Magnetic polyaniline nanocomposites toward toxic hexavalent chromium removal†
Hongbo
Gu
ab,
Sowjanya B.
Rapole
ac,
Jaishri
Sharma
c,
Yudong
Huang
b,
Dongmei
Cao
d,
Henry A.
Colorado
e,
Zhiping
Luo
f,
Neel
Haldolaarachchige
g,
David P.
Young
g,
Bryan
Walters
h,
Suying
Wei
*c and
Zhanhu
Guo
*a
aIntegrated Composites Lab (ICL), Dan F. Smith Department of Chemical Engineering, Lamar University, Beaumont, TX 77710, USA. E-mail: zhanhu.guo@lamar.edu (Z.G.)
bSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin, Heilongjiang 150001, China
cDepartment of Chemistry and Biochemistry, Lamar University, Beaumont, TX 77710, USA. E-mail: suying.wei@lamar.edu (S.W.)
dMaterial Characterization Center, Louisiana State University, Baton Rouge, LA 70803, USA
eMaterials Science and Engineering, University of California, Los Angeles, CA 90066, USA
fDepartment of Chemistry and Physics, Fayetteville State University, Fayetteville, NC 28301, USA
gDepartment of Physics and Astronomy, Louisiana State University, Baton Rouge, LA 70803, USA
hEarth Analytical Sciences, Inc., Beaumont, TX 77705, USA
First published on 17th September 2012
Abstract
The removal of toxic hexavalent chromium (Cr(VI)) from polluted water by magnetic polyaniline (PANI) polymer nanocomposites (PNCs) was investigated. The PNCs were synthesized using a facile surface initiated polymerization (SIP) method and demonstrated unique capability to remove Cr(VI) from polluted solutions with a wide pH range. Complete Cr(VI) removal from a 20.0 mL neutral solution with an initial Cr(VI) concentration of 1.0–3.0 mg L−1 was observed after a 5 min treatment period with a PNC load of 10 mg. The PNC dose of 0.6 g L−1 was found to be sufficient for complete Cr(VI) removal from 20.0 mL of 9.0 mg L−1 Cr(VI) solution. The saturation magnetization was observed to have no obvious decrease after treatment with Cr(VI) solution, and these PNCs could be easily recovered using a permanent magnet and recycled. The Cr(VI) removal kinetics were determined to follow pseudo-first-order behavior with calculated room temperature pseudo-first-order rate constants of 0.185, 0.095 and 0.156 min−1 for the solutions with pH values of 1.0, 7.0 and 11.0, respectively. The Cr(VI) removal mechanism was investigated by Fourier transform infrared spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS) and energy-filter transmission electron microscopy (EFTEM). The results showed that PANI was partially oxidized after treatment with Cr(VI) solution, with Cr(VI) being reduced to Cr(III). The EFTEM observation indicated that the adsorbed Cr(III) had penetrated into the interior of the PNCs instead of simply adsorbing on the PNC surface. This synthesized material was found to be easily regenerated by 1.0 mol L−1p-toluene sulfonic acid (PTSA) or 1.0 mol L−1 hydrochloric acid (HCl) and efficiently reused for further Cr(VI) removal.
1. Introduction
Chromium, one of the heavy metals, is generated from a wide range of anthropogenic sources in effluent streams including the chrome plating, stainless steel, textile dying, pigment, wood preservation, tanning and anti-corrosion fields.1–5 Chromium with an electron configuration of 4s13d5 is a highly reactive and labile element owing to its low energy of high spin configuration. Though Cr(VI) and Cr(III) are the most commonly observed among the chromium compounds, they have a great difference in toxicity and bioactivity. Cr(III), an energetically stable and inert species in the environment, is required for mammals in trace amounts for sugar and lipid metabolism. Being much more labile owing to its high redox potential (Eo = 1.33 V),6 extremely poisonous and notoriously mobile, Cr(VI) has been widely detected in groundwater with a wide range of pH at abandoned industrial sites, causing carcinogenicity, skin lesions and lung cancers upon excessive exposure.7 The US Environmental Protection Agency (EPA) has recommended a maximum limit of 0.1 mg L−1 for total chromium in drinking water to reduce human exposure to toxic chromium.8,9A variety of methods and materials have been developed for environmental cleanup and remediation of chromium from wastewater, such as adsorption,10 electrochemical precipitation,11 ion exchange, reverse osmosis, membrane filtration,9 and biomaterials.12 Although these methods are efficient for chromium removal, the cost is relatively high.13 Reduction of Cr(VI) to Cr(III) is reported to be an effective means to counter the harmful effects of Cr(VI) on the environment14,15 and could be achieved with the oxidation of numerous reductants including zero-valent iron,15,16 divalent iron,17 organic compounds,18 biomaterial,19 hydrogen sulfide20 and hydrogen peroxide.21 The reduction is strongly dependent on the pH value and initial Cr(VI) concentration of the solution.20
As multifunctional materials, conducting polymers have become attractive owing to their wide potential applications including rechargeable batteries,22,23 sensors,24,25 electrochromic devices,26,27 corrosion inhibitors,28 selective ion-transport switchable membranes29,30 and smart structural materials.31–34 Cr(VI) removal with conducting polymers has gained more interest due to its reversibility and high efficiency. For example, complete Cr(VI) removal by electrosynthesized polypyrrole films35 and Cr(VI) removal by polyaniline (PANI) have been reported.36–38 The conjugated PANI has three different structures,39i.e., “leucoemeraldine” (LEB), “emeraldine” (EB) and “pernigraniline” (PB).40 Olad et al.36 have studied the efficiency and kinetics of Cr(VI) removal by various forms of PANI, including film and powder at different oxidation states. Compared with powder form, the film form is difficult to prepare in a large amount in a short time. Meanwhile, films with a smaller specific surface area might have a relatively lower activity with only the external film surface accessible for Cr(VI) removal, which limits the penetration of Cr(VI) into the film interior.35 PANI powders with rough surface may be a good candidate for highly efficient Cr(VI) removal due to their large specific surface area, low cost and easy bulk production.41–43 However, to recycle the PANI powders after Cr(VI) treatment is still a challenge and the reaction mechanisms between PANI and Cr(VI) are still unclear.
Compared with other magnetic nanoparticles (NPs),44 magnetite (Fe3O4) has become more attractive due to its low toxicity, relatively high saturation magnetization (Ms, 92–100 emu g−1 at room temperature)45 and biocompatibility,45 and its admirable applications for sensors,46 catalysts,47 and adsorbents for the removal of hazardous heavy metals.48,49 However, Fe3O4 with a spinel structure composed of a cubic close packed oxygen array50 is easily susceptible to dissolution in acids.9 A protective polymer,51 carbon9,52 or noble metal shell is used to coat magnetic NPs for long-term applications.53 Meanwhile, magnetic NPs enable the recycling of these materials9,54 and the introduced shell, for example carbon, could enhance the Cr(VI) removal.9 Though Bhaumik et al.2 have reported Cr(VI) removal by magnetic polypyrrole (PPy)/Fe3O4 nanocomposites, the removal mechanism was not disclosed.
A great deal of research has focused on the fabrication of PANI/Fe3O4 PNCs using template–free methods55 and in situ polymerization56 in the application of microwave adsorption.57 There is no report of PANI/Fe3O4 PNCs applied to chromium removal so far. In this paper, PANI polymer nanocomposites (PNCs) filled with Fe3O4 NPs were synthesized by using a surface-initiated-polymerization (SIP) method and their function to remove Cr(VI) from wastewater was explored. The effects of initial Cr(VI) concentration, solution pH value and PNC dose on Cr(VI) removal have been studied. The Cr(VI) removal mechanism was interpreted by analysis from FT-IR, XPS, EFTEM and temperature dependent resistivity. The room temperature kinetics were explored by studying the Cr(VI) concentration change with the treatment period. The recycling capability, regeneration and reuse of the PNCs were also investigated.
