Aminobenzoates as building blocks for natural product assembly lines
Christopher T.
Walsh
*,
Stuart W.
Haynes
and
Brian D.
Ames
Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115, USA. E-mail: christopher_walsh@hms.harvard.edu; Fax: +617-432-0483; Tel: +617-432-1715
First published on 8th November 2011
Abstract
Covering: up to the end of September 2011
The ortho-, meta-, and para- regioisomers of aminobenzoate are building blocks for a wide range of microbial natural products. Both the ortho-isomer (anthranilate) and PABA derive from the central shikimate pathway metabolite chorismate while the meta-isomer is not available by that route and starts from UDP-3-aminoglucose. PABA is largely funnelled into folate biosynthesis while anthranilate is the scaffold for biosynthetic elaboration into many natural heterocycles, most notably with its role in indole formation for tryptophan biosynthesis. Anthranilate is also converted to benzodiazepinones, fumiquinazolines, quinoxalines, phenoxazines, benzoxazolinates, quinolones, and phenazines, often with redox enzyme participation. The 5-hydroxy form of 3-aminobenzaote is the starter unit for ansa-bridged rifamycins, ansamitocins, and geldanamycins, whereas regioisomers 2-hydroxy, 4-hydroxy and 2,4-dihydroxy-3-aminobenzoate are key components of antimycin, grixazone, and platencin and platensimycin biosynthesis, respectively. The enzymatic mechanisms for generation of the aminobenzoate regioisomers and their subsequent utilization for diverse heterocycle and macrocycle construction are examined.
 Christopher T. Walsh | Christopher T Walsh, born in 1944, majored in biology at Harvard and completed his PhD in biochemistry in the group of Fritz Lipmann at The Rockefeller University. He was on the MIT Faculty (1972–1987) and since then has been at Harvard Medical School. He served as Chair of the Department of Chemistry at MIT (1982-1978) and of the Department of Biological Chemistry & Molecular Pharmacology at Harvard Medical School (1987–1995). His research interests lie in deciphering the molecular logic and enzymatic machinery of natural product biosynthesis. |
 Stuart W. Haynes | Stuart Haynes (born 1984) graduated from University of Warwick, UK in 2006 with a MChem in chemistry. He went on to obtain his PhD in chemistry from the same institution in 2010 under the supervision of Gregory L. Challis, where he focused on investigating the carbocyclic S. coelicolor metabolite streptorubin B. He is currently a postdoctoral fellow in the group of Christopher T. Walsh at Harvard Medical School, where his research efforts have been targeted towards elucidating the enzymatic logic of fungal alkaloid biosynthesis. |
 Brian D. Ames | Brian Ames (born 1980) attended Brigham Young University, Hawaii, USA where he graduated in 2004 with a B.S. in biochemistry. He obtained his PhD in 2009 under the supervision of Shiou-Chuan (Sheryl) Tsai at the University of California, Irvine where he focused on the structural enzymology of polyketide biosynthesis. He is currently an NIH-sponsored postdoctoral fellow at Harvard Medical School working under the supervision of Christopher T. Walsh. His postdoc research has focused on the enzymology of fungal alkaloid biosynthesis. |
1 Introduction
Aminobenzoates are noncanonical aromatic amino acids. All three isomers, 2-aminobenzoate (anthranilate), 3-aminobenzoate (meta), and 4-aminobenzoate (para-substitution, abbreviation PABA) occur as components of microbial metabolism. Unlike typical α-amino acids, anthranilate, 3-aminobenzoate, and PABA are β-, γ-, and δ-amino acids, respectively. They are nonproteinogenic but their bifunctional nature, which enables ligation to other scaffolds, is utilized in distinct niches in microbial metabolism.All three isomers can be used as chain-initiating building blocks in nonribosomal peptide and in hybrid nonribosomal peptide–polyketide natural product assembly lines. In many instances the amino group of the 2-amino- or 3-aminobenzoyl starter unit can act as an internal nucleophile to generate cyclic lactams. Enzymatic hydroxylation to amino-hydroxybenzoate regioisomers initiates oxidative chemistry, including aminonapthoquinone ring formation in rifamycins, benzoazolinate ring formation in the enediyne C-1027, and dimerization in actinomycins and phenazines.
