indeed, it was found that treatment of 4a with a catalytic amount of TiCl4 (10 mol%) resulted in full conversion to the thio-acetate in 3 h with isolated yields of ca. 86% (Scheme 4). These results therefore support the proposed mechanism and further establish the potential of TiCl4 to mediate the S-t-Bu to S-acetyl exchange reaction. The conversion from 4a to 4b under stoichiometric conditions was completed within 5 s, whereas under catalytic conditions the reaction was finished within 3 h. It should be noted that under these catalytic conditions the catalytic reaction still proceeds faster with TiCl4 then when a stoichiometric quantity of BBr3 is used.
 | ||
| Scheme 4 Thio-ether to thio-acetate conversion as catalysed by TiCl4. | ||
In summary, the method reported herein provides a versatile, mild and selective method compared to existing thio-ether to thio-acetate exchange methods. The use of TiCl4 is more atom economic then the use of BBr3 given that the former can be employed catalytically. Furthermore, conditions using TiCl4 for the exchange tolerate a wider range of functional groups than BBr3-mediated methods, including acetylene groups, which is in contrast to conditions using Br2 that provide only moderate conversion to the thio-acetate.8 The exchange of the t-butyl protecting group for a thio-acetate group in aliphatic thio-ether 11a provides 11b in high yield (Table 1), even though 11a contains a dithienyl ethene photochromic switching unit. The high reaction rate at room temperature implies that the exchange reaction is also able to proceed at low temperature. Indeed, the reaction was found to proceed with full conversion of 4a to 4b within 30 min at −78 °C. Performing the exchange at low temperature opens opportunities to avoid undesirable side-reactions of sensitive substituents.
Conclusions
In conclusion, the conversion from thio-ether to thio-acetate using TiCl4 represents a highly versatile and fast method for a wide range of applications, not least those involving the synthesis of SAM forming thiols.Acknowledgements
Funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035, WRB, BLF), the European Research Council (Advanced Investigator grant no. 227897) and Netherlands organization for Fundamental Research on Materials (FOM, TCP) are acknowledged for financial support.Notes and references
-
(a) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed
; (b) H. Tian and S. Wang, Chem. Commun., 2007, 781 RSC
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed
- J. C. Love, D. B. Wolfe, R. Haasch, M. L. Chabinyc, K. E. Paul, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 2003, 125, 2597 CrossRef CAS PubMed
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407 CrossRef CAS
-
(a) N. Katsonis, T. Kudernac, M. Walko, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Adv. Mater., 2006, 18, 139 CrossRef
- K. Y. Chen, O. Ivashenko, G. T. Carroll, J. Robertus, J. C. M. Kistemaker, G. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219 CrossRef CAS PubMed
- J. Robertus, W. R. Browne and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 354 RSC
- A. Blaszczyk, M. Elbing and M. Mayor, Org. Biolmol. Chem., 2004, 2, 2722 RSC
- S. R. Wasserman, H. Biebuyck and G. M. Whitesides, J. Mater. Res., 1989, 4, 886 CrossRef CAS
- M. Jevric, S. L. Broman and M. B. Nielsen, J. Org. Chem., 2013, 78, 4348 CrossRef CAS PubMed
- J. M. Tour, L.-R. Jones II, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh and S. V. Atre, J. Am. Chem. Soc., 1995, 117, 9529 CrossRef CAS
- T. Itoh and T. Mase, J. Org. Chem., 2006, 71, 2203 CrossRef CAS PubMed
-
(a) N. Stuhr-Hansen, Synth. Commun., 2003, 33, 641 CrossRef CAS
- N. Weibel, A. Blaszczyk, C. von Hänisch, M. Mayor, I. Pobelov, T. Wandlowski, F. Chen and N. Tao, Eur. J. Org. Chem., 2008, 136 CrossRef CAS
- W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem. – Eur. J., 2005, 11, 6430 CrossRef CAS PubMed
- I. A. Aliev, G. A. Kalabin and N. Ghelis, Sulfur Lett., 1991, 12, 123 CAS
- T. Tsuritani, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1999, 40, 8121 CrossRef CAS
- A. Iida, H. Okazaki, T. Misaki, M. Sunagawa, A. Sasaki and Y. Tanabe, J. Org. Chem., 2006, 71, 5380 CrossRef CAS PubMed
- M. Valencic, T. van der Does and E. de Vroom, Tetrahedron Lett., 1998, 39, 1625 CrossRef CAS
- This compound is currently thought to be an adduct 10a-TiClx, as although its 1H-NMR spectrum is similar to that of 10a, its retention on SiO2 TLC plates is significantly different from that of 10a.
- K. Hensen, R. Mayr-Stein, B. Spangenberg, M. Bolte and S. Rühl, Z. Naturforsch., B: Chem. Sci., 2000, 55, 248 CAS
-
(a) R. A. Cutler, R. J. Stenger and C. M. Suter, J. Am. Chem. Soc., 1952, 74, 5475 CrossRef CAS
- H. P. Treffers and L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 1708 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization and spectroscopic data. See DOI: 10.1039/c4ob02120g |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |