Iron-catalyzed/mediated oxidative transformation of C–H bonds
Fan
Jia
and
Zhiping
Li
*
Department of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: zhipingli@ruc.edu.cn
First published on 12th February 2014
Abstract
It has been a long time since C–H bond oxidations first attracted chemists’ attention. In the last several decades, C–H bond oxidation has been extensively investigated and applied in chemistry. Transition-metal catalyzed C–H bond oxidative transformations presents one of the state-of-arts at the frontiers of chemistry. Iron, as a cheap, readily accessible metal, has already shown its unique utility. This review attempts to focus on C–H bond cleavage in oxidative transformation via iron catalysis, as well as applications in synthetic chemistry.
 Fan Jia | Fan Jia was born in Henan, China, in 1988. He received his B.Sc. in applied chemistry from Nanyang Normal University in 2011. In July 2011, he joined Prof. Zhiping Li's group at Renmin University of China for his Master’s work. His current research interests focus on C–H bond oxidative transformations via iron catalysis. |
 Zhiping Li | Zhiping Li began chemistry at Nanjing University of Science and Technology (B.Sc. 1993) and obtained his Ph.D. at Dalian University of Technology in 1999. After postdoctoral research at Peking University (with Professor Zhenfeng Xi, 1999–2000) and Hokkaido University (with Professor Tamotsu Takahashi, 2001–2002), he joined Peking University as an assistant professor. In 2004, he moved to McGill University as a postdoctoral fellow with Professor Chao-Jun Li. He started his independent research work at Renmin University of China as an associate professor in 2006 and has been professor of chemistry from 2009. His research interests include the development of synthetic methodologies, especially focusing on iron-catalyzed oxidative C–H bond transformation and selective C–C bond cleavage, and synthesis of biologically active natural products. |
1 Introduction
Carbon–hydrogen bonds are among the most common chemical bonds in organic molecules.1 The oxidative transformation of such bonds plays a vital role in modern scientific research.2 In nature, many important biochemical processes involve oxidation reactions. Such as oxidations are catalyzed by enzymes within the cells of living organisms. In organic chemistry, generally, oxidation means gain of oxygen and/or loss of hydrogen in an organic substrate.3 Since most oxidation reactions are thermodynamically downhill, achieving high selectivity in such reactions has been extremely challenging. Generally, C–H bond oxidations include oxygenation,4 amination,5 halogenation6 (Fig. 1) and dehydrogenation.7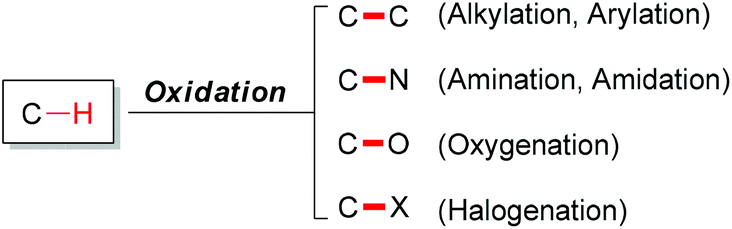 | ||
| Fig. 1 Representative oxidative transformations of C–H bonds. | ||
Over the last several decades, utilizing transition metals as catalysts has played a vital role in the area of C–H activation.8 Chemists tend to use these powerful tools to transform C–H bonds into target functional groups. The vast majority of transition-metal catalyzed C–H oxidation reactions have focused on the transformation of C–H bonds into C–C bonds.9 These powerful methods have been the subject of numerous review articles. Synthetically, the need for the installation of functional groups has led to a great variety of tools for total synthesis of natural products and pharmaceuticals.10
Iron, which is one of the most abundant metals on earth, lies in the first transition series. On account of its electron configuration, iron has a wide range of oxidation states, −II to +VIII, although the potential full oxidation state has never so far been reached. +II and +III are most common oxidation states for iron in general compounds. However, when iron coordinates to π-acidic ligands, such as CO, NO, bipy etc., it can reach 0, −I or −II oxidation states. In terms of positive oxidation states, iron has variable ones, +IV, +V and +VI, but they are all unstable high valent species and always act as oxidants. Based on the natural properties of iron, catalysts based on iron have their unique uses. In the last ten years, iron-catalyzed organic reactions have been widely developed and applied.11 Consequently, a vast number of useful reviews have been written on this fascinating chemistry from different points of view.12
As a cheap, readily accessible and environmentally benign metal, iron-catalyzed C–H bond oxidative transformation mainly results in the formation of C–C bonds, C–N bonds, and C–O bonds. These transformations are the subject of this review, which will focus on types of C–H bonds.
C–H bonds with sp3 hybridisation are commonly divided into unreactive and reactive ones. The former are isolated and not adjacent to heteroatoms, and are termed as aliphatic C–H bonds. Ordinarily, this kind of C–H bond undergoes homolytic cleavage in iron-catalyzed oxidation reactions. Activated sp3 C–H bonds are connected to carbon atoms with other types of hybridisation (sp2 or sp) or adjacent to heteroatoms (such as N or O). This class of C–H bond usually undergoes single electron transfer facilitated by iron followed by hydrogen atom abstraction which results in C–H bond homolytic cleavage (Scheme 1).
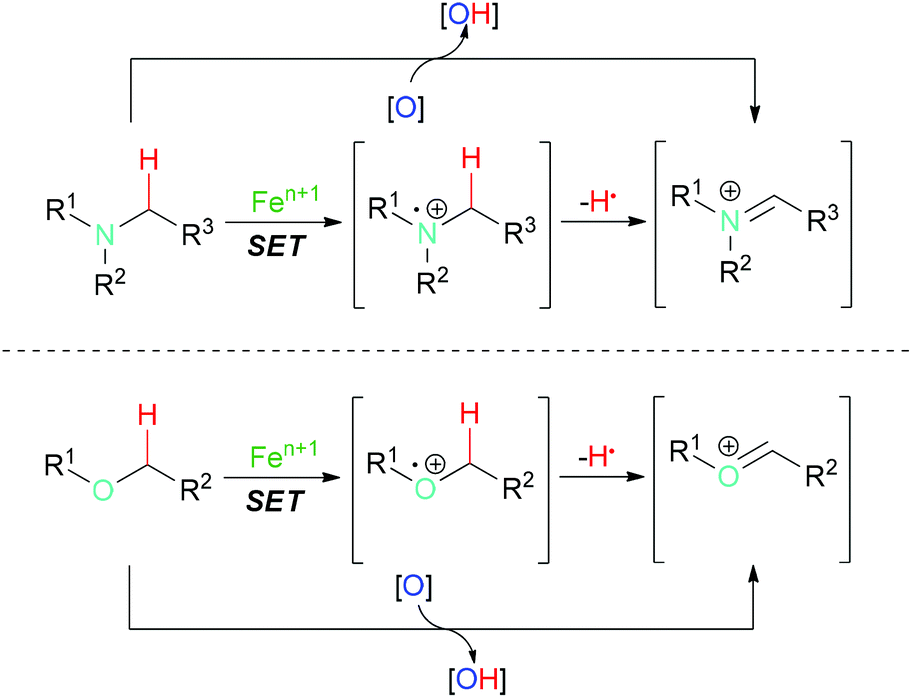 | ||
| Scheme 1 General reaction pathways for reactive sp3 C–H bond homolytic cleavage (adjacent to N or O atom). | ||
C–H bonds with sp2 hybridisation exist in alkenes (belonging to the carbon–carbon double bond), aromatic compounds (carbon located in the aromatic ring) or other organic molecules (such as aldehyde C(O)–H bonds). Such kinds of C–H bond can undergo homolytic cleavage to form a carbon-centered radical or heterolytic cleavage directly to generate a carbon cation (Scheme 2).
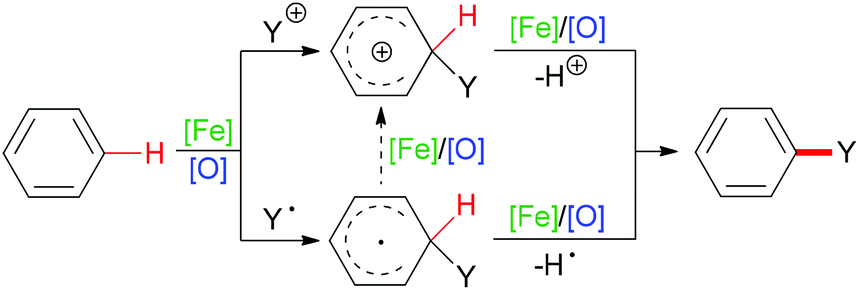 | ||
| Scheme 2 General pathways of sp2 C–H bond cleavage at arenes after addition of cationic group or radical. | ||
2 Oxidative transformation of sp3 C–H bonds
2.1 C–C bond formation
The generation of C–C bonds is one of the most important topics in modern organic synthesis. Many significant contributions for this chemical bond architecture have been made in the last several decades. As a direct way to form C–C bonds, oxidative coupling between sp3 C–H bonds and other types of C–H bonds has become a topical strategy. Cross-dehydrogenative coupling (also called CDC reaction) is a powerful strategy for the construction of C–C bonds (Fig. 2).13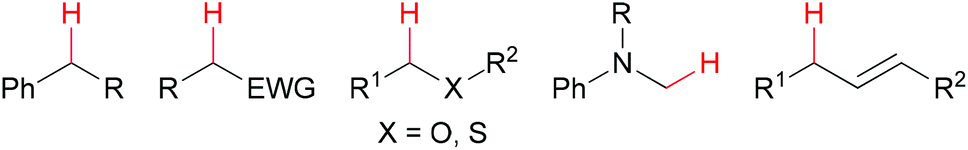 | ||
| Fig. 2 General substrate types for oxidative transformation of sp3 C–H bonds. | ||
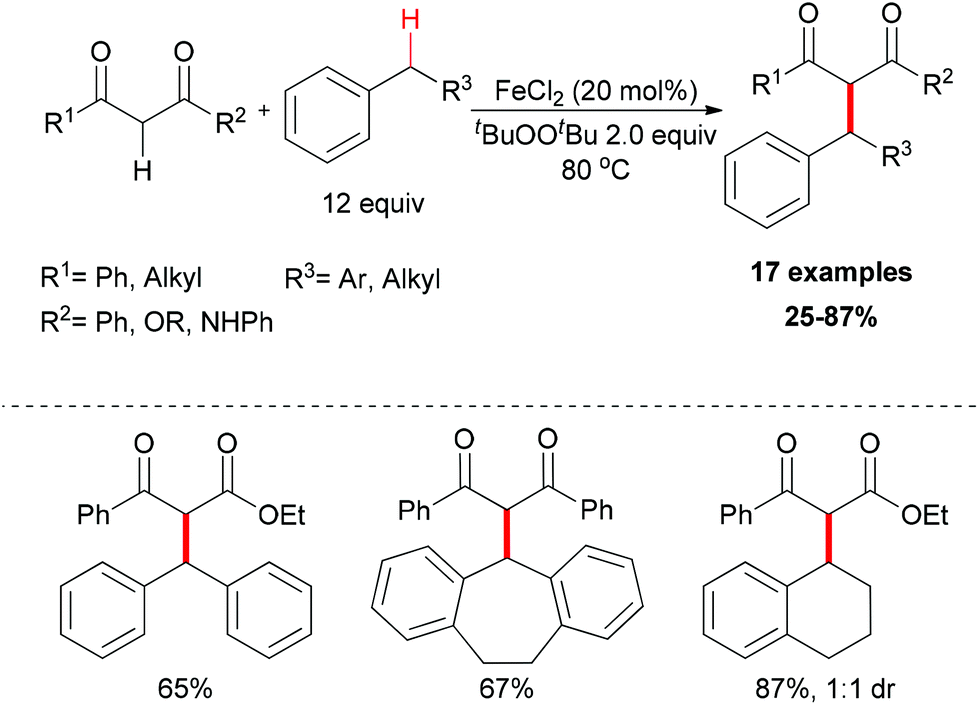 | ||
| Scheme 3 FeCl2-catalyzed benzylic alkylation. | ||
The reaction mechanism can be rationalized by initial homolytic cleavage of the peroxide bond (tBuO–OtBu) of the stoichiometric oxidant di-tert-butyl peroxide by FeCl2 to generate tert-butoxyl radical, which abstracts a hydrogen atom from the benzyl substrate to form a benzylic radical, while the iron(III) compound could react with the dicarbonyl compound leading to a chelate Fe–enolate complex. The benzylic radical could then react with the enolate to form the final product and regenerate the Fe(II) which undergoes the next cycle (Scheme 4). The reaction was also found to proceed efficiently at room temperature, and the coupling product was isolated in 80% yield on extending the reaction time and using diphenylmethane and benzoylacetone as substrates. In the same year, Li and Zhang reported an Fe-catalyzed alkylation of activated methylenes using simple cycloalkanes as substrate in the presence of di-tert-butyl peroxide.16 In contrast to classical Fenton-type initiation,17 the active catalyst can also be the chelate Fe–enolate complex formed at the start from FeCl2 and the dicarbonyl compounds.
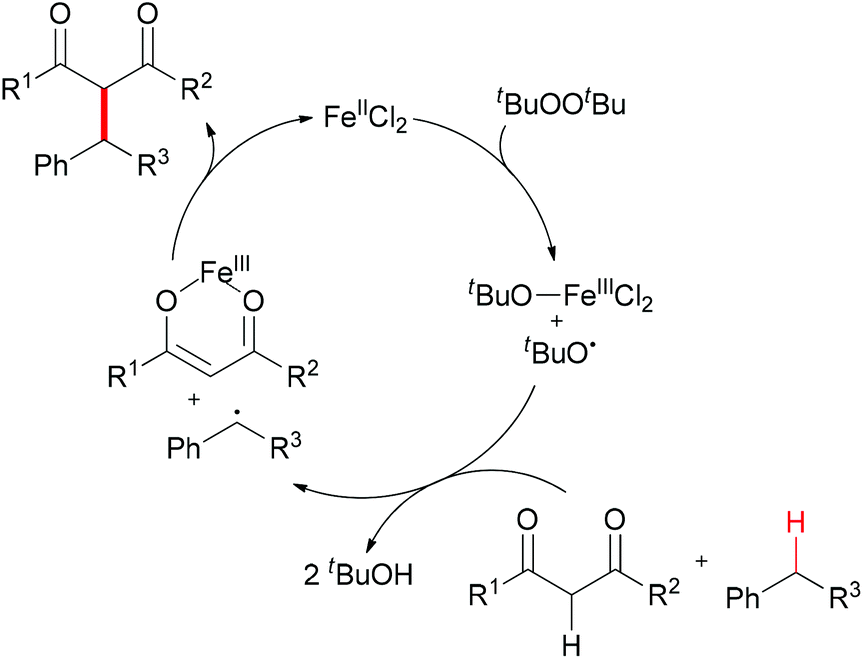 | ||
| Scheme 4 Proposed mechanism for FeCl2-catalyzed benzylic alkylation. | ||
In Li's subsequent studies, simple toluene derivatives were further tested in the oxidative coupling reactions along with 1,3-dicarbonyl compounds.18 Optimisation of the conditions indicated that Fe(OAc)2 was the best catalyst and tert-butyl peroxide was the most efficient oxidant. Under the conditions developed, various toluene derivatives could couple with 1,3-dicarbonyl compounds in moderate to good yields. The authors also performed mechanistic studies and the results suggested that benzylic radical addition to the benzoyl-methana-iron species could occur in the coupling reaction.
The Shi group reported the first example of direct oxidative arylation reactions of benzyl compounds.19 They used a variety of electron-rich aromatic substrates as coupling partners. After catalyst screening, FeCl2 was identified as the best choice. In combination with DDQ as oxidant, the desired oxidative coupling products were smoothly generated (Scheme 5).
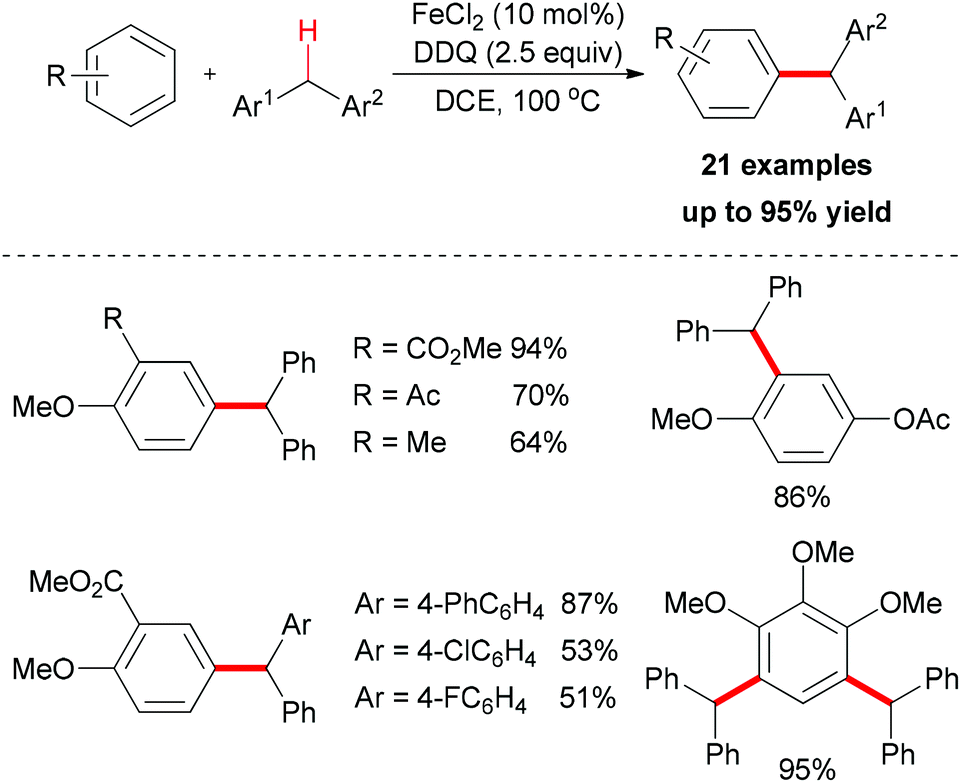 | ||
| Scheme 5 Iron-catalyzed direct oxidative arylation of the benzylic C–H bond. | ||
The authors also conducted mechanistic studies. Based on their experimental results, a possible mechanism was proposed (Scheme 6). The reaction is initiated by a single-electron-transfer process (SET) assisted by the iron salt to generate the benzyl radical, which could be further oxidized to the benzyl cation. The subsequent Friedel–Crafts-type alkylation, followed by abstraction of the proton by the reduced hydroquinone, would release the coupling product and regenerate the catalyst to fulfil the catalytic cycle.
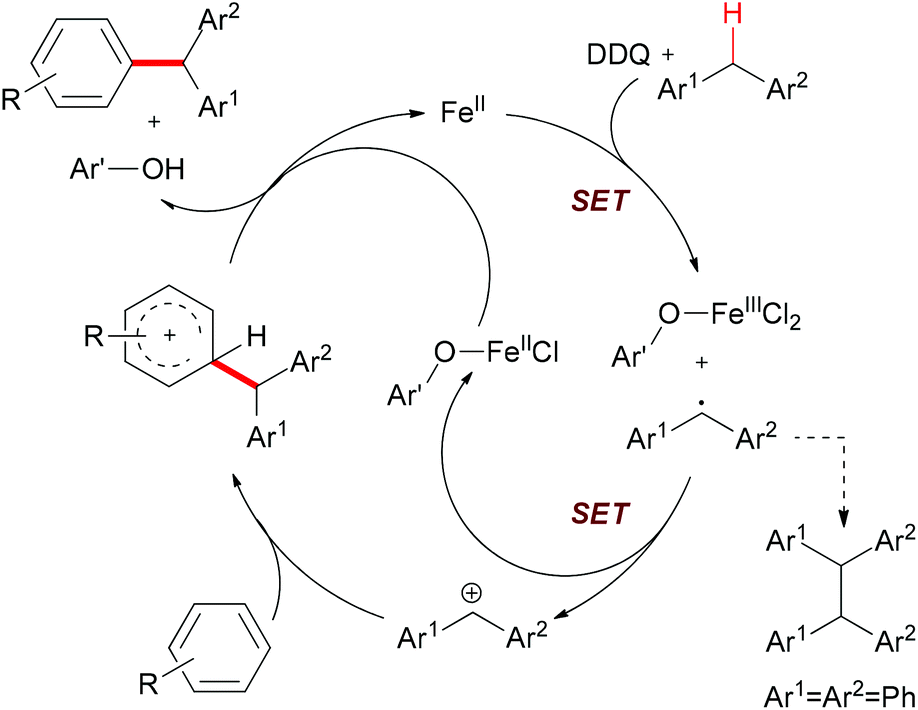 | ||
| Scheme 6 Proposed mechanism of Fe-catalyzed arylation of benzylic C–H bonds. Ar′OH stands for the reduced DDQ. | ||
In the same year, the Shi group and the Gan group expanded their research into benzylic alkylation reactions. They used vinyl acetate as coupling partner to construct sp3 C–C bonds with benzyl compounds.20 After reaction optimization, FeCl2 was identified as the best catalyst and di-tert-butyl peroxide (DTBP) was the most efficient oxidant. Under the best conditions, a series of benzyl compounds could react with vinyl acetate to give coupling products (19 examples, Scheme 7). Mechanistically, the authors proposed a radical pathway and a cationic pathway. However, the experimental results supported the radical mechanism. They also performed an intermolecular isotopic competitive study, and the result (KH/D = 2.4) indicated that a proton abstraction process may be involved in the rate determining step.
