Influenza neuraminidase: A druggable target for natural products
Ulrike
Grienke
a,
Michaela
Schmidtke
b,
Susanne
von Grafenstein
c,
Johannes
Kirchmair
d,
Klaus R.
Liedl
c and
Judith M.
Rollinger
*a
aInstitute of Pharmacy/Pharmacognosy and Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 52c, 6020, Innsbruck, Austria. E-mail: Judith.Rollinger@uibk.ac.at
bDepartment of Virology and Antiviral Therapy, Jena University Hospital, Hans-Knoell-Straße 2, 07745, Jena, Germany
cInstitute of Theoretical Chemistry and Center for Molecular Biosciences, University of Innsbruck, Innrain 52a, 6020, Innsbruck, Austria
dUnilever Centre for Molecular Informatics, Department of Chemistry, Lensfield Road, Cambridge, UK CB2 1EW
First published on 25th October 2011
Abstract
Covering: 2000 to 2011
The imminent threat of influenza pandemics and repeatedly reported emergence of new drug-resistant influenza virus strains demonstrate the urgent need for developing innovative and effective antiviral agents for prevention and treatment. At present, influenza neuraminidase (NA), a key enzyme in viral replication, spread, and pathogenesis, is considered to be one of the most promising targets for combating influenza. Despite the substantial medical potential of NA inhibitors (NAIs), only three of these drugs are currently on the market (zanamivir, oseltamivir, and peramivir). Moreover, sudden changes in NAI susceptibility revealed the urgent need in the discovery/identification of novel inhibitors. Nature offers an abundance of biosynthesized compounds comprising chemical scaffolds of high diversity, which present an infinite pool of chemical entities for target-oriented drug discovery in the battle against this highly contagious pathogen. This review illuminates the increasing research efforts of the past decade (2000–2011), focusing on the structure, function and druggability of influenza NA, as well as its inhibition by natural products. Following a critical discussion of publications describing some 150 secondary plant metabolites tested for their inhibitory potential against influenza NA, the impact of three different strategies to identify and develop novel NAIs is presented: (i) bioactivity screening of herbal extracts, (ii) exploitation of empirical knowledge, and (iii) computational approaches. This work addresses the latest developments in theoretical and experimental research on properties of NA that are and will be driving anti-influenza drug development now and in the near future.
 Ulrike Grienke | Ulrike Grienke studied pharmacy at the University of Innsbruck, Austria (2001–2008), where she already started with her phytochemical investigations on traditional Chinese medicinal plants and evaluation of computational methods (neural networks) for exploring the chemical space relevant for acetylcholinesterase inhibitors. In 2008, she received a PhD scholarship granted by the Austrian Science Fund (FWF), which enabled her to continue her studies on bioactive natural products under the supervision of Prof. Judith M. Rollinger. She has published four research papers, gave four talks at international conferences and is about to finalize her PhD studies. |
 Michaela Schmidtke | Michaela Schmidtke studied biology at the University of Chişinău, Moldova (1980–1985). After receiving her PhD in Antiviral Research from the Faculty of Biology at the Friedrich-Schiller-University, Jena, Germany (1992), she had post-doctorial training at the Institute of Immunology, University Marburg with the focus on coxsackievirus B3-immune cell interactions. Thereafter, she was a postdoctoral fellow at the Department of Virology and Antiviral Therapy, Jena University Hospital, Germany ending up with the habilitation thesis and the venia legendi in Virology. Her research interests focus on the development of novel anti-enteroviral and anti-influenza virus inhibitors communicated in more than 70 papers and patents. |
 Susanne von Grafenstein | Susanne von Grafenstein studied pharmacy at the Ludwig-Maximilians-University, Munich, Germany (2004–2008) and registered as pharmacist in 2009. During her training, she participated in several virtual screening projects at the University of Innsbruck, Austria. There, she joined the research group of Prof. Klaus R. Liedl, starting her PhD studies in 2010. At present her research is focused on the investigation of molecular dynamics and identification of new inhibitors in a project on influenza neuraminidase. |
 Johannes Kirchmair | Johannes Kirchmair studied pharmacy at the University of Innsbruck (1999–2004) and received his PhD degree under the guidance of Prof. Thierry Langer (2007), specializing in virtual screening. He was application scientist at Inte:Ligand GmbH, Vienna, Austria, assistant lecturer at the University of Innsbruck and post-doctoral research fellow with Prof. Klaus R. Liedl and Prof. Klaus-Jürgen Schleifer (BASF SE, Ludwigshafen, Germany). Since 2010 he is working as research assistant with Prof. Robert C. Glen at the Unilever Centre for Molecular Science Informatics, University of Cambridge, United Kingdom. His work is communicated in 35 peer-reviewed papers and book chapters. |
 Klaus R. Liedl | Klaus R. Liedl is head of the Department of Theoretical Chemistry at the Faculty of Chemistry and Pharmacy, University of Innsbruck. After he finished his chemistry and mathematics studies (1989, 1992), he studied theoretical physics and obtained a PhD in chemistry (1995). In 2006 he finished his doctorate in juridical science focusing on intellectual property rights complementing his chemical research as proven by his more than 130 publications in the field. His main research interests are drug development and the understanding and prediction of thermodynamics in the course of biomolecular binding processes and reactions. |
 Judith M. Rollinger | Judith M. Rollinger is Professor of Pharmacognosy at the University of Innsbruck, Austria, member of the Center for Molecular Biosciences Innsbruck, and elected member of the GA Board of Directors (2011–2013). After studying Pharmacy, she received her PhD in 1999 and the venia legendi in Pharmacognosy (2007) at the University of Innsbruck. Her research interests include developing new approaches, e.g. integrating computational techniques and ethno-pharmacological knowhow in natural product research, for rationalized discovery of natural leads targeting neurodegenerative diseases, viral infections and inflammation. Publications (60) resulting from her research appear in highly ranked international journals, as book contributions, and patents. |
1 Introduction
Belonging to the Orthomyxoviridae, influenza A and B are responsible for an acute contagious respiratory infection commonly known as “flu”. The disease appears in waves of seasonal flu characterized by fever, cough, sore throat and myalgia, with the ability to develop severe life-threatening conditions, such as pneumonia.1 Moreover, epidemics as well as pandemics reoccur periodically. Considered a worst-case scenario, the influenza pandemic of 1918 (known as the Spanish Flu) took the lives of an estimated 20 to 40 million people worldwide.2–4Influenza neuraminidase (NA) has been established as a primary drug target for the prophylaxis and treatment of influenza infections, which are still a major threat to human health due to high morbidity and mortality rates, especially when affecting risk groups like children, the old, and people with chronic diseases.5 In contrast to the first approved anti-influenza drugs, the ion channel blockers amantadine (Symmetrel®, approved 1966) and rimantadine (Flumadine®, approved 1993), NA inhibitors (NAIs) are well tolerated and act against influenza A as well as B viruses.6,7 After the introduction of the first NAIs in 1999, interest in the development of drugs targeting this viral enzyme wound down for several years – the market was well served. But, with the outbreak of A/H5N1, the avian influenza (also known as “bird flu”), concerns about a major pandemic started to emerge. The person-to-person transmission of A/H5N1 is limited, but in case these viruses developed efficient human-to-human transmission, estimates show that such a variant may cause a death toll exceeding the devastating Spanish Flu.8 Moreover, in the season of 2007/2008 a naturally oseltamivir-resistant H1N1 mutant emerged suddenly and spread worldwide.9 Awareness rose further with the flu pandemic of 2009 caused by a new A/H1N1 strain (swine-origin influenza H1N1 virus; S–OIV), which was characterized by a high transmission rate but low virulence.10 Though this pandemic developed less critically than anticipated these events highlight the urgent need for novel antiviral therapeutics.11,12
Moreover, with today's very narrow drug portfolio for the prophylaxis and treatment of flu, public health is threatened by the imminent risk of a sudden emerge of drug-resistant viral strains.13–16 Such sudden changes in NAI susceptibility were observed in influenza A viruses circulating worldwide in the last 3 years.17,18
Therefore, twelve years after the launch of Relenza® (zanamivir (1)) and Tamiflu® (oseltamivir (2)) – the two commonly applied/prescribed NAIs worldwide – the search for novel lead structures in the fight against this aggressive pathogen is again an area of intense research.19,20 Emphasis on more sophisticated strategies is needed to overcome shortcomings and drawbacks of currently available drugs and leads, such as side effects and emerging drug resistance.
Nature provides an abundance of structurally diverse chemical scaffolds that represent a rich source for lead structures in drug development.21,22 In the past century various compounds isolated from natural sources have been extensively studied and tested for their anti-viral and anti-influenza activity.23–25 This review focuses on natural products specifically targeting the viral surface protein NA, as do currently available first-line drugs stockpiled worldwide in the case of a pandemic. The survey provides an overview on reported activities as well as an outlook on future prospects.
2 Influenza NA – an established drug target for combating influenza
2.1 Influenza NA function
Influenza NA, a glycoside hydrolase (EC 3.2.1.18) and hemagglutinin (HA) are antigenic glycoproteins on the surface of influenza virions. NA and HA both recognize carbohydrate structures and bind to terminal sialic acid (3) units on the surface of the host cell.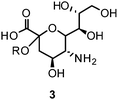
Binding of HA to its receptor 3 initiates viral entry into the host cell. NA also binds 3 and hydrolytically cleaves the α-(2,3) and α-(2,6) glycosidic linkage of terminal 3. Thereby, NA reduces the number of receptor binding sites for HA on host cells as well as on progeny viruses. This allows the mature virus to detach from the host cell during release and prevents self aggregation mediated by HA.26–28 As a consequence, NA plays a critical role for virus spread in infected tissues and enables virus transmission during infection. Due to the antagonistic functions of HA and NA, a balanced interplay between the activity of both viral envelope glycoproteins is essential for efficient entry and release of influenza viruses.29 Additionally, NA's function is related to the mobility of influenza virus within the respiratory tract. NA cleaves the glycan structure of mucus and allows the virus to reach host cells.30–32
2.2 Influenza NA classification
Nine NA subtypes were classified by antigenic characteristics. While all nine subtypes are found in avian hosts, only N1 and N2 subtypes were found in influenza virus strains causing human infections. According to their phylogenetic relationship and structure the nine subtypes split into two families: group-1 includes N1, N4, N5, and N8, whereas group-2 comprises N2, N3, N6, N7, and N9.33 The most prominent structural feature of group-1 NAs is the formation of a pocket located at the 150-loop. Contrary to other structurally determined N1 NAs, the one of the 2009 H1N1 virus lacks the 150-cavity.34,352.3 Influenza NA structure
The structure of an NA entity on the viral surface is composed of four identical subunits forming a homotetramer of around 240 kDa. The tetramer has a mushroom-like shape, where the head is composed of four catalytic domains (Fig. 1A) and the stalk is formed by the extended N-terminal sequences of the four subunits. The stalk amino sequence can vary in length and glycosylation for different subtypes. However, a membrane anchoring helix is conserved for each monomer. In contrast to the stalk region, the head domains – isolated by proteolytic cleavage – are accessible to X-ray crystallography and therefore have been thoroughly investigated within the past 25 years.17,30,36,37 In NA crystals the subunits assemble around a four-fold axis. The typical β-sheet dominated six-blade propeller fold of each subunit was first observed in 1983 by Varghese et al.36 for N2 and is conserved for other subtypes (N9,38,39 N4,33 N6,33 N8,33 N1;33,34,40,41 Influenza B NA42,43). In the centre of each subunit conserved residues form the catalytic active site (Fig. 1B).36,37,44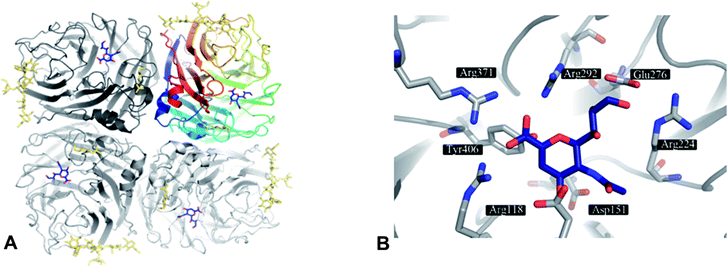 | ||
| Fig. 1 A) NA as tetramer with the inhibitor DANA (4) (blue) bound to the four active sites. The top right NA chain is coloured according to residue number. Glycosylation is indicated in gold. B) Active site of NA subtype N9 (PDB code 1f8b) with 4 (blue). | ||
2.4 Catalytic mechanism
The proposed catalytic mechanism is derived from the binding mode of the endogenous NA ligand 3 to influenza B NA.42 It was further explored by biochemical investigations as well as computational studies on influenza A NA.45–47 Three conserved arginine residues forming a sub-pocket stabilize the orientation of the carboxylate group of 3. In consequence, they direct the pyranoside ring into a boat conformation. Thus, formation of an oxocarbocation intermediate is favoured by this arrangement. The regeneration of the free 3 by addition of a water molecule results in the β-anomer, which subsequently transforms to its favourable α-anomer by mutarotation.45,46 A conserved aspartate is supposed to activate the water molecule and orients it in the optimal position. The cationic transition is proven by deuterium-labelled kinetic experiments.45 Reduced analogues of 3, DANA (2-deoxy-2,3-didehydro-N-acetylneuraminic acid) (4) and FANA (2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid) (5), show micromolar inhibitory activity.48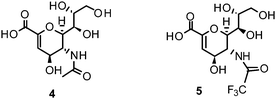
The conformation of the reduced pyranoside ring is similar to the transition state, and therefore the compounds match the binding pocket without undergoing ring deformation (Fig. 1B).46,49,50 The binding of 4 and 5 was crucial for elucidation of the catalytic mechanism. Additionally, they were important as lead compounds for the structure-based development of NAIs.46,47
2.5 Approved NAIs
NAIs show broad spectrum activity against influenza A and B.51 Commonly reported side effects of these drugs cover nausea, vomiting, dizziness, sinusitis, runny or stuffy nose, cough, diarrhea, and headache.1The close correspondence of highly-conserved residues of the active sites of all NAs of influenza A and B viruses renders this enzyme the most promising drug-target for the development of novel lead compounds.42,52,53 Derived from 3, three NAIs have been brought to the market for the prevention and treatment of influenza virus infection. In 1999, zanamivir (Relenza®) was approved by the FDA as inhalative NAI. Currently, a Phase III study of intravenous zanamivirversus oral oseltamivir in adults and adolescents hospitalized with influenza is ongoing.54
Furthermore, the ethyl ester derivative oseltamivir (Tamiflu®) was developed for improved bioavailability, making it the first approved orally absorbed NAI.55Oseltamivir acts as a prodrug, which is turned into the active carboxylate metabolite by hepatic esterases.56 Tamiflu® was stockpiled for treatment of pandemic flu in many countries.57
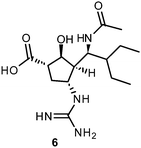
More recently, several cyclopentane and pyrrolidine derivatives have been developed as novel inhibitors of NA. Among those, peramivir (6) (Rapiacta®), an intravenously applied NAI, was first launched in Japan for the treatment of hospitalized and pediatric patients. In the U.S., 6 is currently unapproved but was tolerated under a temporary emergency use authorization by the FDA between October 2009 and June 2010 for hospitalized patients with known or suspected H1N1 influenza infection.58,59 All NAIs need to be administered no later than 36–48 h after manifestation of the symptoms in order to be effective.60
Also dimeric and diversely decorated analogues of zanamivir and oseltamivir have been reported. They in part exhibit enhanced efficacy in prolonging antiviral effects in vivo and are superior in inhibiting virus replication.61–63 Moderate effort has been made so far in designing novel disruptors with a decreased vulnerability toward resistance-conferring mutations, e.g. by selecting residues for ligand anchoring genetically stable due to their contributions to substrate binding.64
2.6 Mechanisms conferring drug resistances in influenza
Changes in proteins of approved anti-influenza targets, i.e. the M2 channel protein as well as the NA, can confer resistance and restrict prevention and treatment of influenza. Ion channel blockers, as well as NAI-resistant influenza viruses, can emerge and spread suddenly.65–70 Based on structural specifics and replication strategies, influenza viruses may evolve by two different ways (i) genetic reassortment, which describes the combination of genome fragments from different virus strains, and (ii) mutations leading to single amino acid exchanges. Reassortment events can be related with pandemic outbreaks in humans, as the immune defence is facing a virus strain with new antigenic characteristics resulting in inadequate immune response. As a result, drug susceptibility can be affected. For example, the H1N1 influenza A strain that emerged 2009 in humans and caused a pandemic showed a new genomic arrangement. While the NA gene and the gene for the matrix protein originate in Eurasian swine strains, the other gene segments have clustered in a previous triple reassortment (human, classical swine, and avian) in a North American porcine lineage of influenza A.71 The M gene segment from Eurasian swine influenza A viruses conferred resistance to ion channel blockers and the NA gene segment from those viruses enables efficient NAI treatment.18 In addition to genetic reassortment, influenza viruses show a high genetic variability within the different gene segments due to point mutations introduced by the viral polymerase that lacks a proof reading capacity. In contrast, the active site of the viral NA was shown to be highly conserved. However, spontaneously emerging point mutations within the inhibitor-binding site of NA were shown to confer oseltamivir and/or zanamivir resistance, e.g. mutations of E119, D198, R152, H274, and R292.51,72 Most of the reported mutations concern active site residues that are in direct contact with the natural substrate upon binding. These residues are highly conserved in the different subtypes and mutations of these residues are often associated with reduced biochemical activity, expression or transmission rates.73,74 A clinically significant case of an in vivo mutation (R152K) causing zanamivir-resistance in influenza B NA was reported in 1998;75 other isolated cases of drug resistance were identified occasionally.51Due to the highly conserved structure of the active site of the viral NA the susceptibility towards NAIs in clinical isolates did not change during the first years of their application.9,17 The situation, however, suddenly changed for the worse when oseltamivir-resistant H1N1 viruses spontaneously emerged and spread globally in the flu season of 2007/2008.6,9,76Oseltamivir-resistance was related to the H274Y mutation. Initially described in a clinical study of oseltamivir, the mutation remained unnoticed at first.77 It occurs at a residue without direct contact with the inhibitor. Results from comparative crystal structure analysis revealed a conformational shift and electrostatic effect induced by the H274Y amino acid exchange.78 The bulkier tyrosine induces a conformational shift of E276. The charged carbonic acid group of E276 is directed to a position directly facing the hydrophobic pentyloxy moiety of oseltamivir. While this mutation knocks out the activity of oseltamivir, zanamivir is still active in the mutant due to its hydrophilic glycerol moiety interacting with E276. The world-wide spread of the resistance indicates that strains carrying this mutation show a good transferability. This is in contrast to preliminary studies of the H274Y mutation.79,80 Bioinformatics analyses of sequence data from extensive surveillance indicate that the circulating oseltamivir-resistant strain does not only show an isolated mutation (H274Y), but is rather accompanied by additional mutations. The point mutation D344N was deemed to be essential for spreading oseltamivir-resistant strains,78 while another study showed H274Y strains spreading without D344N mutation.81 Bloom et al. combined sequence analysis with experimental comparison of NA expression and NA activity. They observed that V234M and R222Q can restore the expression level of a H274Y NA to the wild type level.82
A misbalance in NA/HA activity was also observed as a viral mechanism to overcome NA inhibition in cell culture-based assays. For example, influenza strains treated with NAIs were observed to exhibit mutations in HA e.g. additional glycosylation sites reducing HA affinity to sialic acid. As a result, those strains do not depend on NA activity for virus release and showed no susceptibility to NAIs in MDCK cells.31,83,84 The relevance of these findings for flu treatment is unknown.