2. Experimental section
2.1 Materials
Aniline (C6H7N), ammonium persulfate (APS, (NH4)2S2O8), p-toluene sulfonic acid (PTSA, C7H8O3S) were purchased from Sigma Aldrich. Potassium dichromate (K2Cr2O7) and 1,5-diphenylcarbazide (DPC) were purchased from Alfa Aesar Company. Phosphoric acid (H3PO4, 85 wt%) was obtained from Fisher Scientific. Fe3O4 NPs with an average size of 12 nm were obtained from Nanjing Emperor Nano Material Co., Ltd. All the chemicals were used as received without any further treatment.2.2 Preparation of PANI/Fe3O4 PNCs
PANI/Fe3O4 PNCs with a nanoparticle loading of 30 wt% were fabricated by a SIP method.58 Briefly, Fe3O4 NPs (1.44 g), PTSA (30.0 mmol) and APS (18.0 mmol) were added into 200.0 mL deionized water in an ice-water bath for one hour of sonication. Then the aniline aqueous solution (36.0 mmol in 50.0 mL deionized water) was mixed with the above Fe3O4 nanoparticle suspension and sonicated continuously for an additional hour in an ice-water bath for further polymerization. The product was vacuum filtered and washed with deionized water until the pH was about 7 and was further washed with methanol to remove any possible oligomers. The final PNC powders were dried at 50 °C overnight.2.3 Cr(VI) removal by PANI/Fe3O4 PNCs
The final concentration of Cr(VI) was determined by colorimetric method (ESI†)59 using the obtained standard fitting equation: A = 9.7232 × 10−4C;54 where C is the concentration of Cr(VI) and A is the absorbance obtained from the UV-vis test.The pH value effect on Cr(VI) removal by the synthesized PNCs was investigated by selecting solutions with a pH value of 1.0, 2.0, 3.0, 5.0, 7.0, 9.0, and 11.0. The initial pH value of Cr(VI) solutions was adjusted by NaOH (1.0 mol L−1) and HCl (1.0 mol L−1) with a pH meter (Vernier LabQuest with pH-BTA sensor). The PNCs (10.0 mg) were ultrasonically (Branson 8510) dispersed in 20.0 mL Cr(VI) solutions (4.0 mg L−1) for 5 min. Then this solution was taken out and centrifuged (Fisher Scientific, Centrific 228) for Cr(VI) concentration determination. The effect of initial Cr(VI) concentration on the Cr(VI) removal was investigated by using PNCs (10.0 mg) to treat Cr(VI) solutions (20.0 mL, pH = 7.0) with Cr(VI) concentration varying from 1.0 to 10.0 mg L−1 for 5 min. The effect of the synthesized PNC dose on Cr(VI) removal was studied by using PNCs with loading from 0.25 to 0.75 g L−1 to treat 20.0 mL Cr(VI) neutral solutions with a Cr(VI) concentration of 9.0 mg L−1 for 5 min. For comparison, pure Fe3O4 NPs (10.0 mg) were used to treat 20.0 mL Cr(VI) neutral solution with an initial Cr(VI) concentration of 1.0 mg L−1 for 5 min. For kinetic study, the synthesized PNCs (10.0 mg) were used to treat 20.0 mL Cr(VI) solutions with an initial Cr(VI) concentration of 12.0, 9.0 and 5.0 mg L−1 at pH = 1.0, 7.0 and 11.0 for different treatment periods. The Cr(VI) removal tests were all conducted at room temperature.
The Cr(VI) removal percentage (R%) is calculated using eqn (1):
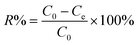 | (1) |
 | (2) |
2.4 Characterization
The chemical structure of the products was obtained by Fourier transform infrared spectroscopy (FT-IR, a Bruker Inc. Vector 22 coupled with an ATR accessory) in the range of 500 to 4000 cm−1 at a resolution of 4 cm−1. The morphologies of the NPs and PNCs were observed on a JSM-6700F system with a JEOL field emission scanning electron microscope (SEM) and were further characterized by a transmission electron microscopy (TEM, FEI Tecnai G2 F20) with a field emission gun, operated at an accelerating voltage of 200 kV. The TEM samples were prepared by drying a drop of ethanol suspension on carbon-coated copper TEM grids.The thermal stability of the PNCs and the nanoparticle weight percentage in the PNCs were determined by thermo-gravimetric analysis (TGA, TA instruments, Q-500) with a heating rate of 10 °C min−1 under an air flow rate of 60 mL min−1 from 25 to 700 °C.
The X-ray photoelectron spectroscopy (XPS) measurements were performed in a Kratos AXIS 165 XPS/AES instrument using monochromatic Al K radiation to see the elemental compositions. The N1s, C1s and Cr2p peaks were deconvoluted into the components consisting of a Gaussian line shape Lorentzian function (Gaussian = 80%, Lorentzian = 20%) on Shirley background.
Brunauer–Emmett–Teller (BET) was used to measure the specific surface area of the PNCs before and after the treatment with Cr(VI). BET adsorption and desorption isotherms were obtained using a surface area analyzer (NOVA 1000 Series, Quantachrome). The as-synthesized PNCs and the PNCs treated with 20.0 mL Cr(VI) solution (4.0 mg L−1) were weighed and placed inside the sample holder cell with a known volume. The refrigerant used was liquid nitrogen placed in a vacuum Dewar at about 77 K and the carrier gas was N2 (ultrahigh purity grade, Airgas).
Inductively coupled plasma optical emission spectrometry (ICP-OES, SPECTRO GENESIS ICP spectrometer) was used to determine the Cr concentration in the solution. The calibration curve of Cr was established using 5 points of calibration (blank, 0.003, 0.03, 3 and 12 ppm). After the calibration, various quality control (QC) samples were used to analyze and determine the accuracy of the calibration. Then the supernatant liquids of the PNCs samples after treatment with Cr(VI) were analyzed to determine the concentration of total Cr in the solution.
The resistivity (ρ) was measured by a standard four-probe method from 50 to 290 K. The PNC powders were pressed in a form of disc pellet with a diameter of 25 mm by applying a pressure of 50 MPa in a hydraulic presser and the average thickness was about 1.0 mm. (The temperature dependent resistivity has been mentioned in the ESI.†) The magnetic properties were investigated in a 9 Tesla Physical Properties Measurement System (PPMS) by Quantum Design at room temperature.
