Chiral spiro iridium catalysts with SpiroPAP ligands: highly efficient for asymmetric hydrogenation of ketones and ketoesters
Xiao-Hui
Yang
,
Jian-Hua
Xie
* and
Qi-Lin
Zhou
*
State Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Collaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300071, China. E-mail: jhxie@nankai.edu.cn; qlzhou@nankai.edu.cn; Fax: +86 22 2350 6177; Tel: +86 22 2350 0011
First published on 16th January 2014
Abstract
This tutorial account describes the design of chiral spiro iridium catalysts bearing spiro pyridine–aminophosphine ligands, and their preparation and application in the asymmetric hydrogenation of ketones and β-aryl-β-ketoesters.
 Xiao-Hui Yang | Xiao-Hui Yang was born in Hebei Province, China, in 1987. She received her BS degree in chemistry from Jiangsu Normal University in 2010. She is now studying at the Institute of Elemento-organic Chemistry under the supervision of Prof. Qi-Lin Zhou in Nankai University. Her research interests focus on catalytic asymmetric catalysis and synthesis of biologically active compounds. |
 Jian-Hua Xie | Jian-Hua Xie was born in 1968. He received his PhD in 2003 under the supervision of Professor Qi-Lin Zhou at the Institute of Elemento-Organic Chemistry, Nankai University. He then worked in the same institute and was promoted to associate professor in 2005 and to full professor in 2010. In 2007, he spent one year as a postdoctoral fellow in Prof. Michael Doyle's group at Maryland University. His research interests focus on asymmetric catalysis and asymmetric synthesis of natural products and chiral drugs. |
 Qi-Lin Zhou | Qi-Lin Zhou obtained his PhD degree at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 1987. After several years of postdoctoral research (with Prof. Klaus Müllen, Prof. Andreas Pfaltz and Prof. Michael Doyle) he started his independent research in East China University of Science and Technology, China in 1996. In 1999, he moved to the Institute of Elemento-organic Chemistry, Nankai University as Cheung Kong Scholar. His research interests include organometallics, asymmetric catalysis and synthesis of biologically active compounds. |
Introduction
Homogeneous catalytic asymmetric hydrogenation is one of the most efficient and convenient methods for the preparation of optically active compounds, and has been applied in the industrial synthesis of pharmaceuticals and agrochemicals.1 Recently we developed a new type of chiral spiro iridium catalysts bearing tridentate spiro pyridine–aminophosphine (abbreviated as SpiroPAP, Fig. 1) ligands and found these chiral iridium catalysts were extremely efficient for the hydrogenation of ketones and β-ketoesters, producing the corresponding chiral alcohols and chiral β-hydroxy esters in excellent yields and enantioselectivities (up to 99.9% ee) with TON's up to millions (ca. 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550000).2 This highly efficient catalytic asymmetric hydrogenation has been successfully applied in the synthesis of pharmaceuticals and pharmaceutical intermediates. In this tutorial account, we describe the design and synthesis of chiral iridium catalysts Ir-(R)-SpiroPAP and their applications in the asymmetric hydrogenation of ketones and β-ketoesters.
550000).2 This highly efficient catalytic asymmetric hydrogenation has been successfully applied in the synthesis of pharmaceuticals and pharmaceutical intermediates. In this tutorial account, we describe the design and synthesis of chiral iridium catalysts Ir-(R)-SpiroPAP and their applications in the asymmetric hydrogenation of ketones and β-ketoesters.