Taken together, the sudden emergence and worldwide spread of NAI-resistant influenza strains may occur despite a highly conserved antiviral target. Based on these prospective findings, identification of novel NAIs breaking resistance is an urgent demand to ensure treatment of future influenza infections.
2.7 Similarities and differences of influenza NA with NA of other biological systems
The enzyme NA is not only specific to viruses but has also been reported as surface protein of other respiratory pathogens, including bacteria e.g. Hemophilus influenzae, Streptococcus pneumoniae, and Pseudomonas aeruginosa.85–87 These bacterial NAs can cleave α-(2,3)-linked 3 from glycoconjugates.26 Their function in pathogenesis is under discussion.27–29,87 The crystal structures of S. pneumoniae and P. aeruginosaNA have been reported.88 The structures revealed similarities with other bacterial NAs e.g. from Salmonella typhimurium, Vibrio cholerae, and Clostridium perfringens, including their interaction with the NAIs 4 and 5 or even striking differences, respectively.89–93 Interestingly, some research groups apply in vitro assays with bacterial NA (e.g. from C. perfringens and V. cholerae) to identify anti-viral agents.94–96 They use commercially available fluorescence (FL) and chemiluminescence (CL) based test systems that work with both viral and bacterial NAs because the substrates used are recognized by viral as well as bacterial NAs.97 Unfortunately, data comparing the two test setups, which would be helpful regarding their validity and significance, is scarcely available. Taking this into account, IC50 values obtained by assays using bacterial NA must be regarded with caution if compared with data obtained by assays using viral NA.In addition to viral and bacterial NAs, there are also mammalian enzymes involved in sialic acid metabolism, including endogenous NA, sialyltransferases, and CMP-synthase. Human NAs (huNAs) can be classified into four groups: intra-lysosomal (NEU1),98,99 cytosolic (NEU2),100,101plasma membrane-bound (NEU3),102,103 and lysosomal or associated with mitochondria (NEU4).104 In some recent studies the putative inhibitory effects of influenza NAIs on related endogenous enzymes have been evaluated.105–107 Though their sequence of amino acids is differing, the structural arrangement of the mammalian NAs is similar to the viral enzymes.108 Inhibition of huNAs is discussed as a reason for side effects of antiviral agents. In vitro studies with established NAIs did not show inhibitory effect on huNAs, which could explain neurological side effects.105 Nevertheless, inhibitors can be accommodated in the sialic acid binding pocket of huNA and corresponding crystal structures are available. This structural information on huNA can be helpful in the development and structural optimization of novel more specific NAIs.107 To avoid cross targeting, amino acids without equivalents in huNA should be preferred as interaction reference point for influenza NAIs.
3 Experimental evaluation of NA inhibition
3.1 Methods used in discovery and functional analysis of first generation NAIs
The first synthetic inhibitors of influenza NA were discovered without the structure of the active site and the detailed cleavage mechanism known. However, function and ligand/substrate of this viral enzyme were already determined. This knowledge allowed on the one hand the establishment of first generation NA inhibitory assays with the phenyl-α-ketoside of N-acetylneuraminic acid or fetuein as substrates to measure enzyme activity and to search for inhibitors.109,110 On the other hand specific synthetic NAIs, 4 and 5 were discovered by using these NA inhibitory assays.110,111 The NA-specific competitive mechanism of inhibition was demonstrated by measuring the inhibition constant Ki. Cell culture-based assays e.g. virus elution from erythrocytes, plaque size reduction, and comparison of inhibition of single step growth cycle and multicycle replication helped to confirm the antiviral activity of 4 and 5.48,110–113Electron microscopy showed that virions grown in the presence of 5 form aggregates near the cell surface.112 The observed virus aggregation proved the hypothesis that 5 directly interacts with the influenza NA and by this manner blocks the enzymatic removal of sialic acid from the virus envelope. As a result, sialylated particles are produced containing the receptor sites for HA of other virus particles. Formation of virus aggregates results in reduction of hemaglutination and infectivity titer. Furthermore, results from these fundamental early experiments have indicated differences between NA of subtype N1 and N2 for the first time.111 The concentration of 5 required for inhibition of N2 was 50–200 times higher than that for N1 influenza A viruses. This implies structural differences that have to be taken into account in the search for new NAIs.About 20 years after the discovery of first specific NAIs, the crystal structure of NA of subtype N2 was solved.114,115 This fuelled the application of computational methods and structure-based design resulting in the development of the highly active N-acetyl-α-D-neuraminic acid (NANA) (8) derivative zanamivir.116 Shortly afterwards, oseltamivir, a second highly active NAI was discovered.55 The biological activity of both compounds was demonstrated in NA inhibitory assays and cell culture-based experiments.
Until now, NA-inhibitory assays, cell culture-based, and structure-based design are used to discover and characterize novel NAIs. However, the methods were adapted in part to automated microtests to save time and material, and to avoid subjective evaluation. They were also refined and standardized.
3.2 Advantages and disadvantages of methods currently applied to identify novel NAIs
Today, functional NA inhibition assays are accepted to be the method of choice in NAI susceptibility surveillance.They are highly standardized, approved in primary screening for novel NAIs for a long time,110,111,117–119 and more predictive of in vivo susceptibility than cell culture-based assays.120 State of the art functional NA enzyme inhibition assays are either based on fluorescence (FL) or chemiluminescence (CL).116,121,122 The substrate for FL assays, 4-methylumbelliferyl-N-acetyl-α-D-neuraminic acid (MUNANA) (7), is cleaved by NA to release 8and the fluorescent compound 4-methylumbelliferone (MU) (9) (Chart 1A).
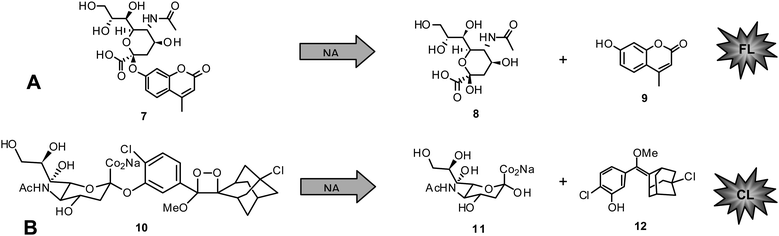 | ||
| Chart 1 A) Fluorescent NA inhibition assay: MUNANA (7) cleaved by NA to give 8 and 4-methylumbelliferone (9). B) Chemiluminescent NA inhibition assay: NA-Star (10) cleaved by NA to give 11 and 12. | ||
The second assay uses the chemiluminescent substrate sodium (2-chloro-5-(4-methoxyspiro{1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.13,7]decan}-4-yl-5-acetamido-3,5-dideoxy-α-D-glycero-D-galacto-2-nonulopyranosid)onate (NA-Star) (10) available as component of NA-Star kit (Tropix, Applied Biosystems, Darmstadt, Germany) (Chart 1B). In the latter case the substrate is enzymatically hydrolyzed to give 11and the respective chemiluminescent product 12. The quantity of FL or CL is negatively proportional to the inhibitory activity of the measured sample. Parameters showing influence on the reliability of generated results include buffer, pH, and substrate concentration in the applied test system.
Comparative studies with both functional inhibition assays were performed with zanamivir and/or oseltamivir. Buxton et al. showed that the use of substrate 10 results in a 67-fold reduction in detection limit in comparison with the fluorogenic substrate 7 from 200 pM to 3 pM NA.121 The IC50 values of zanamivir determined with a panel of influenza A and B viruses were nearly similar in the two NA assays. However, according to two more comprehensive reports on zanamivir and oseltamivir susceptibility of seasonal viruses using standardised commercially kits available now, it was noted that IC50s may vary for the same virus if the NA inhibitory assay was done using substrate 7 and 10.123,124 In both studies, the CL assay results in 2- to 12-times lower IC50 values than the FL assay. The results from NAI susceptibility surveillance studies with FL- as well as CL-based NA-inhibitory assays indicate that virus type, subtype, and strain as well as the NAIs tested have an influence on 50% inhibitory concentrations.69,121,123–125 Based on the proven reproducibility, ease of automation, time required for the assay, and greater sensitivity, the CL assay was selected for NAI susceptibility testing of influenza virus isolates circulation globally. It seems to be also the best choice for antiviral testing.
Cell culture-based assays need to be performed to determine the cytotoxicity of identified NAIs. They can also be applied for confirmation of anti-influenza activity of NAIs but with some limitations. For example, plaque reduction assay that has been traditionally applied for studying the anti-influenza activity of compounds can be used.18,111,113,116,126 Although laborious and time consuming, the drug susceptibility interpretation is generally very reliable. However, the experience from NAI susceptibility testing showed that reduction of plaque number does not necessarily reflect the susceptibility of a given viral NA to the respective inhibitor or the antiviral potential of NAIin vivo.77,113,116,120,127Reduction of plaque number was dependent on the cell line and virus strain used.127,128 Alternatively, plaque size reduction was used as a study parameter because a somewhat better correlation between NA inhibition and antiviral activity was found when the influence of NAIs on plaque size was studied.77,84,127,129 Other procedures currently used for the routine antiviral or virus susceptibility screening include different dye uptake assays.130–132 According to our own experience they work in anti-influenza NAI screening after careful validation under multicycle replication condition. The antiviral activity of NAIs was also confirmed by focus reduction assays18,133 or analyzing virus yield reduction.128,129,131 Using a low multiplicity of infections for most but not all influenza A viruses, a good correlation between enzyme inhibition and virus yield reduction can be observed. Results from assays that facilitate studies on prevention of virus spread in infected cell monolayers under multicycle replication conditions can further underline the antiviral activity of this inhibitor class. Such assays include, for instance, immunohistochemical staining of influenza nucleoprotein84,128 or electron microscopy.51,112,127 In summary, all these culture-based antiviral assays can help to provide evidence for NAI activity against infectious virus. But, the interpretation of results is complicated by several factors unique to NAIs.134 In particular, the validity of results depends on a balanced function of HA and NA. It can be affected negatively by the receptor expression pattern of test cells133,135 as well as changes in the binding efficiency of HA to sialic acid receptors e.g. by additional glycosylation.84,136 As a result, viruses become partially independent from NA function for release and can spread from cell-to-cell, also in the presence of NAI. This explains the observed influence of the virus strain and cell line used on the inhibitory effect in all cell culture-based assays. Therefore, a careful evaluation of cell culture-based assays with approved NAIs considering the impact of test virus and cell line is necessary for successful application of those assays in antiviral research.
Today, NA-directed in silico screening and computational docking studies represent a highly useful method to find new hit compounds from available structure collections and to characterize the putative binding mode of NAIs, respectively.19,137–139 Moreover, knowledge on the dynamics of NA opens new possibilities for the development of next generation NAIs, as summarised below. Potentially active molecules identified or designed by computational approaches can be purchased or synthesised and subsequently evaluated in biological assays. Quantitative structure–activity relationship (QSAR) data gained from experiments can be used to find novel compounds with higher activity and/or compatibility. Increasing knowledge of NAI resistance and mutations enables the consideration of resistant viruses in computational enzyme- as well as cell culture-based approaches. The close interplay between modern computational studies and functional inhibition assays allows for fast discovery and characterization of highly active, resistance-breaking inhibitors of NA.
4 Inhibition of influenza NA by natural compounds
4.1 Evaluation of published data
In the past decade many research teams have demonstrated renewed interest in investigating substances with regard to their NA inhibiting potential. A number of chemically diverse compounds have been identified endowed with this activity. 2634 references (without duplicates) were collected from SciFinder140 (accessed 2011/05/05) using the research topic “influenza neuraminidase” constrained to the publication years (2000–2010) and the document types journal, letter, review, and patent. This answer set was further refined by either the keyword “natural product” (33 ref.) or “compound” (269 ref.). Each category was then analyzed in respect to the frequency between the years 2000 and 2010. Fig. 2 reveals that beside the overall upward trend, the number of literature reports dealing with natural products targeting influenza NA has tremendously increased since 2009. | ||
| Fig. 2 Comparison of number of publications dealing with influenza NA related to the keywords “natural product” (blue) and “compound” (grey) between the years 2000 and 2010. | ||
This tendency might be correlated with the emergence of the H1N1 flu pandemic in 2009, which underlines the urgent need for novel antiviral therapeutics.