3. Results and discussion
3.1 Structure determination of the PANI/Fe3O4 PNCs
Fig. 1 shows the SEM and TEM microstructures of the as-received Fe3O4 NPs and the synthesized PNCs. Compared with pure Fe3O4 NPs, Fig. 1(a), the surface of the Fe3O4 NPs in the PNCs became rougher, Fig. 1(c), and a thin PANI layer was observed surrounding the nanoparticle, Fig. 1(d), indicating that polymerization occurred on the surface of the Fe3O4 NPs.60 The TEM microstructures, Fig. 1(b), show that the average diameter of the as-received Fe3O4 NPs is about 12 nm, which is consistent with the information obtained from the company. The clear lattice fringes observed in Fig. 1(b) and (d) indicate the high crystallization of the Fe3O4 NPs. The selected area electron diffraction (SAED) is introduced to identify the crystal structure of the synthesized PNCs, inset of Fig. 1d. The calculated d-spacing values of 1.92, 2.40, 2.87, 3.73 and 4.04 Å correspond to the (220), (311), (400), (422) and (511) crystallographic planes of the spinel phase Fe3O4.61,62 These indicate that Fe3O4 NPs have been successfully embedded in the PANI without dissolution in acid via the SIP method combining with the ultrasonication. | ||
| Fig. 1 (a) SEM and (b) TEM images of the as-received Fe3O4 NPs; (c) SEM image and (d) HRTEM image and selected area electron diffraction (SAED) pattern (inset picture) of the synthesized PNCs. | ||
In the FT-IR spectrum, Fig. 2(a), the strong absorption peaks at 1560 and 1482 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of NQN (Q = quinoid ring) and N–B–N (B = benzenoid ring), respectively.63 The peak at 1292 cm−1 is related to the C–N stretching vibration of the benzenoid unit. The peaks at 1238 and 674cm−1 are assigned to the C–H stretching vibration of the quinoid rings and the out-of-plane C–H vibration.60,64,65 The peak at around 792 cm−1 is due to the out-of-plane bending of C–H in the substituted benzenoid ring. These characteristic peaks (1560, 1482, 1292, and 1238 cm−1) prove that PANI in the synthesized PNCs appears as the EB form.41 The TGA curve, Fig. 2(b), shows a significant weight loss before 120 °C, which is attributed to the loss of moisture in the synthesized PNCs. There are two-stage weight losses for the samples at 250 and 600 °C, which are due to the elimination of dopant anions and thermal degradation of the PANI chains, respectively.66,67 The calculated initial nanoparticle loading based on the total mass of aniline and NPs is 30 wt%. However, the calculated nanoparticle loading from TGA is 33.1 wt%, Fig. 2(b), which is due to the incomplete polymerization of aniline and is also observed in WO3/PANI PNCs.63
C stretching vibration of NQN (Q = quinoid ring) and N–B–N (B = benzenoid ring), respectively.63 The peak at 1292 cm−1 is related to the C–N stretching vibration of the benzenoid unit. The peaks at 1238 and 674cm−1 are assigned to the C–H stretching vibration of the quinoid rings and the out-of-plane C–H vibration.60,64,65 The peak at around 792 cm−1 is due to the out-of-plane bending of C–H in the substituted benzenoid ring. These characteristic peaks (1560, 1482, 1292, and 1238 cm−1) prove that PANI in the synthesized PNCs appears as the EB form.41 The TGA curve, Fig. 2(b), shows a significant weight loss before 120 °C, which is attributed to the loss of moisture in the synthesized PNCs. There are two-stage weight losses for the samples at 250 and 600 °C, which are due to the elimination of dopant anions and thermal degradation of the PANI chains, respectively.66,67 The calculated initial nanoparticle loading based on the total mass of aniline and NPs is 30 wt%. However, the calculated nanoparticle loading from TGA is 33.1 wt%, Fig. 2(b), which is due to the incomplete polymerization of aniline and is also observed in WO3/PANI PNCs.63
 | ||
| Fig. 2 (a) FT-IR spectrum, (b) TGA curve, (c) C1s and (d) N1s XPS spectra of the synthesized PNCs. | ||
Fig. 2(c) shows the deconvolution of the high-resolution C1s XPS spectra of the synthesized PNCs. The C1s peak from the PNCs is deconvoluted into two major components with peaks at 284.1 and 285.9 eV, which are attributed to C–C or C–H and C–N or CN, respectively.68Fig. 2(d) shows the deconvolution of the corresponding high-resolution N1s XPS spectra. From the properly curve-fitted N1s core-level spectrum, the specific intrinsic redox states of PANI and positive nitrogen spectra could be quantified.69 The N1s XPS spectrum could be deconvoluted into four distinct curves with the peaks locating at 398.1, 399.0, 400.1 and 401.8 eV, respectively. The characteristic binding peaks at about 398.1 and 399.0 eV are attributed to the undoped imine (–N) and undoped amine (–NH–) groups, respectively.70 The peaks at around 400.1 and 401.8 eV are related to the doped imine and amine groups (N+), respectively. Generally, equal proportions of imine and amine N1s components are expected to exist in the undoped EB base form PANI.61 However, the percentages of the area intensity of the undoped imine groups at around 398.1 eV and the undoped amine groups at 399.0 eV are decreased in the EB salt form of PANI doped with acid due to the transformation of the undoped imine and amine groups to the doped imine and amine groups.71,72 Their percentages of the total N1s intensity are 28.4, 38.8, 23.5 and 13.3% for the peaks 398.1, 399.0, 400.1 and 401.8 eV, respectively. Usually, the protonated nitrogen is favorably originated from the imine groups in the PANI chains because the pKa of the imine group is 5.5 compared to 2.5 for the amine group.73 The observed decreased proportion (38.8%) of the undoped amine groups is due to additional protonation (doping) occurring at the amine nitrogen (13.3%).74 The lower binding energy peak at 400.1 eV, Fig. 2(d), is attributed to electron localization through the strong electrostatic interaction between the SO3− functional groups of the doped PTSA and cationic radical nitrogen through space to form five or six-membered rings with energetically favorable configurations.75 These results confirm further that PANI in the synthesized PNCs appears as the EB salt form,68–70 which is consistent with the FT-IR analysis and with the H2SO4 doped PANI synthesized via oxidative polymerization.76
3.2 Cr(VI) removal by PANI/Fe3O4 PNCs
Fig. 3(A) shows the Cr(VI) removal percentage for Cr(VI) solutions with different pH values after treatment with the PNCs for 5 min at room temperature. The Cr(VI) removal percentage by the magnetic PNCs is observed to depend on the pH values. The obvious decrease of the Cr(VI) removal percentage is observed in the neutral solution, whereas an almost complete removal is found in other pH solutions, except a slight decrease in the solution with pH = 11.0. Typically, in acidic solutions, Cr2O72− turns to HCrO4−, which exhibits a very high redox potential (1.33 V) and thus can be easily reduced to Cr(III), eqn (3):37| Cr2O72− + H+ → 2HCrO4− + H2O |
| HCrO4− + 7H+ + 3e− → Cr(III)↓ + 4H2O Eo = 1.33 V | (3) |
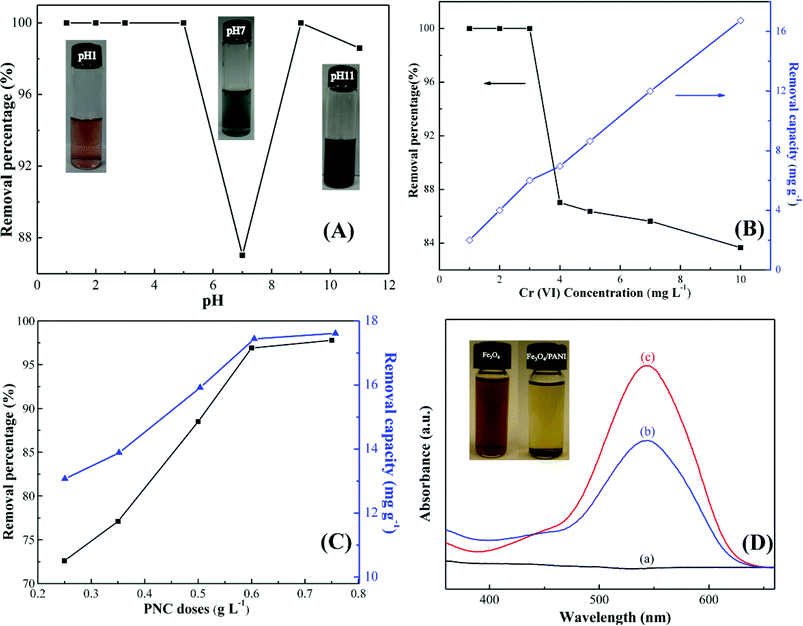 | ||
| Fig. 3 (A) Effect of pH on the removal percentage for 4.0 mg L−1 Cr(VI) solution (20.0 mL) with 10.0 mg PNCs after 5 min treatment period at room temperature; (B) (a) removal percentage and (b) removal capacity of 10.0 mg PNCs for 20.0 mL Cr(VI) neutral solutions with different initial Cr(VI) concentrations after 5 min treatment period at room temperature; (C) (a) removal percentage and (b) removal capacity of different PNC doses for 20.0 mL Cr(VI) neutral solution with an initial concentration of 9.0 mg L−1 after 5 min treatment period at room temperature; (D) (a) blank; (b) 10.0 mg pure Fe3O4 NPs treated with 20.0 mL Cr(VI) neutral solution with initial Cr(VI) concentration of 1.0 mg L−1 after 5 min treatment period and (c) the initial Cr(VI) solution (1.0 mg L−1) blank at room temperature; inset photo is Fe3O4 NPs and PNCs after immersion in 1.0 mol L−1 HCl acid for one hour. | ||
For the PNCs, Cr(VI) removal in acidic solutions arises from the Cr(VI) reduction. However, in alkaline solution, CrO42− is the dominating ion in solution.77 The oxidation ability of this ion is weaker than that of the Cr2O72− ion due to the low redox potential (−0.13 V) and Cr(III) hydroxide is precipitated:20
| CrO42− + 4H2O + 3e− → Cr(OH)3 + 5OH−Eo = −0.13 V | (4) |
In this study, the color change after treatment with Cr(VI) solution with different pH values is observed. For acidic solution, the solution is red and turns to green in the neutral solution, which indicates that different oxidation degree occurred from pH = 1.0 to 7.0, whereas in alkaline solution, the solution is blue, Fig. 3(A). The color change of the solution (pH = 11.0) after treatment with Cr(VI) and doping with phosphoric acid is observed, Fig. S2,† which is due to the dedoping and doping processes. The undoped EB base form of PANI is known to be blue, whereas the doped EB form of PANI is green.40 Specifically, the EB salt form of PANI becomes the EB base form in alkaline solution and after adding acid, the EB base form turns to the EB salt form again.40,43,78 Hence in alkaline solution, the doped acid comes out upon dedoping and reacts with hydroxyl groups. This observed phenomenon in alkaline conditions is different from the prior reported results.37,38,42 For example, decreasing Cr(VI) removal percentage with increasing pH was reported for Cr(VI) removal from solutions with pH > 7 by using PANI nanowires/tubes,42 short chain PANI on jute fibers37 and PANI/polyethylene glycol (PEG) composites.38 However, in neutral solution, the oxidation ability of Cr(VI) is weaker than that in the acidic solution, thus a decreased Cr(VI) removal percentage is observed. Interestingly, it's worth noting that even for the neutral solution, these magnetic PNCs still perform a very high removal percentage of 87.4%, indicating that the synthesized PNCs are suitable for Cr(VI) removal over a wide pH range.