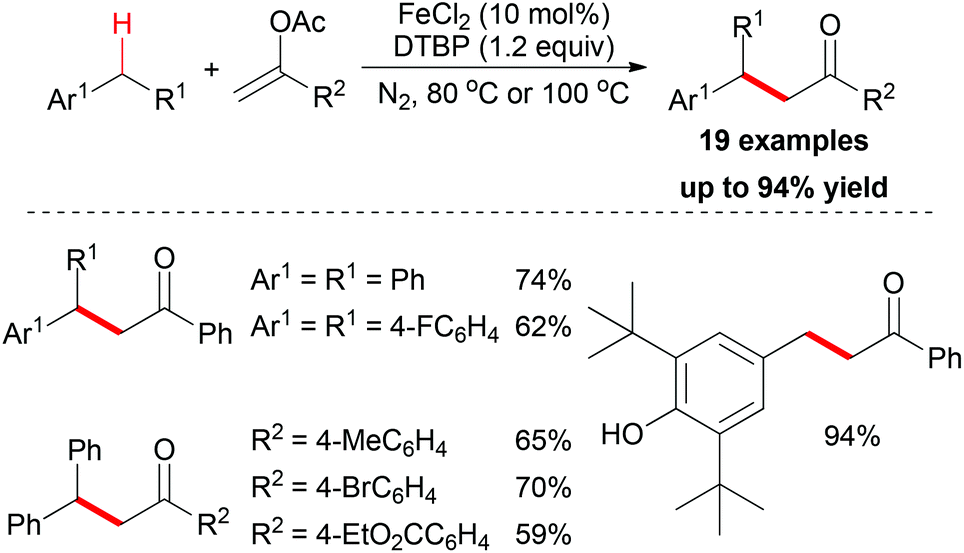 | ||
| Scheme 7 Benzylic alkylation with vinyl acetates via iron catalysis. | ||
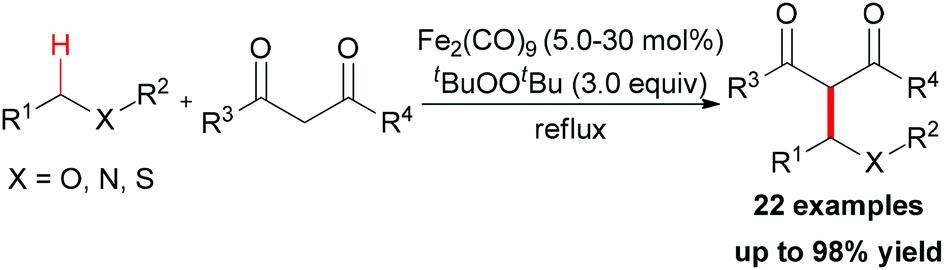 | ||
| Scheme 8 Iron-catalyzed oxidative coupling reactions of 1,3-dicarbonyl compounds. | ||
In Li's subsequent research, they reported an unprecedented dialkylation of the methylene group.22 By using N,N-dimethylaniline as the methylene source and 2 equiv. of 1,3-dicarbonyl compounds, methylene-bridged bis-1,3-dicarbonyl compounds were smoothly constructed in high efficiency. Optimization of the reaction conditions showed that Fe2(CO)9/TBHP was the best catalyst/oxidant combination (Scheme 9).
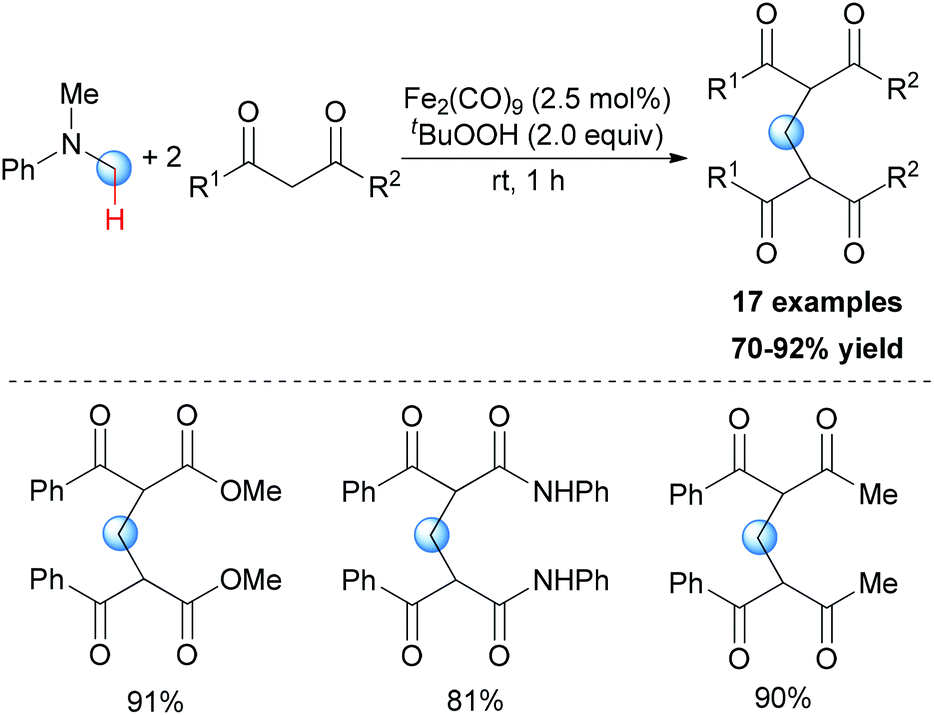 | ||
| Scheme 9 Iron-catalyzed selective oxidation of N-methyl amines. | ||
In Li's proposal, this oxidation reaction likely involves two different pathways (Scheme 10). Based on the previous work, the oxidative coupling intermediate A was formed in the Fe2(CO)9/TBHP system. A can undergo direct SN2 reaction when attacked by the second molecular 1,3-dicarbonyl compound. Also, A may undergo Cope elimination to give the intermediate B followed by Michael addition to generate the final product. Notably, the authors found the formation of formaldehyde in the present transformation by performing the Nash test. This result gave the other potential mechanism which includes reaction between formaldehyde generated in situ and the 1,3-dicarbonyl substrate. However, they could not fully exclude this pathway at that point.
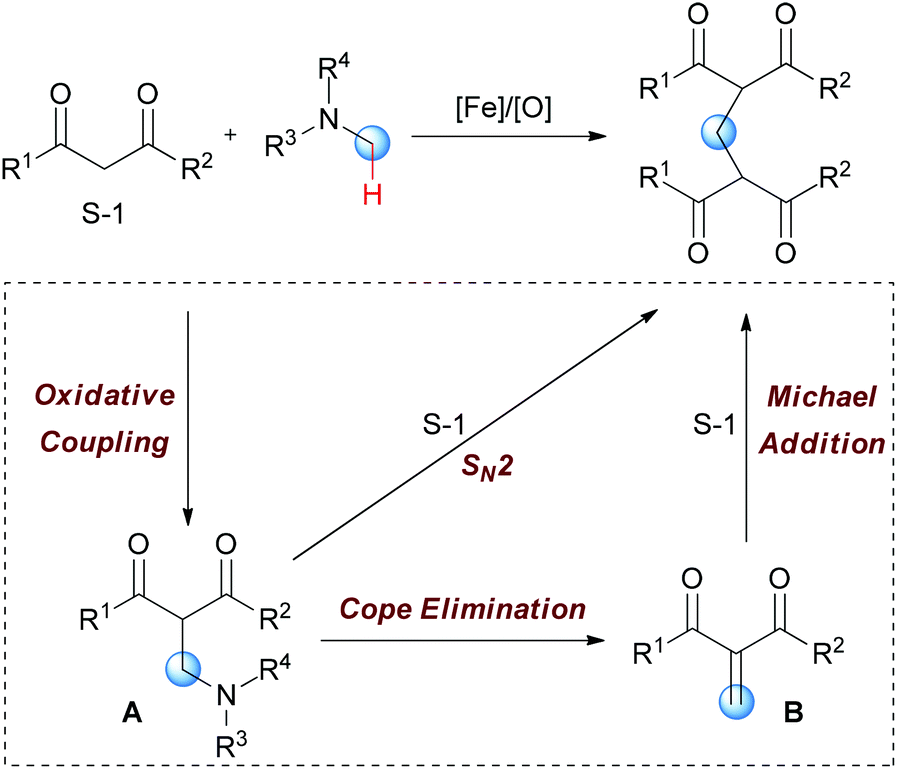 | ||
| Scheme 10 Proposed pathways for iron-catalyzed selective oxidation of N-methyl amines. | ||
Apart from using N,N-dimethylaniline as methylene source, the sp3 C–H bond adjacent to the O-atom of ethers can also undergo oxidative coupling to link indoles. In 2009, Li and co-workers disclosed a one-pot synthetic protocol for constructing symmetric and nonsymmetric 1,1-bis-indolylmethanes via tandem iron-catalyzed C–H bond oxidation and C–O bond cleavage (Scheme 11).23
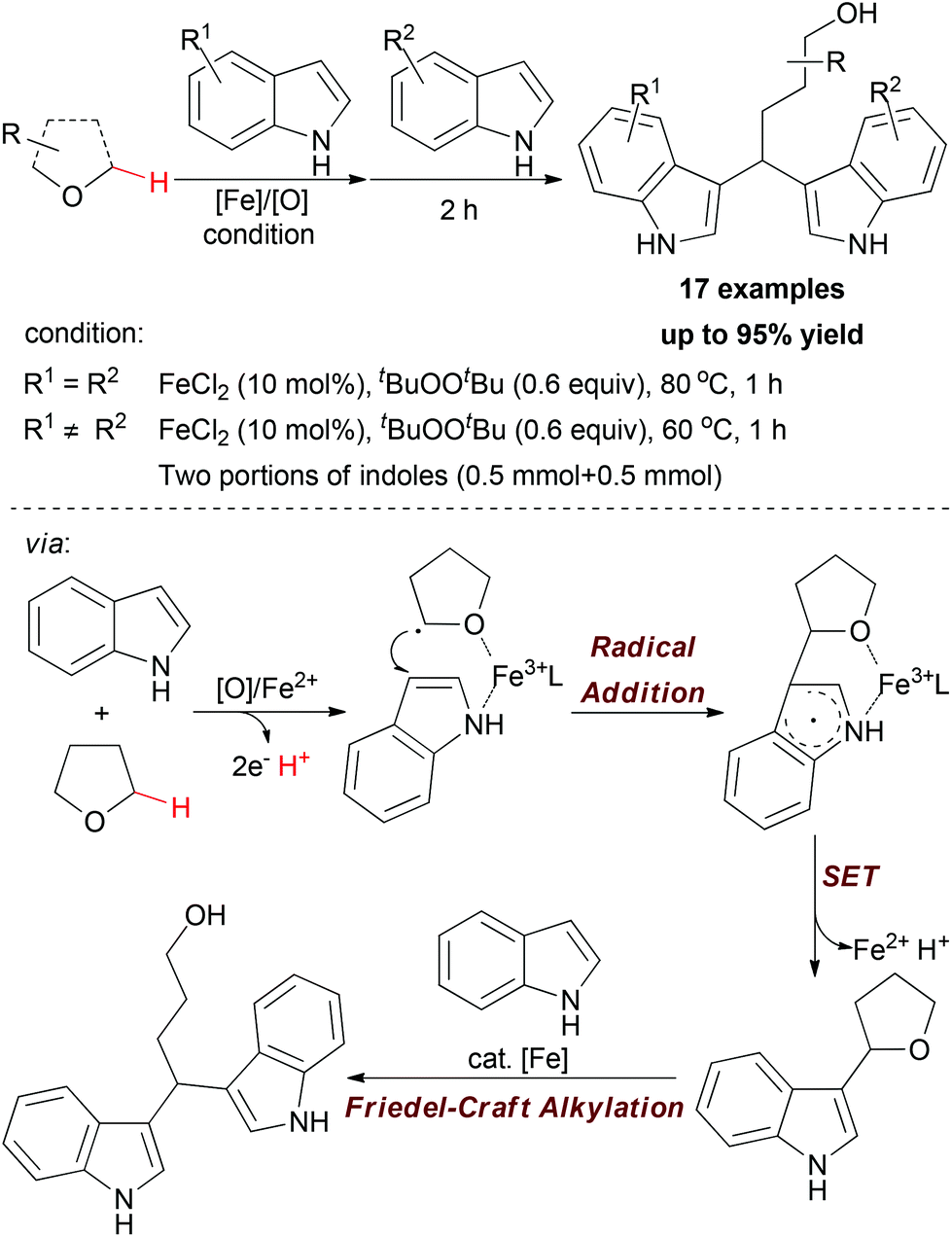 | ||
| Scheme 11 Iron-catalyzed one-pot synthesis of symmetric and nonsymmetric 1,1-bis-indolylmethanes via tandem C–H bond oxidation and C–O bond cleavage. | ||
Amines are known to undergo oxidation to form the iminium ion intermediate, and this process could be utilized in the acylation of indoles. In 2011, Su and co-workers reported this kind of methodology.24 In their research, anilines were used as the carbonyl source to install a benzoyl group onto the 3-position of N–H free indoles through iron-catalyzed oxidative coupling. Under the optimized conditions, several N–H free indoles could be acylated on the 3-position in moderate to good yields (Scheme 12) while the N-alkyl anilines failed in this reaction.
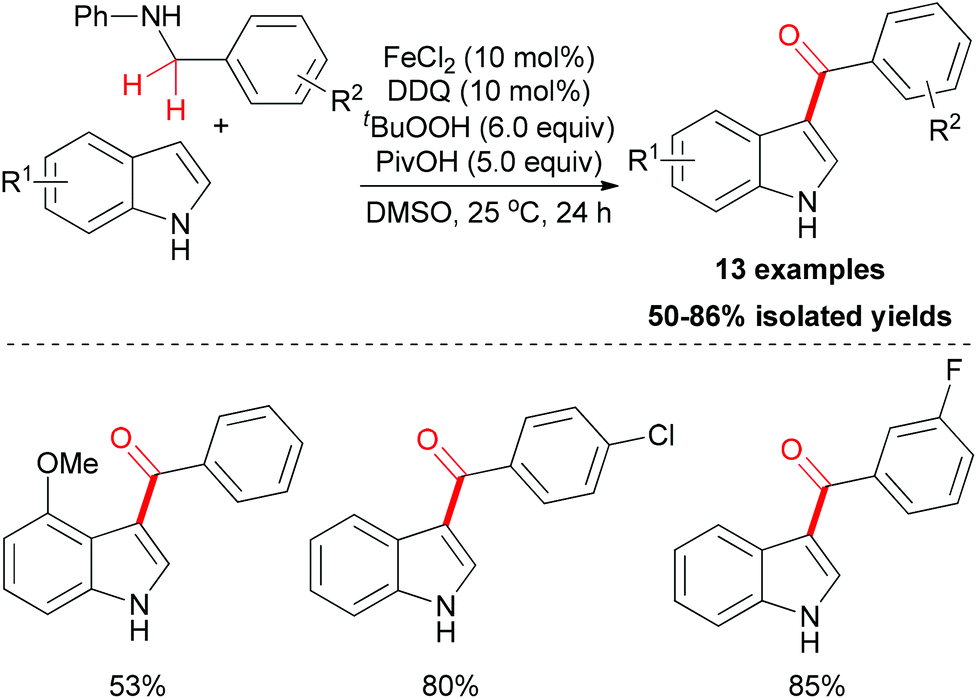 | ||
| Scheme 12 FeCl2-catalyzed acylation of N–H free indoles. | ||
Since 2009, several efficient methods have been developed for the synthesis of oxindoles through oxidative coupling.25 Recently, Li and co-workers disclosed a novel FeCl3-catalyzed oxidative 1,2-alkylarylation of acrylanilides initiated by sp3 C–H bond homolytic cleavage in the presence of TBHP as oxidant (Scheme 13).25a Using this methodology, a series of 3-alkylated oxindoles was constructed. The authors also performed control experiments and UV/Vis titration to explain the role of DBU as a ligand, not a base, but they could not rule out the alkalinity of DBU promoting the reaction. Through the designed control experiments and radical trapping reactions, a radical mechanism was proposed (Scheme 14).
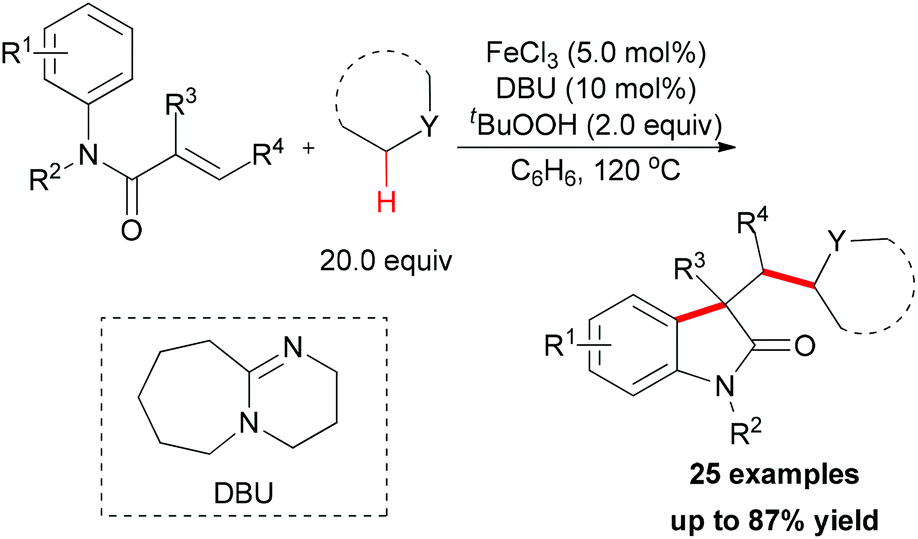 | ||
| Scheme 13 Iron-catalyzed oxidative 1,2-alkylarylation of activated alkenes. | ||
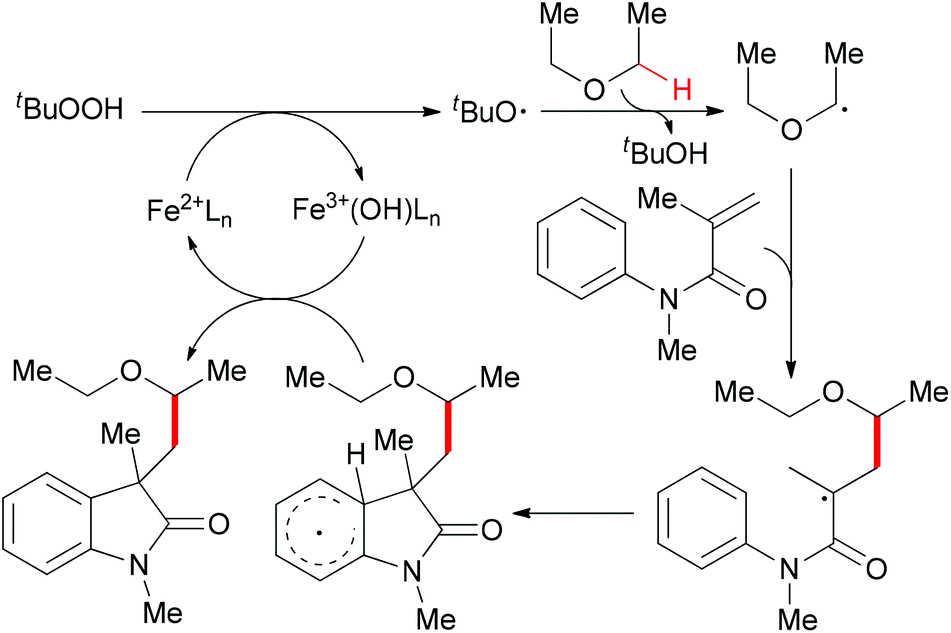 | ||
| Scheme 14 Radical cyclization pathway for oxindole synthesis. | ||
Very recently, You and co-workers reported an efficient route to furnish α-quaternary α-amino acid derivatives.26 In this work, they used a catalytic amount of FeCl3·6H2O and di-tert-butyl peroxide as oxidant. The oxidative sp3 C–H bond coupling exhibited a broad substrate scope for both α-amino acids and nucleophiles as well as good functional group tolerance (Scheme 15). When treated with 2,2,6,6-tetramethylpiperidine oxide (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT), the coupling reaction of ethyl 3-phenyl-2-(picolinamido)propanoate with 1H-indole could be suppressed. On the basis of this experimental result, the authors proposed a possible mechanism involving a single-electron transfer process (Scheme 16).