In the present survey natural products with publicly accessible information concerning their NA inhibiting potential are reviewed. For this purpose SciFinder140 (accessed 2011/01/18) was used to explore the research topic “influenza neuraminidase”. Search results were constrained to the publication years 2000 till present, and to the document types journal, letter, review, and patent (2761 ref. without duplicates). For further analysis, this set was organized by the subject category “organisms” (1639 ref.). Plant species or keywords related to natural products were then selected manually (21 ref.). In an additional query approach, not to neglect insufficiently tagged literature, further research topics such as “influenza sialidase”, “influenza neuraminidase inhibitor”, and “influenza sialidase inhibitor” were explored. Furthermore, primarily obtained search results were refined by using chemical structure classes as keywords. In a final step, false positive records, such as IC50 values obtained by the use of bacterial instead of viral NA assays, were sorted out.
For a clear presentation of the pool of information gained, an in-depth evaluation was performed considering only reported results of NA inhibition assays of pure compounds. This procedure was accompanied by difficulties, because not all research groups use the same viral strain or isolate, assay type, positive control or concentration unit. Table 1 provides a compilation of essential information on natural compounds tested for NA inhibition. Comprised data is ordered alphabetically by compound structure class and aims at providing insight not only into influenza NA inhibiting compounds and applied assay types, but also the distribution of NA-inhibiting secondary metabolites between plant species and families, respectively. In this context it needs to be emphasized that apart from natural compounds with significant NA inhibiting effects, also those showing only weak or no activity on the respective target have been taken into account. We decided to include reported negative test results due to their potential value for future work, viz. (i) to avoid a retesting of inactive compounds, (ii) to validate hypotheses (e.g. computational models), and (iii) to enable the establishment of SARs.
For the sake of clarity, a set of three distinct influenza viral subtypes, i.e.H1N1, H3N2, and H9N2, were selected to provide an overview on reported NA inhibiting activities, without going further into detail by specifying the viral strain and isolate, respectively. In case recombinant H1N1 (rvH1N1) has been used, the corresponding IC50 values are marked with the letter b (b) in Table 1. If compounds have been tested on several strains or isolates of one viral subtype, an activity range is given. For standardization reasons, IC50 values originally published in μg mL−1 or ng mL−1 have been converted to μM or nM and are marked with the letter a (a). References to primary literature are provided.
4.2 Compound classes and sources of natural NAIs
Published data from NAIs from synthetic chemistry and natural sources have roughly been classified by Zhang et al. in 2010.141 The survey of our literature search focusing on natural products (Table 1) allowed categorizing influenza NA inhibiting natural compounds in detail. Due to already mentioned difficulties, i.e. heterogeneous test systems, a threshold between effective and ineffective NAIs needed to be drawn. In this review, compounds with IC50 values >100 μM obtained by FL assays and >50 μM obtained by CL assays were considered inactive. Fig. 3 provides the distribution by percentage of chemical classes of reported NA inhibiting natural compounds. Among the most prominent scaffolds are flavonoids and (oligo)stilbenes. Other chemical classes investigated for their potential on influenza NA are not present in this diagram, e.g.monoterpenes and triterpenes, because they could not reach the set threshold.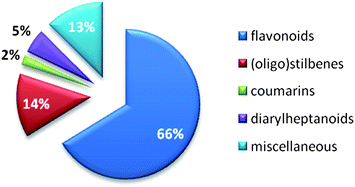 | ||
| Fig. 3 Percentage of natural product compound classes showing inhibition of influenza NA (publication years: 2000 until present). | ||
4.3 Phenolic compounds as NAIs
Natural product research dealing with influenza NAIs focuses mainly on the biological activity of substituted phenyl-benzopyranes, so called flavonoids, which are widespread in the plant kingdom. In particular, species belonging to the Fabaceae family have been intensively studied.142–145Flavonoid constituents are categorized into ten major sub-groups, i.e.aurones, biflavonoids, catechins, chalcones, flavanones, flavanonols, flavans, flavones, flavonoles, and isoflavones.In the context of druglikeness or druggability, flavonoids represent an interesting compound class of low-molecular weight chemical entities accompanied with mostly well-know bioavailability and innocuousness.146 Various research groups working with these polyphenolic compounds studied their SARs, allowing the establishment of an NA activity order according to the involved functional groups and scaffolds. Liu et al. investigated 25 commonly occurring flavonoids and record the following order of NA inhibiting potency: aurones > flavon(ol)es > isoflavones > flavanon(ol)es and flavan(ol)es.142 Efforts to categorize NA inhibitory activity measured by a standard FL assay resulted in the classification of three groups: (i) good (IC50 < 40 μM), (ii) moderate (IC50 40–80 μM), and (iii) weak (IC50 > 80 μM) inhibitors.142 Thus, essential functional groups for good NA inhibiting effects of flavonoids are hydroxyl groups in position 4′ and 7, an oxo group in position 4, and a double bond between position 2 and 3. Concerning the aurone subtypes, essential functionalities are a hydroxyl group in position 6, an oxo group in position 3, and a double bond between position 2 and the phenylidene moiety.142 The presence of a bulky sugar moiety distinctly reduces the NA inhibitory potential due to an assumed sterical hindrance.142 Recently, these findings were incorporated in a theoretical computational approach by Mercader et al. to predict NA inhibiting activity based on quantitative structure–activity relationships (QSAR) exploiting already reported activities of flavonoids including relevant 2D and 3D descriptors.147
Although Liu and co-workers ranked substances belonging to the structure class of aurones (IC50 between 22.0 and 72.0 μM; H1N1) as most potent NAIs among flavonoid derivatives,142 no further studies on the NA inhibiting properties of these compounds have been published to the knowledge of the authors.
Biflavonoids like ginkgetin (13) with an IC50 of 97.09 μM (H1N1), isolated from Ginkgo biloba and Cephalotaxus harringtonia, hinokiflavone (14) (IC50 77.63 μM; H1N1), and 4′-O-methylochnaflavone (15) (IC50 2.01–3.54 μM; H1N1) have successfully been identified as potential NAIs.19,147,148
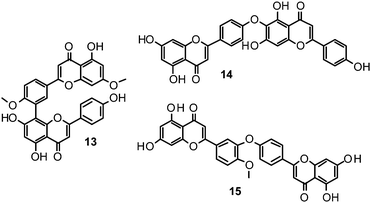
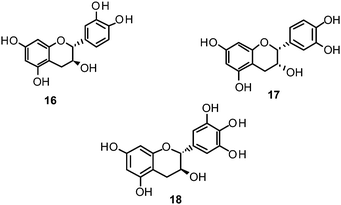
Catechin (16) and its derivatives epicatechin (17) and gallocatechin (18) were found to exert no or only weak NA inhibition.142,149
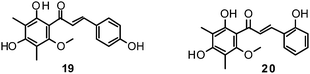
Fourteen C-methylated flavonoids (chalcones, flavanones, isoflavones, and one flavanonol) from Cleistocalyx operculatus were found to have NA inhibiting properties, with (E)-4,2′,4′-trihydroxy-6′-methoxy-3′,5′-dimethylchalcone (19) (IC50 3.31–20.45 μM) and 2,2′,4′-trihydroxy-6′-methoxy-3′,5′-dimethylchalcone (20) (IC50 2.55–28.12 μM) showing the strongest effects against NA from the novel influenza H1N1 (WT) and oseltamivir-resistant H1N1 (H274Y mutant), respectively.150
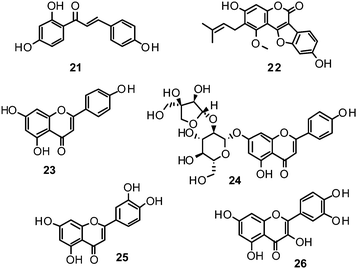
Ryu et al. reported the isolation and NA inhibition evaluation of 18 polyphenols from the methanol extract of the roots of Glycyrrhiza uralensis.143 The investigated compounds include chalcones, (prenylated) flavonoids, (prenylated) coumarins and one phenylbenzofuran. Among the chalcones, isoliquiritigenin (21) exhibited the most potent inhibitory activity, with an IC50 value of 9.0 μM towards recombinant influenza virus A (rvH1N1).143,151 Most favourable effects against rvH1N1 were demonstrated for the coumestan glycyrol (22) (IC50 3.1 μM), which is characterized by a condensed furan ring in contrast to classical coumarins lacking this feature. In addition, the presence of a free hydroxyl group in position 7 and an isoprene residue in position 6 seem to be essential functionalities for these scaffolds.143
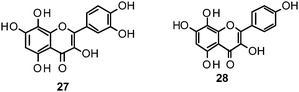
Flavones, such as apigenin (23), apiin (24), and luteolin (25), have also proven significant activities against influenza NA with IC50 values between 25.92 and 33.71 μM (H1N1).152 Jeong and co-workers investigated five flavonols isolated from the roots of Rhodiola rosea and determined their NA inhibiting properties.153 For in-depth SARs they compared the activities to a further nine available flavonoids. Regarding the inhibitory effects towards rvH1N1 quercetin (26), gossypetin (27), and herbacetin (28) exhibited the most promising activities (IC50 2.2, 2.6, and 8.8 μM).
Within the sub-group of reviewed isoflavones, only daidzein (29) and genistein (30) displayed mentionable inhibitory activities against NA with IC50 values of 37.1 and 77.1, respectively (H1N1).142
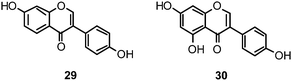
Recently, (oligo)stilbenes isolated from Vitis amurensis and Gnetum pendulum were reported to exert anti-viral activities by targeting influenza NA. Among the most potent constituents were (+)-viniferol C (31) (IC50 8.94–61.16 μM; H1N1) and gnetupendin B (32) (IC50 13.14 μM; H1N1).154,155
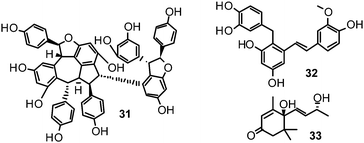
In addition, various compounds not belonging to the mentioned chemical classes or sub-groups have been investigated on their NA-inhibiting properties. One example is represented by vomifoliol (33), a 2-cyclohexen-1-one derivative, isolated from Chaenomeles speciosa showing an IC50 of 10.39 μM (H1N1).156
4.4 Isoprenoids – a compound class showing no effect on NA
As discussed before, reports on compounds with experimentally determined inactivity on influenza NA have also been regarded as valuable input for future research efforts. Three monoterpenes from the essential oil of Melaleuca alternifolia (Myrtaceae) and five triterpenes occurring in Chaenomeles speciosa (Rosaceae) were evaluated for their inhibitory potential on NA. All tested compounds of this structural class did not exhibit any inhibitory activity on NA.156,1574.5 Extracts with promising NA inhibitory activity
Publications dealing with promising NA inhibiting plant extracts have appeared rather infrequently during the last decade. The majority of research groups seem to preferentially elucidate bioactivity and mechanism of action of isolated pure compounds instead of extracts, allowing for publication in higher-ranked journals. Another aspect might be that extracts representing multi-component mixtures are rather tested in general antiviral assays, such as viral replication tests, than in more specific assays directly evaluating the NA activity. Nevertheless, eastern medicines, such as, especially, the traditional Chinese medicine (TCM), describe well-defined anti-influenza preparations composed of one or more herbs instead of individual compounds. In most cases the active principle(s) and mechanism of action is unknown or not fully understood. A review published in 2006 by Wang et al. deals in detail with TCM-derived anti-influenza agents from plants.158 This issue was also the subject of a recently published review by Geet al. describing the importance of four TCM anti-flu prescriptions in the fight against the 2009 H1N1 pandemic in China.159 In a very recent study, water extracts from 439 traditional Chinese medicinal plants were investigated for their NA inhibiting activities in vitro. Among the five extracts analyzed in vivo using a mouse model, the extract of Melia toosendan significantly increased the survival rate of H1N1 viral infected mice.1605 Strategies for the discovery of NA inhibiting natural compounds
Researchers in the field of phytochemistry are faced with a nearly unmanageable quantity of structurally diverse natural products as only the estimated number of plant species on earth is about 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.161 Given such issues the search for novel innovative natural leads resembles looking for a needle in a haystack. In the very recent analysis of Newman et al., approved drugs for all kind of diseases were statistically evaluated in respect to what structures led to the design of the compounds subsequently approved and what origins they have.162 In the case of antiviral agents, 80% of 46 entities registered in the last 29 years (1981–2010), can be classified as natural product-botanicals, synthetic but natural product mimics, natural product pharmacophores, or a combination of the latter two.162Oseltamivir is one of the success stories of anti-influenza drug synthesis having its roots in nature: the abundant plant constituents, quinic acid (34) and shikimic acid (35), are used as starting materials for the development of oseltamivir.55 Both natural compounds are intermediates in the biosynthesis of essential aromatic amino acids and also precursors to lignans and phenols.163,164
000.161 Given such issues the search for novel innovative natural leads resembles looking for a needle in a haystack. In the very recent analysis of Newman et al., approved drugs for all kind of diseases were statistically evaluated in respect to what structures led to the design of the compounds subsequently approved and what origins they have.162 In the case of antiviral agents, 80% of 46 entities registered in the last 29 years (1981–2010), can be classified as natural product-botanicals, synthetic but natural product mimics, natural product pharmacophores, or a combination of the latter two.162Oseltamivir is one of the success stories of anti-influenza drug synthesis having its roots in nature: the abundant plant constituents, quinic acid (34) and shikimic acid (35), are used as starting materials for the development of oseltamivir.55 Both natural compounds are intermediates in the biosynthesis of essential aromatic amino acids and also precursors to lignans and phenols.163,164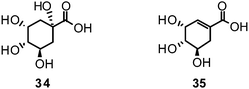
They accumulate to some extent, primarily in gymnosperms and woody dicotyledons. The most important source of 35 has been found in the genus Illicium, where it was first isolated from I. religiosum in 1885.165 Star anise (I. verum) turned out as very rich source of 35, and was accordingly used in large amounts to yield this much valued precursor for the semi-synthesis of oseltamivir.166
In order to rationalize the access to novel natural NA inhibiting leads, which otherwise would be dependent on serendipity, three different strategies have been applied so far: (i) in vitro screening of natural compounds or extracts followed by bioassay-guided fractionation and isolation, (ii) strategies based on the exploitation of empirical knowledge, and (iii) computational approaches. Also the combination of heuristic approaches (e.g. ligand or structure based) with empirical ones might be favourable.
5.1 In vitro screening of extracts–Bioassay-guided fractionation and isolation
Random selection of natural material followed by an unbiased in vitro screening allows first activity categorizations of multi-component mixtures.167,168 Although the identity of the individual compounds responsible for the measured effect cannot be elucidated in this way, promising natural starting material can be selected for further processing. Extracts displaying desirable activities are in most cases subjected to a bioassay-guided fractionation with the goal to identify the active principle(s). In the course of this process the individual bioactive components responsible for the given effects can ideally be traced down to a molecular level, allowing structure elucidation.168 This strategy represents one of the classical lead finding methods especially applied by academic research groups. At a first glance this approach is more straightforward than laborious isolation and structure elucidation of single components without knowledge of proven bioactivity of the mother sample. However, for this strategy experience is of pivotal importance to avoid pitfalls, e.g. assay-disturbing constituents.A respective screening strategy was applied by Lee and co-workers who investigated crude extracts from 260 plant species on their inhibiting potential towards NA of C. perfringens.96 These results provide highly valuable data for further selection and processing of promising plant material e.g. in the course of bioassay-guided fractionations.