Fig. 3(B) shows the Cr(VI) removal percentage and removal capacity from neutral solutions with different initial Cr(VI) concentrations after treatment with PNCs for 5 min. The synthesized PNCs with a weight of 10.0 mg are observed to be able to treat 20.0 mL Cr(VI) solution in the concentration range 1.0–3.0 mg L−1 with a 100% removal. Although for solutions with Cr(VI) concentration higher than 3.0 mg L−1, the removal percentage is observed to decrease with increasing the initial Cr(VI) concentration due to the overoxidation and degradation of PANI arising from the high oxidative environment of the concentrated Cr(VI),79 the removal percentage is still very high (about 84% for 10.0 mg L−1 initial Cr(VI) concentration). The removal percentage decreases sharply with increasing initial Cr(VI) concentration from 3.0 to 4.0 mg L−1 and then decreases slightly with higher initial Cr(VI) concentration. However, the removal capacity increases from 2.0 to 16.73 mg g−1 for solutions with an initial Cr(VI) concentration from 1.0 to 10.0 mg L−1, which is due to the different oxidizing ability of solutions of different initial Cr(VI) concentration. Therefore, the synthesized PNCs are capable of complete Cr(VI) removal in neutral solutions with an initial Cr(VI) concentration ranging from 1.0–3.0 mg L−1.
Fig. 3(C) shows the Cr(VI) removal percentage and removal capacity with different PNC doses. Both removal percentage and capacity are observed to increase with increasing the PNC dose due to the increased surface area of the available magnetic PNCs.80 At low PNC dose (<0.6 g L−1), the Cr(VI) removal percentage increases, reaches an almost constant value with PNC doses higher than 0.6 g L−1, and does not increase further even when more PNCs are added. Thus, the PNC dose of 0.6 g L−1 is enough for Cr(VI) removal from 20.0 mL solution contaminated with 9.0 mg L−1 initial Cr(VI) concentration, indicating that the synthesized PNCs have a good removal performance even for a high Cr(VI) concentration.
Fig. 3(D) shows the UV-vis absorption of the Cr(VI) solution after treatment with pure Fe3O4 NPs (10.0 mg) for comparison. As expected, pure Fe3O4 NPs also exhibit Cr(VI) removal by adsorption81 and can remove toxic Cr(VI) from the aqueous solution with a low removal percentage of only 14.95%, which is even lower than that previously reported (22%).81 1 mol L−1 HCl is also used to evaluate the stability of pure Fe3O4 NPs and the synthesized PNCs in acidic solution, inset of Fig. 3(D). The pure Fe3O4 NPs dissolved in acid, which turned to red-yellow. However, the PNCs were not dissolved and the green color of the solution was from the doped PANI. These results confirm that Fe3O4 NPs coated with PANI have an improved stability against acid etching.
3.3 Cr(VI) removal mechanisms
The Cr(VI) removal mechanism by magnetic PNCs was explored by FT-IR, XPS, EFTEM and temperature dependent resistivity. Fig. 4 shows the FT-IR spectra of the PNCs before and after treatment with Cr(VI) solutions with different initial Cr(VI) concentration. The major changes in the spectra between treated and untreated PNCs are observed in the region 1000 and 1600 cm−1.82 The strong absorption peak at 1565 cm−1 corresponds to the CC stretching vibration of the NQN, and the peak at 1485 cm−1 is related to the ring stretching of N–B–N.63 Moreover, these two bands in the treated PNCs have slightly shifted (about 6–10 cm−1) to lower frequency compared with those of the untreated PNCs. The peak centered at 1238 cm−1 is due to the C–H single stretching vibration bonds of the quinoid ring and it becomes stronger after treatment with Cr(VI). Furthermore, the peak at around 791 cm−1 is attributed to the out-of-plane bending of C–H in the substituted benzenoid ring60 and has shifted to 794 and 796 cm−1 after Cr(VI) treatment. A strong peak at 1178 cm−1 appears in the treated PNCs, exhibiting a similar change upon full oxidation, which has also been observed in prior reports82,83 and is related to the carbon-nitrogen vibration.83 However, only one shoulder at the same peak position is observed in the untreated PNCs. Similar results are also noticed in the PNCs treated with Cr(VI) solutions having different pH values, Fig. S5.† The FT-IR spectra are consistent with the change of oxidation from the EB form78 to the PB structure82 after Cr(VI) treatment.