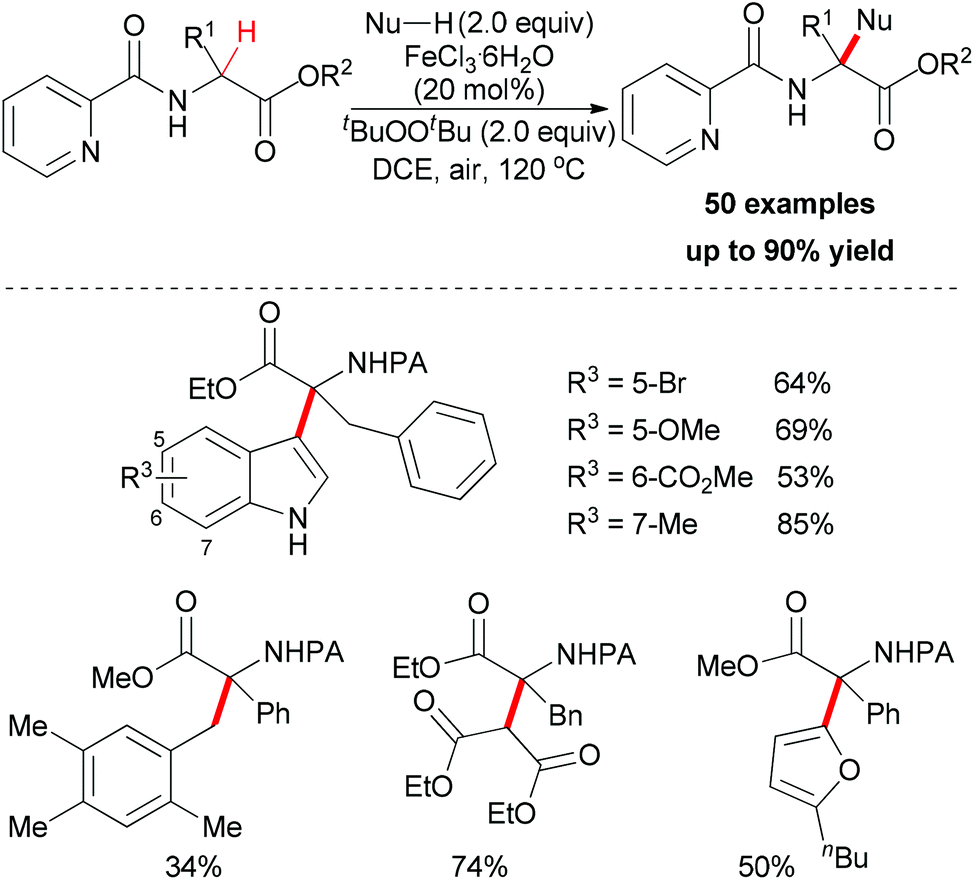 | ||
| Scheme 15 Fe(III)-catalyzed oxidative functionalization of α-sp3 C–H bonds of α-tertiary α-amino acid esters. | ||
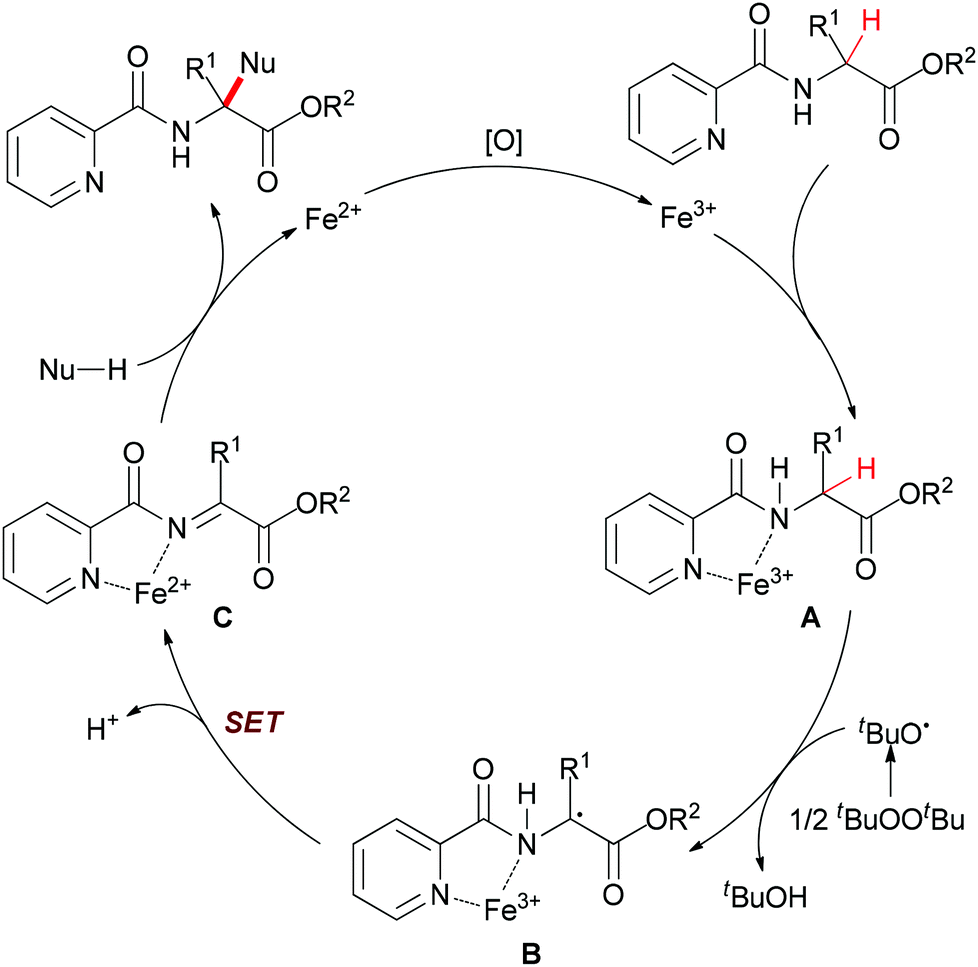 | ||
| Scheme 16 Possible mechanism of the α-sp3 C–H bond functionalization of α-substituted α-amino acid esters. | ||
In their proposal (Scheme 16), 2-picolinamido α-tertiary amino acid ester coordinates with FeIII to yield the intermediate A. Then the tert-butoxyl radical (tBuO˙) generated from di-tert-butyl peroxide (DTBP) abstracts the α-hydrogen atom of A to form the radical intermediate B. Next, the radical species B undergoes an intramolecular single-electron transfer (SET) process to give the α-ketimine intermediate C. Subsequently, the coordination of the picolinamido group with FeIII species activates the α-ketimine and facilitates the addition of the nucleophile to intermediate C which results in releasing the desired α-quaternary α-amino acid ester product. The authors also performed the model reaction under N2 atmosphere and the desired product was generated in low yield (39% yield compared with 90% yield under the standard condition). This result indicated that the FeII released is reoxidized to FeIII by air27 as well as DTBP28 to fulfil the catalytic cycle.
2.2 C–N bond formation
Oxidative C–H bond transformations to construct C–N bonds are important processes in the synthesis of nitrogen-containing compounds. In 1982, Breslow and Gellman reported the amidation of cyclohexane using PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs as a stoichiometric oxidant.29 After their seminal report, many developed alternative nitrogen sources showed their utilities, such as chloramines-T,30 bromamines-T,31 and tosyloxycarbamates.32 However the direct installation of nitrogen in oxidative C–H bond transformation process still remains challenge.
NTs as a stoichiometric oxidant.29 After their seminal report, many developed alternative nitrogen sources showed their utilities, such as chloramines-T,30 bromamines-T,31 and tosyloxycarbamates.32 However the direct installation of nitrogen in oxidative C–H bond transformation process still remains challenge.
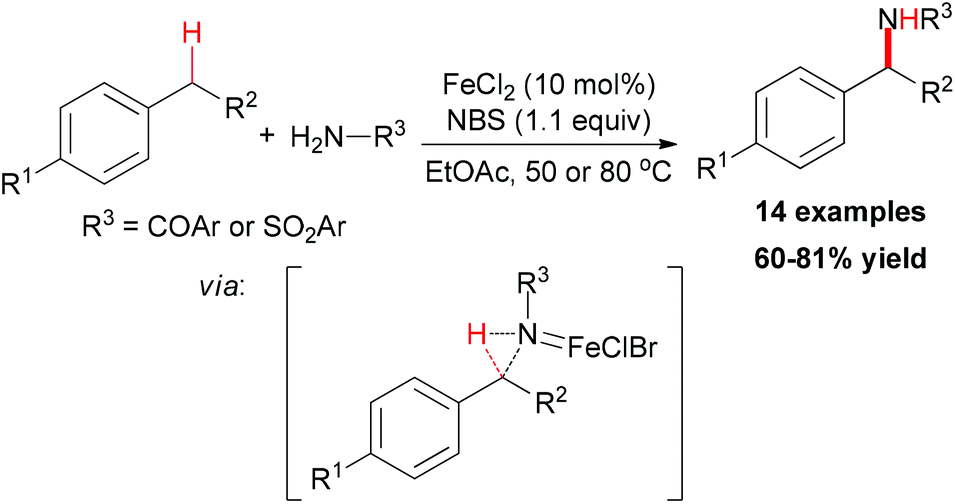 | ||
| Scheme 17 Iron-catalyzed benzylic sp3 C–H bond amidation. | ||
In 2011, Chen and Qiu reported a direct C–N coupling method between imidazoles and benzylic compounds through iron-catalyzed oxidative transformation of sp3 C–H bonds (Scheme 18, top).34 The reaction utilized the inexpensive FeCl2/DTBP combination which is suitable for the oxidative coupling of a series of benzylic sp3 C–H bonds with imidazoles. In Chen's subsequent research, they extended their strategy into N-alkylation of azoles via oxidative cleavage of a sp3 C–H bond adjacent to the N-atom in amides and sulphonamides (Scheme 18, bottom).35 Under the optimized conditions, a wide range of amides and sulphonamides could be used as substrates for N-alkylation of azoles.
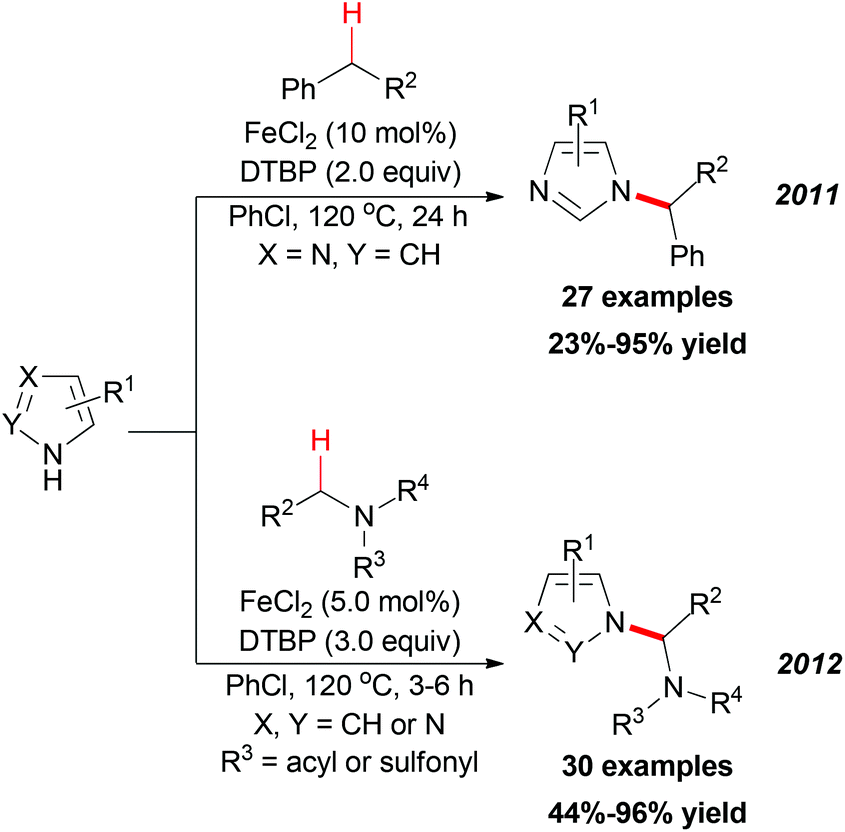 | ||
| Scheme 18 Iron-catalyzed sp3 C–H bond oxidation and C–N bond formation. | ||
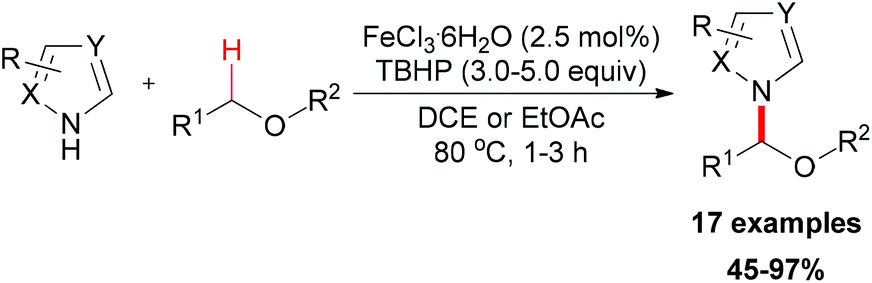 | ||
| Scheme 19 Iron-catalyzed N-alkylation of azoles. | ||
The proposed mechanism (Scheme 20) showed that TBHP decomposed into a tert-butoxyl radical and a hydroxyl anion in the presence of the ferrous catalyst (step a). Deprotonation of the azole gave the anion species A (step b). On the other hand, hydrogen abstraction of the C–H bond adjacent to an oxygen atom afforded B, which could be trapped by TEMPO, and followed by ferric oxidation to generate oxonium ion C (step c). Finally, the nucleophilic addition of A to C provided the desired coupling product (step d). Overall, the Fe2+–Fe3+ redox process played a key role in the present C–N bond formation reaction, in the reductive heterolytic cleavage of the O–O bond in the peroxide (step a) and the oxidation of the carbon-centered radical to oxonium (step c).
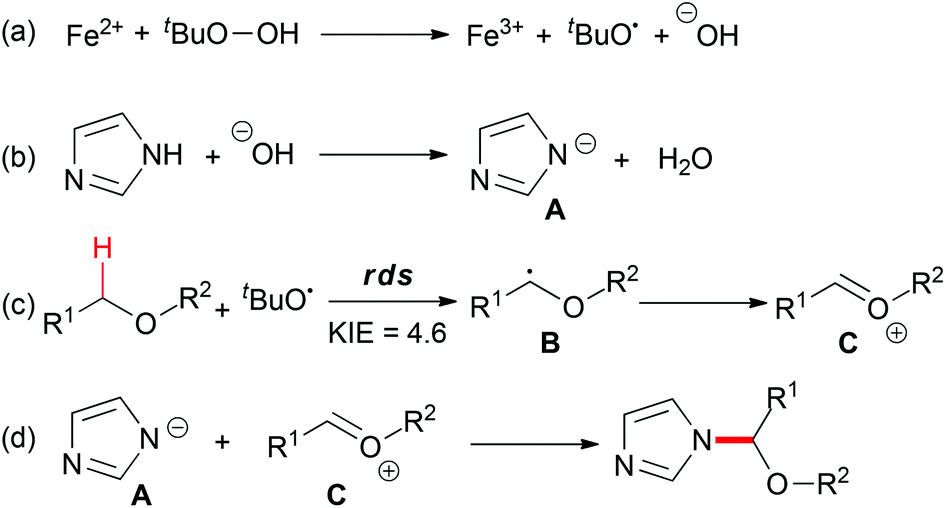 | ||
| Scheme 20 Plausible pathways for the iron-catalyzed N-alkylation of azoles. | ||
2.3 C–O bond formation
The direct oxygenation of sp3 C–H bonds presents a powerful approach to alcohol products,1 which is one of the most important organic synthetic intermediates.As a chemical model for cytochrome P450 mono-oxygenase,39 porphyrin-based iron catalysts show their powerful utilities in aliphatic C–H bond oxidations (Fig. 3). This oxidation chemistry has been broadly reviewed.38,40 In contrast, iron-based non-heme complexes also have been developed for alkane hydroxylations, but the mechanism of such reactions remains the subject of debate.
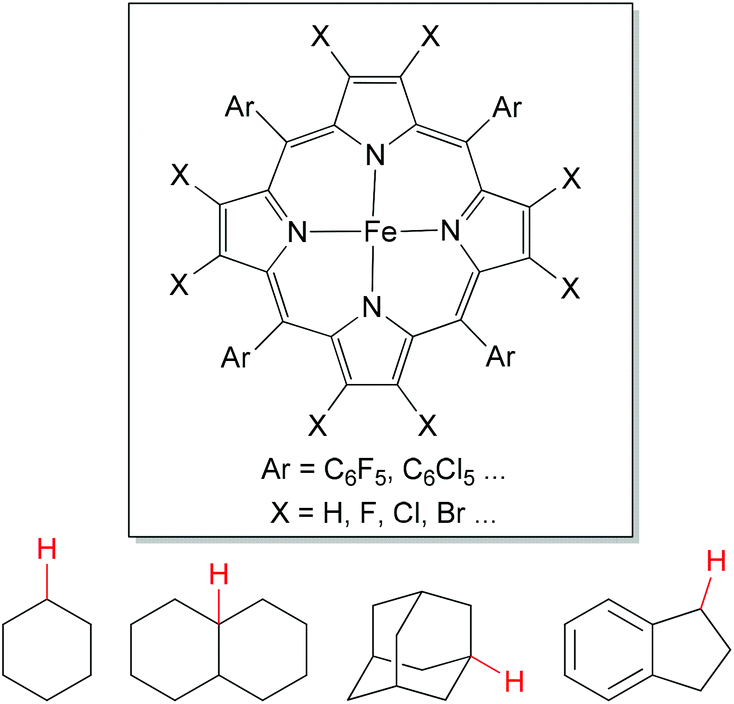 | ||
| Fig. 3 General structures for porphyrin-based iron catalyst and benchmark substrates for direct oxygenation of sp3 C–H bonds. | ||
In 1983, Barton introduced a particular type of aliphatic oxidation reaction using an iron catalyst (termed as Gif chemistry). After that, large numbers of studies focused on elucidating mechanistic and kinetic details rather than on using the concept for synthetic applications.41 Moreover, Gif chemistry also provoked a long-term mechanistic controversy.
In the synthetic field, the White group made a series of contributions on aliphatic C–H bond oxidations.42
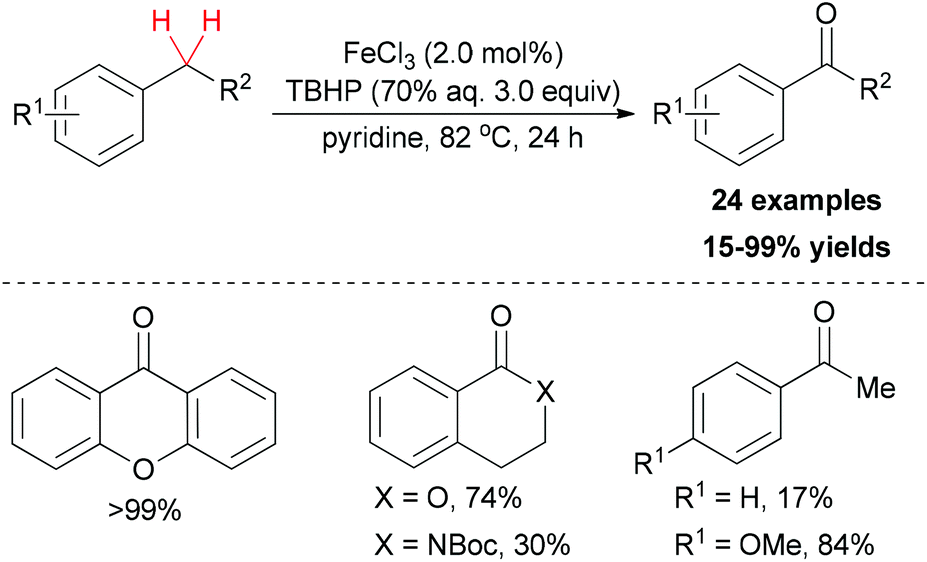 | ||
| Scheme 21 Iron-catalyzed benzylic oxidation with aqueous tert-butyl hydroperoxide. | ||
In addition to using hydroperoxide as oxidant in sp3 C–H bond oxidation/oxygenation, dioxygen could also act as terminal oxidant in such reactions. In 2012, Maes and co-workers developed a sustainable oxidation method for the synthesis of aryl(di)azinyl ketones.44 After optimization of the conditions, FeCl2·4H2O showed the best catalytic properties among other simple iron salts. Under the best conditions, a series of substrates bearing both electron-donating and electron-withdrawing groups could be oxidized to the corresponding ketones in moderate to good yields (Scheme 22).
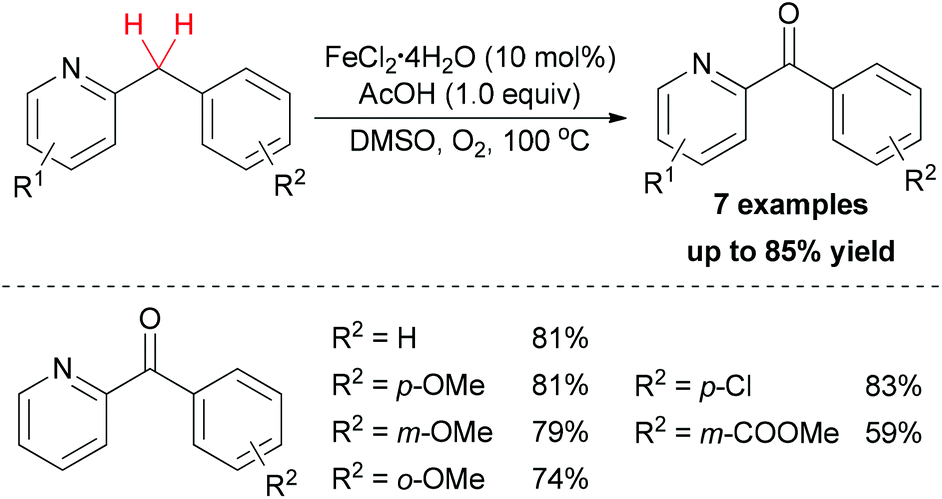 | ||
| Scheme 22 Oxidation of the methylene group of aryl(di)azinyl-methanes. | ||
In contrast to using environmentally unfriendly organic solvents, water used as reaction medium has become an attractive topic in green chemistry.45 Recently, Novák and co-workers reported a benzylic oxidation in water.46 They used a solution of sodium dodecylsulphate (SDS) to build up the iron–surfactant nanocomposite catalytic system. The simple iron(III) salts (Fe2(SO4)3 and FeCl3) were used as catalyst source and aqueous TBHP (70 wt% aq.) as a cheap oxidant. Under the optimized reaction conditions, a series of benzylic compounds were smoothly transformed to ketones (30 examples, 21–99% yield).
In 2012, the Jiao group reported an iron-facilitated oxidative dehydrogenative C–O bond formation reaction.47 They used aryl propargyl azides as substrates which were generated in situ followed by the oxidative coupling with carboxylic acids (Scheme 23). Iron salts were investigated for their activities and FeCl2 was the most effective catalyst. The oxidative coupling reaction was performed smoothly using DDQ as oxidant in DCE solvent (28 examples, up to 83% yield). Attempting to use other oxidants and solvents were not successful. This methodology also showed its useful synthetic applications, as the reaction products are useful synthons for a wide range of synthetic targets such as 4,5-disubstituted-1,2,3-triazoles, 3-alkoxyenals, and benzotriazoles.