In contrast, para-aminobenzoate is known to be an essential building block for folate coenzyme biosynthesis and resultant DNA biosynthesis but is utilized only sparingly in hybrid NRPS–polyketide synthase (PKS) pathways. The 1,4-relation of amino and carboxylate groups in PABA is suitable for linear condensation products but not the cyclic outcomes seen with 2-amino- or 3-aminobenzoate building blocks. (Fig. 1)
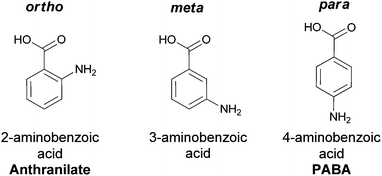 | ||
| Fig. 1 The three aminobenzoate regioisomers: anthranilate (ortho), 3-aminobenzoate (meta), 4-aminobenzoate (para). | ||
2 Generation of the aminobenzoate isomers during metabolism
2.1 Routing chorismate to 2-aminobenzoate and 4-aminobenzoate
o-Aminobenzoate (anthranilate) and p-aminobenzoate (PABA) both arise from enzymatic transformation of the dihydroaromatic enolpyruvyl-containing framework of chorismate to aromatic amino acid-based molecular scaffolds. Chorismate is famously the substrate for the 3,3-sigmatropic rearrangement catalyzed by chorismate mutase to yield prephenate,1–4 a proximal precursor of both phenylalanine and tyrosine in proteinogeneic aromatic amino acid biosynthesis in microbes (Fig. 2).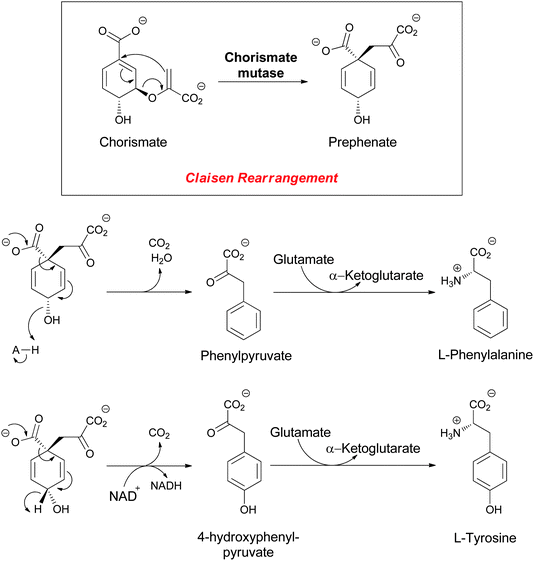 | ||
| Fig. 2 The enzymatic conversion of chorismate to prephenate, precursor to the proteinogenic amino acids L-phenylalanine (Phe) and L-tyrosine (Tyr). | ||
Chorismate can also be rerouted to either hydroxylated or aminated products by a related family of five chorismate-utilizing enzymes (Fig. 3). Anthranilate synthase generates the ortho-amino benzoate,5 while aminodeoxychorismate synthase produces the nascent product on the way to PABA in the folate biosynthetic pathway.6 Isochorismate synthase adds water instead of nascent ammonia on the way to 2,3-dihydroxybenzoate in the pathway to the enterobactin siderophore in gram negative enteric bacteria.7 The fourth enzyme, isochorismate pyruvate lyase (salicylate synthase), again uses water as a nucleophile to generate an ortho-substituted benzoate, in this case salicylate, which is used by plants as part of alarmone systems and by bacteria as an iron-chelating moiety in siderophores, such as yersiniabactin (Fig. 4).8–10 The fifth enzyme, chorismate lyase (UbiC), catalyzes aromatization with loss of pyruvate across carbons 2 and 3 to yield 4-hydroxybenzoate, which is a precursor to ubiquinone coenzymes.11–13 Studies on isochorismate pyruvate lyase show transfer of the hydrogen (deuterium) removed from C2 of isochorismate to C3 of the pyruvate product, consistent with a concerted pericyclic (but asynchronous) transition state for the aromatization to salicylate.14 The possibility that the other three enzymes also catalyze the 1,2-elimination of pyruvyl ethers by pericyclic processes has been suggested.15
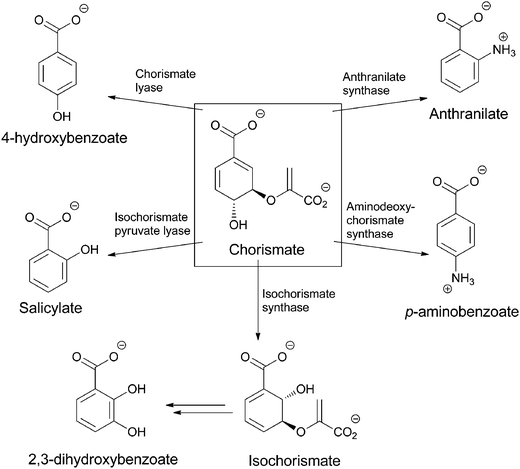 | ||
| Fig. 3 The routing of chorismate to five amino and hydroxybenzoates in microbial metabolism: anthranilate, PABA, salicylate, 2,3-dihydroxybenzoate, 4-hydroxybenzoate. | ||
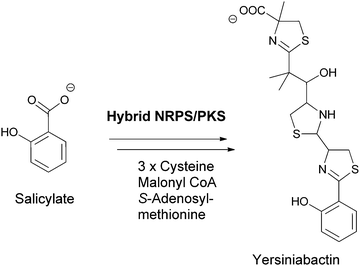 | ||
| Fig. 4 Salicylate as a building block for phenolic ligands for iron chelation in siderophores, exemplified by yersiniabactin assembly. | ||
Anthranilate synthase and PABA synthase generally have two subunits, one for processing the chorismate substrate for regiospecific amination, and the other acting as a glutaminase to release nascent ammonia from the γ-carboxamide of glutamine on demand.8 The nascent ammonia may travel down a tunnel from the glutaminase active site to the chorismate amination site. Regioselective addition of NH3 must be controlled by the orientation of the chorismate cosubstrate. In the case of anthranilate synthase, one can formulate the attack as a doubly vinylogous nucleophilic substitution (SN2′′) (Fig. 5) with net displacement of the 3-OH as NH3 adds to C5 of chorismate. The resultant aminodeoxyisochorismate then undergoes aromatizing elimination of the C5H and the C4 enolpyruvyl group to yield anthranilate (perhaps via a pericyclic transition state).