Many other research groups successfully identified novel NAIs based on such large scale anti-influenza screening campaigns; e.g. Dao et al. discovered 14 bioactive C-methylated flavonoids (Table 1) from Cleistocalix operculatus buds by using an anti-influenza screening approach.150
In the course of a project by the Chinese Academy of Medical Sciences more than 10000 plants were tested.152 Among them a pronounced NA inhibiting effect was observed for the herb extract of Elsholtzia rugulosa, which prompted Liu et al. to further investigate this plant and isolate constituents responsible for its activity, e.g.23 (H3N2; IC50 28.90 μM) and 25 (H3N2; IC50 32.63 μM) (Table 1).152
5.2 Exploitation of empirical knowledge
This procedure takes advantage of ethnopharmacological hints about beneficial anti-influenza effects, mainly of herbal remedies as well as reports on anti-influenza or anti-viral multi-component mixtures or even individual compounds of not yet identified mechanism of action.Concerning toxicity and bioavailability, naturally derived remedies and their ingredients used by humans for decades, centuries or even millennia are supposed to be safer and more efficacious than substances missing this background.167 In particular, preparations with ethnomedical background might be favourable compared to synthetic compounds evoking from combinatorial chemistry.167 Natural compounds occupy an even larger and more complex chemical space compared to synthetic compounds.169
Some extracts (e.g. from Agrimonia pilosa,170Echinacea purpurea,171 and Prunus mume172) or multi-component mixtures (e.g.polyphenol fractions from Punica granatum173 and secoiridoid glucosides from Ligustrum lucidum174) have already shown a significant reduction of virus-induced cytopathic effects and general anti-viral or anti-influenza activity. These phenomenological effects often observed in folk medicine give valuable hints for tracing activity on a target specific level exploring a possible impact on influenza NA. Concerning viral infections, a great number of preparations containing natural materials have shown bioactivity with often complementary and overlapping antiviral mechanisms of action.24,25 In most cases therapy with these simply prepared multi-component mixtures of complex chemical composition is intrinsically tied to traditional medicines, i.e.TCM, Ayurveda or indigenous medicines.167 Many of the contained secondary metabolites, possibly active principles, have never been examined chemically or biochemically using modern medical knowledge. Phytochemical and pharmacological work performed with ethnomedical anti-viral agents mainly from plants revealed a variety of active metabolites from different chemical classes, e.g.stilbenoids from Gnetum pendulum,155C-methylated flavonoids from Cleistocalix operculatus,150 and polyphenols from Glycyrrhiza uralensis.143
Successful identification of novel bioactive leads from the follow-up of phenomenological effects or ethnopharmacological hints is based on the selection of the correct starting material, its medicinal preparation, the kind of application, and the pharmacological profile.175 This strategy, however, is not a guarantee to discover constituents acting on the supposed target. Reported positive effects in flu treatment, regardless if obtained from clinical trials or empirical knowledge handed down to the present, can only give clues for further research efforts. One might not know if therapy is successful due to NA inhibition or if improvement of health conditions is simply triggered by a stimulation of the immune system. Furthermore, other anti-influenza targets, e.g. the non-structural NS1A protein, the nucleoprotein, and the viral polymerase should not be disregarded.176
5.3 Exploiting influenza NA inhibition by computer-guided approaches
Computational strategies to identify novel NAIs can be classified in ligand-based, structure-based or combined approaches, according to the data and methods employed.177 Given the accessibility of influenza NA to X-ray crystallography, structure-based methods have been playing a major role in the development of NAIs.178Zanamivir, the first approved inhibitor of influenza NA resulted from a highly successful optimization campaign during which affinity to the target was increased by 5000-fold, driven by computational analyses using the computer software GRID.179Molecular mechanics (MM) calculations based on demanding molecular dynamics (MD) simulations were applied to estimate the binding energy of potential optimisations during the development of zanamivir.46 However, ligand-based methods have also been employed and successfully identified, e.g., NSC 404789 (36), a uronic acid derivative as an inhibitor of influenza NA (IC50 1.8 μM).180
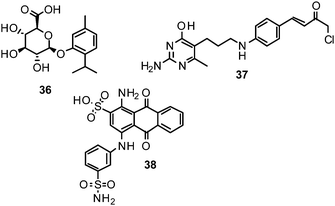
Protein–ligand docking is one of the most popular structure-based in silico methods. It is particularly useful for identifying bioactive compounds and for deriving the potential binding mode of small organic molecules to their target. Using a docking-based virtual screening approach, NSC 89853 (37) was recently identified as a bulky NAI for N1. Docking studies suggest that this compound requires an open-form binding site to be accommodated.181
Employing MD simulations of influenza NA, Amaro et al. observed extended conformational shifts of the 150- and the 430-loop.182,183,184 It appears that the binding site of influenza NA can open to a much larger extent than anticipated from prior X-ray structure analyses, which can be systematically explored for the development of more potent, chemically diverse inhibitors. In this context, computational analyses of multiple target conformations of H5N1 NA revealed novel protein hot spots proximate to the sialic acid binding region that could serve as potent anchor points for small organic inhibitors.185
In general, docking programs consider the target structure to be rigid. Some algorithms allow including a certain degree of target flexibility during the docking process; however, far-reaching conformational arrangements are not sampled by most of the conventional docking algorithms. Ensemble-based docking has turned out to be a particularly powerful approach to address highly flexible targets in virtual screening. Thereby, a collection of representative protein conformations (as observed during the MD simulations or in X-ray crystallographic experiments) is generated and, subsequently, virtual screening is performed on each of these structures. Promising candidate molecules are then selected from all individual hit lists in order to cover a large proportion of the biomolecule's phase space. In the case of influenza NA, it has become increasingly popular to sample target flexibility employing MD simulations to generate a number of representative protein conformations (conformational ensemble) for docking in order to obtain superior performance.186
Inspired by the observations of a highly flexible 150-loop, which has been initially observed in X-ray structures, several novel scaffolds for inhibitors targeting extended conformations of the influenza NA have previously been reported.137 The fact that the loop region was found to be more flexible in the apo form of group-1 compared to group-2 NAs, renders it an interesting structural characteristic that could be exploited for rational design of novel NAIs.33 Rudrawar et al. reported the first experimental observation of derivatives of 3 stabilising the 150-loop of group-1 influenza NA in its open conformation,187 a proof of concept for the rational design of NAIs selectively targeting group-1 NAs.
Among novel scaffolds that were identified by taking these aspects into account, initial in vitro testing confirmed the predicted activity of several of them, e.g. NSC 117079 (38).188NA conformations derived from MD simulations were also used to deduce analogues of zanamivir and oseltamivir with predicted superior activities.189
Recently, we have identified novel lead structures of NAIs from plants.118 Based on promising literature data on the inhibiting potential of a methanol extract of Alpinia katsumadai (Zingiberaceae) towards NA from C. perfringens,96 the seed extract of this plant was phytochemically investigated to track down the active principle(s). Six constituents were isolated, with four diarylheptanoids showing in vitro influenza NA inhibitory activity in a CL based enzyme inhibition assay within the low micromolar range against human influenza virus A/PR/8/34 of subtype H1N1. Katsumadain A (39) (IC50 1.05 ± 0.42 μM) was identified as the most promising compound (Table 1). This bulky diarylheptanoid, characterized by a monocyclic α-pyrone moiety and an additional phenyl group also inhibited the NA of four H1N1 swine influenza viruses with IC50 values between 0.59 and 1.64 μM, whereas the NA of an oseltamivir-resistant human H1N1 isolate was only weakly affected.

In addition to 39, four active compounds (40–43) comprising simple diarylheptanoid skeletons with two peripheral phenyl groups connected via a heptyl chain were used for SAR analyses.
The potential binding mode of 39 was resolved using protein–ligand docking. 39 is unlikely to bind to influenza NA in a target conformation observed in X-ray experiments due to its large molecular volume. MD simulations were performed to derive conformations of influenza NA. A representative frame of a well-populated cluster of the MD trajectory could be identified that allowed the establishment of favourable interaction patterns between the ligand and the protein. Overall a very good surface complementarity was observed. In particular, the extended conformation of the 430- and the 245-loop appeared to be essential for the accommodation of the ligand.
The proposed binding mode of 39 was taken as a starting point for shape-focused screening to identify novel active molecules from the NCI database (Developmental Therapeutics Program, DTP, National Cancer Institute, U.S.) and all five selected compounds showed activities in the very low micromolar range.19 Four of them, i.e.quercetin 5,7,3′,4′-tetramethyl ether (44), artocarpin (45), 15, and 1-(5-hydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-2-phenyl-ethanone (46) are flavonoid derivatives (Table 1). 45, the most active compound identified, inhibits oseltamivir-sensitive H1N1 strains with an IC50 range of 0.18–0.30 μM.19 Interestingly, this compound also exhibits significant activity on an oseltamivir-resistant H1N1 strain (IC50 0.55 μM).19Fig. 4 depicts a docking pose of 45 and the suggested binding interactions with amino acid residues from the NA protein backbone.
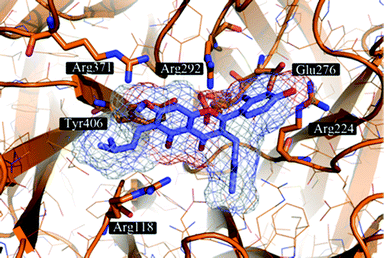 | ||
| Fig. 4 Artocarpin (45) (carbons lavender, oxygens red) docked to a representative frame of the MD simulation of influenza NA. The docking pose suggests the formation of hydrogen bonding interactions with several Arg and Glu residues present in the binding site. Hydrophobic contacts are formed with Arg224 and Tyr406. | ||
6 Conclusions and future perspectives
The threat of influenza pandemics claiming utterly devastating death tolls and the small number of effective drugs to fight this pathogen, including the emergence of resistances, are the main driving forces behind the search for innovative NAIs.Within the past three years, scientific communities all over the world have shown renewed interest in the search for novel anti-viral agents from plant origin. Natural compounds are widely recognized as privileged structures, trimmed by evolutionary processes to interact with macromolecular targets. However, working with them requires expertise and might be paved with challenges, viz. legal access to correctly identified starting material, analytics, isolation from multi-component mixtures (extracts), purification, structure determination and chemical complexity, just to name some aspects. Natural product research is accordingly highly multifaceted and interdisciplinary, and asks for target-oriented strategies exploiting their bioactivities. This might be of relevance for outsourcing (parts of) lead discovery campaigns focusing on natural products from industry to academic groups, as happened in the past decades. Concerning antiviral research and in particular anti-influenza agents, input from ethnopharmacological knowhow and phenomenological proofs about antiviral effects are highly valuable hints for the selection of promising natural materials. In combination with heuristic approaches based on previous structural knowledge about the target's binding site and its ligands, optimal starting points are available to further rationalize the efficient search for improved NAIs. This survey aims at bundling these data by summarising the recently reported discoveries on influenza NA and the mechanism of resistance to common NAIs together with available information on natural products tested for their influenza NA-inhibiting potential (∼150 compounds). It is anticipated that the recent findings of the enzyme flexibility and thereby extended binding pocket of the influenza NA, as well as the progress in computational methods exploiting these findings will fuel natural product research activities for effective and innovative drugs to fight influenza.