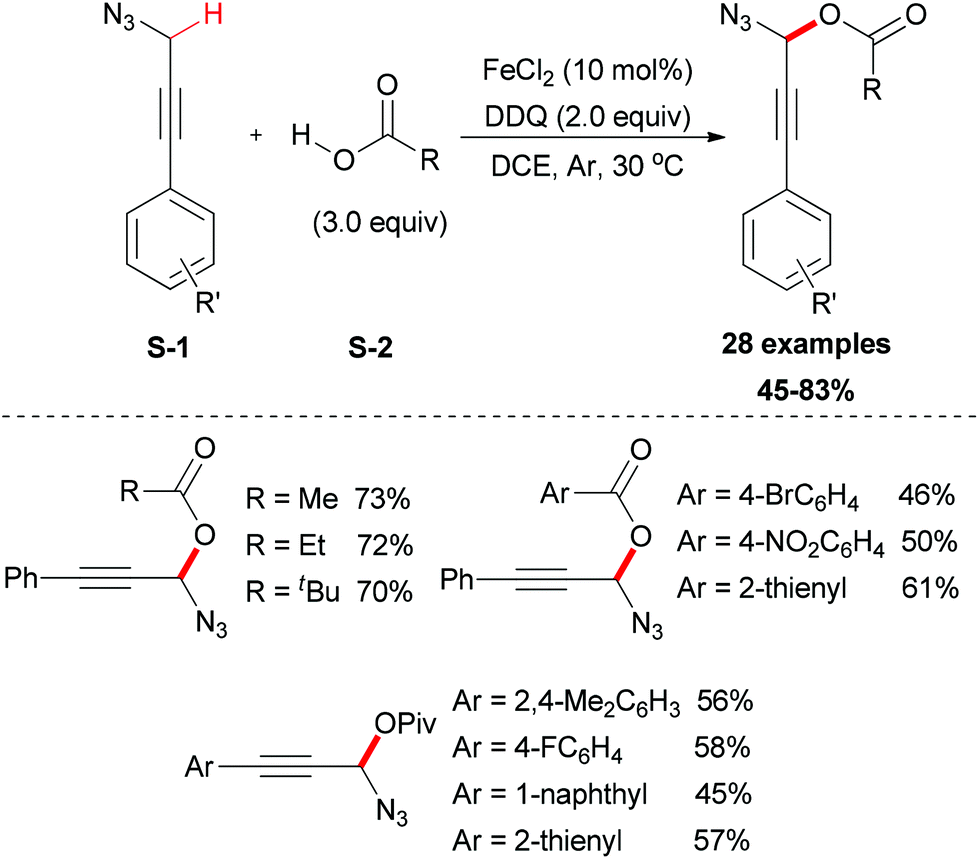 | ||
| Scheme 23 Iron-facilitated oxidative dehydrogenative C–O bond formation. | ||
The authors proposed a tentative mechanism shown in Scheme 24. Initially, substrate S-1 undergoes hydrogen abstraction through iron-facilitated single-electron transfer (SET) with DDQ to form the radical species A, which may be stabilized by the azido group. Then the radical species A was further oxidized to give the aryl propargyl cation B. Next, nucleophilic attack of cation B by carboxylic acid (S-2) gave the desired product with regeneration of the catalyst.
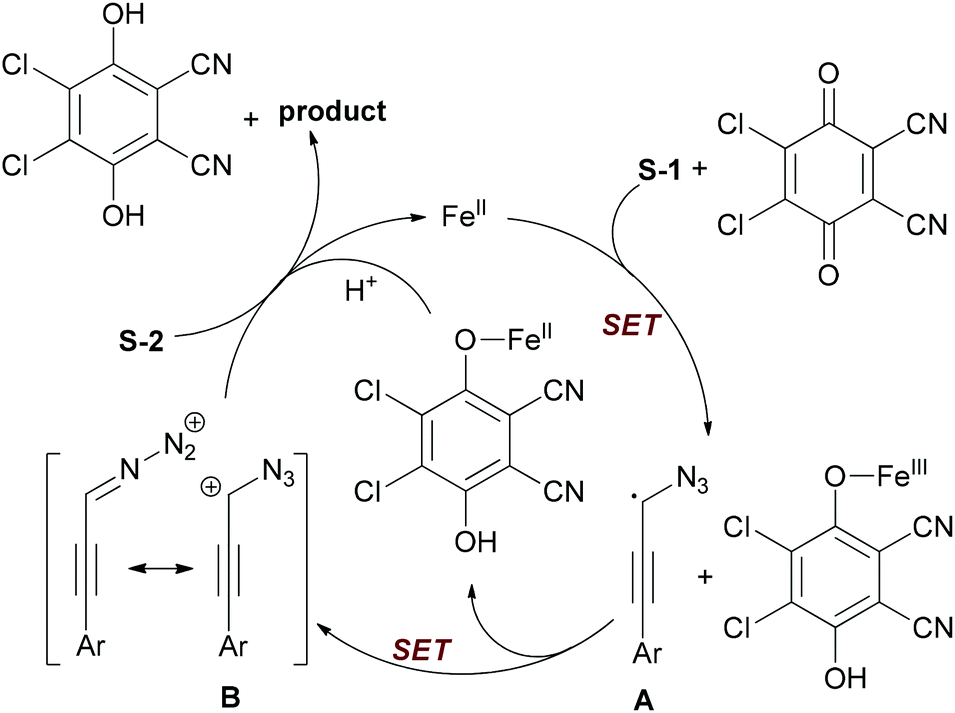 | ||
| Scheme 24 Proposed mechanism of iron-catalyzed oxidative dehydrogenative C–O bond formation. | ||
Instead of C–C and C–N bond formation through oxidative transformation of a C–H bond adjacent to heteroatom, in 2011 Liang and co-workers reported a C–O bond formation by iron(II)-catalyzed oxidation of sp3 C–H bonds adjacent to a nitrogen atom of unprotected arylureas with tert-butyl hydroperoxide (TBHP) in water (Scheme 25).48 The authors separated and identified an N-tert-butylperoxylated intermediate B under the standard reaction conditions, which indicated that the peroxyl radical could react with the aminyl radical derived from oxidation of the arylurea. The elimination of the tert-butylperoxy radical generated a new carbon-centered radical C via 1,4-hydrogen atom transfer (1,4-HAT),49 which was trapped by tert-butanol to give α-tert-butoxylated urea D and regenerated TBHP. Finally, a hydroxyl group was introduced to the other α-position of D via hydrogen abstraction by TBHP and the final product was released (Scheme 26).
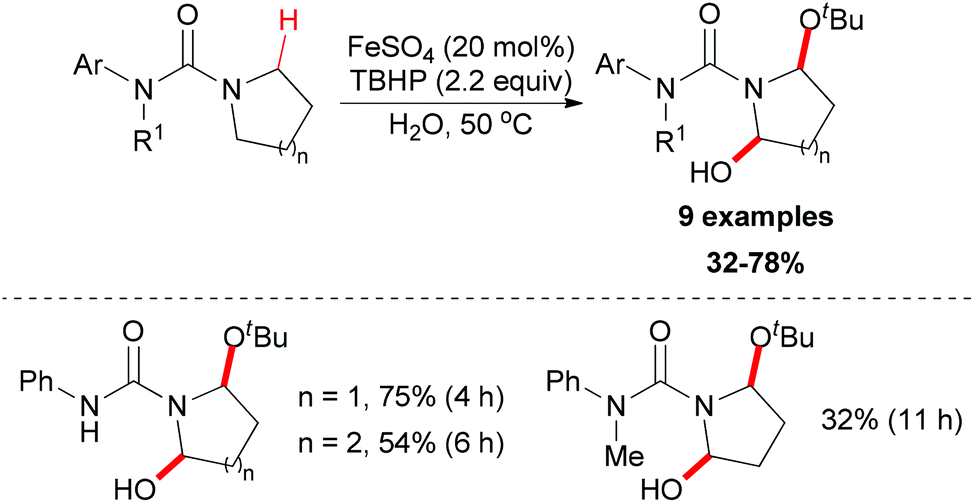 | ||
| Scheme 25 Iron-catalyzed oxidation of sp3 C–H bonds adjacent to a nitrogen atom of unprotected arylureas. | ||
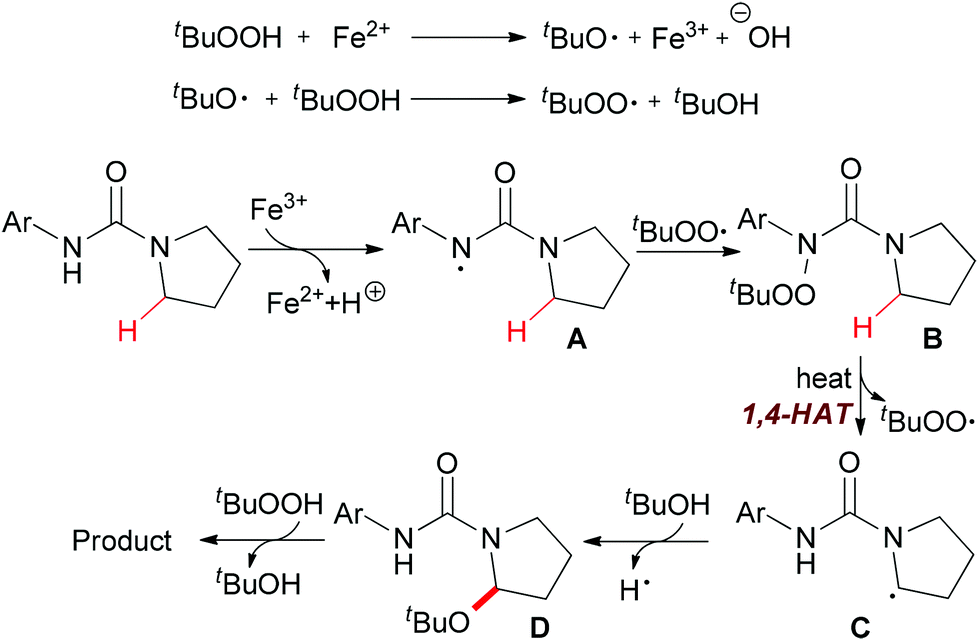 | ||
| Scheme 26 Proposed mechanism for iron-catalyzed oxidation of sp3 C–H bonds adjacent to a nitrogen atom. | ||
In 2012, Urabe and co-workers reported an iron-promoted C–H bond oxygenation reaction for the synthesis of tert-butyl peroxyacetals. Under the optimized conditions, a series of tert-butyl peroxyacetals was synthesized in good to excellent yields (Scheme 27).50 Mechanistically, the authors proposed two possible pathways to explain the formation of tert-butyl peroxyacetals (Scheme 28). The initiation step generating a tert-butyl peroxy radical may start efficiently in the presence of Fe catalyst. Then the peroxy radical abstracts a hydrogen atom on the ether substrate to form the benzyl radical. This benzyl radical could go through two pathways to form the final product.
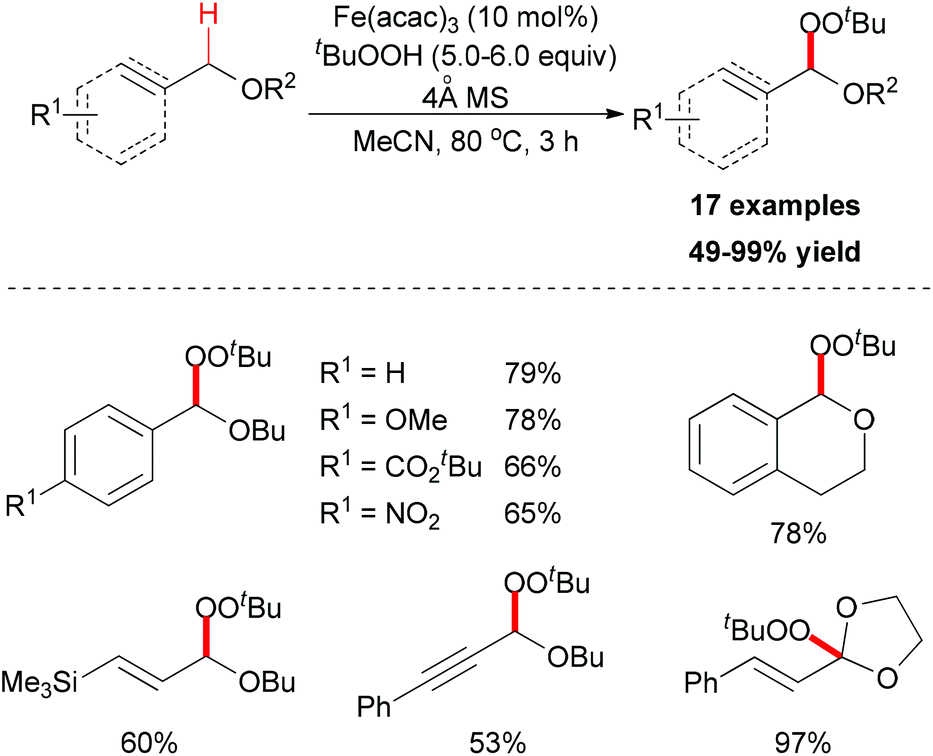 | ||
| Scheme 27 Synthesis of tert-butyl peroxyacetals via iron-catalyzed sp3 C–H bond oxidative functionalization. | ||
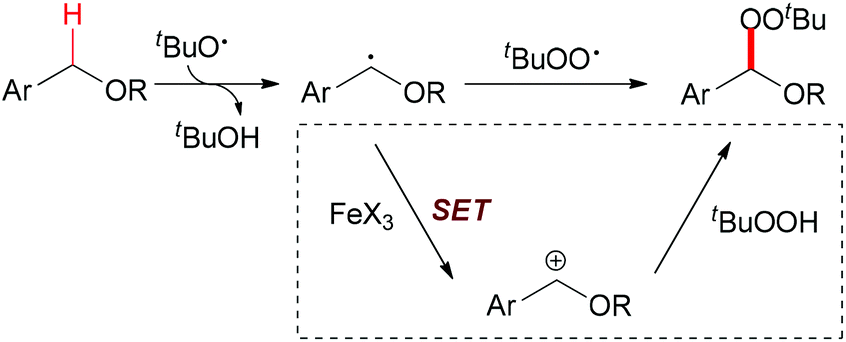 | ||
| Scheme 28 Nucleophilic attack and radical coupling pathways. | ||
3 Oxidative transformations of sp2 C–H bonds
3.1 C–C bond formation
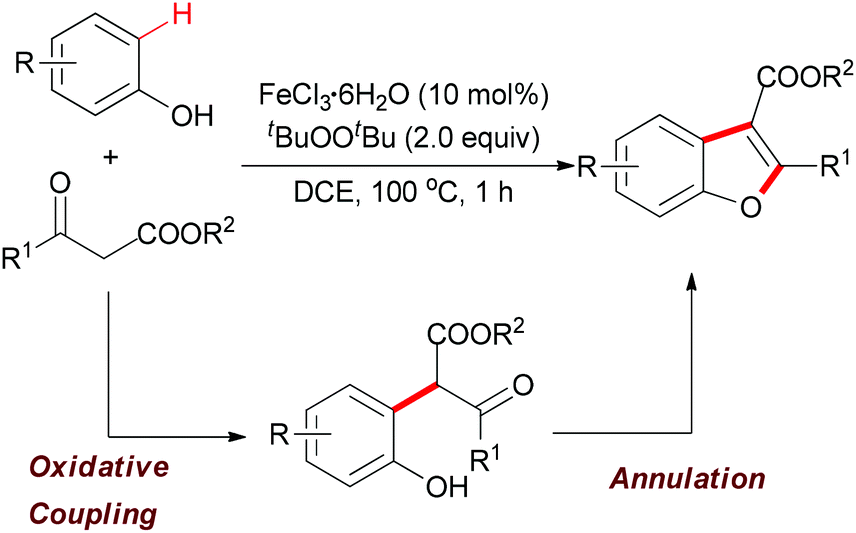 | ||
| Scheme 29 Iron-catalyzed tandem oxidative coupling and annulation to construct polysubstituted benzofurans. | ||
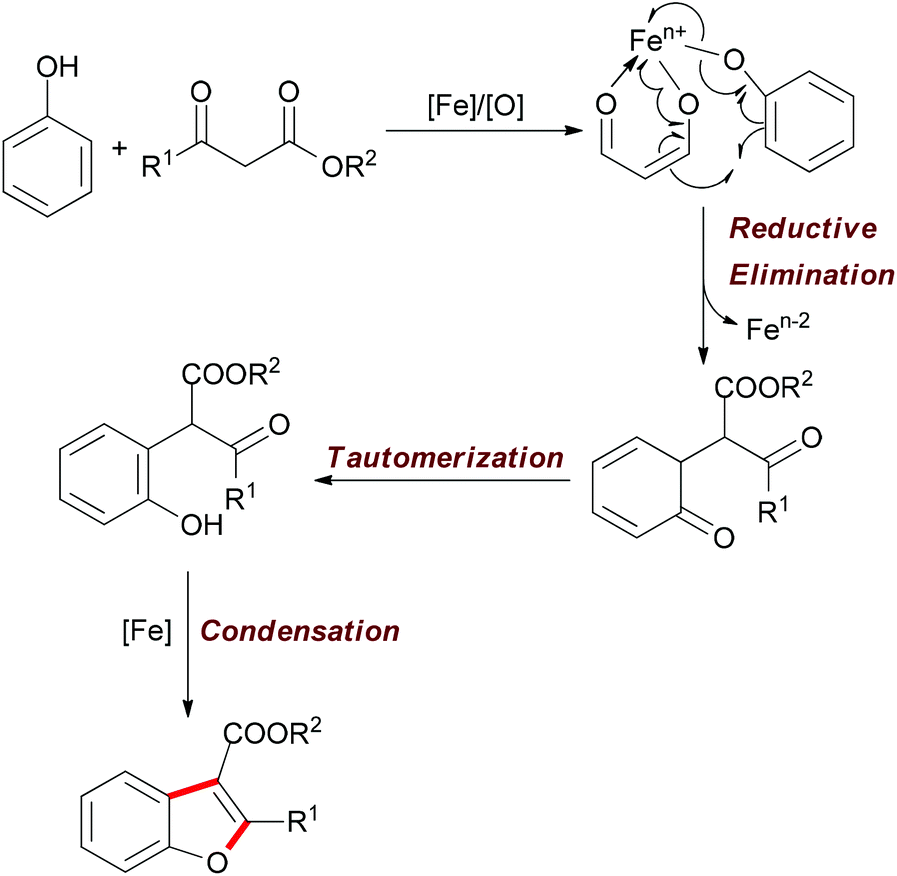 | ||
| Scheme 30 Tentative mechanism of the iron-catalyzed oxidative reaction of phenols and β-keto esters. | ||
The above methodology presented an efficient approach to the synthesis of polysubstituted benzofurans. Such heterocyclic motifs are important structural units and widely found in biological and medical compounds. Recently, Pappo and co-workers applied this concept in the total synthesis of coumestrol.52 Based on their previous work53 and the Li group's pioneering work,51 the oxidative coupling reaction occurred successfully by using ethyl 2-(2,4-dimethoxybenzoyl)acetate and 3-methoxyphenol under the modified reaction conditions. The desired benzofuran was obtained in 61% yield (gram-scale yield). The transformation of benzofuran to coumestrol was carried out by using a one-pot protocol and the natural product was smoothly constructed. Notably, the present method for the total synthesis of coumestrol can be scaled up to 10 mmol and 59% overall yield was achieved in only two steps (Scheme 31).
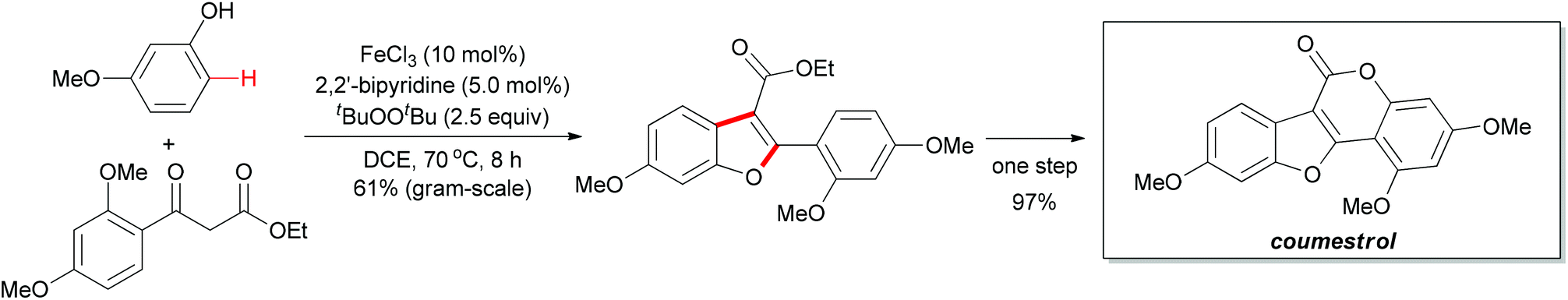 | ||
| Scheme 31 Total synthesis of coumestrol via oxidative coupling between phenol and β-keto ester. | ||
The sp2 carbon adjacent to OH group in β-naphthols could also act as nucleophile in an decarboxylative coupling reaction. In 2009, Li and co-workers disclosed the first example of iron-catalyzed intermolecular decarboxylative coupling reaction.54 Proline derivatives and β-naphthols could couple smoothly under the reaction conditions developed (Scheme 32). The tentative mechanism showed that the iron-coordinated imine ion was the key intermediate in the coupling process, which was then attacked by the β-naphthol followed by releasing the coupling product.