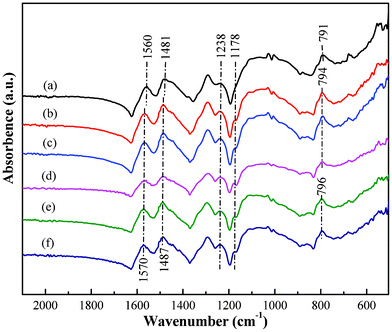 | ||
| Fig. 4 FT-IR spectra of the PNCs (a) before and after treatment with Cr(VI) solution with an initial Cr(VI) concentration of (b) 1.0, (c) 2.0, (d) 3.0, (e) 4.0 and (f) 5.0 mg L−1. | ||
High resolution XPS spectrum can be used to confirm the Cr element chemical state. Generally, for the Cr2p XPS spectrum, the characteristic bindings at 577.0–578.0 eV and 586.0–588.0 eV correspond to Cr(III) and the characteristic binding energy peaks for the Cr(VI) are at 580.0–580.5 and 589.0–590.0 eV.84Fig. 5(a) shows the Cr2p spectra of the PNCs after treatment with 20.0 mL neutral solution with an initial Cr(VI) concentration of 5.0 mg L−1 for 5 min at room temperature. The binding energy peaks of Cr2p are observed to be located at around 575.2 and 584.9 eV, which confirms that the adsorbed Cr is in the form of Cr(III) and the peaks have shifted slightly compared with the literature reports.84 The former arises from the Cr2p3/2 orbital and the latter from the Cr2p1/2 orbital.85 The presence of Cr(III) implies that the synthesized PNCs have reduced the Cr(VI) ions to Cr(III) ions. The Cr(III) composition obtained from XPS spectra increases with increasing initial Cr(VI) concentration, for example, from 0.27% for the solution with an initial Cr(VI) concentration of 3.0 mg L−1 to 0.75% for the solution with an initial Cr(VI) concentration of 5.0 mg L−1. Fig. 5(b) shows the N1s XPS spectra of the PNCs after treatment with Cr(VI). The N1s peak has been deconvoluted into three major components at 397.3, 398.2 and 400.6 eV, which are attributed to the imine group (–N), amine group (–NH–) and protonated nitrogen (N+), respectively.70 These peaks have shifted to a lower binding energy compared with those of the as-synthesized PNCs, Fig. 2(d), indicating the interaction between Cr(III) and PANI, which is also observed in the PPy–TiO2 PNCs.86 Their percentages of the total N1s intensity are 64.9, 27.5 and 7.6% for the peaks at 397.3, 398.2 and 400.6 eV, respectively. The observed decreased proportion (27.5%) of amine groups indicates that most EB form PANI has been oxidized after treatment with Cr(VI) solution (initial Cr(VI) concentration: 5.0 mg L−1).76
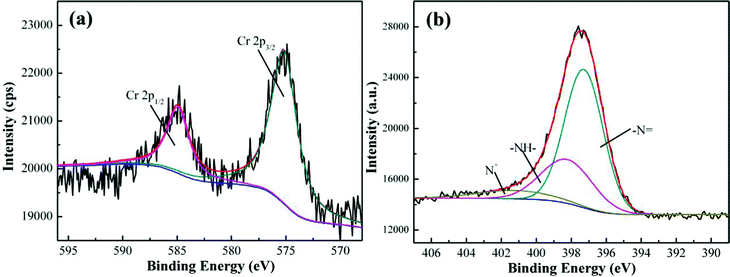 | ||
| Fig. 5 (a) Cr 2p XPS spectra and (b) N1s XPS spectra of the PNCs after treatment with 20.0 mL Cr(VI) neutral solution (4.0 mg L−1) for 5 min at room temperature. | ||
EFTEM was conducted on the samples to further investigate the 2-d elemental distribution in the PNCs after treatment with Cr(VI) solution. The brighter area in the elemental EFTEM mapping images represents a higher concentration of the corresponding element.54Fig. 6 shows the zero-loss image, and elemental maps of Fe, O, C, N, S, and Cr in different colors to verify the element position within the whole PNCs. Fig. 6(b) and (c) depict the Fe and O element maps, which are shown in red and blue, respectively, and come from Fe3O4 NPs. C and N element maps, shown in green and yellow, Fig. 6(d) and (e), are from PANI. The S element map in purple, Fig. 6(f), is from the doped acid PTSA. These pictures clearly show that the Fe3O4 NPs are inside the PNCs and PANI exists surrounding the Fe3O4 NPs. Fig. 6(g) displays the distribution of Cr element, which is inside the PNCs and is mainly distributed in the PANI and the Fe3O4-PANI interface. The summation of Fe, C, O, N, S and Cr, Fig. 6(i), gives all the elemental distributions, confirming that after reduction of Cr(VI) to Cr(III), the Cr(III) penetrated into the synthesized PNCs instead of adsorption on the PNC surface. The BET results show an average specific surface area of 39.26 m2 g−1 for the as-synthesized PNCs, whereas the specific surface area of the PNCs after treated with Cr(VI) is decreased to 30.24 m2 g−1, which is due to the Cr(III) penetration into the treated PNCs and is consistent with EFTEM results. The temperature dependent resistivity was measured and the results show that some EB salt form exists in the treated samples and the resistivity of the samples is increased dramatically after treatment with Cr(VI), Fig. S6.†
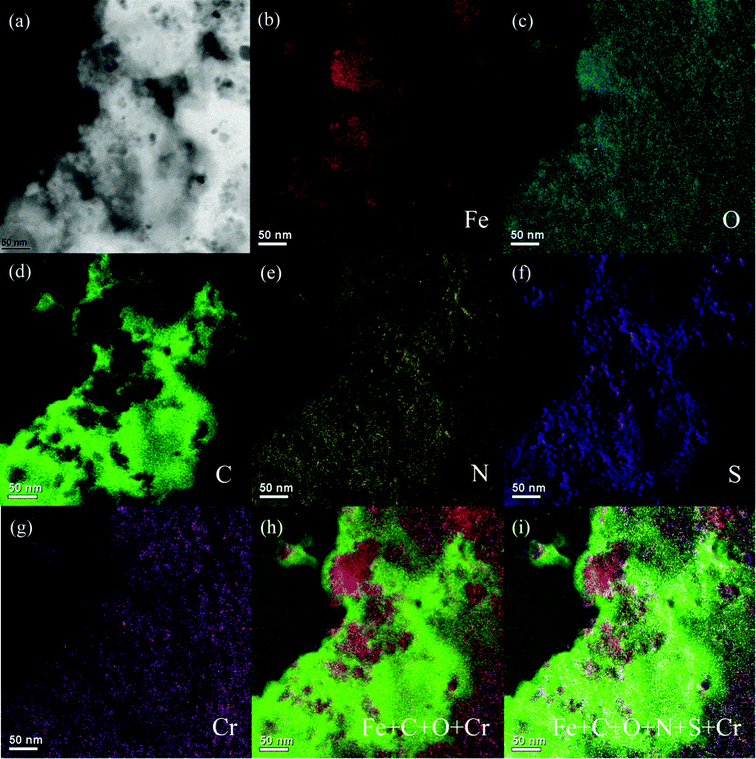 | ||
| Fig. 6 EFTEM of the PNCs after 5 min treatment with 20.0 mL Cr(VI) solution (pH = 1.0; 4.0 mg L−1): (a) zero-loss image, (b) Fe map, (c) O map, (d) C map, (e) N map, (f) S map, (g) Cr map, (h) Fe + C + O + Cr map, and (i) Fe + C + O + N + S + Cr map. | ||
All the aforementioned analysis confirms that the EB salt form PANI in the synthesized PNCs is partially oxidized to the PB structure after treatment with Cr(VI) and Cr(VI) is converted into Cr(III). The ICP-OES test is used to confirm whether all the Cr(III) has been adsorbed. The ICP-OES test can only provide the concentration of the elements in the solution, but it can not provide the chemical valence state of elements. The Cr concentration in the solution is 0.4998 mg L−1 after treated with solution with an initial Cr(VI) concentration of 3.8922 mg L−1. This concentration should be the concentration of Cr(III) in the solution due to the complete removal of Cr(VI) from the solution by the PNCs, which is confirmed by colorimetric method. This result also confirms that most of Cr(III) has come into the PNCs as mentioned above. The proposed mechanism of Cr(VI) removal by PNCs is shown in Scheme 1.