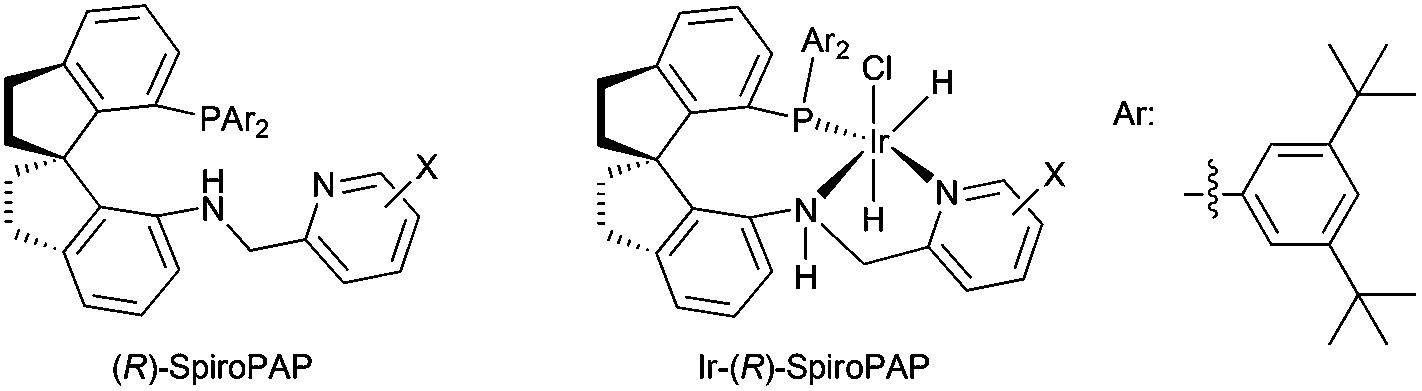 | ||
| Fig. 1 Chiral spiro pyridine–aminophosphine ligands and their iridium complexes. | ||
Results and discussion
Since Noyori and co-workers reported that chiral RuCl2(diphosphine)(diamine) complexes were highly efficient catalysts for the asymmetric hydrogenation of ketones,3 research on the catalytic asymmetric hydrogenation of ketones has been focused on the development of chiral ruthenium catalysts bearing diphosphine and diamine ligands.4 Recently, we developed a new type of chiral spiro ligand containing phosphine and amine moieties, named as SpiroAP (Fig. 2), and found their iridium complexes were highly active for the asymmetric hydrogenation of ketones.5 The turnover frequencies (TOF) of the hydrogenation were as high as 37000 h−1, however, the turnover numbers (TON) reached only 10000.5b The Ir-SpiroAP catalyst was unstable under a hydrogen atmosphere and quickly transformed into an inactive iridium dihydride species with two SpiroAP ligands. To prohibit the coordination of the second SpiroAP ligand to the iridium atom of the catalyst, we introduced an additional coordinating pyridine moiety to the SpiroAP, making it a tridentate ligand named SpiroPAP.2a This modification led us to develop a novel chiral spiro iridium catalyst with exceptionally high stability and activity for the hydrogenation of ketones.
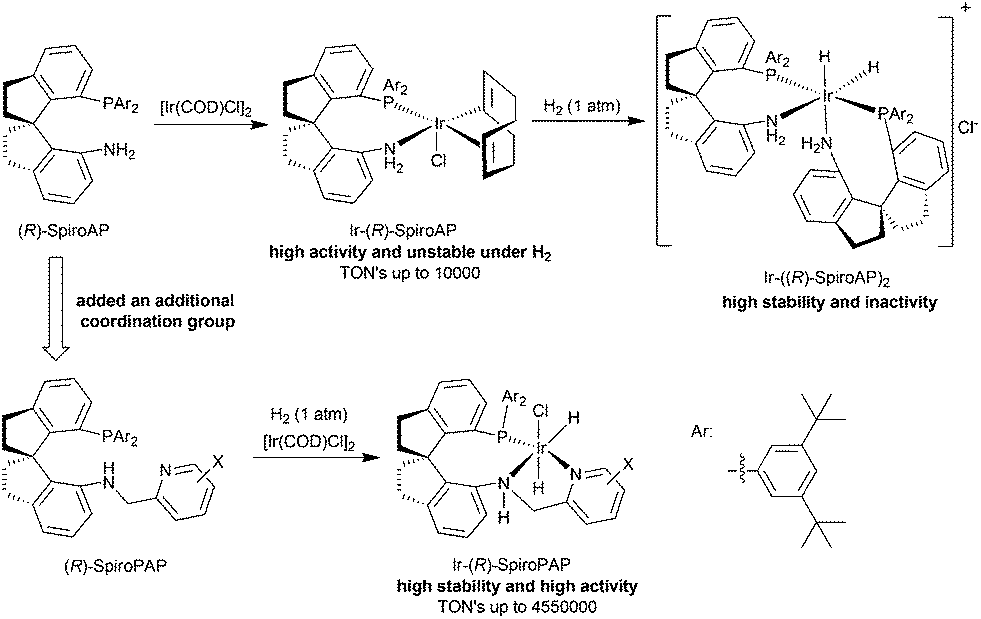 | ||
| Fig. 2 The design and synthesis of chiral spiro pyridine–aminophosphine ligands and their iridium complexes. | ||
The Ir-(R)-SpiroPAP catalyst with 3,5-tert-butyl groups on the P-phenyl rings and a 3-methyl group on the pyridinyl ring was demonstrated to be the best choice.2a Under the optimal reaction conditions (0.02 mol% Ir-(R)-SpiroPAP, [KOtBu] = 0.02 M; [substrate] = 2.1 M, 10 atm of initial H2 pressure, in anhydrous EtOH at room temperature for 20 min to 4 hours), a series of alkyl aryl ketones were hydrogenated to chiral alcohols in quantitative yields with 96–99.9% ee (Fig. 3). This is an extremely efficient asymmetric hydrogenation and the TON's are up to millions. For example, in the asymmetric hydrogenation of acetophenone the catalyst loading can be reduced to 0.0001 mmol% (S/C = 1000000) and 100% conversion