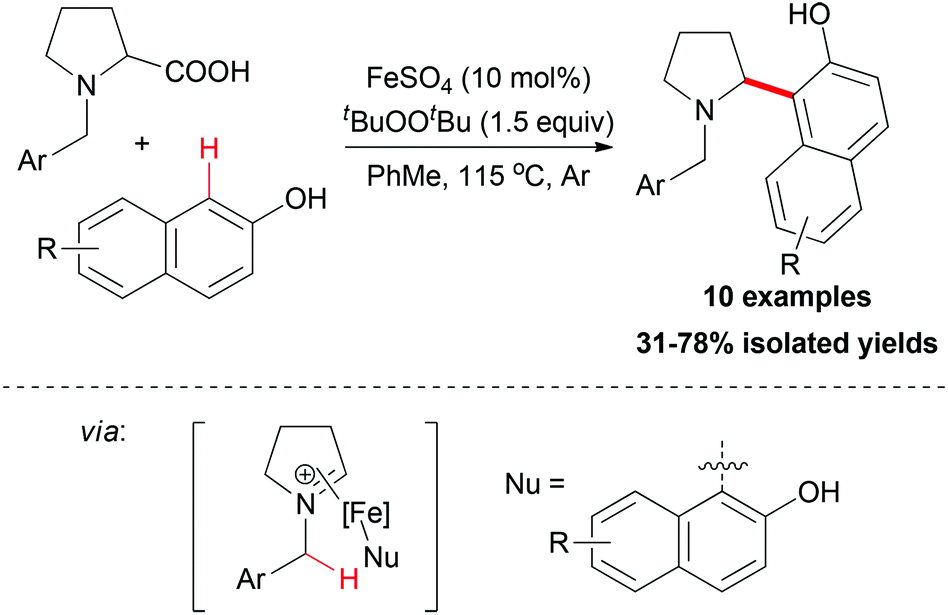 | ||
| Scheme 32 Iron-catalyzed decarboxylative coupling of proline derivatives with β-naphthols. | ||
Decarboxylative couplings have recently emerged as a promising concept for bond formation reactions.55 However, such an area using simple and easily handed iron salts as catalyst is still undeveloped. As such, iron-based catalysts might play a role in future studies on decarboxylative chemistry.
The sp2 C–H bonds located on the aromatic rings of phenol and catechol derivatives are known to undergo oxidation reactions by using simple iron salts, especially FeCl3.12i This kind of reaction usually generates the undesirable homo-coupled products, higher-molecular-weight polymers or C,O-connected phenol portions. However, this traditional process could be altered when olefins and external oxidants were introduced into the reaction. Recently, Lei and co-workers reported a novel FeCl3-catalyzed oxidative coupling reaction of phenols and olefins.56 Under the optimized conditions, the highly selective oxidative coupling/cyclization reaction occurred at room temperature (Scheme 33).
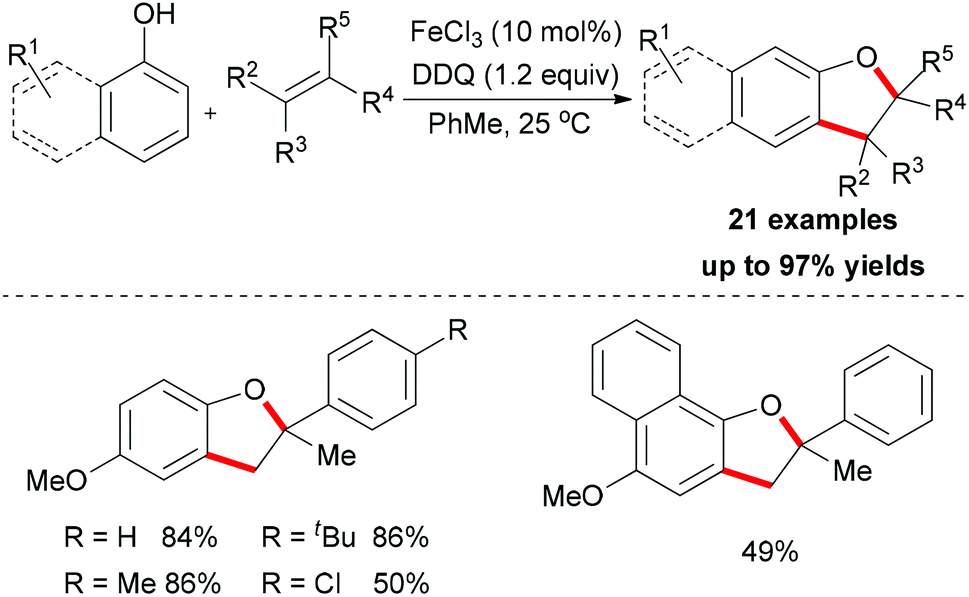 | ||
| Scheme 33 Iron-catalyzed oxidative radical cross coupling–cyclization between phenols and olefins. | ||
Based on the radical trapping experiment, EPR and operando IR studies, the authors proposed a radical pathway (Scheme 34). First, DDQ oxidized the phenol to generate the corresponding phenol radical I.57 As a Lewis acid, FeCl3 was more likely to coordinate with the O-atom which could stabilize the resonance structure of the phenol as well as increase the activity of the radical II to react with alkenes. The resulting radical III58 underwent H-atom abstraction by the HDDQ radical to form the final product.
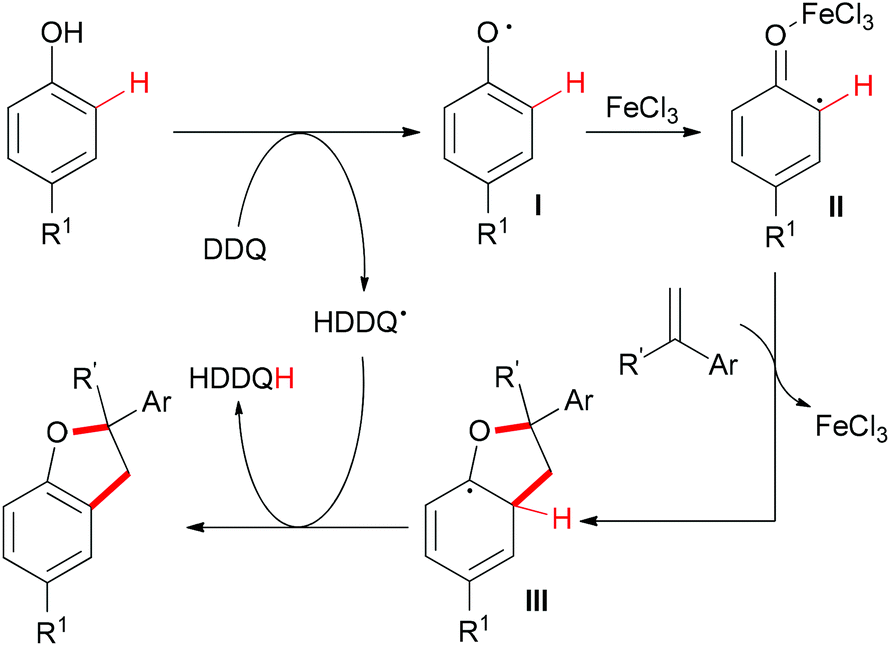 | ||
| Scheme 34 Proposed mechanism for the synthesis of dihydrobenzofuran. | ||
Meantime, Pappo and co-workers disclosed another strategy to construct the 2,3-dihydrobenzofuran motif (Scheme 35).59 The present reaction is sensitive to both the iron source and the reaction concentration. By utilizing FeCl2 as catalyst, the tandem coupling reaction occurred. However, when FeCl3·6H2O was used as catalyst in a dilute solution, the desired 2,3-dihydrobenzofuran formed. The former result could be explained by the formation of BINOL via homo-coupling of 2-naphthol followed by a rapid oxidative/addition dearomatization reaction with styrene under the same conditions. Using anhydrous FeCl3 (20 mol%) as catalyst, the furan product was isolated in low yield, which indicated that water might play a role in the proton transfer process of the reaction.51
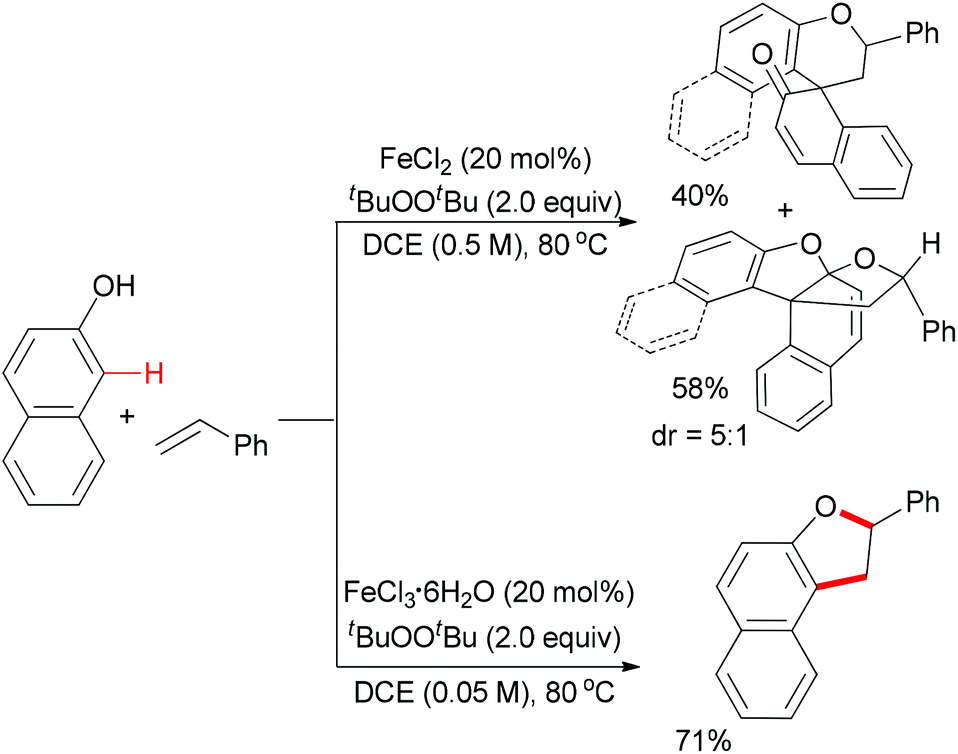 | ||
| Scheme 35 Iron-catalyzed oxidative cross-coupling of phenols and alkenes. | ||
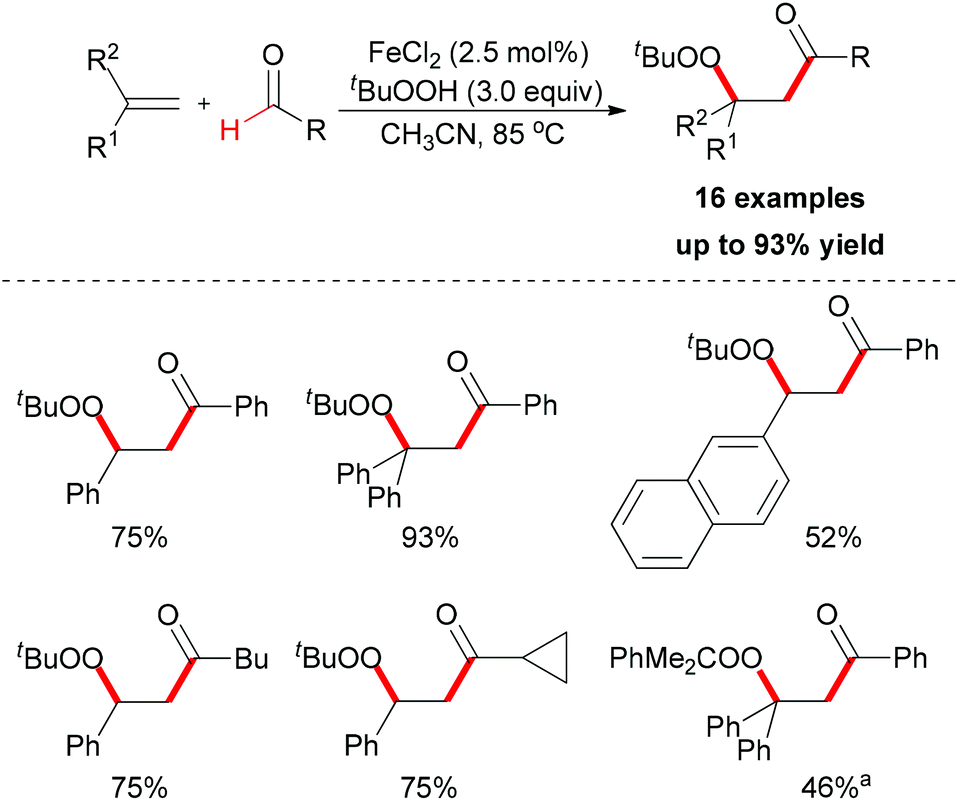 | ||
| Scheme 36 Iron-catalyzed carbonylation–peroxidation of alkenes. aUsing PhMe2COOH instead of TBHP as oxidant. | ||
When TEMPO was introduced into the reaction, the formation of the β-peroxy ketone product was completely suppressed while the TEMPO-adduct aldehyde was isolated in quantitative yield. This experimental result indicated that the acyl radical61 was generated under the standard conditions while the reaction is unlikely to have involved a cationic pathway.62Scheme 37 shows a tentative pathway for iron-catalyzed carbonylation–peroxidation reaction. Alkyloxy and alkylperoxy radicals were generated from the FeCl2/TBHP system, followed by hydrogen abstraction by the oxyl radical via sp2 C–H bond homolytic cleavage. The final β-peroxy ketone product was formed after radical addition and then radical coupling reaction. In the subsequent research, Li and co-workers expended this concept to the synthesis of α-ester-β-keto peroxides.63
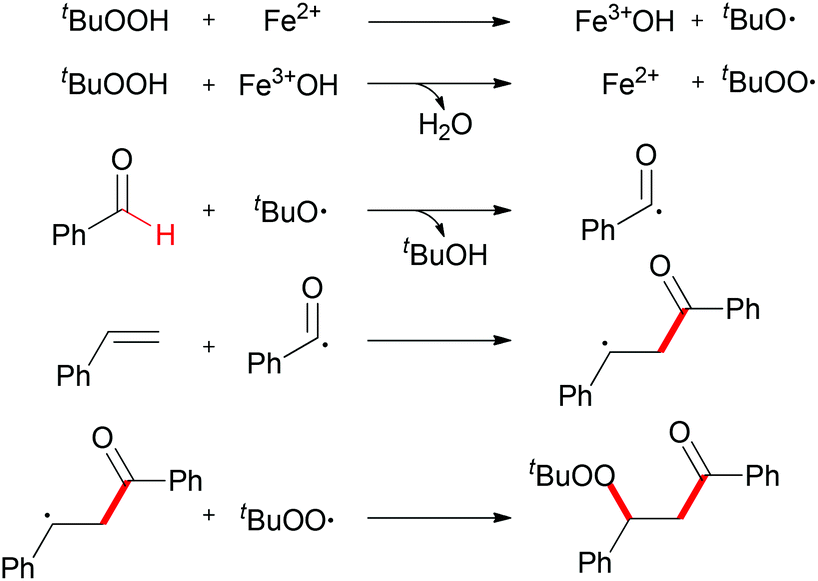 | ||
| Scheme 37 A proposed mechanism for iron-catalyzed carbonylation–peroxidation. | ||
Recently, after reporting of carbonylation–peroxidation of alkenes, Li and co-workers disclosed an iron-catalyzed carbonylation–arylation of activated alkenes (Scheme 38).64 Although a 68% yield of the desired product was obtained in the absence of a catalyst,25j using FeCl3 as catalyst is beneficial to the efficiency of the reaction. However, other metal salts (such as CuCl2 and CoCl2) showed low catalytic ability. Alkene substrates bearing alkyl or aryl protecting groups on the nitrogen were excellent for this transformation, and both α-substituted olefins and internal olefins were compatible with the reaction conditions. Many aldehyde substrates undergo hydrogen atom abstraction by an oxy radical initiated with the combined FeCl3/TBHP system via sp2 C–H bond homolytic cleavage. The resulting acyl radical, which is nucleophilic in nature, is known to add more easily to electron-deficient alkenes than normal alkenes.65 Then the radical generated underwent an intramolecular 5-exo-trig cyclization66 followed by the oxidation step to form the final oxindole.
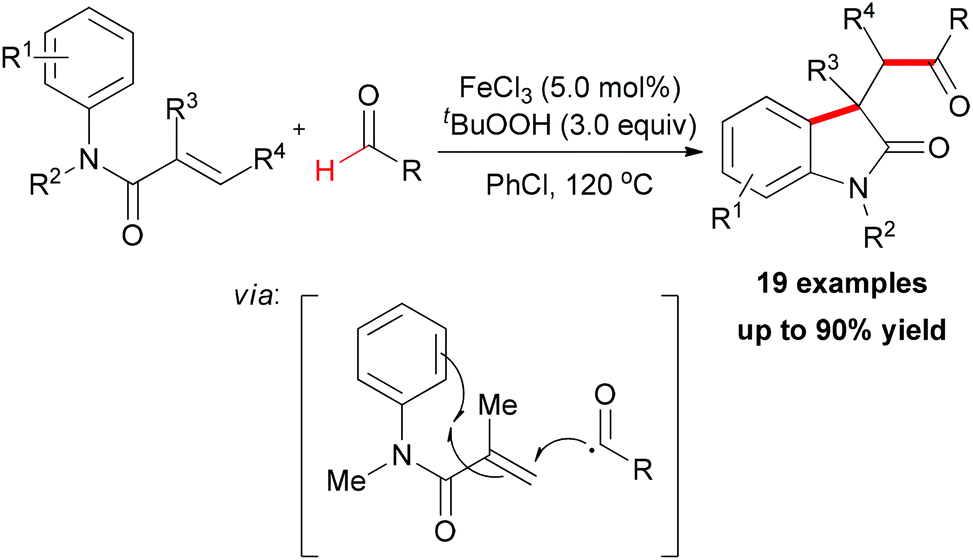 | ||
| Scheme 38 Iron-catalyzed carbonylation–arylation of alkenes. | ||
Recently, the Studer group reported a synthetic methodology for constructing fluorenones and xanthones (Scheme 39).67 The screening of conditions indicated that FeCp2 was the best radical initiator and tert-butyl peroxide was the most efficient oxidant. Under the conditions developed, various readily available ortho-formyl biphenyls and ortho-formyl biphenyl ethers were tested. In contrast, radical chains were shorter and a higher iron salt loading was necessary in the fluorenone synthesis. Therefore, lower yields were achieved.
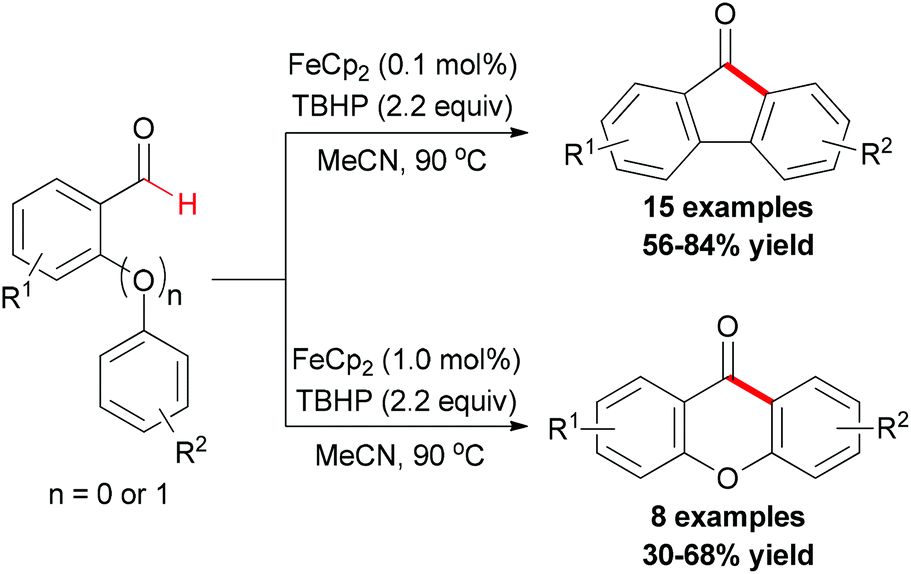 | ||
| Scheme 39 Iron-catalyzed CDC reaction via base promoted homolytic aromatic substitution (BHAS). | ||
Mechanistically, the author proposed a base promoted homolytic aromatic substitution (BHAS) pathway (Scheme 40).68 Initiation occurred by reducing TBHP with Fe(II) to generate the tert-butoxyl radical and Fe(III) complex. The tert-butoxyl radical underwent hydrogen atom abstraction from the aldehyde to give the acyl radical, which then attacked the arene to form the cyclohexadienyl radical. Deprotonation with the basic hydroxide anion resulted in forming the biaryl radical anion. Then this radical anion reduced TBHP by single-electron transfer (SET) to give the final product and a tert-butoxyl radical which underwent the next radical chain reaction.