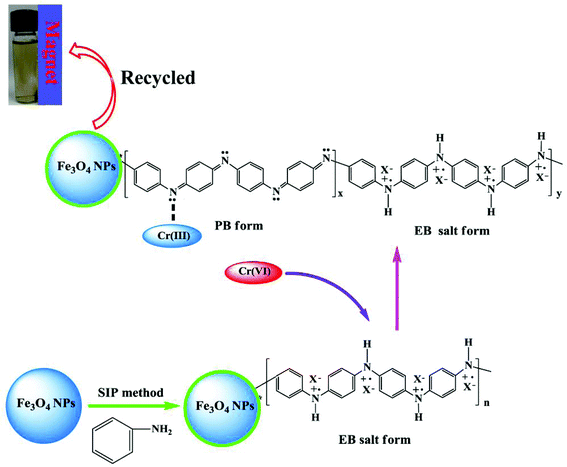 | ||
| Scheme 1 Proposed mechanism of Cr(VI) removal by magnetic PANI nanocomposites (X represents doped acid PTSA). | ||
3.4 Kinetics
The Cr(VI) concentration change in the solution with pH = 1.0 after treating for different time periods is shown in Fig. 7(a). The Cr(VI) concentration in the solution decreases from 12.0 to 0.23 mg L−1 for a treatment period of 12 min and after that it reaches the equilibrium. As compared to the 12 min equilibrium time for the PNCs, the PANI powder has been reported to require 10–12 min,36 activated carbon required 3 h,87 polypyrrole/Fe3O4 magnetic nanocomposites required 30 min,2 oxidized multiwalled carbon nanotubes required 280 h,88 nanocrystalline akaganeite (iron(III) oxide-hydroxide/chloride mineral) required 1 h.89 Thus, the synthesized EB PANI/Fe3O4 nanocomposites show fairly good performance in Cr(VI) removal. | ||
| Fig. 7 (a) Cr(VI) concentration change of 10.0 mg PNCs after treatment with 20.0 mL Cr(VI) solution (pH = 1.0; 12.0 mg L−1) with different treatment periods at room temperature; (b) corresponding kinetic plot in pH = 1.0 Cr(VI) solution; (c) kinetic plot after treatment with 20.0 mL Cr(VI) solution of pH = 7.0, 9.0 mg L−1 and (d) pH = 11.0, 5.0 mg L−1 at room temperature. | ||
The representative kinetics (reduction rate of Cr(VI) by the PNCs) in the pH = 1.0 Cr(VI) solution are obtained, Fig. 7(b). For the PNC system, this one-stage behavior is given by the oxidation of PANI and reduction of Cr(VI), eqn (5):
| 2HCrO4− (aq) + 14H+(aq) + 3PANI2+ (s) → 2Cr3+ (aq) + 3PANI4+(s) + 8H2O (l) | (5) |
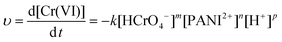 | (6) |
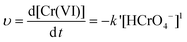 | (7) |
The kinetics in the Cr(VI) solutions with pH = 7.0 and 11.0 was also investigated, Fig. 7(c) and (d), and is observed to obey the pseudo-first-order behavior with a correlation coefficient R2 of 0.98258 and 0.98816 for the pH = 7.0 and 11.0 solutions, respectively. The typical values of the pseudo-first-order rate constants obtained from the slope are 0.095 and 0.156 min−1 for solutions with pH = 7.0 and 11.0, respectively. The rate constants of the three different pH solutions obey the following decreasing relationship: pH1.0 > pH11.0 > pH7.0. This result is consistent with the effect of pH values on Cr(VI) removal; the oxidative ability of the Cr(VI) is stronger in acidic solutions and weaker in neutral and basic solutions. The pseudo-first-order behavior is also reported in the reduction of Cr(VI) by PPy,35 PPy-coated carbon substrate,92 hydrogen peroxide,21 iron93 and PANI films.90
3.5 Magnetic properties of the recycled EB PANI/Fe3O4 PNCs
To investigate the recycling of the PNCs, the magnetic properties were also measured before and after Cr(VI) treatment. Fig. 8 shows the room temperature magnetic hysteresis loops. For both PNCs before and after treated with Cr(VI), Ms is not reached even at a high magnetic field and is determined by the extrapolated Ms obtained from the intercept of M–H−1 at high field.94,95 The calculated Ms of the PNCs is 24.24 emu g−1, and the calculated Ms for the treated PNCs is almost the same with a value of 24.09 emu g−1. The coercivity (coercive force, Hc) is observed to be 0 Oe in both samples, indicating a superparamagnetic behavior. These observed similar magnetic properties have demonstrated that PANI has effectively protected the Fe3O4 NPs from dissolution, consistent with the acid test, the inset of Fig. 3(D). These PNC powders after treatment with Cr(VI) could still be attracted by a permanent magnet, which is essential for the recycling of the powders after treatment with Cr(VI).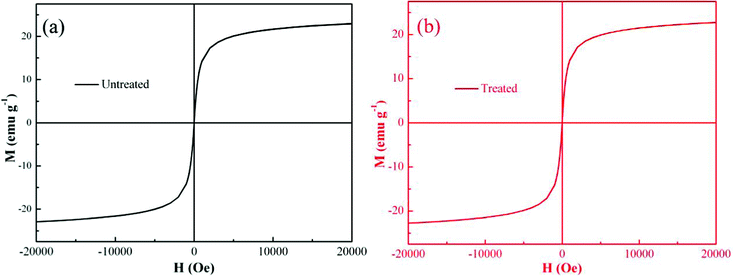 | ||
| Fig. 8 Room-temperature magnetic hysteresis loops of (a) the synthesized PNCs, and (b) the PNCs after 5 min treatment with 20.0 mL Cr(VI) solution (pH = 1.0; 4.0 mg L−1). | ||
3.6 Regeneration and reusage of EB PANI/Fe3O4 PNCs
The recovery of Cr from PNCs, and the regeneration and reusage of the PNCs were also investigated. A sample of the PANI/Fe3O4 PNCs already treated with initial Cr(VI) concentration of 3.8922 mg L−1 was used for the test. The sample was mixed with 1.0 mol L−1 PTSA solution (or 1.0 mol L−1 HCl solution) for 5 min under sonication, then the solution was taken out for the ICP-OES test and the recycled powders were washed with DI-water until the pH was about 7. Then the sample was dried at 50 °C overnight. The regenerated sample was used to treat a solution with initial Cr(VI) concentration of 3.8922 mg L−1. The result showed that the Cr(VI) was completely removed by the regenerated PNCs, indicating that this nanocomposite material is easily reproduced and can be efficiently reused. The ICP-OES result shows that the Cr concentration in the solution is 1.1835 mg L−1, indicating that some Cr(III) (there is no Cr(VI) in the acid solution as confirmed by the colorimetric analysis) has been dedoped by PTSA.4. Conclusion
The feasibility of Cr(VI) removal by SIP method synthesized magnetic PANI/Fe3O4 nanocomposites is demonstrated. These nanocomposites can rapidly and efficiently remove Cr(VI) in a simple one stage process from aqueous solutions with a wide pH range. The initial Cr(VI) concentration is found to have a significant effect on Cr(VI) removal, which is 1.0–3.0 mg L−1 for the complete removal of 20.0 mL Cr(VI) neutral solution after 5 min treatment, and the PNC dose of 0.6 g L−1 is enough for Cr(VI) removal from 20.0 mL solution contaminated with 9.0 mg L−1 initial Cr(VI) concentration. FT-IR, XPS and temperature dependent resistivity results show that the Cr(VI) removal by the synthesized PNCs follows one stage Cr(VI) reduction by PANI and the reduced Cr(III) adsorbs in the PNCs as verified by XPS analysis. The PANI is partially converted into its oxidized state after treatment with Cr(VI). The resistivity of the PNCs increases dramatically after treatment with Cr(VI). The EFTEM results show that the adsorbed Cr(III) has penetrated into the interior of the PNCs instead of simple adsorption on the surface of the PNCs. The kinetics of the process obey pseudo-first-order behavior with respect to Cr(VI). The typical values for the pseudo-first-order rate constants are 0.185, 0.095 and 0.156 min−1 for the solutions with pH values of 1.0, 7.0 and 11.0, respectively. The magnetic properties of the synthesized PNCs have no obvious decrease after treatment with Cr(VI), indicating that the presence of the magnetic Fe3O4 NPs favors the recycling of the PNC powders and PANI has effectively protected the Fe3O4 NPs from dissolution. These synthesized PNCs are easily regenerated by 1.0 mol L−1 PTSA or 1.0 mol L−1 HCl and the removal efficiency of Cr(VI) by the recycled PNCs has no obvious decrease.Acknowledgements
This project is financially supported by the National Science Foundation-Chemical and Biological Separation under the EAGER program (CBET 11-37441) managed by Dr Rosemarie D. Wesson. Some financial support from Texas Hazardous Waste Research Center is kindly acknowledged. D. P. Young acknowledges support from the NSF under Grant No. DMR 10-05764. H. Gu acknowledges the support from China Scholarship Council (CSC) program. The authors thank Earth Analytical Sciences, Inc. for the ICP-OES test.References
- A. A. Hasin, S. J. Gurman, L. M. Murphy, A. Perry, T. J. Smith and P. H. E. Gardiner, Environ. Sci. Technol., 2010, 44, 400–405 CrossRef.