with 98% ee was obtained under 50 atm of initial H2 pressure at room temperature for 30 hours. On further reduction of the catalyst loading to 0.00002 mol% (S/C = 5000000) the hydrogenation gave 91% conversion under an initial 100 atm of H2 pressure at room temperature for 15 days. The enantioselectivity of the reaction remained at 98% ee, showing that the catalyst was very stable.
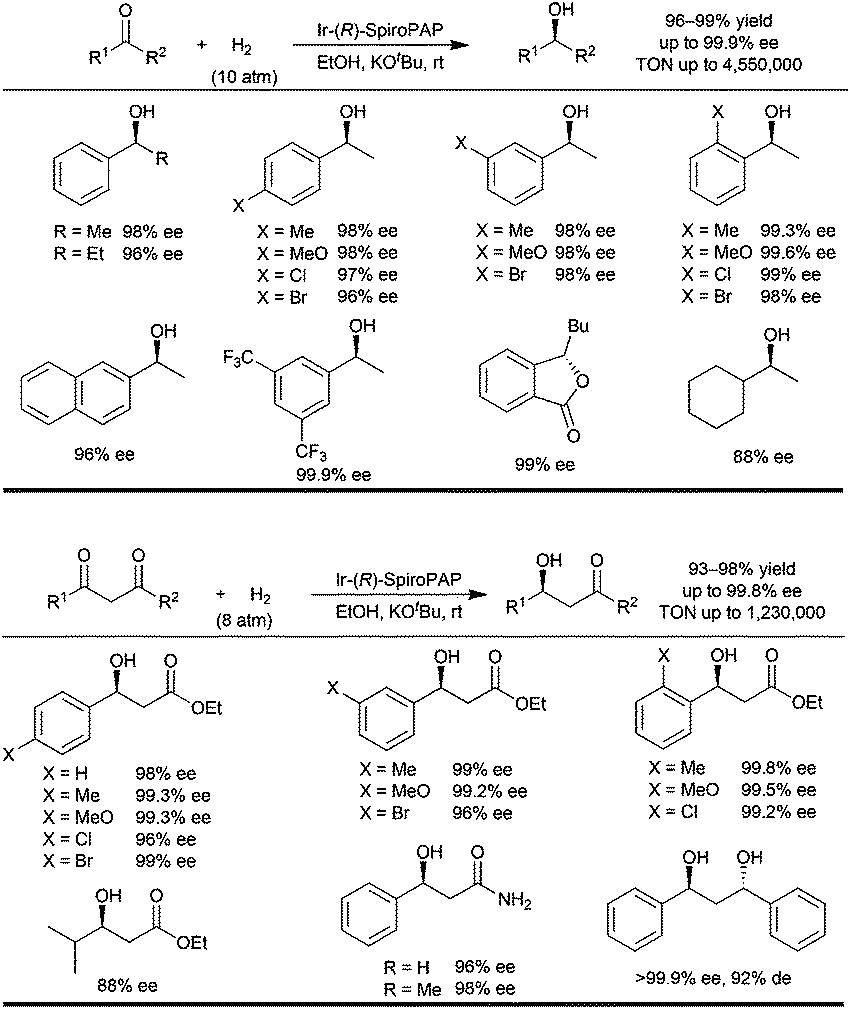 | ||
| Fig. 3 The asymmetric hydrogenation of ketones and ketoesters with an Ir-(R)-SpiroPAP catalyst. | ||
Encouraged by these results we investigated the catalysts Ir-(R)-SpiroPAP for the hydrogenation of functionalized ketones such as β-ketoesters. Previous reports in the literature have shown that the chiral RuX2(diphosphine) (X = Cl or Br) complexes are by far the most popular catalysts for the hydrogenation of β-ketoesters,4 and the RuCl2(diphosphine)(diamine) complexes are inert for this hydrogenation.3a The major reason for the inertness may be that the RuCl2(diphosphine)(diamine) catalyst needs a strong base such as KOtBu to activate it. However, the strong base enolizes the β-ketoester substrate instead of activating the catalyst. Because the aromatic N–H of the catalysts Ir-SpiroPAP is more acidic than the aliphatic N–H of the catalysts RuCl2(diphosphine)(diamine), the catalysts Ir-SpiroPAP can be activated by a relatively weak base such as the enolate salt of a β-ketoester. We evaluated the catalysts Ir-SpiroPAP for the hydrogenation of β-ketoesters and found they are also highly efficient for the hydrogenation of the challenging substrates β-aryl-β-ketoesters.
Under the optimal reaction conditions (0.1 mol% Ir-(R)-SpiroPAP, [KOtBu] = 0.02 M; [substrate] = 1 M, 8 atm of initial H2 pressure, in anhydrous EtOH at room temperature for 25 min to 4 hours), a range of β-aryl-β-ketoesters were hydrogenated by the catalyst Ir-(R)-SpiroPAP to yield the corresponding chiral β-hydroxy esters in 93–98% yields with 96–99.8% ee (Fig. 3).2b The TON's of these hydrogenations were up to millions. For example, when the catalyst loading was reduced to 0.001 mol% (S/C = 100000), the hydrogenation of ethyl 3-oxo-3-phenylpropanoate under 50 atm of initial H2 pressure at room temperature for 19 hours afforded