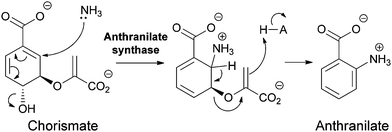 | ||
| Fig. 5 The conversion of chorismate to anthranilate by anthranilate synthase: nascent ammonia, derived from in situ hydrolysis of glutamine, acts as a SN2′′ nucleophile to produce aminodeoxyisochorismate that then undergoes aromatization by loss of the elements of pyruvate in the enzyme active site. | ||
Analogously, PABA arises from chorismate by a two subunit aminating catalyst that makes the 4-amino-4-deoxychorismate (ADC) and a separate lyase that carries out the aromatizing elimination of pyruvate to generate PABA (Fig. 6). It appears that the ADC synthase mechanism also uses a SN2′′ mechanism but that the initial nucleophile is an active site Lys-NH2 rather than cosubstrate NH3.16 The resultant adduct can then be attacked by NH3 in a second SN2′′ substitution, now generating 4-amino-4-deoxychorismate. This diffuses out of the active site and is aromatized by the partner lyase enzyme.
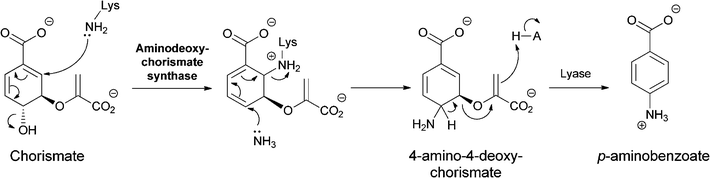 | ||
| Fig. 6 The conversion of chorismate to 4-amino-4-deoxychorismate (ADC) by two sequential SN2′′ aminations, the first from an active site lysine, the second from nascent NH3. Aromatization of ADC to 4-aminobenzoate (PABA) is catalyzed by a separate enzyme ADC lyase. | ||
2.2 The aminoshikimate pathway to 3-amino-5-hydroxybenzoate (AHBA)
The 3-aminobenzoate scaffold (meta-isomer) is not available by comparable SN2′′ chemistry from chorismate. The widely utilized form of biologically generated meta aminobenzoate is 3-amino-5-hydroxybenzoate, which is the starter unit for the ansa-bridged molecules, including rifamycin and ansamitocin family members. Floss and colleagues demonstrated this substituted aminobenzoate arises by reactions in the aminoshikimate pathway encoded by a clustered gene set first described in studies of rifamycin metabolism in Amycolaptosis mediterranei.17–21 The amino group is installed during reductive transamination of UDP-3-ketoglucose and in a series of steps yields 3-amino-3-deoxyfructose-6-P. A series of careful in vitro and in vivo experiments have implicated the action of a transketolase (rif-orf15) in the conversion of 3-amino-3-deoxyfructose-6-P to the unstable aldimine 1-imino erythrose-4-P. This highly reactive intermediate is then shuttled to RifH, which catalyses the subsequent capture with PEP to yield 4-amino-3,4-dideoxy-D-arabinoheptulosonic-7-P, in which the NH2 is now in the form of a stable amine linkage.22,23 The amino DHAP undergoes enzymatic cyclisation and processing to aminodehydroquinate and then aminodehydroshikimate before a final dehydration generates the aromatized 3-amino-5-hydroxybenzoate, acronym AHBA (Fig. 7).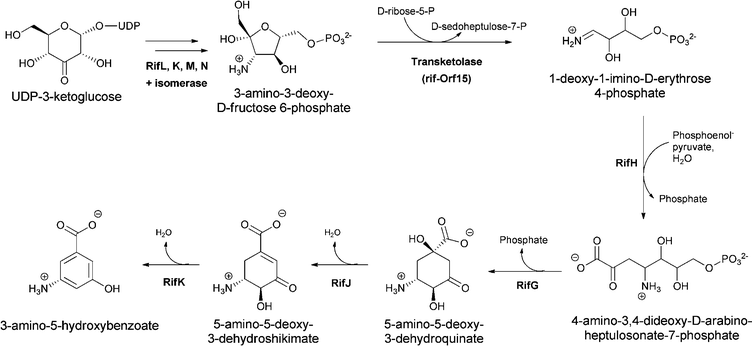 | ||
| Fig. 7 3-amino-5-hydoxybenzoate (AHBA) formation via the aminoshikimate pathway, starting from 3-amino-fructose-6-P and proceeding via amino-deoxyheptuloarabinose-7-P and aminodehydroshikimate. | ||
3 Anthranilate as a building block