| Compound name | No. | Natural source | Family | NA inhibition IC50 [μM] | Assay type | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| H1N1 | H3N2 | H9N2 | ||||||
| a For clarity purposes IC50 values originally published in μg mL−1 or ng mL−1 have been converted to μM or nM, respectively. b rvH1N1 = recombinant H1N1. c n. g.: not given. d Os: oseltamivir. e Os ac: oseltamivir acid. f Neu5Ac2en: 2-deoxy-2,3-dehydro-N-acetylneuraminic acid. g MA: 8-methoxyapigenin. | ||||||||
| Coumarins | ||||||||
| glycyrin | 47 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 10% at 200b μM | n. g. | n. g. | FL | 143 |
| (Os: 1.59b nM) | ||||||||
| licopyranocoumarin | 48 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 10% at 200b μM | n. g. | n. g. | FL | 143 |
| (Os: 1.59b nM) | ||||||||
| Coumestanes | ||||||||
| glycyrol | 22 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 3.1b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| isoglycyrol | 49 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 92.4b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| Diarylheptanoids | ||||||||
| (3S)-1,7-diphenyl-(6E)-6-hepten-3-ol | 40 | Alpinia katsumadai Hayata | Zingiberaceae | 4.13 (Os: 0.13 nM) | n. g. | n. g. | CL | 118 |
| (3S,5S)-trans-3,5-dihydroxy-1,7-diphenyl-1-heptene | 41 | Alpinia katsumadai Hayata | Zingiberaceae | 29.75 (Os: 0.13 nM) | n. g. | n. g. | CL | 118 |
| (E,E)-5-hydroxy-1,7-diphenyl-4,6-heptadien-3-one | 42 | Alpinia katsumadai Hayata | Zingiberaceae | 4.67 (Os: 0.13 nM) | n. g. | n. g. | CL | 118 |
| alnustone | 43 | Alpinia katsumadai Hayata | Zingiberaceae | 6.10 (Os: 0.13 nM) | n. g. | n. g. | CL | 118 |
| katsumadain A | 39 | Alpinia katsumadai Hayata | Zingiberaceae | 0.59–1.05 (Os: 0.06–0.13 nM) | n. g. | n. g. | CL | 118 |
| Flavonoids and related structure types | ||||||||
| Aurones | ||||||||
| (2E)-4,6-dihydroxy-2-[(4-hydroxyphenyl) methylene]-3(2H)-benzofuranone | 50 | n. g. | n. g. | 25.6 (Os ac: 15 nM) | 22.3 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| (2E)-6-hydroxy-2-(phenylmethylene)-3(2H)-benzofuranone | 51 | n. g. | n. g. | 72.0 (Os ac: 15 nM) | 73.3 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| (2E)-6-hydroxy-2-[(4-hydroxyphenyl)methylene]-3(2H)-benzofuranone | 52 | n. g. | n. g. | 22.0 (Os ac: 15 nM) | 22.1 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| sulfuretin | 53 | n. g. | n. g. | 29.6 (Os ac: 15 nM) | 27.7 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| Biflavonoids | ||||||||
| 4′-O-methylochnaflavone | 15 | Ochna sp.; Lonicera japonica Thunb. | Ochnaceae; Caprifoliaceae | 2.01–3.54 (Os: 0.12–0.23 nM) | n. g. | n. g. | CL | 19 |
| ginkgetin | 13 | Ginkgo biloba L.; Cephalotaxus harringtonia Knight ex J.Forbes | Ginkgoaceae; Cephalotaxaceae | 97.09a (MA: 32.57a μM) | 17.26a (MA: 29.81a μM) | n. g. | n. g. | 147,148 |
| hinokiflavone | 14 | n. g. | n. g. | 77.63a (Os: 1.66 nM) | n. g. | n. g. | n. g. | 147 |
| Catechins | ||||||||
| catechin | 16 | n. g. | n. g. | >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142 |
| epicatechin | 17 | Gouania obtusifolia Vent. ex Brongn. | Rhamnaceae | >100 (Os ac: 15 nM); 5.49 (Neu5Ac2en: 44.47 μM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142,149 |
| gallocatechin | 18 | Gouania obtusifolia Vent. ex Brongn. | Rhamnaceae | 10.22 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| Chalcones | ||||||||
| (E)-4,2′,4′-trihydroxy-6′-methoxy-3′,5′-dimethylchalcone | 19 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 3.31–20.45 (Os: 12.29–95.22 nM) | n. g. | 18.57 (Os: 10.34 nM) | FL | 150 |
| 2,2′,4′-trihydroxy-6′-methoxy-3′,5′-dimethylchalcone | 20 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 2.55–28.12 (Os: 12.29–95.22 nM) | n. g. | 23.12 (Os: 10.34 nM) | FL | 150 |
| 2′,4′-dihydroxy-3′-methyl-6′-methoxychalcone | 54 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 26.02–93.77 (Os: 12.29–95.22 nM) | n. g. | 66.48 (Os: 10.34 nM) | FL | 150 |
| 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone | 55 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 5.03–35.23 (Os: 12.29–95.22 nM) | n. g. | 21.74 (Os: 10.34 nM) | FL | 150 |
| 2′-methoxyisoliquiritigenin | 56 | Glycyrrhiza uralensis Fisch. ex DC.; Caesalpinia sappan L. | Fabaceae | 24.3b (Os: 1.59b nM); 54.02a (Os ac: 3.52a nM) | 64.38a (Os ac: 1.65a nM) | n. g. | FL | 143,144 |
| 5-prenylbutein | 57 | Glycyrrhiza inflata Batalin; Erythrina addisoniae Hutch. & Dalziel | Fabaceae | 76.01a (Os: 16.42a - 127.21a nM); 64.43a (Os: 127.21a nM) | n. g. | 60.14a (Os: 15.81a nM); 104.30a (Os: 15.81a nM) | FL | 145,151 |
| abyssinone VI | 58 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | 70.23a (Os: 127.21a nM) | n. g. | 65.23a (Os: 15.81a nM) | FL | 145 |
| echinatin | 59 | Glycyrrhiza inflata Batalin | Fabaceae | 8.18a - 21.46a (Os: 16.42a - 127.21a nM) | n. g. | 21.09a (Os: 15.81a nM) | FL | |
| isoliquiritigenin | 21 | Glycyrrhiza inflata Batalin; Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 13.35a - 32.82a (Os: 16.42a - 127.21a nM); 9.0b (Os: 1.59b nM) | n. g. | 37.81a (Os: 15.81a nM) | FL | 143,151 |
| isoliquiritin | 60 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 124.0b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| isoliquiritin apioside | 61 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 12.9b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| kanzonol C | 62 | Glycyrrhiza inflata Batalin | Fabaceae | 192.06a (Os: 16.42a - 127.21a nM) | n. g. | 134.93a (Os: 15.81a nM) | FL | |
| licoagrochalcone A | 63 | Glycyrrhiza inflata Batalin; Erythrina addisoniae Hutch. & Dalziel | Fabaceae | 159.05a (Os: 16.42a - 127.21a nM); 66.31a (Os: 127.21a nM) | n. g. | 175.48a (Os: 15.81a nM); 61.75a (Os: 15.81a nM) | FL | 145,151 |
| licochalcone A | 64 | Glycyrrhiza inflata Batalin | Fabaceae | 12.41a - 56.41a (Os: 16.42a - 127.21a nM) | n. g. | 53.13a (Os: 15.81a nM) | FL | |
| licochalcone D | 65 | Glycyrrhiza inflata Batalin | Fabaceae | 80.76a (Os: 16.42a - 127.21a nM) | n. g. | 99.35a (Os: 15.81a nM) | FL | |
| licochalcone F | 66 | Glycyrrhiza inflata Batalin | Fabaceae | 106.32a (Os: 16.42a - 127.21a nM) | n. g. | 118.82a (Os: 15.81a nM) | FL | |
| sappanchalcone | 67 | Caesalpinia sappan L. | Fabaceae | 49.95a (Os ac: 3.52a nM) | 74.05a (Os ac: 1.65a nM) | n. g. | FL | 144 |
| Flavanones | ||||||||
| (2S)-2,7-dihydroxy-5-methoxy-6,8-dimethylflavanone | 68 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 282.58 (Os: 95.22 nM) | n. g. | 284.78 (Os: 10.34 nM) | FL | 150 |
| (2S)-5,7-dihydroxy-6,8-dimethylflavanone | 69 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 333.59 (Os: 95.22 nM) | n. g. | 319.12 (Os: 10.34 nM) | FL | 150 |
| (2S)-5-hydroxy-7-methoxy-6,8-dimethylflavanone | 70 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | >500 (Os: 95.22 nM) | n. g. | >500 (Os: 10.34 nM) | FL | 150 |
| (2S)-6-formyl-8-methyl-7-O-methylpinocembrin | 71 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 268.59 (Os: 95.22 nM) | n. g. | 256.25 (Os: 10.34 nM) | FL | 150 |
| (2S)-7-hydroxy-5-methoxy-8-methylflavanone | 72 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | >500 (Os: 95.22 nM) | n. g. | >500 (Os: 10.34 nM) | FL | 150 |
| (2S)-8-formyl-5-hydroxy-7-methoxy-6-methylflavanone | 73 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 289.94 (Os: 95.22 nM) | n. g. | 298.91 (Os: 10.34 nM) | FL | 150 |
| (2S)-8-methylpinocembrin | 74 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | >500 (Os: 95.22 nM) | n. g. | >500 (Os: 10.34 nM) | FL | 150 |
| (S)-naringenin | 75 | n. g. | n. g. | >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142 |
| hesperidin | 76 | n. g. | n. g. | >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142 |
| liquiritigenin | 77 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 46.8b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| liquiritin | 78 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 82.3b (Os: 1.59b nM); >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142,143 |
| liquiritin apioside | 79 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 18.2b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| Flavanonoles | ||||||||
| (2S,3S)-2,3-trans-5,7-dihydroxy-6,8-dimethyldihydroflavonol | 80 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 374.67 (Os: 95.22 nM) | n. g. | 367.87 (Os: 10.34 nM) | FL | 150 |
| engeletin | 81 | Gouania obtusifolia Vent. ex Brongn. | Rhamnaceae | 5.44 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| Flavans | ||||||||
| 7,4′-di-O-galloyltricetinflavan | 82 | n. g. | n. g. | 49.14a (Os: 1.66 nM) | n. g. | n. g. | FL | 147 |
| 7-O-galloyltricetinflavan | 83 | n. g. | n. g. | 35.49a (Os: 1.66 nM) | n. g. | n. g. | FL | 147 |
| Flavones | ||||||||
| 8-methoxyapigenin | 84 | n. g. | n. g. | 43.80a (Os: 1.66 nM) | n. g. | n. g. | FL | 147 |
| apigenin | 23 | Elsholtzia rugulosa Hemsl. | Lamiaceae | 31.63a (Os ac: 16.53a nM); 33.4b(Os: 1.59b nM) | 28.90a (Os ac: 3.52a nM) | n. g. | FL | 142,147,152,153 |
| apiin | 24 | Elsholtzia rugulosa Hemsl. | Lamiaceae | 25.92a (Os ac: 16.53a nM) | 44.89a (Os ac: 3.52a nM) | n. g. | FL | 152 |
| artocarpin | 45 | Artocarpus heterophyllus Lam. & other A. species | Moraceae | 0.18–0.30 (Os: 0.12–0.23 nM) | n. g. | n. g. | CL | 19 |
| cosmosiin | 85 | n. g. | n. g. | 46.9b (Os: 1.59b nM) | n. g. | n. g. | 153 | |
| chrysin | 86 | n. g. | n. g. | 45.7 (Os ac: 15 nM) | 33.36 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| dinatin | 87 | n. g. | n. g. | 46.3 (Os ac: 15 nM) | 26.0 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| galuteolin | 88 | Elsholtzia rugulosa Hemsl. | Lamiaceae | 47.35a (Os ac: 16.53a nM) | 52.23a (Os ac: 3.52a nM) | n. g. | FL | 142,147,152 |
| luteolin | 25 | Elsholtzia rugulosa Hemsl. | Lamiaceae | 33.68a (Os ac: 16.53a nM); 11.0b(Os: 1.59b nM) | 32.63a (Os ac: 3.52a nM) | n. g. | FL | 142,147,152,153 |
| luteolin 3′-glucuronyl acid methyl ester | 89 | Elsholtzia rugulosa Hemsl. | Lamiaceae | 44.42a (Os ac: 16.53a nM) | 50.97a (Os ac: 3.52a nM) | n. g. | FL | 152 |
| scutellarin | 90 | n. g. | n. g. | 50.6 (Os ac: 15 nM) | 47.3 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| vitexin | 91 | n. g. | n. g. | 46.5 (Os ac: 15 nM) | 45.1 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| Flavonoles | ||||||||
| astragalin | 92 | n. g. | n. g. | 38.4b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| gossypetin | 27 | n. g. | n. g. | 2.6b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| herbacetin | 28 | Rhodiola rosea L. | Crassulaceae | 8.9b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| isoquercitrin | 93 | Ziziphus cambodiana Pierre | Rhamnaceae | 9.87 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| kaempferol | 94 | Rhodiola rosea L. | Crassulaceae | 11.2b (Os: 1.59b nM); 58.6 (Os ac: 15 nM) | 38.1 (Os ac: 3.2 nM) | n. g. | FL | 142,147,153 |
| kaempferol 3-O-α-L-rhamnoside | 95 | Gouania obtusifolia Vent. ex Brongn. | Rhamnaceae | 5.47 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| kumatakenin | 96 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 36.4b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| leucoside | 97 | n. g. | n. g. | 42.21a (Os: 1.66 nM) | n. g. | n. g. | n. g. | 147 |
| licoflavonol | 98 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 20.6b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| linocinamarin | 99 | n. g. | n. g. | 44.2b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| myricetin | 100 | n. g. | n. g. | 82.6 (Os ac: 15 nM) | 46.2 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| nicotiflorin | 101 | n. g. | n. g. | 31.7b (Os: 1.59 nM) | n. g. | n. g. | 153 | |
| quercetin | 26 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | 58.4 (Os ac: 15 nM); 2.2b (Os: 1.59b nM) | 87.6 (Os ac: 3.2 nM) | n. g. | FL | 142,147,153,156 |
| quercetin 3-O-β-xylopyranosyl-(12)-O-α-rhamnopyranoside | 102 | Ziziphus cambodiana Pierre | Rhamnaceae | 4.82 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| quercetin 5,7,3′,4′-tetramethyl ether | 44 | Sambucus nigra L. | Adoxaceae | 0.39–1.09 (Os: 0.12–0.23 nM) | n. g. | n. g. | CL | 19,190 |
| quercitrin | 103 | Gouania obtusifolia Vent. ex Brongn.; Ziziphus cambodiana Pierre | Rhamnaceae | 7.89 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| rhamnetin | 104 | Caesalpinia sappan L. | Fabaceae | 48.69a (Os ac: 3.52a nM) | 76.20a (Os ac: 1.65a nM) | n. g. | FL | 144 |
| rhamnocitrin | 105 | n. g. | n. g. | 51.5 (Os ac: 15 nM) | 83.9 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| rhodiolinin | 106 | Rhodiola rosea L. | Crassulaceae | 10.3b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| rhodionin | 107 | Rhodiola rosea L. | Crassulaceae | 32.2b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| rhodiosin | 108 | Rhodiola rosea L. | Crassulaceae | 56.5b (Os: 1.59b nM) | n. g. | n. g. | FL | 153 |