- M. Bhaumik, A. Maity, V. V. Srinivasu and M. S. Onyango, J. Hazard. Mater., 2011, 190, 381–390 CrossRef CAS.
- M. A. Omole, V. A. Okello, V. Lee, L. Zhou, O. A. Sadik, C. Umbach and B. Sammakia, ACS Catal., 2011, 1, 139–146 CrossRef CAS.
- D. Sazou, M. Kourouzidou and E. Pavlidou, Electrochim. Acta, 2007, 52, 4385–4397 CrossRef CAS.
- L. Hsu, S. A. Masuda, K. H. Nealson and M. Pirbazari, RSC Adv., 2012, 2, 5844–5855 RSC.
- P. A. Kumar and S. Chakraborty, J. Hazard. Mater., 2009, 162, 1086–1098 CrossRef CAS.
- S. S. Wise, F. Shaffiey, C. LaCerte, C. E. C. Goertz, J. L. Dunn, F. M. D. Gulland, A.-M. Aboueissa, T. Zheng Sr. and J. P. Wise, Aquat. Toxicol., 2009, 91, 329–335 CrossRef CAS.
- Y. Xu and D. Zhao, Water Res., 2007, 41, 2101–2108 CrossRef CAS.
- D. Zhang, S. Wei, C. Kaila, X. Su, J. Wu, A. B. Karki, D. P. Young and Z. Guo, Nanoscale, 2010, 2, 917–919 RSC.
- O. S. Amuda, A. A. Giwa and I. A. Bello, Biochem. Eng. J., 2007, 36, 174–181 CrossRef CAS.
- N. Kongsricharoern and C. Polprasert, Water Sci. Technol., 1995, 31, 109–117 CrossRef CAS.
- D. Park, Y. S. Yun, H. W. Lee and J. M. Park, Bioresour. Technol., 2008, 99, 1141–1147 CrossRef CAS.
- J. Hu, G. Chen and I. M. C. Lo, Water Res., 2005, 39, 4528–4536 CrossRef CAS.
- H. Cui, M. Fu, S. Yu and M. K. Wang, J. Hazard. Mater., 2011, 186, 1625–1631 CrossRef CAS.
- K. C. K. Lai and I. M. C. Lo, Environ. Sci. Technol., 2008, 42, 1238–1244 CrossRef CAS.
- K. M. Sirk, N. B. Saleh, T. Phenrat, H. J. Kim, B. Dufour, J. Ok, P. L. Golas, K. Matyjaszewski, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2009, 43, 3803–3808 CrossRef CAS.
- M. Pettine, I. Barra, L. Campanella and F. J. Millero, Water Res., 1998, 32, 2807–2813 CrossRef CAS.
- P. R. Wittbrodt and C. D. Palmer, Environ. Sci. Technol., 1995, 29, 255–263 CrossRef CAS.
- D. Park, Y. S. Yun and J. M. Park, Environ. Sci. Technol., 2004, 38, 4860–4864 CrossRef CAS.
- C. Kim, Q. Zhou, B. Deng, E. C. Thornton and H. Xu, Environ. Sci. Technol., 2001, 35, 2219–2225 CrossRef CAS.
- M. Pettine, L. Campanella and F. J. Millero, Environ. Sci. Technol., 2002, 36, 901–907 CrossRef CAS.
- A. G. MacDiarmid, L. S. Yang, W. S. Huang and B. D. Humphrey, Synth. Met., 1987, 18, 393–398 CrossRef CAS.
- D. Saikia, Y. H. Chen, Y. C. Pan, J. Fang, L. D. Tsai, G. T. K. Fey and H. M. Kao, J. Mater. Chem., 2011, 21, 10542–10551 RSC.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS.
- J. Zhu, H. Gu, Z. Luo, N. Haldolaarachige, D. P. Young, S. Wei and Z. Guo, Langmuir, 2012, 28, 10246–10255 CrossRef CAS.
- J. Zhu, S. Wei, M. Alexander Jr., T. D. Dang, T. C. Ho and Z. Guo, Adv. Funct. Mater., 2010, 20, 3076–3084 CrossRef CAS.
- J. Zhu, S. Wei, M. Alexander Jr., D. Cocke, T. C. Ho and Z. Guo, J. Mater. Chem., 2010, 20, 568–574 RSC.
- B. Wessling and J. Posdorfer, Electrochim. Acta, 1999, 44, 2139–2147 CrossRef CAS.
- M. Nishizawa, V. P. Menon and C. R. Martin, Science, 1995, 268, 700–702 CAS.
- H. Cui, Q. Li, Y. Qian, R. Tang, H. An and J. Zhai, Water Res., 2011, 45, 5736–5744 CrossRef CAS.
- J. Zhu, X. Zhang, N. Haldolaarachchige, Q. Wang, Z. Luo, J. Ryu, D. P. Young, S. Wei and Z. Guo, J. Mater. Chem., 2012, 22, 4996–5005 RSC.
- H. Wei, X. Yan, Y. Li, S. Wu, A. Wang, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 4500–4510 CAS.
- X. Zhang, J. Zhu, N. Haldolaarachchige, J. Ryu, D. P. Young, S. Wei and Z. Guo, Polymer, 2012, 53, 2109–2120 CrossRef CAS.
- X. Zhang, S. Wei, N. Haldolaarachchige, H. A. Colorado, Z. Luo, D. P. Young and Z. Guo, J. Phys. Chem. C, 2012, 116, 15731–15740 CAS.
- R. Senthurchelvan, Y. Wang, S. Basak and K. Rajeshwar, J. Electrochem. Soc., 1996, 143, 44–51 CrossRef CAS.
- A. Olad and R. Nabavi, J. Hazard. Mater., 2007, 147, 845–851 CrossRef CAS.
- P. A. Kumar, S. Chakraborty and M. Ray, Chem. Eng. J., 2008, 141, 130–140 CrossRef CAS.
- M. R. Samani, S. M. Borghei, A. Olad and M. J. Chaichi, J. Hazard. Mater., 2010, 184, 248–254 CrossRef CAS.
- A. G. MacDiarmid, S. K. Manohar, J. G. Masters, Y. Sun, H. Weiss and A. J. Epstein, Synth. Met., 1991, 41, 621–626 CrossRef CAS.
- T. Ogoshi, Y. Hasegawa, T. Aoki, Y. Ishimori, S. Inagi and T.-a Yamagishi, Macromolecules, 2011, 44, 7639–7644 CrossRef CAS.
- N. R. Chiou and A. J. Epstein, Adv. Mater., 2005, 17, 1679–1683 CrossRef CAS.
- X. Guo, G. T. Fei, H. Su and D. Z. Li, J. Phys. Chem. C, 2011, 115, 1608–1613 CAS.
- J. Huang and R. B. Kaner, Chem. Commun., 2006, 367–376 RSC.
- S. Wei, Q. Wang, J. Zhu, L. Sun, H. Lin and Z. Guo, Nanoscale, 2011, 3, 4474–4502 RSC.
- P. Hu, S. Zhang, H. Wang, D. Pan, J. Tian, Z. Tang and A. A. Volinsky, J. Alloys Compd., 2011, 509, 2316–2319 CrossRef CAS.
- T. Atarashi, T. Imai and J. Shimoiizaka, J. Magn. Magn. Mater., 1990, 85, 3–6 CrossRef CAS.