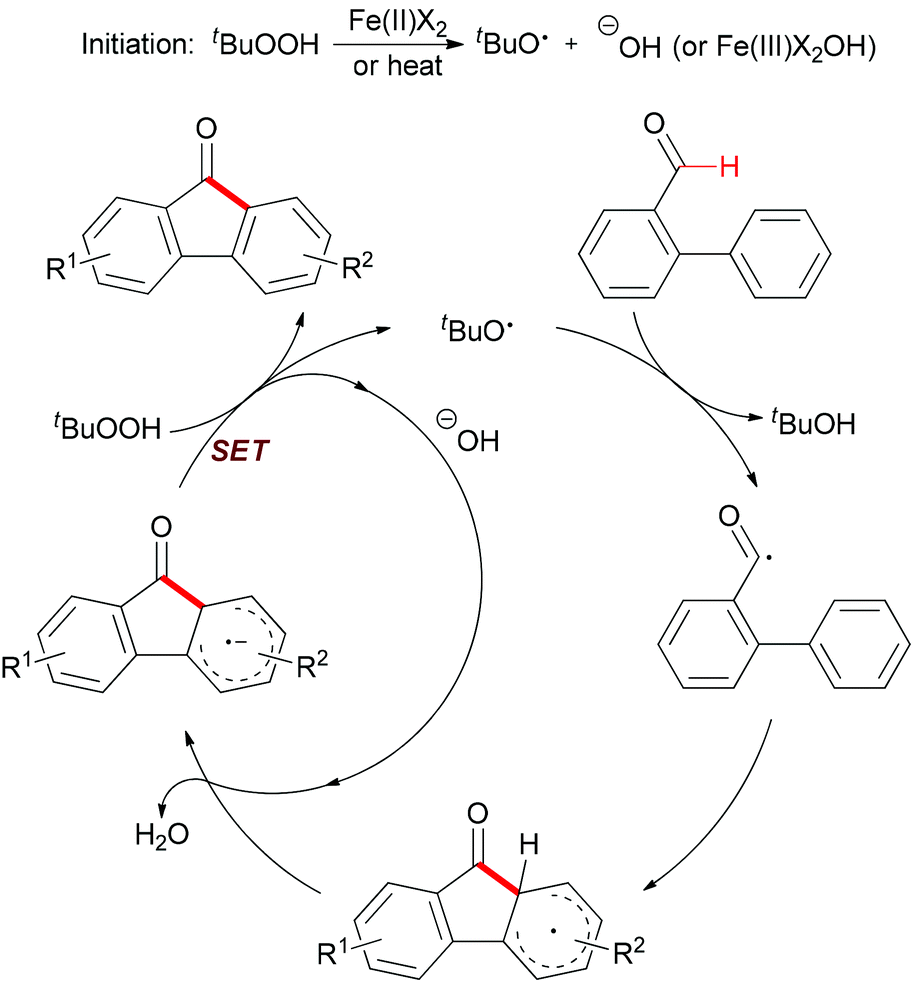 | ||
| Scheme 40 Suggested mechanism for BHAS reaction. | ||
In Studer's subsequent research, they applied this BHAS concept in constructing 6-aroylated phenanthridines (Scheme 41).69 Readily available 2-isocyanobiphenyls and aromatic aldehydes were used as substrates. By introducing small amounts of ferric chloride as radical initiator and TBHP as an efficient oxidant, a series of 6-aroylated phenanthridines were smoothly constructed through a radical cascade reaction. In the radical initiation step, the Fe(III) chloride was first reduced to Fe(II) chloride through ligand exchange to give FeCl2OOtBu species. This iron–peroxyl intermediate underwent homolytic Fe–O bond cleavage to form FeCl2 and the tert-peroxyl radical which could also abstract a H-atom from the aldehyde. It is known that an aliphatic acyl radical can undergo decarbonylation reaction to generate an alkyl radical. Interestingly, when valeraldehyde was used as acyl radical precursor in this reaction, no 6-butylphenanthridine product was observed and the target product was formed in relatively low yield (39%). However, when cyclohexanecarbaldehyde was introduced as substrate, the decarbonylated 6-cyclohexylphenanthridine was observed along with the 6-acylated product (44% combined yield, ratio 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :43).
:43).
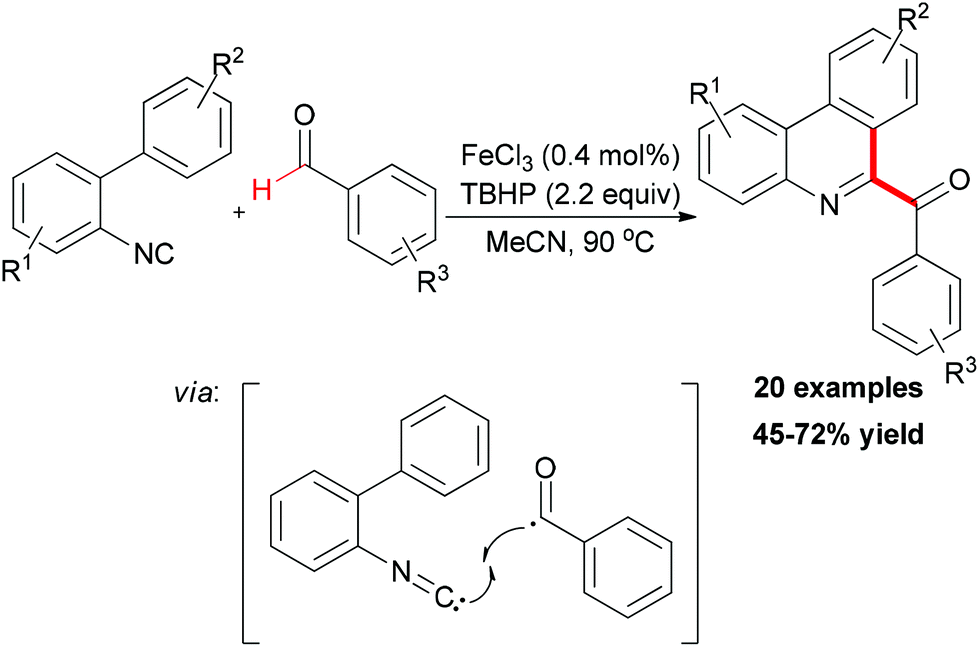 | ||
| Scheme 41 Synthesis of 6-aroylated phenanthridines via BHAS. | ||
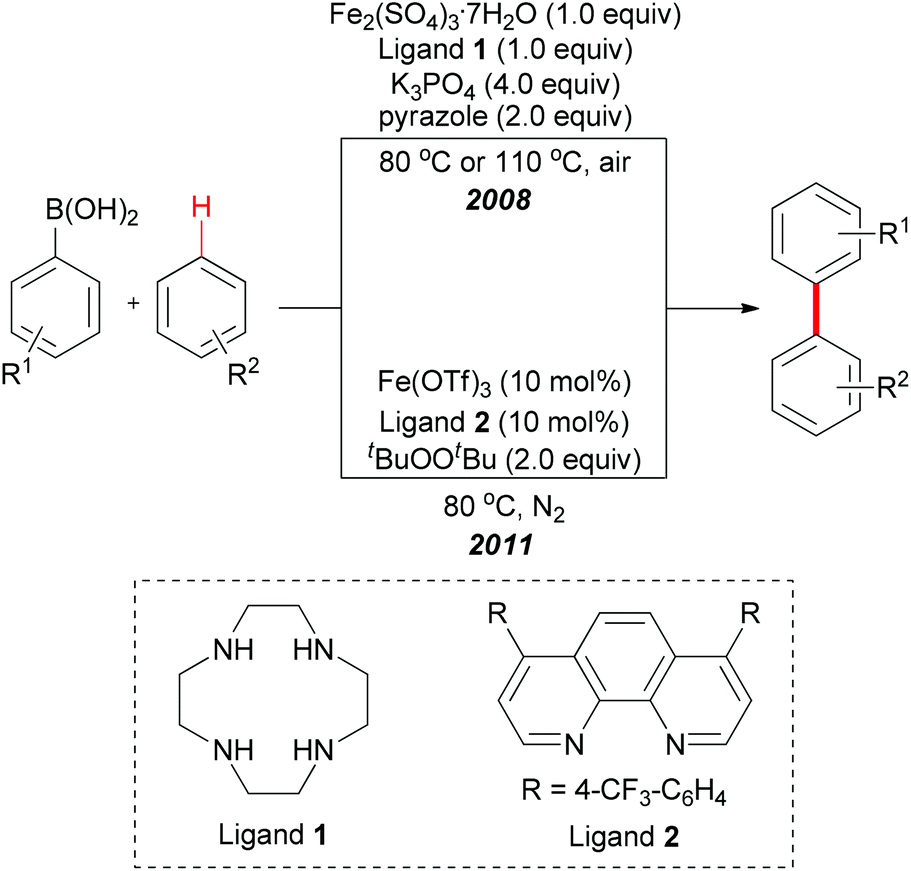 | ||
| Scheme 42 Iron-mediated/catalyzed direct arylation of unactivated arenes. | ||
In Yu's subsequent research, they extended their study by utilizing electron-rich and electron-deficient heteroarenes (pyrrole and pyridine) as substrates.72 Coupling reactions could successfully occur with the arylboronic acids as coupling partners and FeC2O4·2H2O/MCPA ligand as catalytic system (Scheme 43). When pyrroles were used as substrates, the amount of iron catalyst could be decreased to 20 mol% by employing O2 as indispensable oxidant. It worth noted that the arylation selectively occurred at the C2 position of pyrroles.
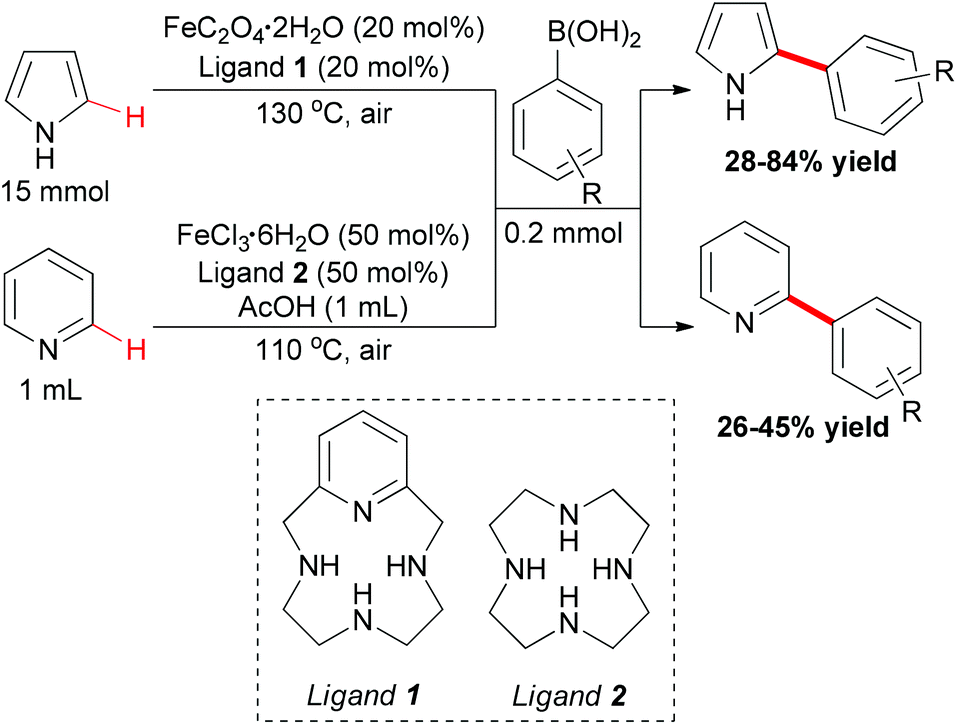 | ||
| Scheme 43 Iron-catalyzed ortho-arylation of pyrrole and pyridine. | ||
Mechanistically, the authors proposed that the oxoiron complex might be the active species in the present reaction, and DFT calculations indicated the C–H activation through σ-bond metathesis. The full catalytic cycle is shown in Scheme 44.
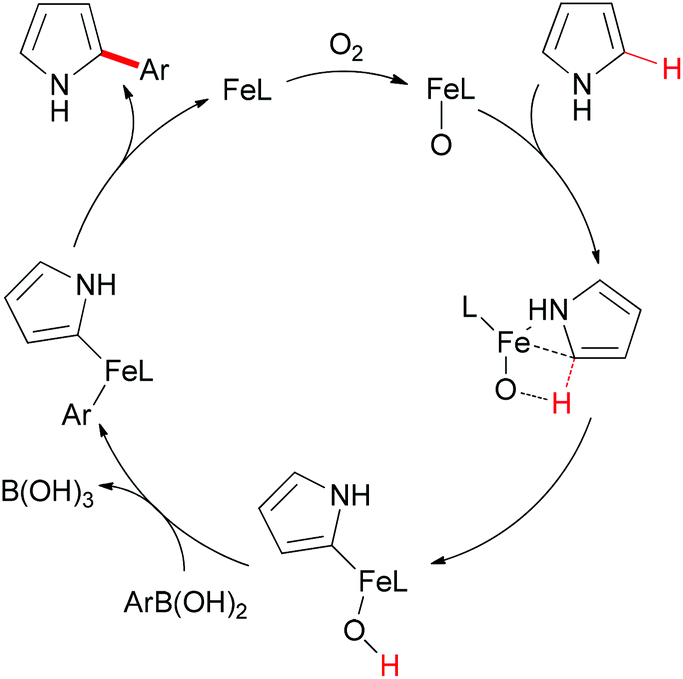 | ||
| Scheme 44 Proposed catalytic cycle of the iron-catalyzed direct Suzuki–Miyaura coupling. (L = ligand). | ||
In 2008, Wang and co-workers reported an iron-mediated oxidative cyclization for the total synthesis of (±)-tylophorine, (±)-deoxytylophorinine and (±)-antofine.73 Using a mixture of E and Z isomers SM-a, SM-b and SM-c as starting materials to undergo the oxidative coupling by utilizing 3.5 equiv. of FeCl3, the desired phenanthrene products were smoothly synthesized (Scheme 45). In the authors’ later report, they proposed a tentative mechanism involving a radical initiation and heterolytic cleavage of the sp2 C–H bond on the phenyl ring.74
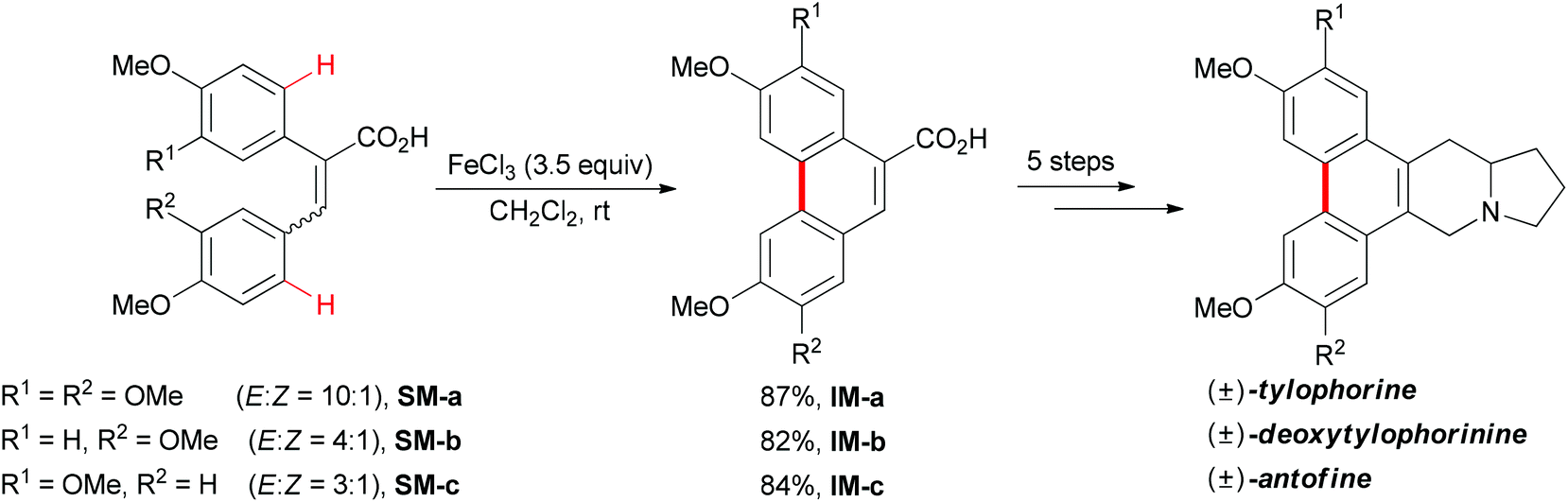 | ||
| Scheme 45 Total syntheses of (±)-tylophorine, (±)-deoxytylophorinine and (±)-antofine via oxidative phenolic coupling. | ||
In 2009, the Itami group and the Wünsch group reported an iron-catalyzed oxidative coupling of heteroarenes with methylamines by using Fe(II) as catalyst and pyridine N-oxide as oxidant.75 The oxidative coupling reaction could be used for the intermolecular coupling of thiophenes, furans, and indoles with methylamines (Scheme 46). Although the detailed mechanism was still unknown, the authors proposed a tentative mechanism involving the iron-bound iminium species which could undergo electrophilic substitution on the thiophene moiety (Scheme 47).10a
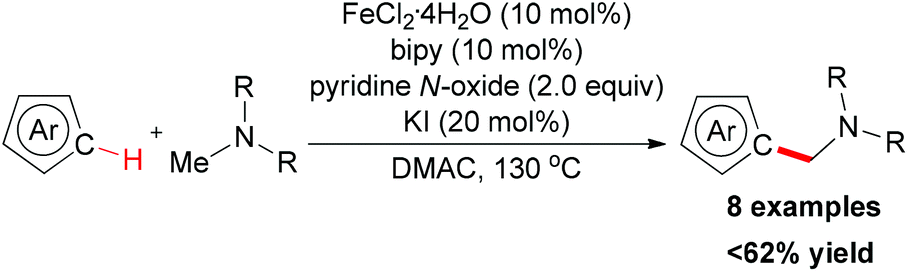 | ||
| Scheme 46 Iron-catalyzed oxidative coupling of heteroarenes and methylamines. | ||
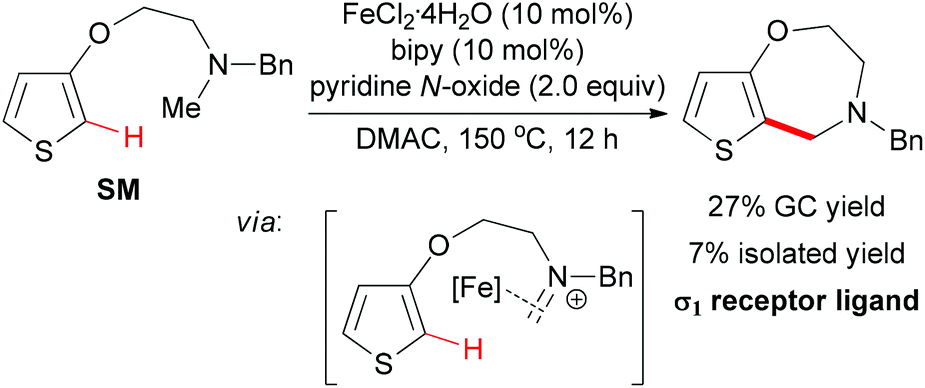 | ||
| Scheme 47 Intramolecular oxidative coupling for constructing new σ1-receptor ligands. | ||
When intramolecular oxidative coupling was performed using thiophene SM as substrate, the benzazepine-like bicyclic nitrogen heterocycle was formed, albeit in low yield (Scheme 47). The authors also identified the coupling product has a good binding affinity toward the σ1 receptor protein. The difficulty of cyclizing seven-membered rings may be one of the reasons for the low efficiency.
In 2010, Liang and co-workers reported an iron catalyzed direct intramolecular oxidative coupling reaction for the synthesis of indoles (Scheme 48).76 In their studies, the best catalyst was FeCl3 and the highly active Cu(OAc)2·CuCl2 was used as oxidant.
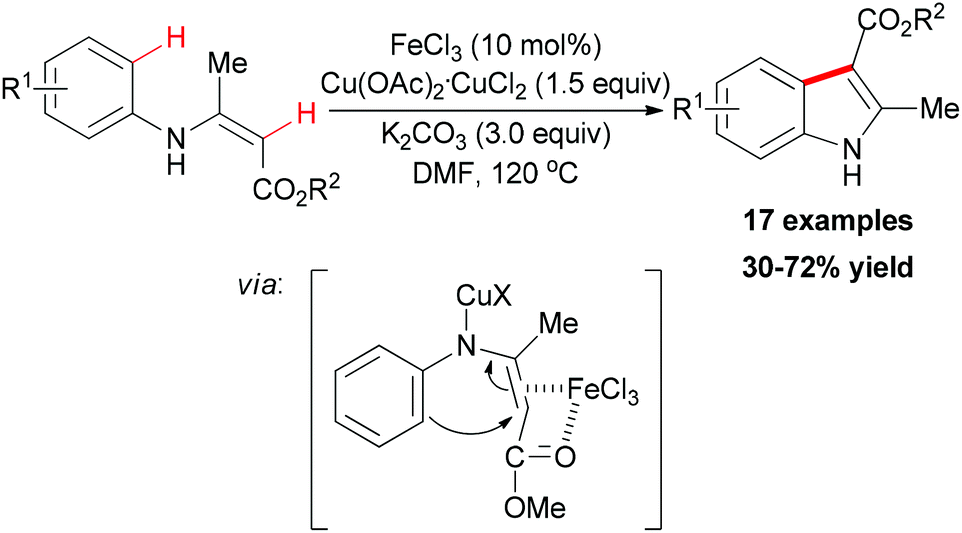 | ||
| Scheme 48 FeCl3-catalyzed direct oxidative coupling for the preparation of indoles. | ||
Direct arylation of aryl C–H bonds is not limited to using arylboronic acids as coupling partners. In recent years, the iron-catalyzed direct arylation of aryl C–H bonds with organometallic reagents has been developed (Scheme 49). The Nakamura group has made a significant contribution in this area.77 In 2008, they reported the first iron-catalyzed direct arylation of aryl C–H bonds (Scheme 49, top).77k In the initial study, they used organozinc reagents generated in situ from aryl Grignard reagents and ZnCl2 in the presence of the proper ligand. The extensive optimizations showed that Fe(acac)3/phen/DCIB was the best catalytic system. In this reaction, the substrate arene displaying a Lewis-basic directing group could be regioselectively functionalized at remarkably low reaction temperatures through chelation control. It is noteworthy that the proper ligand was essential for this coupling reaction and the oxidant dihalide was converted to the corresponding olefin.