| rutin | 109 | n. g. | n. g. | 52.2 (Os ac: 15 nM); 34.4b (Os: 1.59b nM) | 87.7 (Os ac: 3.2 nM) | n. g. | FL | 142,147,153 |
| Isoflavones | ||||||||
| 7-hydroxy-5-methoxy-6,8-dimethylflavone | 110 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 414.32 (Os: 95.22 nM) | n. g. | 362.87 (Os: 10.34 nM) | FL | 150 |
| 7-hydroxy-5-methoxy-6,8-dimethylisoflavone | 111 | Cleistocalyx operculatus (Roxb.) Merr. & L.M.Perry | Myrtaceae | 430.51 (Os: 95.22 nM) | n. g. | 391.01 (Os: 10.34 nM) | FL | 150 |
| corylin | 112 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | >115a (Os: 127.21a nM) | n. g. | >115a (Os: 15.81a nM) | FL | 145 |
| daidzein | 29 | n. g. | n. g. | 37.1 (Os ac: 15 nM) | 26.6 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| erysenegalensein M | 113 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | >115a (Os: 127.21a nM) | n. g. | >115a (Os: 15.81a nM) | FL | 145 |
| erythraddison A | 114 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | >115a (Os: 127.21a nM) | n. g. | >115a (Os: 15.81a nM) | FL | 145 |
| formononetin | 115 | n. g. | n. g. | >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142 |
| genistein | 30 | n. g. | n. g. | 77.1 (Os ac: 15 nM) | 134.4 (Os ac: 3.2 nM) | n. g. | FL | 142,147 |
| glyasperin C | 116 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 20% at 200b μM (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| glyasperin D | 117 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 20% at 200b μM (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| isowighteone | 118 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | >115a (Os: 127.21a nM) | n. g. | >115a (Os: 15.81a nM) | FL | 145 |
| licorisoflavan A | 119 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 30% at 200b μM (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| ononin | 120 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 30% at 200b μM (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| sophoricoside | 121 | n. g. | n. g. | >100 (Os ac: 15 nM) | >100 (Os ac: 3.2 nM) | n. g. | FL | 142 |
| wighteone | 122 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | >115a (Os: 127.21a nM) | n. g. | >115a (Os: 15.81a nM) | FL | 145 |
| (Oligo)stilbenes | ||||||||
| (+)-ampelopsin A | 123 | Vitis amurensis Rupr. | Vitaceae | 215.30–261.60 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| (+)-viniferol C | 31 | Vitis amurensis Rupr. | Vitaceae | 8.94–61.16 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| amurensin G | 124 | Vitis amurensis Rupr. | Vitaceae | 38.15–42.47 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| amurensin-K | 125 | Vitis amurensis Rupr. | Vitaceae | 14.43–48.44 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| erythraddison B | 126 | Erythrina addisoniae Hutch. & Dalziel | Fabaceae | 29.30a (Os: 127.21a nM) | n. g. | 23.94a (Os: 15.81a nM) | FL | 145 |
| gnetin D | 127 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 30.18a (Os ac: 3.52a nM) | 38.26a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| gnetulin | 128 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 36.15a (Os ac: 3.52a nM) | 41.20a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| gnetupendin B | 32 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 13.14a (Os ac: 3.52a nM) | 24.71a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| isorhapontigenin | 129 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 35.62a (Os ac: 3.52a nM) | 63.50a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| napalensinol B | 130 | Vitis amurensis Rupr. | Vitaceae | 30.42–52.30 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| piceid | 131 | Vitis amurensis Rupr. | Vitaceae | 110.25–150.81 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| shegansu B | 132 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 32.07a (Os ac: 3.52a nM) | 40.43a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| trans-resveratrol | 133 | Gnetum pendulum C.Y.Cheng | Gnetaceae | 79.74a (Os ac: 3.52a nM) | 64.84a (Os ac: 1.65a nM) | n. g. | FL | 155 |
| trans-vitisin B | 134 | Vitis amurensis Rupr. | Vitaceae | 23.89–66.73 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| trans-ε-viniferin | 135 | Vitis amurensis Rupr. | Vitaceae | 88.54–173.79 (Os: 3.89–70.88 nM) | n. g. | n. g. | FL | 154 |
| Isoprenoid compounds | ||||||||
| Monoterpenes | ||||||||
| terpinen-4-ol | 136 | Melaleuca alternifolia Cheel | Myrtaceae | not effective | n. g. | n. g. | FL | 157 |
| terpinolene | 137 | Melaleuca alternifolia Cheel | Myrtaceae | not effective | n. g. | n. g. | FL | 157 |
| α-terpineol | 138 | Melaleuca alternifolia Cheel | Myrtaceae | not effective | n. g. | n. g. | FL | 157 |
| Sesquiterpene | ||||||||
| trans,trans-farnesol | 139 | Alpinia katsumadai Hayata | Zingiberaceae | 81.40 (Os: 0.13 nM) | n. g. | n. g. | CL | 118 |
| Triterpenes | ||||||||
| 3-O-acetylursolic acid | 140 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| 3β-acetoxyurs-11-en-13β,28-olide | 141 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| masilinic acid | 142 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| oleanolic acid | 143 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| speciosaperoxide | 144 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| tormentic acid | 145 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| ursolic acid | 146 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| Phenylpropanoids & analogues | ||||||||
| (6S,7E,9R)-6,9-dihydroxy-4,7-megastigmadien-3-one 9-O-[β-D-xylopyranosyl(1→6)-glucopyranoside] | 147 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| 1-(5-hydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-2-phenyl-ethanone | 46 | n. g. | n. g. | 1.33–2.21 (Os: 0.12–0.23 nM) | n. g. | n. g. | CL | 19 |
| 1,4-dicaffeoylquinic acid | 148 | Cynara scolymus L.; Arnica montana L.; Echinacea purpurea (L.) Moench | Asteraceae | 17.0 (Os: 0.12–0.23 nM) | n. g. | n. g. | CL | 19 |
| 3,4-dihydroxybenzoic acid | 149 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | 8.24a (Os: 0.99a μM) | n. g. | n. g. | FL | 156 |
| gallic acid | 150 | Mangifera odorata Griff. | Anacardiaceae | >2600 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| iriflophenone 2-O-β-glucoside | 151 | Mangifera odorata Griff. | Anacardiaceae | >110 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| iriflophenone 3-C-β-glucoside | 152 | Mangifera odorata Griff. | Anacardiaceae | 80.71 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| licocoumarone | 153 | Glycyrrhiza uralensis Fisch. ex DC. | Fabaceae | 27.8b (Os: 1.59b nM) | n. g. | n. g. | FL | 143 |
| mangiferin | 154 | Gouania obtusifolia Vent. ex Brongn.; Mangifera odorata Griff. | Rhamnaceae; Anacardiaceae | 0.82 (Neu5Ac2en: 44.47 μM) | n. g. | n. g. | FL | 149 |
| roseoside | 155 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | not effective | n. g. | n. g. | FL | 156 |
| vomifoliol | 33 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | 10.39a (Os: 0.99 μM) | n. g. | n. g. | FL | 156 |
| Miscellaneous | ||||||||
| 1-[5-(2-formylfuryl)methyl] hydrogen 1-hydroxyethane-1,2-dicarboxylate | 156 | Prunus mume Siebold & Zucc. | Rosaceae | 1640.0a (Os: 1.50a nM) | n. g. | n. g. | FL | 191 |
| 2-hydroxy-2-[(5-formyl-2-furanyl)methyl]-1,2,3-propanetricarboxylic acid ester | 157 | Prunus mume Siebold & Zucc. | Rosaceae | 1620.0a (Os: 1.50a nM) | n. g. | n. g. | FL | 191 |
| 2-hydroxy-4-[(5-formyl-2-furanyl)methyl]-butanedioic acid ester | 158 | Prunus mume Siebold & Zucc. | Rosaceae | 713.0a (Os: 1.50a nM) | n. g. | n. g. | FL | 191 |
| 5-hydroxymethyl-2-formylfuran | 159 | Prunus mume Siebold & Zucc. | Rosaceae | 33400.0a (Os: 1.50a nM) | n. g. | n. g. | FL | 191 |
| brazilein | 160 | Caesalpinia sappan L. | Fabaceae | 93.22a (Os ac: 3.52a nM) | 86.54a (Os ac: 1.65a nM) | n. g. | FL | 144 |
| brazilin | 161 | Caesalpinia sappan L. | Fabaceae | 69.51a (Os ac: 3.52a nM) | 50.65a (Os ac: 1.65a nM) | n. g. | FL | 144 |
| methyl 3-hydroxybutanedioic ester | 162 | Chaenomeles speciosa (Sweet) Nakai | Rosaceae | 12.83a (Os: 0.99a μM) | n. g. | n. g. | FL | 156 |
| mumefural | 163 | Prunus mume Siebold & Zucc. | Rosaceae | 207.0a (Os: 1.50a nM) | n. g. | n. g. | FL | 191 |
| protosappanin A | 164 | Caesalpinia sappan L. | Fabaceae | 94.40a (Os ac: 3.52a nM) | 78.97a (Os ac: 1.65a nM) | n. g. | FL | 144 |
| spinosulfate A | 165 | unidentified pycnidial fungus | n. g. | 0.5 (Neu5Ac2en: 0.9 μM) | n. g. | n. g. | FL | 192 |
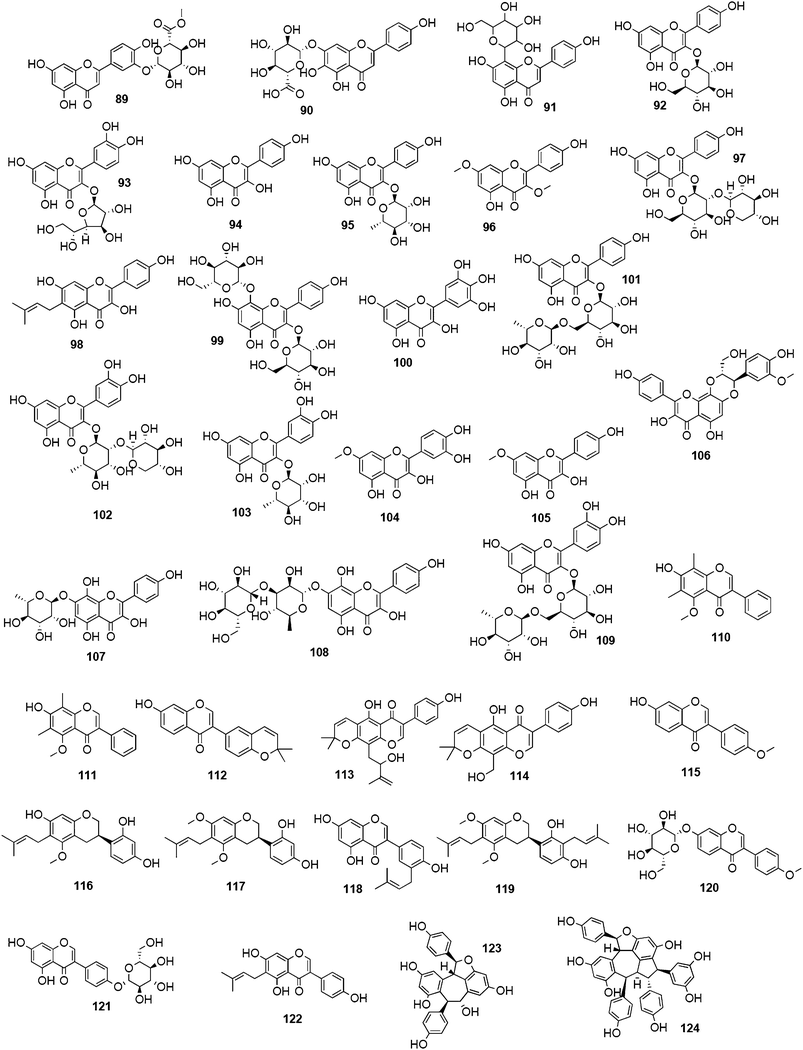
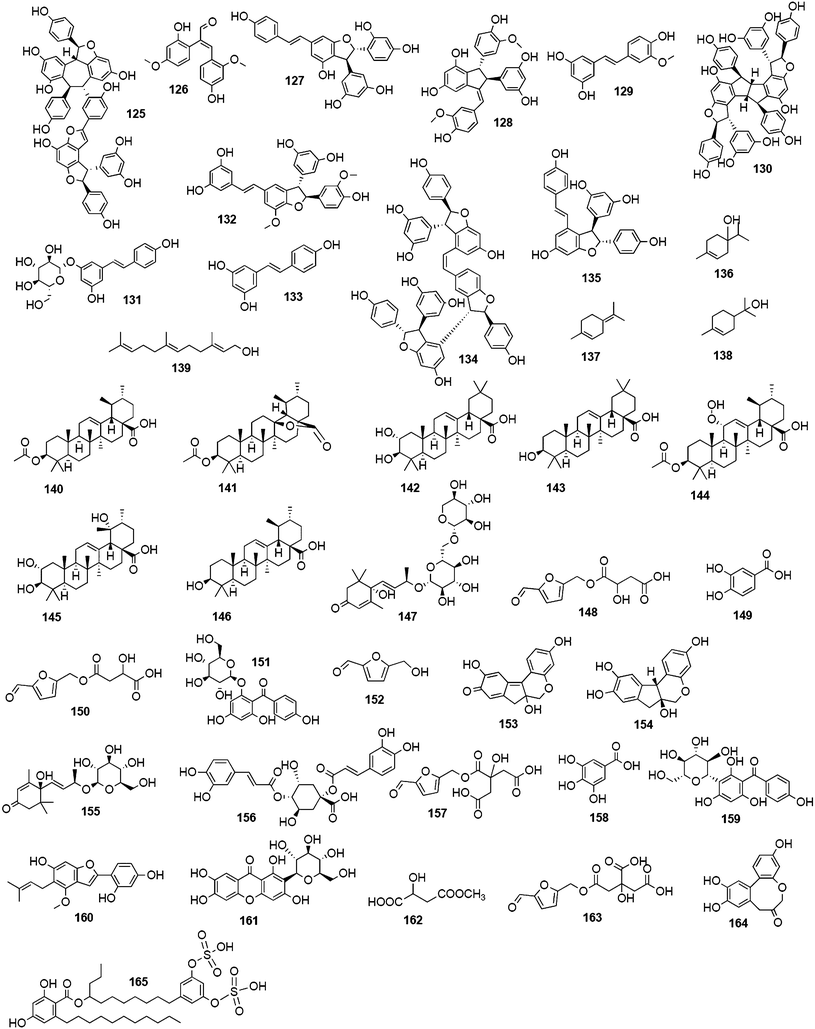
7 References
- Centers for Disease Control and Prevention, http://www.cdc.gov/flu/about/qa/disease.htm, accessed April 2011.
- T. M. Tumpey, C. F. Basler, P. V. Aguilar, H. Zeng, A. Solorzano, D. E. Swayne, N. J. Cox, J. M. Katz, J. K. Taubenberger, P. Palese and A. Garcia-Sastre, Science, 2005, 310, 77–80 CrossRef CAS.
- C. F. Basler, Infect. Disord.: Drug Targets, 2007, 7, 282–293 CrossRef CAS.
- J. K. Taubenberger and D. M. Morens, Emerg. Infect. Dis., 2006, 12, 15–22 Search PubMed.
- M. J. Memoli, D. M. Morens and J. K. Taubenberger, Drug Discovery Today, 2008, 13, 590–595 CrossRef CAS.
- A. Moscona, N. Engl. J. Med., 2005, 353, 1363–1373 CrossRef CAS.
- F. G. Hayden, Antiviral Res., 2006, 71, 372–378 CrossRef CAS.
- R. G. Webster and E. J. Walker, Am. Sci., 2003, 91, 122–129 CrossRef.
- A. Meijer, A. Lackenby, O. Hungnes, B. Lina, S. van-der-Werf, B. Schweiger, M. Opp, J. Paget, J. van-de-Kassteele, A. Hay and M. Zambon, Emerging Infect. Dis., 2009, 15, 552–560 CrossRef.
- F. S. Dawood, S. Jain, L. Finelli, M. W. Shaw, S. Lindstrom, R. J. Garten, L. V. Gubareva, X. Xu, C. B. Bridges and T. M. Uyeki, N. Engl. J. Med., 2009, 360, 2605–2615 CrossRef.
- K. Das, J. M. Aramini, L. C. Ma, R. M. Krug and E. Arnold, Nat. Struct. Mol. Biol., 2010, 17, 530–538 CAS.
- G. Neumann, T. Noda and Y. Kawaoka, Nature, 2009, 459, 931–939 CrossRef CAS.
- P. A. Reece, J. Med. Virol., 2007, 79, 1577–1586 CrossRef CAS.
- M. A. Rameix-Welti, F. Agou, P. Buchy, S. Mardy, J. T. Aubin, M. Veron, S. van der Werf and N. Naffakh, Antimicrob. Agents Chemother., 2006, 50, 3809–3815 CrossRef CAS.
- E. van der Vries, F. F. Stelma and C. A. Boucher, N. Engl. J. Med., 2010, 363, 1381–1382 CrossRef CAS.
- J. Cinatl, Jr., M. Michaelis and H. W. Doerr, Med. Microbiol. Immunol., 2007, 196, 203–212 CrossRef.
- K. Bauer, M. Richter, P. Wutzler and M. Schmidtke, Antivir. Res., 2009, 82, 34–41 CrossRef CAS.
- L. V. Gubareva, A. A. Trujillo, M. Okomo-Adhiambo, V. P. Mishin, V. M. Deyde, K. Sleeman, H. T. Nguyen, T. G. Sheu, R. J. Garten, M. W. Shaw, A. M. Fry and A. I. Klimov, Antiviral Therapy, 2010, 15, 1151–1159 CrossRef CAS.
- J. Kirchmair, J. M. Rollinger, K. R. Liedl, N. Seidel, A. Krumbholz and M. Schmidtke, Future Med. Chem., 2011, 3, 437–450 CrossRef CAS.
- Y. Liu, F. Jing, Y. Xu, Y. Xie, F. Shi, H. Fang, M. Li and W. Xu, Bioorg. Med. Chem., 2011, 19, 2342–2348 CrossRef CAS.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS.
- P. Ertl, S. Roggo and A. Schuffenhauer, J. Chem. Inf. Model., 2008, 48, 68–74 CrossRef CAS.
- M. Roxas and J. Jurenka, Altern. Med. Rev., 2007, 12, 25–48 Search PubMed.
- A. J. Vlietinck, T. De Bruyne and D. A. Vanden Berghe, Curr. Org. Chem., 1997, 1, 307–344 CAS.
- P. Cos, D. Vanden Berghe, T. De Bruyne and A. J. Vlietinck, Curr. Org. Chem., 2003, 7, 1163–1180 CrossRef CAS.
- S. J. King, K. R. Hippe and J. N. Weiser, Mol. Microbiol., 2006, 59, 961–974 CrossRef CAS.
- S. Manco, F. Hernon, H. Yesilkaya, J. C. Paton, P. W. Andrew and A. Kadioglu, Infect. Immun., 2006, 74, 4014–4020 CrossRef CAS.
- C. J. Orihuela, G. Gao, K. P. Francis, J. Yu and E. I. Tuomanen, J. Infect. Dis., 2004, 190, 1661–1669 CrossRef CAS.
- H. H. Tong, L. E. Blue, M. A. James and T. F. DeMaria, Infect. Immun., 2000, 68, 921–924 CrossRef CAS.
- J. Z. Gong, W. F. Xu and J. Zhang, Curr. Med. Chem., 2007, 14, 113–122 CrossRef CAS.
- R. Wagner, M. Matrosovich and H. D. Klenk, Rev. Med. Virol., 2002, 12, 159–166 CrossRef CAS.
- M. N. Matrosovich, T. Y. Matrosovich, T. Gray, N. A. Roberts and H. D. Klenk, J. Virol., 2004, 78, 12665–12667 CrossRef CAS.
- R. J. Russell, L. F. Haire, D. J. Stevens, P. J. Collins, Y. P. Lin, G. M. Blackburn, A. J. Hay, S. J. Gamblin and J. J. Skehel, Nature, 2006, 443, 45–49 CrossRef CAS.
- Q. Li, J. Qi, W. Zhang, C. J. Vavricka, Y. Shi, J. Wei, E. Feng, J. Shen, J. Chen, D. Liu, J. He, J. Yan, H. Liu, H. Jiang, M. Teng, X. Li and G. F. Gao, Nat. Struct. Mol. Biol., 2010, 17, 1266–1268 CAS.
- C. J. Vavricka, Y. Liu, Q. Li, Y. Shi, Y. Wu, Y. P. Sun, J. X. Qi and G. F. Gao, Chin. Sci. Bull., 2011, 56, 1747–1752 CrossRef CAS.
- J. N. Varghese, W. G. Laver and P. M. Colman, Nature, 1983, 303, 35–40 CrossRef CAS.
- P. M. Colman, J. N. Varghese and W. G. Laver, Nature, 1983, 303, 41–44 CrossRef CAS.
- A. T. Baker, J. N. Varghese, W. G. Laver, G. M. Air and P. M. Colman, Proteins: Struct., Funct., Genet., 1987, 2, 111–117 CrossRef CAS.
- J. N. Varghese, V. C. Epa and P. M. Colman, Protein Sci., 1995, 4, 1081–1087 CrossRef CAS.