- D. M. Huang, D. B. Cao, Y. W. Li and H. Jiao, J. Phys. Chem. B, 2006, 110, 13920–13925 CrossRef CAS.
- V. K. Gupta, S. Agarwal and T. A. Saleh, Water Res., 2011, 45, 2207–2212 CrossRef CAS.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS.
- E. Lima Jr., A. J. Brandl, A. D. Arelaro and G. F. Goya, J. Appl. Phys., 2006, 99, 083908 CrossRef.
- J. L. Wilson, P. Poddar, N. A. Frey, H. Srikanth, K. Mohomed, J. P. Harmon, S. Kotha and J. Wachsmuth, J. Appl. Phys., 2004, 95, 1439–1443 CrossRef CAS.
- S. R. Rudge, T. L. Kurtz, C. R. Vessely, L. G. Catterall and D. L. Williamson, Biomaterials, 2000, 21, 1411–1420 CrossRef CAS.
- J. Zhu, H. Gu, S. B. Rapole, Z. Luo, S. Pallavkar, N. Haldolaarachchige, T. J. Benson, T. C. Ho, J. Hopper, D. P. Young, S. Wei and Z. Guo, RSC Adv., 2012, 2, 4844–4856 RSC.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS.
- Z. Zhang and M. Wan, Synth. Met., 2003, 132, 205–212 CrossRef CAS.
- H. Wang, R. Wang, L. Wang and X. Tian, Colloids Surf., A, 2011, 384, 624–629 CrossRef CAS.
- C. Yang, J. Du, Q. Peng, R. Qiao, W. Chen, C. Xu, Z. Shuai and M. Gao, J. Phys. Chem. B, 2009, 113, 5052–5058 CrossRef CAS.
- H. Gu, Y. Huang, X. Zhang, Q. Wang, J. Zhu, L. Shao, N. Haldolaarachchige, D. P. Young and S. Wei, Polymer, 2012, 53, 801–809 CrossRef CAS.
- M. Gardner and S. Comber, Analyst, 2002, 127, 153–156 RSC.
- J. Zhu, S. Wei, L. Zhang, Y. Mao, J. Ryu, N. Haldolaarachige, D. P. Young and Z. Guo, J. Mater. Chem., 2011, 21, 3952–3959 RSC.
- J. Zhang, J. Chen and Z. Wang, Mater. Lett., 2007, 61, 1629–1632 CrossRef CAS.
- J. Sun, S. Zhou, P. Hou, Y. Yang, J. Weng, X. Li and M. Li, J. Biomed. Mater. Res., Part A, 2007, 80A, 333–341 CrossRef CAS.
- J. Zhu, S. Wei, L. Zhang, Y. Mao, J. Ryu, A. B. Karki, D. P. Young and Z. Guo, J. Mater. Chem., 2011, 21, 342–348 RSC.
- P. Mavinakuli, S. Wei, Q. Wang, A. B. Karki, S. Dhage, Z. Wang, D. P. Young and Z. Guo, J. Phys. Chem. C, 2010, 114, 3874–3882 CAS.
- M. Mahmoudi, A. Simchi, M. Imani, A. S. Milani and P. Stroeve, J. Phys. Chem. B, 2008, 112, 14470–14481 CrossRef CAS.
- S. Xuan, Y. X. J. Wang, K. C. F. Leung and K. Shu, J. Phys. Chem. C, 2008, 112, 18804–18809 CAS.
- X. Feng, C. Mao, G. Yang, W. Hou and J. Zhu, Langmuir, 2006, 22, 4384–4389 CrossRef CAS.
- B. C. Beard and P. Spellane, Chem. Mater., 1997, 9, 1949–1953 CrossRef CAS.
- J. P. Pouget, M. E. Jozefowicz, A. J. Epstein, X. Tang and A. G. MacDiarmid, Macromolecules, 1991, 24, 779–789 CrossRef CAS.
- E. T. Kang, K. G. Neoh and K. L. Tan, Prog. Polym. Sci., 1998, 23, 277–324 CrossRef CAS.
- A. A. Qaiser, M. M. Hyland and D. A. Patterson, J. Phys. Chem. B, 2011, 115, 1652–1661 CrossRef CAS.
- X. L. Wei, M. Fahlman and A. J. Epstein, Macromolecules, 1999, 32, 3114–3117 CrossRef CAS.
- S. Tan and D. Bélanger, J. Phys. Chem. B, 2005, 109, 23480–23490 CrossRef CAS.
- M. G. Han and S. S. Im, Polymer, 2000, 41, 3253–3262 CrossRef CAS.
- J. Yue and A. J. Epstein, Macromolecules, 1991, 24, 4441–4445 CrossRef CAS.
- Y. Chen, E. T. Kang, K. G. Neoh, S. L. Lim, Z. H. Ma and K. L. Tan, Colloid Polym. Sci., 2001, 279, 73–76 CAS.
- Y. Li, B. Gao, T. Wu, D. Sun, X. Li, B. Wang and F. Lu, Water Res., 2009, 43, 3067–3075 CrossRef CAS.
- J. Stejskal, P. Kratochvíl and A. D. Jenkins, Polymer, 1996, 37, 367–369 CrossRef CAS.
- A. Mirmohseni and A. Oladegaragoze, J. Appl. Polym. Sci., 2002, 85, 2772–2780 CrossRef CAS.
- R. Zhang, H. Ma and B. Wang, Ind. Eng. Chem. Res., 2010, 49, 9998–10004 CrossRef CAS.
- P. Yuan, D. Liu, M. Fan, D. Yang, R. Zhu, F. Ge, J. Zhu and H. He, J. Hazard. Mater., 2010, 173, 614–621 CrossRef CAS.
- Y. Cao, Synth. Met., 1990, 35, 319–332 CrossRef CAS.
- J. Tang, X. Jing, B. Wang and F. Wang, Synth. Met., 1988, 24, 231–238 CrossRef CAS.
- D. Park, Y. S. Yun and J. M. Park, J. Colloid Interface Sci., 2008, 317, 54–61 CrossRef CAS.
- B. A. Manning, J. R. Kiser, H. Kwon and S. R. Kanel, Environ. Sci. Technol., 2007, 41, 586–592 CrossRef CAS.
- S. Wei, M. Pallavi, W. Qiang, C. Daniel, A. Ramesh, M. Yuanbing, H. Neel, P. Y. David and Z. Guo, J. Electrochem. Soc., 2011, 158, K205–K212 CrossRef CAS.
- S. Babel and T. A. Kurniawan, Chemosphere, 2004, 54, 951–967 CrossRef CAS.
- J. Hu, C. Chen, X. Zhu and X. Wang, J. Hazard. Mater., 2009, 162, 1542–1550 CrossRef CAS.
- N. K. Lazaridis, D. N. Bakoyannakis and E. A. Deliyanni, Chemosphere, 2005, 58, 65–73 CrossRef CAS.
- S. T. Farrell and C. B. Breslin, Environ. Sci. Technol., 2004, 38, 4671–4676 CrossRef CAS.
- J. W. Moore, R. G. Pearson and A. A. Frost, Kinetics and mechanism, Wiley, 1961 Search PubMed.
- F. J. Rodríguez, S. Gutiérrez, J. G. Ibanez, J. L. Bravo and N. Batina, Environ. Sci. Technol., 2000, 34, 2018–2023 CrossRef.
- M. J. Alowitz and M. M. Scherer, Environ. Sci. Technol., 2002, 36, 299–306 CrossRef CAS.
- D. Zhang, A. B. Karki, D. Rutman, D. P. Young, A. Wang, D. Cocke, T. H Ho and Z. Guo, Polymer, 2009, 50, 4189–4198 CrossRef CAS.
- Z. Guo, L. L.Henry, V. Palshin and E. J. Podlaha, J. Mater. Chem., 2006, 16, 1772–1777 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21991c |
| This journal is © The Royal Society of Chemistry 2012 |