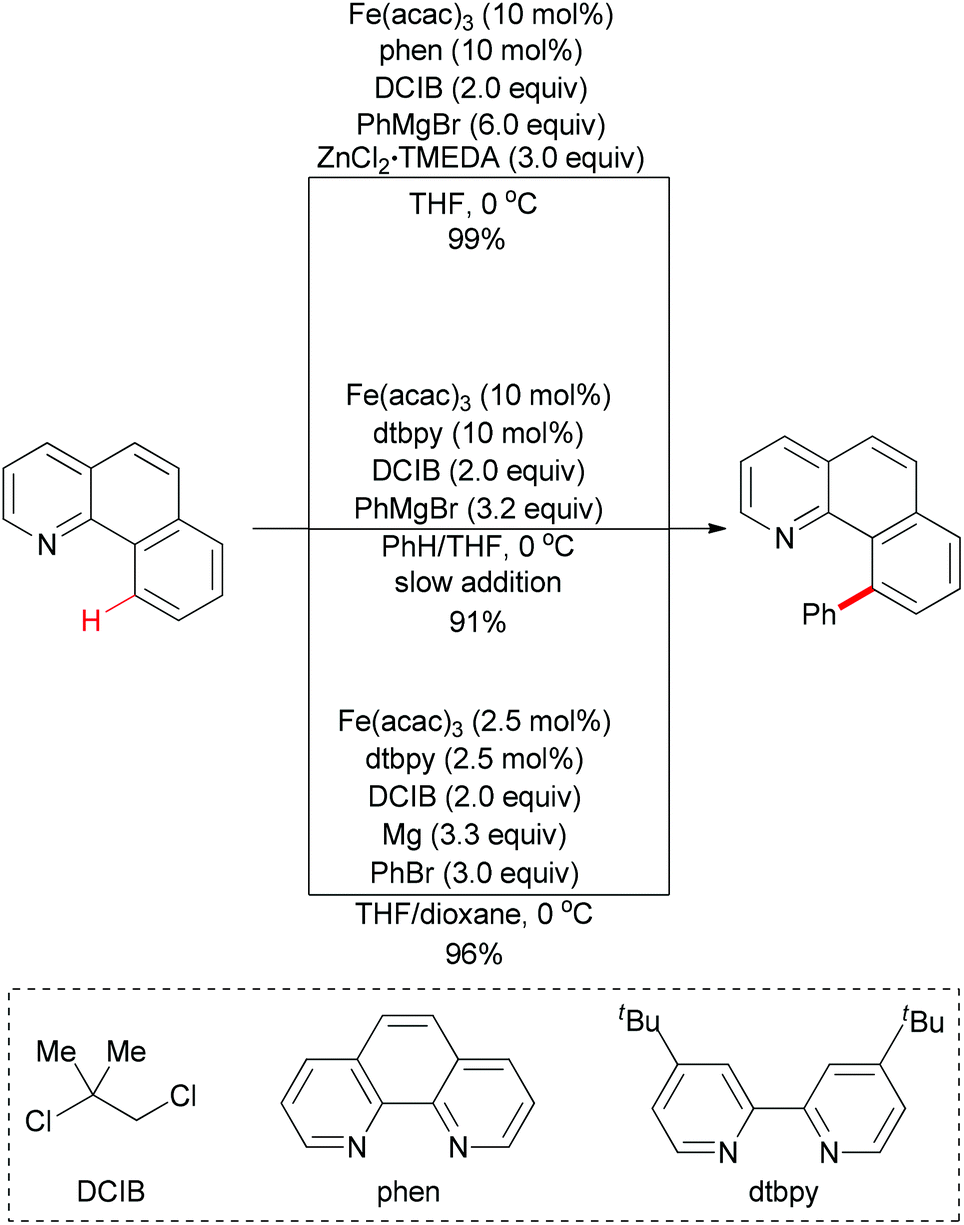 | ||
| Scheme 49 Iron-catalyzed direct phenylation of α-benzo-quinoline through directed C–H bond activation. | ||
Obviously, the above reaction has an unattractive feature, that is, it requires the use of large amounts of the zinc salt (3 equiv.) and the aryl Grignard reagent (6 equiv.) for the in situ generation of the reactive arylzinc reagent. In Nakamura's subsequent research, they successfully overcome this disadvantage. The key elements of the development were the use of an aromatic co-solvent, such as chlorobenzene and benzene, and the slow addition of the Grignard reagent (Scheme 49, middle).77f
In contrast with using organozinc regents or Grignard reagents, metallic magnesium could also be utilized in such coupling reaction. The use of a 1:1 mixture of tetrahydrofuran and 1,4-dioxane is essential for this C–H bond activation reaction (Scheme 49, bottom).77e
3.2 C–N bond formation
NTs/PhINNs as the nitrogen source.78 They used FeCl2/pyridine as the in situ formed precatalyst and the reaction was accomplished in moderate to excellent yields (Scheme 50). The mechanistic studies showed that [Fe(py)4Cl2] was formed in situ and thus facilitated the insertion of a putative iron–nitrene/imido group to the formylic C–H bond of aldehyde.
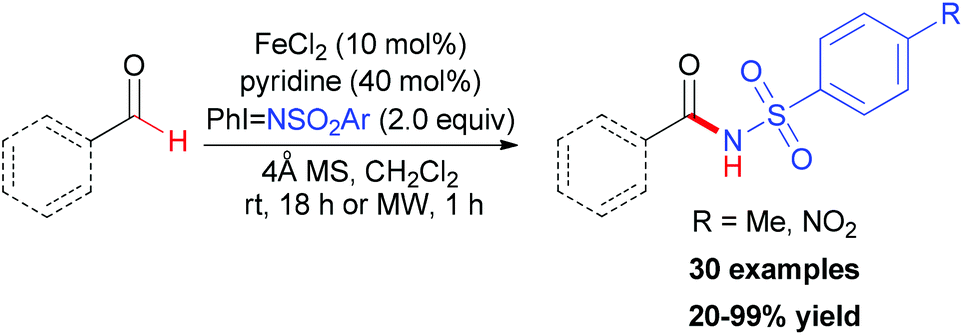 | ||
| Scheme 50 Iron(II)-catalyzed amidation of aldehydes with iminoiodinanes. | ||
In 2012, De Luca and co-workers reported an iron-catalyzed amidation of aldehydes with N-chloroamines (Scheme 51).79 In this methodology, they gave a new example of coupling of an acyl radical and a nitrogen-centered radical. FeCl3·6H2O is the best choice while FeCl2·4H2O shows almost the same efficiency in this amidation reaction. Introducing a radical trapping regent, such as TEMPO resulted in the formation of a TEMPO-adduct aldehyde product. This result indicated that the acyl radical may be generated in the initiation step via homolytic sp2 C–H bond cleavage in aldehyde substrates.
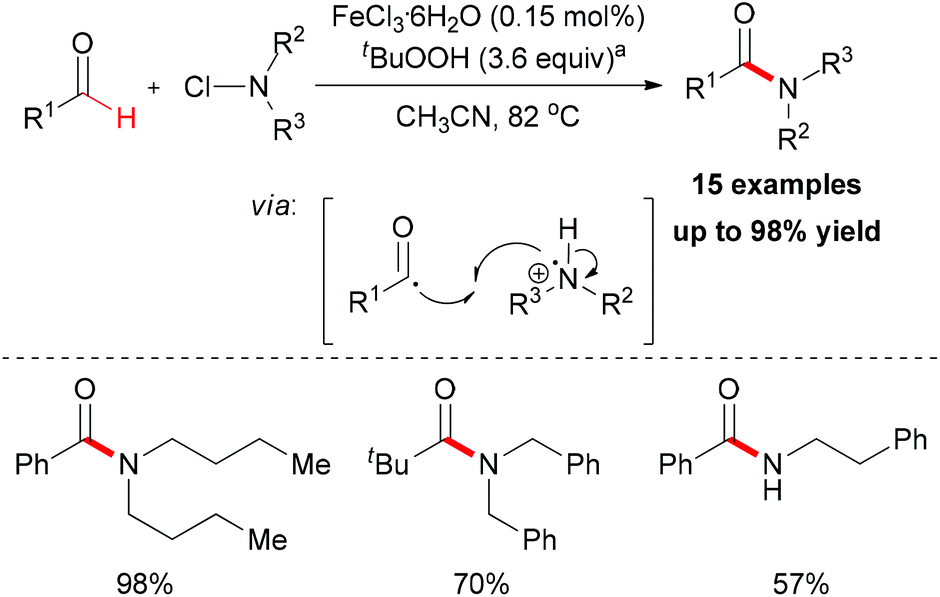 | ||
| Scheme 51 Amidation of aldehydes with N-chloroamines. a70 wt% solution in water. | ||
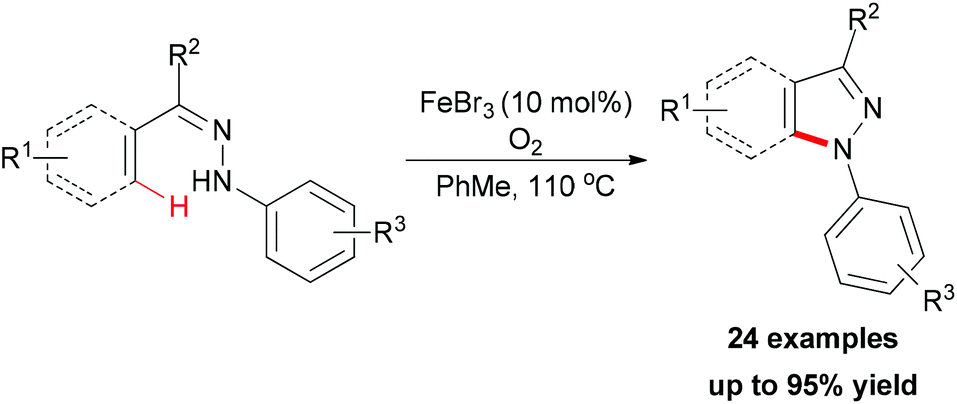 | ||
| Scheme 52 FeBr3/O2 mediated intramolecular sp2 C–H amination. | ||
More recently, Maes and co-workers reported iron-catalyzed sp2 C–H amination for the construction of C8–N9 annulated purines.81 Catalyst screening indicated that FeCl2·4H2O was the best choice, while FeCl3·6H2O gave a similar result. However, copper salts showed low catalytic ability. Using O2 as oxidant, a series of easily accessible 5-(pyridin-2-ylamino)pyrimidine-2,4(1H,3H)-dione substrates underwent direct amination reaction to form the substituted pyrido[1,2-e]purines (Scheme 53). The addition of TEMPO to the cyclization reaction gave a significant reduction in conversion suggesting that radicals might be involved in the reaction mechanism. Based on the experimental results and previous reports, a catalytic cycle was proposed and is shown in Scheme 53.
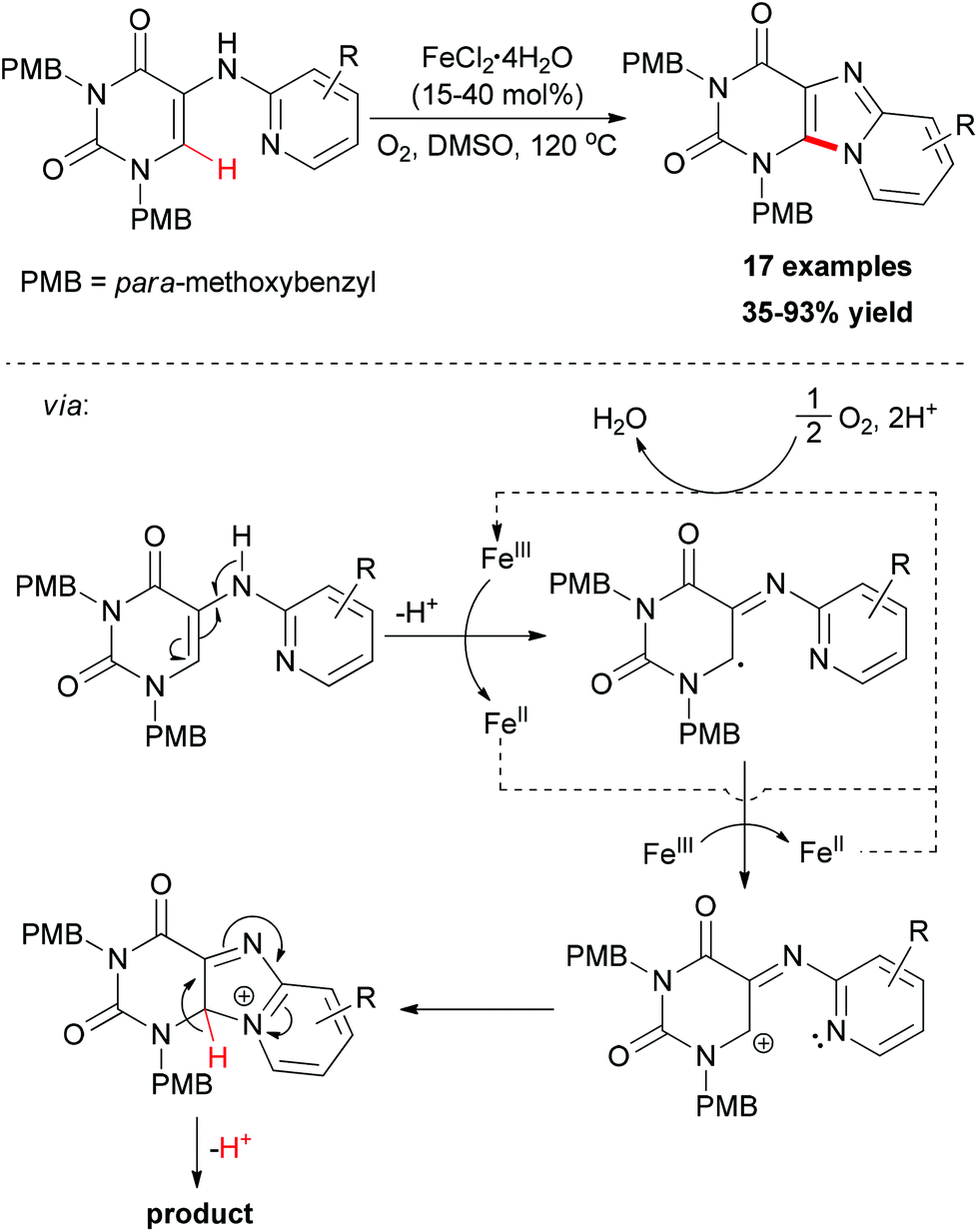 | ||
| Scheme 53 Iron-catalyzed sp2 C–H amination for construction of C8–N9 annulated purines. | ||
Maiti and co-workers reported an important methodology for constructing nitroolefins via a predictably selective nitration of olefins with Fe(NO3)3·9H2O and TEMPO (Scheme 54).82 Screening of the conditions indicated that Fe(NO3)3·9H2O was the best nitrating agent along with catalytic TEMPO. Notably, the nature of the solvent had a significant effect on the nitration reaction as less/nonpolar solvents were found to be a better choice compared to polar solvents.
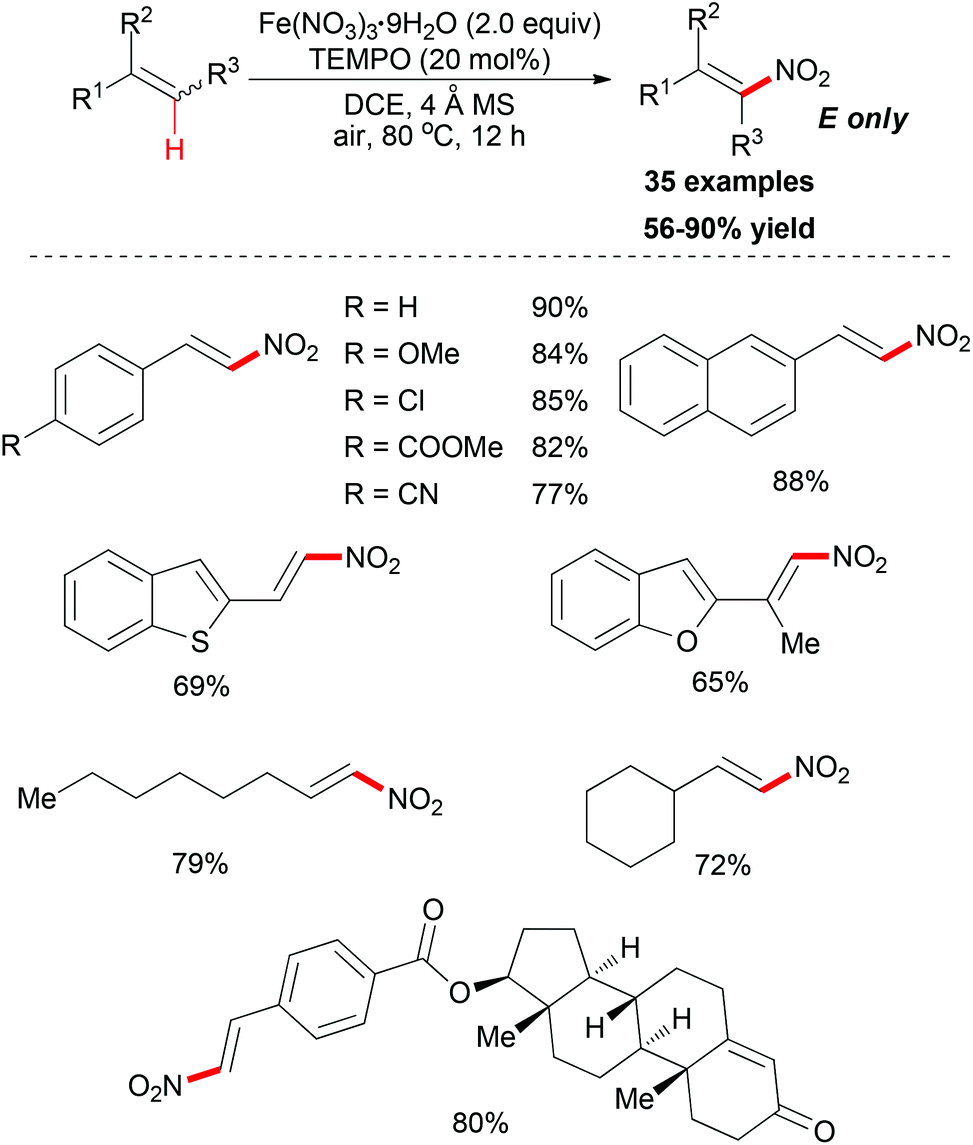 | ||
| Scheme 54 Iron-mediated selective nitration of olefins. | ||
Under the conditions developed, a wide variety of aromatic, aliphatic, and heteroaromatic olefins smoothly underwent nitration in regio- and stereoselective manner. Synthetically, the present reaction provided nitro-olefins in preparatively useful yields with excellent E-selectivity. Based on the authors’ previous work,83 two tentative pathways were proposed (Scheme 55). Initially, the nitro radical (NO2˙) could be generated from Fe(NO3)3·9H2O under thermal conditions.84 Then the nitro radical would react with the olefin at the less-hindered side to form the carbon-centered radical. This nitroalkane radical could be transformed to the final product via two possible pathways. In path 1, TEMPO can directly abstract a hydrogen radical, which released the nitro-olefin. In path 2, TEMPO acts as a radical trapping regent after the formation of carbon-centered radical followed by oxidation to generate the final product.
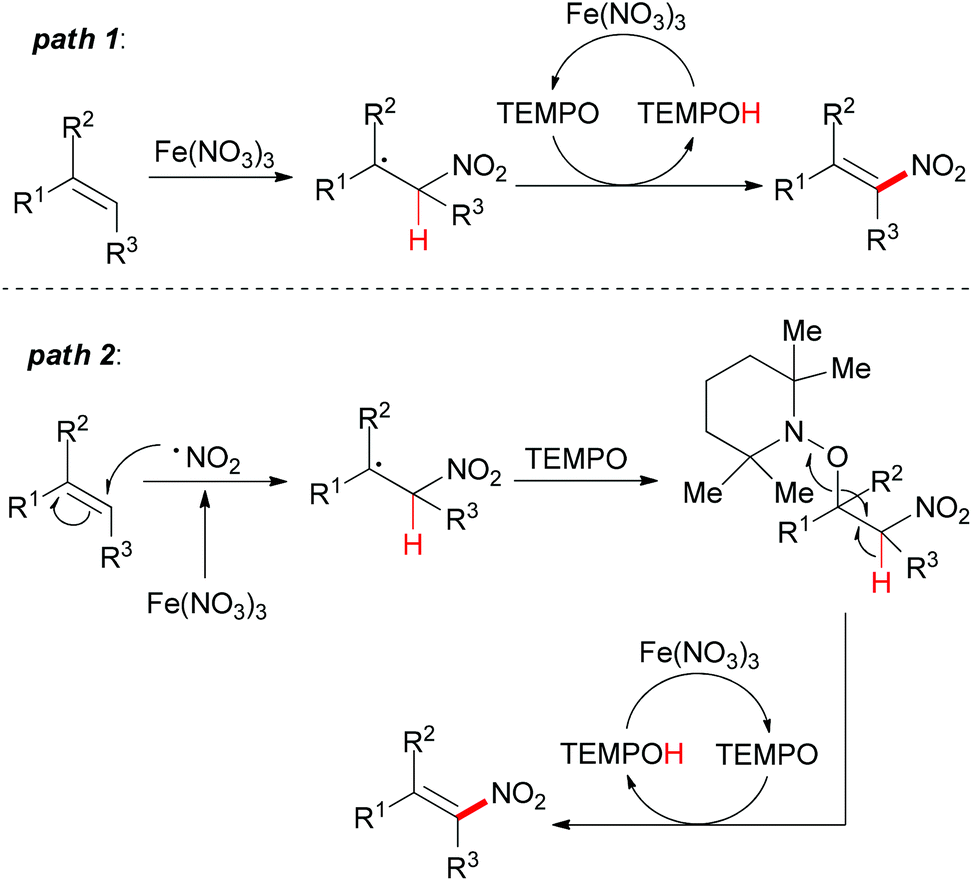 | ||
| Scheme 55 Proposed pathways for stereoselective nitration. | ||
3.3 C–O bond formation
Direct oxidation of arenes to phenols is a difficult reaction. One reason is that many metal complexes capable of arene sp2 C–H bond activation could not survive under the oxidizing conditions. In addition, because phenols are more electron-rich than the substrate arene, over-oxidation is also a complicated problem.One hundred years ago, in 1900, the first example of the direct hydroxylation of arenes to phenols was reported.85 Benzene was oxidized to phenol under a mixture of FeSO4 and hydrogen peroxide (Fenton's reagent).17 After this seminal report, many optimized systems were developed. However, no significant progress was made in the synthetic field.