- P. J. Collins, L. F. Haire, Y. P. Lin, J. F. Liu, R. J. Russell, P. A. Walker, J. J. Skehel, S. R. Martin, A. J. Hay and S. J. Gamblin, Nature, 2008, 453, 1258–U1261 CrossRef CAS.
- X. J. Xu, X. Y. Zhu, R. A. Dwek, J. Stevens and I. A. Wilson, J. Virol., 2008, 82, 10493–10501 CrossRef CAS.
- W. P. Burmeister, R. W. Ruigrok and S. Cusack, EMBO J., 1992, 11, 49–56 CAS.
- A. J. Oakley, S. Barrett, T. S. Peat, J. Newman, V. A. Streltsov, L. Waddington, T. Saito, M. Tashiro and J. L. McKimm-Breschkin, J. Med. Chem., 2010, 53, 6421–6431 CrossRef CAS.
- P. M. Colman, P. A. Hoyne and M. C. Lawrence, J. Virol., 1993, 67, 2972–2980 CAS.
- A. K. Chong, M. S. Pegg, N. R. Taylor and M. von Itzstein, Eur. J. Biochem., 1992, 207, 335–343 CrossRef CAS.
- N. R. Taylor and M. von Itzstein, J. Med. Chem., 1994, 37, 616–624 CrossRef CAS.
- M. von Itzstein, W. Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. Van Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron and C. R. Penn, Nature, 1993, 363, 418–423 CrossRef CAS.
- P. Meindl, G. Bodo, P. Palese, J. Schulman and H. Tuppy, Virology, 1974, 58, 457–463 CrossRef CAS.
- W. P. Burmeister, B. Henrissat, C. Bosso, S. Cusack and R. W. Ruigrok, Structure, 1993, 1, 19–26 CrossRef CAS.
- P. Bossart-Whitaker, M. Carson, Y. S. Babu, C. D. Smith, W. G. Laver and G. M. Air, J. Mol. Biol., 1993, 232, 1069–1083 CrossRef CAS.
- L. V. Gubareva, Virus Res., 2004, 103, 199–203 CrossRef CAS.
- D. Kobasa, S. Kodihalli, M. Luo, M. R. Castrucci, I. Donatelli, Y. Suzuki, T. Suzuki and Y. Kawaoka, J. Virol., 1999, 73, 6743–6751 CAS.
- P. M. Colman and C. W. Ward, Curr. Top. Microbiol. Immunol., 1985, 114, 177–255 CAS.
- Clinical Trials – A service of the U.S. National Institues of Health, http://clinicaltrials.gov/ct2/show/NCT01231620?term=zanamivir+oseltamivir&rank=2, accessed April 2011.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 CrossRef CAS.
- W. Li, P. A. Escarpe, E. J. Eisenberg, K. C. Cundy, C. Sweet, K. J. Jakeman, J. Merson, W. Lew, M. Williams, L. Zhang, C. U. Kim, N. Bischofberger, M. S. Chen and D. B. Mendel, J. Antimicrob. Chemother., 1998, 42, 647–653 CrossRef CAS.
- G. Laver and E. Garman, Science, 2001, 293, 1776–1777 CrossRef CAS.
- US Food and Drug Administration, http://www.fda.gov, accessed 2011.
- D. Birnkrant and E. Cox, N. Engl. J. Med., 2009, 361, 2204–2207 CrossRef CAS.
- L. V. Gubareva, L. Kaiser and F. G. Hayden, Lancet, 2000, 355, 827–835 CrossRef CAS.
- S. J. Macdonald, K. G. Watson, R. Cameron, D. K. Chalmers, D. A. Demaine, R. J. Fenton, D. Gower, J. N. Hamblin, S. Hamilton, G. J. Hart, G. G. A. Inglis, B. Jin, H. T. Jones, D. B. McConnell, A. M. Mason, V. Nguyen, I. J. Owens, N. Parry, P. A. Reece, S. E. Shanahan, D. Smith, W. Y. Wu and S. P. Tucker, Antimicrob. Agents Chemother., 2004, 48, 4542–4549 CrossRef CAS.
- S. J. Macdonald, R. Cameron, D. A. Demaine, R. J. Fenton, G. Foster, D. Gower, J. N. Hamblin, S. Hamilton, G. J. Hart, A. P. Hill, G. G. Inglis, B. Jin, H. T. Jones, D. B. McConnell, J. McKimm-Breschkin, G. Mills, V. Nguyen, I. J. Owens, N. Parry, S. E. Shanahan, D. Smith, K. G. Watson, W. Y. Wu and S. P. Tucker, J. Med. Chem., 2005, 48, 2964–2971 CrossRef CAS.
- M. Yamashita, T. Tomozawa, M. Kakuta, A. Tokumitsu, H. Nasu and S. Kubo, Antimicrob. Agents Chemother., 2009, 53, 186–192 CrossRef CAS.
- H. X. Liu, X. J. Yao, C. Q. Wang and J. A. Han, Mol. Pharmaceutics, 2010, 7, 894–904 CrossRef CAS.
- R. A. Bright, D. K. Shay, B. Shu, N. J. Cox and A. I. Klimov, JAMA, J. Am. Med. Assoc., 2006, 295, 891–894 CrossRef CAS.
- V. M. Deyde, X. Xu, R. A. Bright, M. Shaw, C. B. Smith, Y. Zhang, Y. Shu, L. V. Gubareva, N. J. Cox and A. I. Klimov, J. Infect. Dis., 2007, 196, 249–257 CrossRef CAS.
- A. Lackenby, O. Hungnes, S. G. Dudman, A. Meijer, W. J. Paget, A. J. Hay and M. C. Zambon, Euro. Surveill., 2008, 13 CAS :pii=8026. Available online: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8026.
- A. Moscona, N. Engl. J. Med., 2009, 360, 953–956 CrossRef CAS.
- T. G. Sheu, V. M. Deyde, M. Okomo-Adhiambo, R. J. Garten, X. Xu, R. A. Bright, E. N. Butler, T. R. Wallis, A. I. Klimov and L. V. Gubareva, Antimicrob. Agents Chemother., 2008, 52, 3284–3292 CrossRef CAS.
- T. G. Sheu, A. M. Fry, R. J. Garten, V. M. Deyde, T. Shwe, L. Bullion, P. J. Peebles, Y. Li, A. I. Klimov and L. V. Gubareva, J. Infect. Dis., 2011, 203, 13–17 CrossRef CAS.
- R. J. Garten, C. T. Davis, C. A. Russell, B. Shu, S. Lindstrom, A. Balish, W. M. Sessions, X. Y. Xu, E. Skepner, V. Deyde, M. Okomo-Adhiambo, L. Gubareva, J. Barnes, C. B. Smith, S. L. Emery, M. J. Hillman, P. Rivailler, J. Smagala, M. de Graaf, D. F. Burke, R. A. M. Fouchier, C. Pappas, C. M. Alpuche-Aranda, H. Lopez-Gatell, H. Olivera, I. Lopez, C. A. Myers, D. Faix, P. J. Blair, C. Yu, K. M. Keene, P. D. Dotson, D. Boxrud, A. R. Sambol, S. H. Abid, K. S. George, T. Bannerman, A. L. Moore, D. J. Stringer, P. Blevins, G. J. Demmler-Harrison, M. Ginsberg, P. Kriner, S. Waterman, S. Smole, H. F. Guevara, E. A. Belongia, P. A. Clark, S. T. Beatrice, R. Donis, J. Katz, L. Finelli, C. B. Bridges, M. Shaw, D. B. Jernigan, T. M. Uyeki, D. J. Smith, A. I. Klimov and N. J. Cox, Science, 2009, 325, 197–201 CrossRef CAS.
- J. L. McKimm-Breschkin, Antiviral Res., 2000, 47, 1–17 CrossRef CAS.
- L. V. Gubareva, M. J. Robinson, R. C. Bethell and R. G. Webster, J. Virol., 1997, 71, 3385–3390 CAS.
- H. L. Yen, L. M. Herlocher, E. Hoffmann, M. N. Matrosovich, A. S. Monto, R. G. Webster and E. A. Govorkova, Antimicrob. Agents Chemother., 2005, 49, 4075–4084 CrossRef CAS.
- L. V. Gubareva, M. N. Matrosovich, M. K. Brenner, R. C. Bethell and R. G. Webster, J. Infect. Dis., 1998, 178, 1257–1262 CrossRef CAS.
- D. Weinstock and G. Zuccotti, JAMA, J. Am. Med. Assoc., 2009, 301, 1066–1069 CrossRef CAS.
- L. V. Gubareva, L. Kaiser, M. N. Matrosovich, Y. Soo-Hoo and F. G. Hayden, J. Infect. Dis., 2001, 183, 523–531 CrossRef CAS.
- P. J. Collins, L. F. Haire, Y. P. Lin, J. Liu, R. J. Russell, P. A. Walker, S. R. Martin, R. S. Daniels, V. Gregory, J. J. Skehel, S. J. Gamblin and A. J. Hay, Vaccine, 2009, 27, 6317–6323 CrossRef CAS.
- J. A. L. Ives, J. A. Carr, D. B. Mendel, C. Y. Tai, R. Lambkin, L. Kelly, J. S. Oxford, F. G. Hayden and N. A. Roberts, Antiviral Res., 2002, 55, 307–317 CrossRef CAS.
- M. L. Herlocher, R. Truscon, S. Elias, H. L. Yen, N. A. Roberts, S. E. Ohmit and A. S. Monto, J. Infect. Dis., 2004, 190, 1627–1630 CrossRef CAS.
- M. A. Rameix-Welti, V. Enouf, F. Cuvelier, P. Jeannin and S. van der Werf, PLoS Pathog., 2008, 4, 5 Search PubMed.
- J. D. Bloom, L. I. Gong and D. Baltimore, Science, 2010, 328, 1272–1275 CrossRef CAS.
- J. L. McKimm-Breschkin, T. J. Blick, A. Sahasrabudhe, T. Tiong, D. Marshall, G. J. Hart, R. C. Bethell and C. R. Penn, Antimicrob. Agents Chemother., 1996, 40, 40–46 CAS.
- K. Bauer, R. Dürrwald, M. Schlegel, K. Pfarr, N. Wiesener, H.-M. Dahse, P. Wutzler and M. Schmidtke, Med. Microbiol. Immun., 2011 DOI:10.1007/s00430-011-0206-1.
- R. T. Kelly, S. Farmer and D. Greiff, J. Bacteriol., 1967, 94, 272–273 CAS.
- M. M. Pettigrew, K. P. Fennie, M. P. York, J. Daniels and F. Ghaffar, Infect. Immun., 2006, 74, 3360–3365 CrossRef CAS.
- G. Soong, A. Muir, M. I. Gomez, J. Waks, B. Reddy, P. Planet, P. K. Singh, Y. Kaneko, M. C. Wolfgang, Y. S. Hsiao, L. Tong and A. Prince, J. Clin. Invest., 2006, 116, 2297–2305 CrossRef CAS.
- Y. S. Hsiao, D. Parker, A. J. Ratner, A. Prince and L. Tong, Biochem. Biophys. Res. Commun., 2009, 380, 467–471 CrossRef CAS.
- S. Crennell, E. Garman, G. Laver, E. Vimr and G. Taylor, Structure, 1994, 2, 535–544 CrossRef CAS.
- A. Gaskell, S. Crennell and G. Taylor, Structure, 1995, 3, 1197–1205 CrossRef CAS.
- S. L. Newstead, J. A. Potter, J. C. Wilson, G. Xu, C. H. Chien, A. G. Watts, S. G. Withers and G. L. Taylor, J. Biol. Chem., 2008, 283, 9080–9088 CrossRef CAS.
- G. Xu, X. Li, P. W. Andrew and G. L. Taylor, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2008, 64, 772–775 CrossRef.
- G. Xu, J. A. Potter, R. J. Russell, M. R. Oggioni, P. W. Andrew and G. L. Taylor, J. Mol. Biol., 2008, 384, 436–449 CrossRef CAS.
- Y. B. Ryu, M. J. Curtis-Long, J. W. Lee, J. H. Kim, J. Y. Kim, K. Y. Kang, W. S. Lee and K. H. Park, Bioorg. Med. Chem., 2009, 17, 2744–2750 CrossRef CAS.
- Y. B. Ryu, M. J. Curtis-Long, J. H. Kim, S. H. Jeong, M. S. Yang, K. W. Lee, W. S. Lee and K. H. Park, Bioorg. Med. Chem. Lett., 2008, 18, 6046–6049 CrossRef CAS.
- C.-H. Lee, S.-I. Kim, K.-B. Lee, Y.-C. Yoo, S.-Y. Ryu and K.-S. Song, Arch. Pharmacal Res., 2003, 26, 367–374 CrossRef CAS.
- M. C. Mann, R. J. Thomson, J. C. Dyason, S. McAtamney and M. von Itzstein, Bioorg. Med. Chem., 2006, 14, 1518–1537 CrossRef CAS.
- E. Bonten, A. van der Spoel, M. Fornerod, G. Grosveld and A. d'Azzo, Genes Dev., 1996, 10, 3156–3169 CrossRef CAS.
- A. V. Pshezhetsky, C. Richard, L. Michaud, S. Igdoura, S. Wang, M. A. Elsliger, J. Qu, D. Leclerc, R. Gravel, L. Dallaire and M. Potier, Nat. Genet., 1997, 15, 316–320 CrossRef CAS.
- E. Monti, A. Preti, E. Rossi, A. Ballabio and G. Borsani, Genomics, 1999, 57, 137–143 CrossRef CAS.
- E. Monti, A. Preti, C. Nesti, A. Ballabio and G. Borsani, Glycobiology, 1999, 9, 1313–1321 CrossRef CAS.
- E. Monti, M. T. Bassi, N. Papini, M. Riboni, M. Manzoni, B. Venerando, G. Croci, A. Preti, A. Ballabio, G. Tettamanti and G. Borsani, Biochem. J., 2000, 349, 343–351 CrossRef CAS.
- T. Wada, Y. Yoshikawa, S. Tokuyama, M. Kuwabara, H. Akita and T. Miyagi, Biochem. Biophys. Res. Commun., 1999, 261, 21–27 CrossRef CAS.
- E. Monti, M. T. Bassi, R. Bresciani, S. Civini, G. L. Croci, N. Papini, M. Riboni, G. Zanchetti, A. Ballabio, A. Preti, G. Tettamanti, B. Venerando and G. Borsani, Genomics, 2004, 83, 445–453 CrossRef CAS.
- K. Hata, K. Koseki, K. Yamaguchi, S. Moriya, Y. Suzuki, S. Yingsakmongkon, G. Hirai, M. Sodeoka, M. von Itzstein and T. Miyagi, Antimicrob. Agents Chemother., 2008, 52, 3484–3491 CrossRef CAS.
- Y. Li, H. Cao, H. Yu, Y. Chen, K. Lau, J. Qu, V. Thon, G. Sugiarto and X. Chen, Mol. BioSyst., 2011, 7, 1060–1072 RSC.
- L. M. Chavas, R. Kato, N. Suzuki, M. von Itzstein, M. C. Mann, R. J. Thomson, J. C. Dyason, J. McKimm-Breschkin, P. Fusi, C. Tringali, B. Venerando, G. Tettamanti, E. Monti and S. Wakatsuki, J. Med. Chem., 2010, 53, 2998–3002 CrossRef CAS.
- L. M. Chavas, C. Tringali, P. Fusi, B. Venerando, G. Tettamanti, R. Kato, E. Monti and S. Wakatsuki, J. Biol. Chem., 2005, 280, 469–475 CAS.
- D. Aminoff, Biochem. J., 1961, 81, 384–392 CAS.
- P. Meindl, G. Bodo, J. Lindner and P. Palese, Z. Naturforsch. B., 1971, 26, 792–797 CAS.
- J. L. Schulman and P. Palese, Virology, 1975, 63, 98–104 CrossRef CAS.