the chiral ethyl 3-hydroxy-3-phenylpropanoate in 98% yield with 98% ee. The catalyst loading can be further reduced to as low as 0.00067% mol% (S/C = 1500000) and the hydrogenation reaction gave 98% ee and 82% conversion (TON = 1230000), however the reaction needed a longer time (96 hours) and higher H2 pressure (100 atm, initial, at 50 °C). The chiral iridium catalysts with a tridentate spiro pyridine–aminophosphine ligand (SpiroPAP) showed the highest TON's for hydrogenations with molecular catalysts.6
The chiral iridium dihydride catalysts with spiro pyridine–aminophosphine SpiroPAP ligand were easily prepared by the reaction of [Ir(COD)Cl]2 with a small excess of SpiroPAP ligands under ambient H2 pressure at room temperature for 1 hour. The light yellow powder obtained by removing the solvent under reduced pressure can be directly used without further purification. The catalysts Ir-(R)-SpiroPAP are stable in air for several days and can be stored under inert atmosphere for several months without losing activity and selectivity, which is extremely convenient for manipulating the hydrogenation. However, the catalysts Ir-(R)-SpiroPAP are unstable in solution, losing activity in air in few hours. The hydrogenations of ketones and β-ketoesters using Ir-SpiroPAP were also easy to perform, even at low catalyst loading. However, to ensure the reproducibility of the aforementioned hydrogenations in the laboratory, the solvents and the liquid substrates are preferably distilled, but there is no need to degas them prior to use. This is mainly because the reactions use very small amounts of catalyst (usually less than 2.0 mg) and any impurities, especially acidic impurities contained in the solvents or liquid substrates, may lead to the deactivation of the catalyst.
This iridium-catalysed asymmetric hydrogenation reaction is practically useful. Zhejiang Jiuzhou Pharmaceutical Co. has applied this reaction for the industrial scale-up preparation of rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer's disease.7 The asymmetric hydrogenation of 3-hydroxyacetophenone could be performed on a 25 kg-scale at S/C = 100000 by using an Ir-(S)-SpiroPAP catalyst. (R)-3-(1-Hydroxyethyl)phenol was produced in 91% yield with 96% ee.