In 2010, the Beller group disclosed the first example of iron-catalyzed selective oxidation of the sp2 C–H bond of arenes and phenols.86 Under two types of three component catalytic system (FeCl3·6H2O–H2Pydic–amine = 1/1/2.2), oxidation of 2-methyl-naphthalene and TMP (2,3,6-trimethylphenol) took place in 55% and 77% yield (Scheme 56), respectively. This oxidation reaction offered an important method for the synthesis of vitamin E intermediates and vitamin K3.
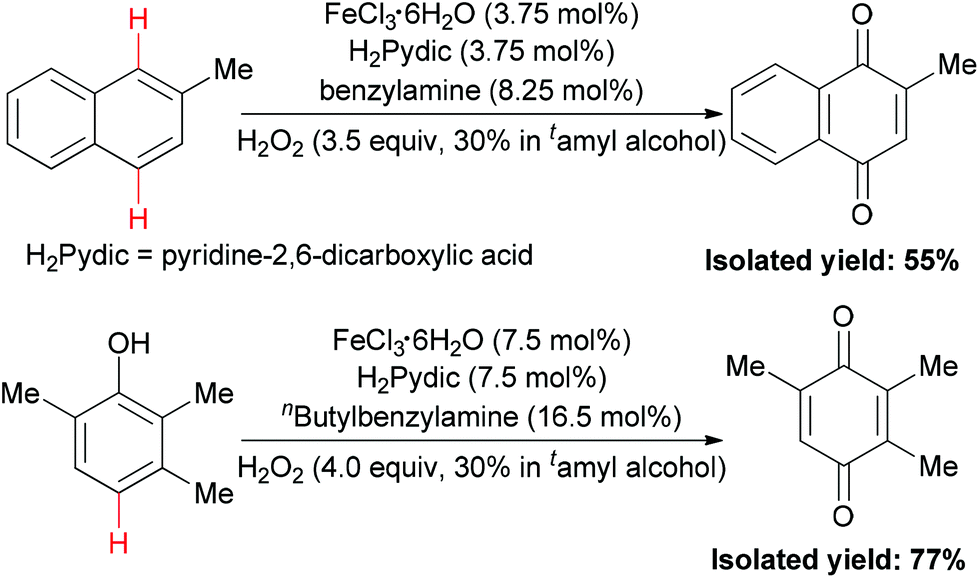 | ||
| Scheme 56 Iron-catalyzed selective oxidation of arenes and phenols with hydrogen peroxide. | ||
4 Future challenges
Although many significant developments have been reported in iron-catalyzed/mediated C–H bond oxidative transformation, this attractive field still holds many challenges. The problems can generally be divided into three aspects: reactivity, selectivity and mechanistic studies.87Due to the strength of most C–H bonds, reactivity in their oxidative transformation becomes a very important challenge. Iron, as discussed above, has already shown its unique power in such transformations, but a more reactive catalyst and lower catalyst loading are much needed in this area.
Generally, most organic molecules contain C–H bonds with different chemical environments. The target C–H bond should be selectively transformed, leaving the other C–H bonds unchanged. Moreover, because most oxidation reactions are thermodynamically downhill, avoiding over-oxidation may be a potential challenge. As such, the regioselectivity and chemoselectivity may continue to attract organic chemists’ attention. In addition, the synthesis of chirality-containing compounds will encourage chemists to find more methods to transform C–H bonds in high stereoselectivity.
As mentioned above, the mechanisms of many oxidation processes are still unclear. Further research towards the design of a more powerful catalytic cycle should be based on mechanistic studies. The combination of iron-catalyst/oxidant may also play a vital role in the design of new reactions.
Acknowledgements
Financial support by the National Science Foundation of China (21072223, 21272267).Notes and references
- G. Dyker, Handbook of C-H Transformations: Applications in Organic Synthesis, Wiley-VCH, 2004 Search PubMed.
- For selected books, please see: (a) J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, 2nd edn, 2005 Search PubMed; (b) M. Hudlucky, Oxidations in Organic Chemistry, ACS Monograph Series, American Chemical Society, Washington, 1990 Search PubMed; (c) R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds: Mechanistic Principles and Synthetic Methodology Including Biochemical Processes, Academic Press, New York, 1981 Search PubMed. For selected recent reviews on some important oxidation processes, please see: (d) M. Sun, J. Zhang, P. Putaj, V. Caps, F. Lefebvre, J. Pelletier and J.-M. Basset, Chem. Rev., 2014, 114, 981 CrossRef CAS PubMed; (e) R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421 CrossRef PubMed; (f) N. Keller, M.-N. Ducamp, D. Robert and V. Keller, Chem. Rev., 2013, 113, 5029 CrossRef CAS PubMed. For selected review on transition metal catalyzed C–H bond oxidative transformations, please see: (g) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology (the “Gold Book”)—oxidation.
- For selected reviews on transition metal catalyzed C–O bond formation reactions, please see: T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed.
- For selected reviews on transition metal catalyzed C–N bond formation reactions, please see: (a) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (b) P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510 CrossRef CAS PubMed; (c) H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518 CrossRef CAS PubMed.
- For selected review on C–X bond formation reactions, please see: A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS PubMed.
- For selected reviews on transition metal catalyzed dehydrogenation reactions, please see: (a) J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed.
- For selected reviews on transition-metal catalyzed C–H activations, please see: (a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (b) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (c) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (d) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS; (e) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; (f) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed; (g) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (h) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (i) W. Jones and F. Fehe, Acc. Chem. Res., 1989, 22, 91 CrossRef CAS.
- For selected recent reviews on transition-metal catalyzed C–C bond formation reactions, please see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (c) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed.
- For selected reviews on C–H activation in total synthesis, please see: (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- For text books, please see: (a) B. Plietker, Iron Catalysis:Fundamentals and Applications, Topics in Organometallic Chemistry, vol. 33, 2011 Search PubMed; (b) B. Plietker, Iron Catalysis in Organic Chemistry: Reactions and Applications, Wiley-VCH, 2008 Search PubMed.
- For selected reviews and highlights on iron-catalyzed organic reactions, please see: (a) M. D. Greenhalgh and S. P. Thomas, Synlett, 2013, 531 CAS; (b) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (c) J. E. M. N. Klein and B. Plietker, Org. Biomol. Chem., 2013, 11, 1271 RSC; (d) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243 RSC; (e) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (f) O. G. Mancheño, Angew. Chem., Int. Ed., 2011, 50, 2216 CrossRef PubMed; (g) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (h) L.-X. Liu, Curr. Org. Chem., 2010, 14, 1099 CrossRef CAS; (i) A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730 RSC; (j) W. M. Czaplik, M. Mayer, J. Cvengrošm and A. J. von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (k) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed; (l) E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341 CrossRef CAS; (m) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed; (n) A. Correa, O. G. Manchenõ and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (o) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed.
- For the concept of CDC reaction, please see: C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- For selected reviews on cross-dehydrogenative C–H bond functionalizations, please see: (a) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (b) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC; (c) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253 RSC; (d) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (e) L. Yang and H. Huang, Catal. Sci. Technol., 2012, 2, 1099 RSC; (f) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC; (g) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (h) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353 CrossRef CAS; (i) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (j) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC; (k) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS PubMed; (l) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935 CAS; (m) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS PubMed; (n) P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS.
- Z. Li, L. Cao and C.-J. Li, Angew. Chem., Int. Ed., 2007, 46, 6505 CrossRef CAS PubMed.
- Y. Zhang and C.-J. Li, Eur. J. Org. Chem., 2007, 4654 CrossRef CAS.
- For general reviews on Fenton chemistry, please see: (a) S. Goldstein and D. Meyerstein, Acc. Chem. Res., 1999, 32, 547 CrossRef CAS; (b) P. A. MacFaul, D. D. M. Wayner and K. U. Ingold, Acc. Chem. Res., 1998, 31, 159 CrossRef CAS; (c) C. Walling, Acc. Chem. Res., 1998, 31, 155 CrossRef CAS; (d) D. T. Sawyer, A. Sobkowiak and T. Matsushita, Acc. Chem. Res., 1996, 29, 409 CrossRef CAS. For recent development, please see: (e) R. D. Bach and O. Dmitrenko, J. Org. Chem., 2010, 75, 3705 CrossRef CAS PubMed.
- S. Pan, J. Liu, Y. Li and Z. Li, Chin. Sci. Bull., 2012, 57, 2382 CrossRef CAS.
- Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS PubMed.
- C.-X. Song, G.-X. Cai, T. R. Farrell, Z.-P. Jiang, H. Li, L.-B. Gan and Z.-J. Shi, Chem. Commun., 2009, 6002 RSC.
- Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS PubMed.
- H. Li, Z. He, X. Guo, W. Li, X. Zhao and Z. Li, Org. Lett., 2009, 11, 4176 CrossRef CAS PubMed.
- X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848 CrossRef CAS PubMed.
- W. Wu and W. Su, J. Am. Chem. Soc., 2011, 133, 11924 CrossRef CAS PubMed.
- For selected recent examples of synthesis of oxindoles, please see: (a) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; (b) Y.-M. Li, M. Sun, H.-L. Wang, Q.-P. Tian and S.-D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed; (c) M.-B. Zhou, C.-Y. Wang, R.-J. Song, Y. Liu, W.-T. Wei and J.-H. Li, Chem. Commun., 2013, 49, 10817 RSC; (d) Y. Meng, L.-N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540 RSC; (e) S.-L. Zhou, L.-N. Guo, H. Wang and X.-H. Duan, Chem.–Eur. J., 2013, 19, 12970 CrossRef CAS PubMed; (f) X. Li, X. Xu, P. Hu, X. Xiao and C. Zhou, J. Org. Chem., 2013, 78, 7343 CrossRef CAS PubMed; (g) X.-H. Wei, Y.-M. Li, A.-X. Zhou, T.-T. Yang and S.-D. Yang, Org. Lett., 2013, 15, 4158 CrossRef CAS PubMed; (h) P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Chem.–Eur. J., 2013, 19, 14039 CrossRef CAS PubMed; (i) H. Wang, L.-N. Guo and X.-H. Duan, Adv. Synth. Catal., 2013, 355, 2222 CrossRef CAS; (j) M.-B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W. T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690 RSC; (k) W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, J. Org. Chem., 2013, 78, 12202 CrossRef CAS PubMed.
- K. Li, G. Tan, J. Huang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12942 CrossRef CAS PubMed.
- M. O. Ratnikov, X. Xu and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 9475 CrossRef CAS PubMed.
- (a) N. Takasu, K. Oisaki and M. Kanai, Org. Lett., 2013, 15, 1918 CrossRef CAS PubMed; (b) E. Shirakawa, N. Uchiyama and T. Hayashi, J. Org. Chem., 2011, 76, 25 CrossRef CAS PubMed.
- R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400 RSC.
- (a) R. Bhuyan and K. M. Nicholas, Org. Lett., 2007, 9, 3957 CrossRef CAS PubMed; (b) M. R. Fructos, S. Trofimenko, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2006, 128, 11784 CrossRef CAS PubMed.
- (a) J. D. Harden, J. V. Ruppel, G.-Y. Gao and X. P. Zhang, Chem. Commun., 2007, 4644 RSC; (b) G.-Y. Gao, J. D. Harden and X. P. Zhang, Org. Lett., 2005, 7, 3191 CrossRef CAS PubMed.
- (a) H. Lebel and K. Huard, Org. Lett., 2007, 9, 639 CrossRef CAS PubMed; (b) H. Lebel, K. Huard and S. Lectard, J. Am. Chem. Soc., 2005, 127, 14198 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2008, 10, 1863 CrossRef CAS PubMed.
- Q. Xia, W. Chen and H. Qiu, J. Org. Chem., 2011, 76, 7577 CrossRef CAS PubMed.
- Q. Xia and W. Chen, J. Org. Chem., 2012, 77, 9366 CrossRef CAS PubMed.
- S. Pan, J. Liu, H. Li, Z. Wang, X. Guo and Z. Li, Org. Lett., 2010, 12, 1932 CrossRef CAS PubMed.
- R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437 RSC.
- (a) D. Mansuy, Coord. Chem. Rev., 1993, 125, 129 CrossRef CAS; (b) B. Meunier, Chem. Rev., 1992, 92, 1411 CrossRef CAS.
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS PubMed; (b) S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532 CrossRef CAS PubMed.
- (a) A. A. Shteinman, Russ. Chem. Bull., 2001, 50, 1795 CrossRef CAS; (b) G. B. Maravin, M. V. Avdeev and E. I. Bagrii, Pet. Chem., 2001, 40, 3 Search PubMed.
- For selected reviews on Gif Chemistry, please see: (a) P. Stavropoulos, R. Çelenligil-Çetin and A. E. Tapper, Acc. Chem. Res., 2001, 34, 745 CrossRef CAS PubMed; (b) U. Schuchardt, M. J. D. M. Jannini, D. T. Richens, M. C. Guerreiro and E. V. Spinacé, Tetrahedron, 2011, 57, 2685 CrossRef; (c) D. H. R. Barton and B. Hu, Pure Appl. Chem., 1997, 69, 1941 CrossRef CAS; (d) D. H. R. Barton, Chem. Soc. Rev., 1996, 237 RSC; (e) M. J. Perkins, Chem. Soc. Rev., 1996, 229 RSC; (f) D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504 CrossRef CAS.
- For selected reviews, please see: (a) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (b) M. A. Bigi, P. Liu, L. Zou, K. N. Houk and M. C. White, Synlett, 2012, 2768 CAS; For recent contributions on Fe-catalyzed C–H oxidations by the White group, please see: (c) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052 CrossRef CAS PubMed; (d) M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721 CrossRef CAS PubMed; (e) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036 CrossRef CAS PubMed; (f) M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216 CrossRef CAS PubMed; (g) M. S. Chen and M. C. White, Science, 2010, 327, 566 CrossRef CAS PubMed; (h) N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078 CrossRef CAS; (i) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed.
- M. Nakanishi and C. Bolm, Adv. Synth. Catal., 2007, 349, 861 CrossRef CAS.
- J. De Houwer, K. A. Tehrani and B. U. W. Maes, Angew. Chem., Int. Ed., 2012, 51, 2745 CrossRef CAS PubMed.
- C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed.
- F. Szabó, B. Pethő, Z. Gonda and Z. Novák, RSC Adv., 2013, 3, 4903 RSC.
- T. Wang, W. Zhou, H. Yin, J.-A. Ma and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 10823 CrossRef CAS PubMed.
- Y. Wei, H. Ding, S. Lin and F. Liang, Org. Lett., 2011, 13, 1674 CrossRef CAS PubMed.
- For an example of 1,4-hydrogen transfer, please see: M. Journet and M. Malacria, Tetrahedron Lett., 1992, 33, 1893 CrossRef CAS.
- S. Iwata, T. Hata and H. Urabe, Adv. Synth. Catal., 2012, 354, 3480 CrossRef CAS.
- X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387 CrossRef CAS PubMed.
- U. A. Kshirsagar, R. Parnes, H. Goldshtein, R. Ofir, R. Zarivach and D. Pappo, Chem.–Eur. J., 2013, 19, 13575 CrossRef CAS PubMed.
- R. Parnes, U. A. Kshirsagar, A. Werbeloff, C. Regev and D. Pappo, Org. Lett., 2012, 14, 3324 CrossRef CAS PubMed.
- H.-P. Bi, W.-W. Chen, Y.-M. Liang and C.-J. Li, Org. Lett., 2009, 11, 3246 CrossRef CAS PubMed.
- For selected reviews on decarboxylative coupling reactions, please see: (a) L. J. Gooßen and K. Gooßen, Top. Organomet. Chem.: Inventing Reactions, Springer, 2013, vol. 44, p. 121 Search PubMed; (b) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC; (c) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS; (d) N. Rodríguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (e) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (f) L. J. Gooßen, F. Collet and K. Gooßen, Isr. J. Chem., 2010, 50, 617 CrossRef; (g) L. J. Gooßen, K. Gooßen, N. Rodríguez, M. Blanchot, C. Linder and B. Zimmermann, Pure Appl. Chem., 2008, 80, 1725 CrossRef; (h) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (i) S.-L. You and L.-X. Dai, Angew. Chem., Int. Ed., 2006, 45, 5246 CrossRef CAS PubMed.
- Z. Huang, L. Jin, Y. Feng, P. Peng, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7151 CrossRef CAS PubMed.
- D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153 CrossRef CAS PubMed.
- J. J. Scepaniak, A. M. Wright, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 19350 CrossRef CAS PubMed.
- U. A. Kshirsagar, C. Regev, R. Parnes and D. Pappo, Org. Lett., 2013, 15, 3174 CrossRef CAS PubMed.
- W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756 CrossRef CAS PubMed.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- (a) J. F. Van Humbeck, S. P. Simonovich, R. R. Knowles and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10012 CrossRef CAS PubMed; (b) T. H. Graham, C. M. Jones, N. T. Jui and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 16494 CrossRef CAS PubMed; (c) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124 CrossRef CAS PubMed.
- K. Liu, Y. Li, X. Zheng, W. Liu and Z. Li, Tetrahedron, 2012, 68, 10333 CrossRef CAS.
- F. Jia, K. Liu, H. Xi, S. Lu and Z. Li, Tetrahedron Lett., 2013, 54, 6337 CrossRef CAS.
- (a) H.-S. Dang and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1996, 2201 RSC; (b) H.-S. Dang and B. P. Roberts, J. Chem. Soc., Perkin Trans. 1, 1998, 67 RSC.
- K. Gilmore and I. V. Alabugin, Chem. Rev., 2011, 111, 6513 CrossRef CAS PubMed.
- S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928 CrossRef CAS PubMed.
- For the concept of BHAS, please see: A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed.
- D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef CAS PubMed.
- J. Wen, J. Zhang, S.-Y. Chen, J. Li and X.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 8897 CrossRef CAS PubMed.
- N. Uchiyama, E. Shirakawa, R. Nishikawa and T. Hayashi, Chem. Commun., 2011, 47, 11671 RSC.
- J. Wen, S. Qin, L.-F. Ma, L. Dong, J. Zhang, S.-S. Liu, Y.-S. Duan, S.-Y. Chen, C.-W. Hu and X.-Q. Yu, Org. Lett., 2010, 12, 2694 CrossRef CAS PubMed.
- K.-L. Wang, M.-Y. Lü, Q.-M. Wang and R.-Q. Huang, Tetrahedron, 2008, 64, 7504 CrossRef CAS.
- K. Wang, M. Lü, A. Yu, X. Zhu and Q. Wang, J. Org.Chem., 2009, 74, 935 CrossRef CAS PubMed.
- M. Ohta, M. P. Quick, J. Yamaguchi, B. Wünsch and K. Itami, Chem.–Asian J., 2009, 4, 1416 CrossRef CAS PubMed.
- Z. H. Guan, Z.-Y. Yan, Z.-H. Ren, X. Y. Liu and Y.-M. Liang, Chem. Commun., 2010, 46, 2823 RSC.
- (a) S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed; (b) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030 CrossRef CAS PubMed; (c) M. Sekine, L. Ilies and E. Nakamura, Org. Lett., 2013, 15, 714 CrossRef CAS PubMed; (d) L. Ilies, E. Konno, Q. Chen and E. Nakamura, Asian J. Org. Chem., 2012, 1, 142 CrossRef CAS; (e) L. Ilies, M. Kobayashi, A. Matsumoto, N. Yoshikai and E. Nakamura, Adv. Synth. Catal., 2012, 354, 593 CrossRef CAS; (f) N. Yoshikai, S. Asako, T. Yamakawa, L. Ilies and E. Nakamura, Chem.–Asian J., 2011, 6, 3059 CrossRef CAS PubMed; (g) L. Ilies, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 7672 CrossRef CAS PubMed; (h) A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 6557 CrossRef CAS PubMed; (i) N. Yoshikai, A. Mieczkowski, A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 132, 5568 CrossRef CAS PubMed; (j) N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Synlett, 2010, 313 CAS; (k) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS PubMed.
- T. M. U. Ton, C. Tejo, S. Tania, J. W. W. Chang and P. W. H. Chan, J. Org. Chem., 2011, 76, 4894 CrossRef CAS PubMed.
- A. Porcheddu and L. De Luca, Adv. Synth. Catal., 2012, 354, 2949 CrossRef CAS.
- T. Zhang and W. Bao, J. Org. Chem., 2013, 78, 1317 CrossRef CAS PubMed.
- J. Maes, T. R. M. Rauws and B. U. W. Maes, Chem.–Eur. J., 2013, 19, 9137 CrossRef CAS PubMed.
- T. Naveen, S. Maity, U. Sharma and D. Maiti, J. Org. Chem., 2013, 78, 5949 CrossRef CAS PubMed.
- (a) S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355 CrossRef CAS PubMed; (b) S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Chem. Commun., 2013, 49, 5286 RSC.
- T. Taniguchi, T. Fujii and H. Ishibashi, J. Org. Chem., 2010, 75, 8126 CrossRef CAS PubMed.
- C. F. Cross, E. J. Bevan and T. Herberg, Ber., 1900, 33, 2015 CrossRef CAS.
- K. Möller, G. Wienhöfer, K. Schöder, B. Join, K. Junge and M. Beller, Chem.–Eur. J., 2010, 16, 10300 CrossRef PubMed.
- For selected recent contributions on mechanism studies of oxidative coupling reactions, please see: (a) M. O. Ratnikov and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 1549 CrossRef CAS PubMed; (b) B. Schweitzer-Chaput, A. Sud, Á. Pintér, S. Dehn, P. Schulze and M. Klussmann, Angew. Chem., Int. Ed., 2013, 52, 13228 CrossRef CAS PubMed; (c) E. Böβ, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317 CrossRef PubMed; (d) E. Böβ, D. Sureshkumar, A. Sud, C. Wirtz, C. Farès and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106 CrossRef PubMed.
| This journal is © the Partner Organisations 2014 |