- P. Palese and R. W. Compans, J. Gen. Virol., 1976, 33, 159–163 CrossRef CAS.
- P. Palese, J. L. Schulman, G. Bodo and P. Meindl, Virology, 1974, 59, 490–498 CrossRef CAS.
- P. M. Colman, W. R. Tulip, J. N. Varghese, P. A. Tulloch, A. T. Baker, W. G. Laver, G. M. Air and R. G. Webster, Philos. Trans. R. Soc. London, Ser. B, 1989, 323, 511–518 CrossRef CAS.
- J. N. Varghese and P. M. Colman, J. Mol. Biol., 1991, 221, 473–486 CrossRef CAS.
- J. M. Woods, R. C. Bethell, J. A. Coates, N. Healy, S. A. Hiscox, B. A. Pearson, D. M. Ryan, J. Ticehurst, J. Tilling and S. M. Walcott, et al. , Antimicrob. Agents Chemother., 1993, 37, 1473–1479 CAS.
- P. Chand, P. L. Kotian, A. Dehghani, Y. El-Kattan, T. H. Lin, T. L. Hutchison, Y. S. Babu, S. Bantia, A. J. Elliott and J. A. Montgomery, J. Med. Chem., 2001, 44, 4379–4392 CrossRef CAS.
- U. Grienke, M. Schmidtke, J. Kirchmair, K. Pfarr, P. Wutzler, R. Durrwald, G. Wolber, K. R. Liedl, H. Stuppner and J. M. Rollinger, J. Med. Chem., 2010, 53, 778–786 CrossRef CAS.
- V. P. Mishin, F. G. Hayden and L. V. Gubareva, Antimicrob. Agents Chemother., 2005, 49, 4515–4520 CrossRef CAS.
- J. M. Barnett, A. Cadman, D. Gor, M. Dempsey, M. Walters, A. Candlin, M. Tisdale, P. J. Morley, I. J. Owens, R. J. Fenton, A. P. Lewis, E. C. Claas, G. F. Rimmelzwaan, R. De Groot and A. D. Osterhaus, Antimicrob. Agents Chemother., 2000, 44, 78–87 CrossRef CAS.
- R. C. Buxton, B. Edwards, R. R. Juo, J. C. Voyta, M. Tisdale and R. C. Bethell, Anal. Biochem., 2000, 280, 291–300 CrossRef CAS.
- M. Potier, L. Mameli, M. Bélisle, L. Dallaire and S. B. Melancon, Anal. Biochem., 1979, 94, 287–296 CrossRef CAS.
- H. T. Nguyen, T. G. Sheu, V. P. Mishin, A. I. Klimov and L. V. Gubareva, Antimicrob. Agents Chemother., 2010, 54, 3671–3677 CrossRef CAS.
- N. T. Wetherall, T. Trivedi, J. Zeller, C. Hodges-Savola, J. L. McKimm-Breschkin, M. Zambon and F. G. Hayden, J. Clin. Microbiol., 2003, 41, 742–750 CrossRef CAS.
- A. C. Hurt, I. G. Barr, G. Hartel and A. W. Hampson, Antiviral Res., 2004, 62, 37–45 CrossRef CAS.
- F. G. Hayden, K. M. Cote and R. G. Douglas, Jr, Antimicrob. Agents Chemother., 1980, 17, 865–870 CAS.
- L. V. Gubareva, M. S. Nedyalkova, D. V. Novikov, K. G. Murti, E. Hoffmann and F. G. Hayden, J. Gen. Virol., 2002, 83, 2683–2692 CAS.
- K. Bauer, C. Schrader, J. Suess, P. Wutzler and M. Schmidtke, Antiviral Res., 2007, 75, 219–226 CrossRef CAS.
- N. A. Ilyushina, E. A. Govorkova, T. E. Gray, N. V. Bovin and R. G. Webster, PLoS Pathog., 2008, 4, e1000043 Search PubMed.
- M. Schmidtke, U. Schnittler, B. Jahn, H.-M. Dahse and A. Stelzner, J. Virol. Methods, 2001, 95, 133–143 CrossRef CAS.
- D. F. Smee, J. H. Huffman, A. C. Morrison, D. L. Barnard and R. W. Sidwell, Antimicrob. Agents Chemother., 2001, 45, 743–748 CrossRef CAS.
- W. Watanabe, K. Konno, K. Ijichi, H. Inoue, T. Yokota and S. Shigeta, J. Virol. Methods, 1994, 48, 257–265 CrossRef CAS.
- M. Matrosovich, T. Matrosovich, J. Carr, N. A. Roberts and H. D. Klenk, J. Virol., 2003, 77, 8418–8425 CrossRef CAS.
- M. Tisdale, Rev. Med. Virol., 2000, 10, 45–55 CrossRef CAS.
- S. Hatakeyama, Y. Sakai-Tagawa, M. Kiso, H. Goto, C. Kawakami, K. Mitamura, N. Sugaya, Y. Suzuki and Y. Kawaoka, J. Clin. Microbiol., 2005, 43, 4139–4146 CrossRef CAS.
- V. P. Mishin, D. Novikov, F. G. Hayden and L. V. Gubareva, J. Virol., 2005, 79, 12416–12424 CrossRef CAS.
- L. S. Cheng, R. E. Amaro, D. Xu, W. W. Li, P. W. Arzberger and J. A. McCammon, J. Med. Chem., 2008, 51, 3878–3894 CrossRef CAS.
- J. Zhang, Q. Wang, H. Fang, W. Xu, A. Liu and G. Du, Bioorg. Med. Chem., 2008, 16, 3839–3847 CrossRef CAS.
- Q. Zhang, J. Yang, K. Liang, L. Feng, S. Li, J. Wan, X. Xu, G. Yang, D. Liu and S. Yang, J. Chem. Inf. Model., 2008, 48, 1802–1812 CrossRef CAS.
- SciFinder, http://www.cas.org/products/sfacad/index.html, accessed 2011.
- Q. Zhang, Q. J. Zhao, R. S. Xiong, J. F. Li and J. S. Shen, Yaoxue Xuebao (Acta Pharmaceutica Sinica), 2010, 45, 289–299 CAS.
- A.-L. Liu, H.-D. Wang, S. M. Lee, Y.-T. Wang and G.-H. Du, Bioorg. Med. Chem., 2008, 16, 7141–7147 CrossRef CAS.
- Y. B. Ryu, J. H. Kim, S.-J. Park, J. S. Chang, M.-C. Rho, K.-H. Bae, K. H. Park and W. S. Lee, Bioorg. Med. Chem. Lett., 2010, 20, 971–974 CrossRef CAS.
- A.-L. Liu, S.-H. Shu, H.-L. Qin, S. M. Y. Lee, Y.-T. Wang and G.-H. Du, Planta Med., 2009, 75, 337–339 CrossRef CAS.
- P. H. Nguyen, M. Na, T. T. Dao, D. T. Ndinteh, J. T. Mbafor, J. Park, H. Cheong and W. K. Oh, Bioorg. Med. Chem. Lett., 2010, 20, 6430–6434 CrossRef CAS.
- A. E. Robles-Sardin, A. V. Bolanos-Villar, G. A. Gonzalez-Aguilar and L. A. de la Rosa, Fruit Vegetable Phytochem., 2010, 155–175 CAS.
- A. G. Mercader and A. B. Pomilio, Eur. J. Med. Chem., 2010, 45, 1724–1730 CrossRef CAS.
- K. Miki, T. Nagai, K. Suzuki, R. Tsujimura, K. Koyama, K. Kinoshita, K. Furuhata, H. Yamada and K. Takahashi, Bioorg. Med. Chem. Lett., 2007, 17, 772–775 CrossRef CAS.
- X. Li, T. Ohtsuki, S. Shindo, M. Sato, T. Koyano, S. Preeprame, T. Kowithayakorn and M. Ishibashi, Planta Med., 2007, 73, 1195–1196 CrossRef CAS.
- T.-T. Dao, B.-T. Tung, P.-H. Nguyen, P.-T. Thuong, S.-S. Yoo, E.-H. Kim, S.-K. Kim and W.-K. Oh, J. Nat. Prod., 2010, 73, 1636–1642 CrossRef CAS.
- T. T. Dao, P. H. Nguyen, H. S. Lee, E. Kim, J. Park, S. Il Lim and W. K. Oh, Bioorg. Med. Chem. Lett., 2011, 21, 294–298 CrossRef CAS.
- A.-L. Liu, B. Liu, H.-L. Qin, S. M. Lee, Y.-T. Wang and G.-H. Du, Planta Med., 2008, 74, 847–851 CrossRef CAS.
- H. J. Jeong, Y. B. Ryu, S.-J. Park, J. H. Kim, H.-J. Kwon, J. H. Kim, K. H. Park, M.-C. Rho and W. S. Lee, Bioorg. Med. Chem., 2009, 17, 6816–6823 CrossRef CAS.
- T. N. A. Nguyen, T. T. Dao, B. T. Tung, H. Choi, E. Kim, J. Park, S.-I. L. Lim and W. K. Oh, Food Chem., 2010, 124, 437–443 CrossRef.
- A.-L. Liu, F. Yang, M. Zhu, D. Zhou, M. Lin, S. M.-Y. Lee, Y.-T. Wang and G.-H. Du, Planta Med., 2010, 76, 1874–1876 CrossRef CAS.
- L. Zhang, Y.-X. Cheng, A.-L. Liu, H.-D. Wang, Y.-L. Wang and G.-H. Du, Molecules, 2010, 15, 8507–8517 CrossRef CAS.
- A. Garozzo, R. Timpanaro, A. Stivala, G. Bisignano and A. Castro, Antiviral Res., 2011, 89, 83–88 CrossRef CAS.
- X. Wang, W. Jia and A. Zhao, Phytother. Res., 2006, 20, 335–341 CrossRef CAS.
- H. Ge, Y. F. Wang, J. Xu, Q. Gu, H. B. Liu, P. G. Xiao, J. Zhou, Y. Liu, Z. Yang and H. Su, Nat. Prod. Rep., 2010, 27, 1758–1780 RSC.
- L. Tian, Z. Wang, H. Wu, S. Wang, Y. Wang, J. Xu, L. Wang, F. Qi, M. Fang, D. Yu and X. Fang, J. Ethnopharmacol., 2011, 137, 534–542 CrossRef.
- Botanic Gardens Conservation International, http://www.bgci.org/ourwork/1521, accessed April 2011.
- D. J. Newman, Abstracts of Papers, 241st ACS National Meeting & Exposition, Anaheim, CA, United States, March 27–31, 2011, 2011, MEDI–170 Search PubMed.
- A. Boudet, Phytochemistry, 1973, 12, 363–370 CrossRef CAS.
- B. Moeller and K. Herrmann, Phytochemistry, 1983, 22, 477–481 CrossRef CAS.
- S. Jiang and G. Singh, Tetrahedron, 1998, 54, 4697–4753 CrossRef CAS.
- V. Farina and J. D. Brown, Angew. Chem., Int. Ed., 2006, 45, 7330–7334 CrossRef CAS.
- D. S. Fabricant and N. R. Farnsworth, Environ. Health Perspect. Suppl., 2001, 109, 69–75 CAS.
- J. M. Rollinger, T. Langer and H. Stuppner, Curr. Med. Chem., 2006, 13, 1491–1507 CrossRef CAS.
- R. J. Quinn, A. R. Carroll, N. B. Pham, P. Baron, M. E. Palframan, L. Suraweera, G. K. Pierens and S. Muresan, J. Nat. Prod., 2008, 71, 464–468 CrossRef CAS.
- W.-J. Shin, K.-H. Lee, M.-H. Park and B.-L. Seong, Microbiol. Immunol., 2010, 54, 11–19 CrossRef CAS.
- S. Pleschka, M. Stein, R. Schoop and J. B. Hudson, Virol. J., 2009, 6, :197, DOI:10.1186/1743-422X-6-197.
- S. Yingsakmongkon, D. Miyamoto, N. Sriwilaijaroen, K. Fujita, K. Matsumoto, W. Jampangern, H. Hiramatsu, C. T. Guo, T. Sawada, T. Takahashi, K. Hidari, T. Suzuki, M. Ito, Y. Ito and Y. Suzuki, Biol. Pharm. Bull., 2008, 31, 511–515 CAS.
- M. Haidari, M. Ali, S. W. Casscells, III and M. Madjid, Phytomedicine, 2009, 16, 1127–1136 CrossRef CAS.
- S. C. Ma, Z. D. He, X. L. Deng, P. P. But, V. E. Ooi, H. X. Xu, S. H. Lee and S. F. Lee, Chem. Pharm. Bull., 2001, 49, 1471–1473 CrossRef CAS.
- J. M. Rollinger and M. Schmidtke, Med. Res. Rev., 2011, 31, 42–92 CrossRef CAS.
- R. M. Krug and J. M. Aramini, Trends Pharmacol. Sci., 2009, 30, 269–277 CrossRef CAS.
- J. Kirchmair, S. Distinto, K. R. Liedl, P. Markt, J. M. Rollinger, D. Schuster, G. M. Spitzer and G. Wolber, Infect. Disord. Drug Targets, 2011, 11, 64–93 CAS.
- M. von Itzstein, J. C. Dyason, S. W. Oliver, H. F. White, W. Y. Wu, G. B. Kok and M. S. Pegg, J. Med. Chem., 1996, 39, 388–391 CrossRef CAS.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- A. M. Abu Hammad and M. O. Taha, J. Chem. Inf. Model., 2009, 49, 978–996 CrossRef CAS.
- J. H. An, D. C. W. Lee, A. H. Y. Law, C. L. H. Yang, L. L. M. Poon, A. S. Y. Lau and S. J. M. Jones, J. Med. Chem., 2009, 52, 2667–2672 CrossRef CAS.
- R. E. Amaro, D. D. Minh, L. S. Cheng, W. M. Lindstrom Jr, A. J. Olson, J. H. Lin, W. W. Li and J. A. McCammon, J. Am. Chem. Soc., 2007, 129, 7764–7765 CrossRef CAS.
- R. E. Amaro, X. Cheng, I. Ivanov, D. Xu and J. A. McCammon, J. Am. Chem. Soc., 2009, 131, 4702–4709 CrossRef CAS.
- R. E. Amaro, R. V. Swift, L. Votapka, W. W. Li, R. C. Walker and R. M. Bush, Nat. Comm., 2011, 2, 388 CrossRef.
- M. R. Landon, R. E. Amaro, R. Baron, C. H. Ngan, D. Ozonoff, J. A. McCammon and S. Vajda, Chem. Biol. Drug Des., 2008, 71, 106–116 CAS.
- R. E. Amaro and W. W. Li, Curr. Top. Med. Chem., 2010, 10, 3–13 CrossRef CAS.
- S. Rudrawar, J. C. Dyason, M. A. Rameix-Welti, F. J. Rose, P. S. Kerry, R. J. Russell, S. van der Werf, R. J. Thomson, N. Naffakh and M. von Itzstein, Nat. Commun., 2010, 1, 1–7 CrossRef CAS.
- B. K. Mai, M. H. Viet and M. S. Li, J. Chem. Inf. Model., 2010, 50, 2236–2247 CrossRef CAS.
- J. D. Durrant and J. A. McCammon, Comput. Biol. Chem., 2010, 34, 97–105 CrossRef CAS.
- B. Roschek, Jr, R. C. Fink, M. D. McMichael, D. Li and R. S. Alberte, Phytochemistry, 2009, 70, 1255–1261 CrossRef.
- N. Sriwilaijaroen, A. Kadowaki, Y. Onishi, N. Gato, M. Ujike, T. Odagiri, M. Tashiro and Y. Suzuki, Food Chem., 2011, 127, 1–9 CrossRef CAS.
- M. Shibazaki, K. Tanaka, K. Nagai, M. Watanabe, S. Fujita, K. Suzuki, G. Okada and T. Yamamoto, J. Antibiot., 2004, 57, 812–815 CAS.
| This journal is © The Royal Society of Chemistry 2012 |