Conclusion
In summary, we have developed a new strategy for preventing the deactivation of the chiral iridium catalysts Ir-SpiroAP by introducing an additional coordinating group, which leads to the development of the extremely efficient chiral spiro catalysts Ir-SpiroPAP. These chiral spiro iridium catalysts are highly efficient for the hydrogenation of both simple ketones and β-ketoesters, offering the corresponding chiral alcohols in excellent yields and enantioselectivities with TON's as high as 4550000. The high stability, high activity and high enantioselectivity of the catalysts Ir-SpiroPAP, and the easy performance of the reaction, make this asymmetric hydrogenation practically useful.
Practical procedure
The typical procedures for the preparation of catalyst Ir-SpiroPAP and the asymmetric hydrogenation of acetophenone at S/C = 1000000 are described in Fig. 4.
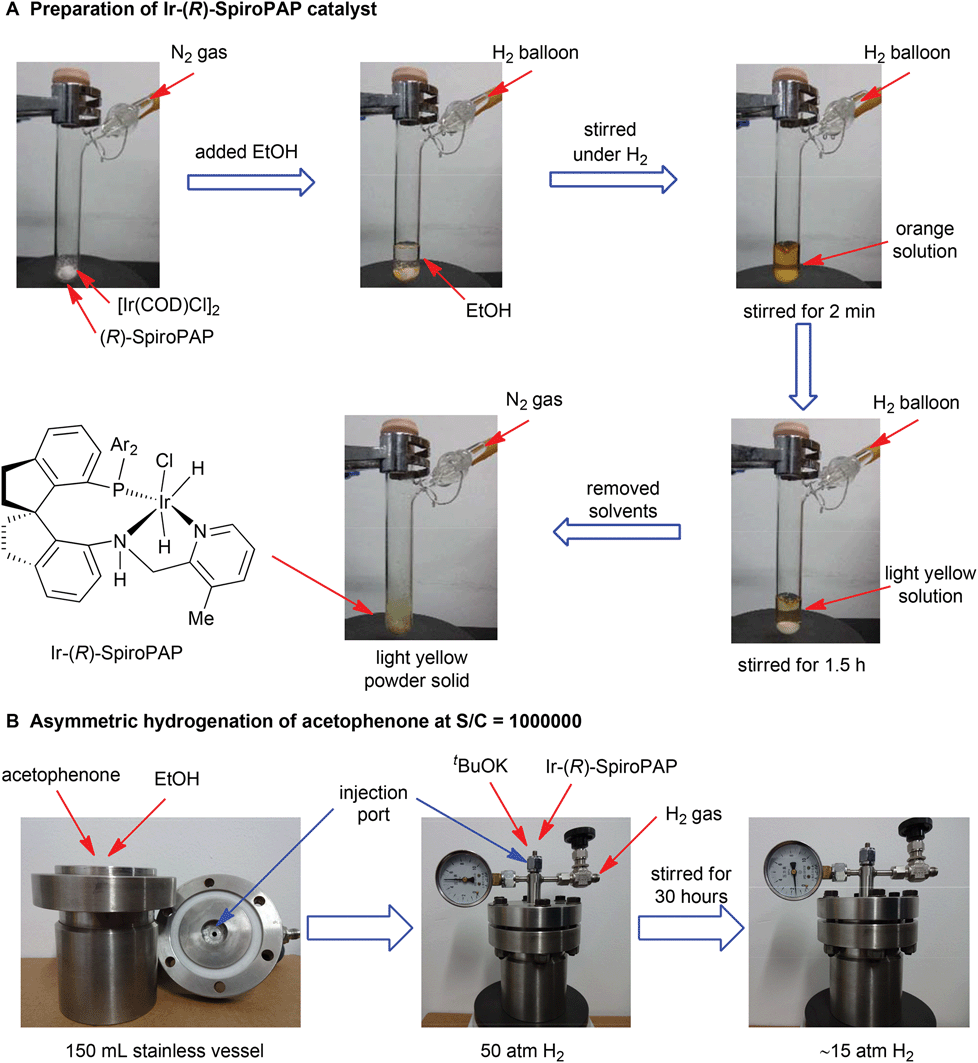 | ||
| Fig. 4 The preparation of Ir-SpiroPAP catalyst and asymmetric hydrogenation of acetophenone at S/C = 1000000. | ||
Preparation of Ir-(R)-SpiroPAP
A dry Schlenk tube (25 mL) was charged with 70.5 mg of (R)-SpiroPAP (0.094 mmol) and 30 mg of [Ir(COD)Cl]2 (0.045 mmol) and was purged with H2 three times. After addition of 6 mL EtOH (dried by refluxing over magnesium turnings and distilled under nitrogen), the reaction mixture was stirred at room temperature. The colour of the reaction solution changed from orange to light yellow after stirring for 1.5 hours. The solvent was removed under reduced pressure and the solid was dried under vacuum for 5 h to yield Ir-(R)-SpiroPAP as a light yellow powder in quantitative yield. The obtained Ir-(R)-SpiroPAP was used directly without further purification, and usually stored under inert atmosphere at room temperature.Asymmetric hydrogenation of acetophenone at S/C = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 000000
000000
To a stainless steel vessel (150 mL) were added 18.0 g (150 mmol) of acetophenone (purified by distillation over anhydrous K2CO3 to remove acidic substances), 20 mL anhydrous EtOH, a solution of t-BuOK (280 mg, 2.5 mmol) in 5 mL anhydrous EtOH and a solution of Ir-(R)-SpiroPAP (1.0 mL, 0.15 μmol mL−1, 0.15 μmol) in anhydrous EtOH via an injection port under nitrogen atmosphere. The vessel was then closed and quickly purged with hydrogen gas three times. After the vessel was pressurized to 50 atm with H2 gas the reaction mixture was stirred at room temperature (25–30 °C) for 30 hours until no obvious hydrogen pressure drop was observed (∼15 atm). After releasing the hydrogen pressure carefully, a small amount of the reaction mixture was filtered through a short silica gel column, and the filtrate was analyzed by GC to determine the conversion (100% conv.). The solvent of the reaction mixture was removed under reduced pressure and the residue was purified by flash chromatography to yield (S)-1-phenylethanol as a colourless liquid (17.9 g, 98% yield). The ee value of the product was 98% ee (S) determined by GC equipped with a chiral column Supelco β-DEX™ 225 (GC conditions: N2, 2 mL min−1; injection temp., 250 °C; initial column temperature, 100 °C; progress rate, 1 °C min−1; final column temperature, 160 °C; tR(R) = 9.77 min; tR(S) = 9.98 min).
Acknowledgements
We thank the National Natural Science Foundation of China, the National Basic Research Program of China (973 Program) (2012CB821600), and the “111” project (B06005) of the Ministry of Education of China for financial support.Notes and references
- (a) T. Ohkuma, M. Kitamura and R. Noyori, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-Interscience, New York, 2nd edn, 2000, pp. 1–110 Search PubMed; (b) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (d) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin, 2012, 70, 1427 Search PubMed.
- (a) J.-H. Xie, X.-Y. Liu, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7329 CrossRef CAS PubMed; (b) J.-H. Xie, X.-Y. Liu, X.-H. Yang, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 201 CrossRef CAS PubMed.
- (a) T. Ohkuma, H. Ooka, S. Hashiguchi, I. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (b) H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703 CrossRef CAS; (c) B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed.
- T. Ohkuma and R. Noyori, in Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, p. 1105 Search PubMed.
- (a) J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS PubMed; (b) J.-B. Xie, J.-H. Xie, X.-Y. Liu, Q.-Q. Zhang and Q.-L. Zhou, Chem.–Asian J., 2011, 6, 899 CrossRef CAS PubMed.
- N. Arai and T. Ohkuma, Chem. Rec., 2012, 12, 284 CrossRef CAS PubMed.
- P.-C. Yan, G.-L. Zhu, J.-H. Xie, X.-D. Zhang, Q.-L. Zhou, Y.-Q. Li, W.-H. Shen and D.-Q. Che, Org. Process Res. Dev., 2013, 17, 307 CrossRef CAS.
| This journal is © the Partner Organisations 2014 |