3.1 Tryptophan biosynthesis
The most well-known set of enzymatic transformations of anthranilate occur in primary microbial metabolism as anthranilate forms the core of the tryptophan scaffold in a set of steps that are still remarkable even though they have been known for half a century. N-ribosylation of the nucleophilic amino group of anthranilate with the biological ribosylating agent 5-phosphoribose-1-pyrophosphate proceeds to the ring-opened 3-hydroxy-1-iminium ion that isomerizes via the enol to the 1-amino-3-keto-species that is the substrate for the enzyme indole-3-glycerol phosphate synthase.24 This enzyme uses the electron pair on the exocyclic nitrogen as a neighboring group to facilitate attack of the π electrons of the adjacent aryl double bond on the ketone and generate the 6-5 bicyclic 3-hydroxy iminium adduct. Loss of CO2 then occurs with the iminium acting as an electron sink, before a final dehydration driven by gain of conjugation in the final heteroaromatic bicyclic indole ring yields the five membered ring double bond (Fig. 8).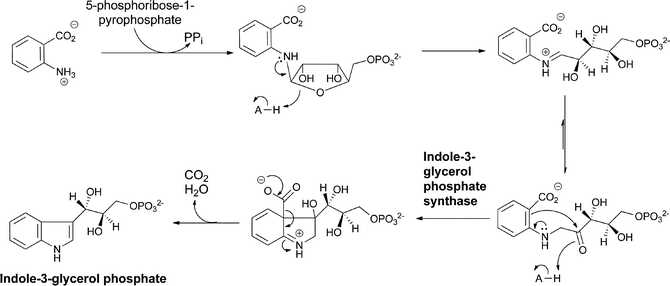 | ||
| Fig. 8 The conversion of anthranilate to indole-3-glycerol-P in tryptophan biosynthesis via N-phosphoribosylation and cyclodehydration. | ||
The further processing of indole-3-glycerol-P to tryptophan by tryptophan synthase has been exhaustively characterized and involves generation of nascent free indole in the enzyme's active site and reaction of the beta carbon of that indole as nucleophile on an electrophilic aminoacryl-PLP species generated by dehydration of cosubstrate serine (Fig. 9).24
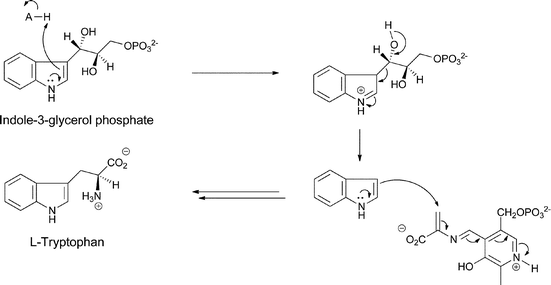 | ||
| Fig. 9 The processing of indoleglycerol-3-P to tryptophan by tryptophan synthase. | ||
The anthranilate scaffold has undergone decarboxylation on the way to Trp and so provides the benzene ring and the nitrogen in the final bicyclic indole heterocycle. Tryptophan is in turn a building block for thousands of indole-containing natural products and in this sense anthranilate is a pervasive scaffolding element across a huge swath of natural product space.
3.2 Anthranilate as a chain-initiating substrate in fungal nonribosomal peptide assembly
Anthranilate is also a building block for natural products independent from the generation of the indole ring of tryptophan. In particular, the 6,7 bicyclic benzodiazepinone scaffold of fungal metabolites, such as acetylaszonalenin (Fig. 10), arises from the utilization of anthranilate as the starter unit for a bimodular nonribosomal peptide synthetase (NRPS) enzyme (AnaPS).25 Additionally, the 6-6-6 tricylic quinazolinones framework characteristic of the fumiquinazolines, widely produced by Aspergillus fumigatus strains (all 40 strains so far cultivated and examined for production have been shown to be producers), is also derived from anthranilate.26 Fumiquinazoline F is produced by a trimodular NRPS starting from anthranilate, Trp, and Ala (Fig. 10).27,28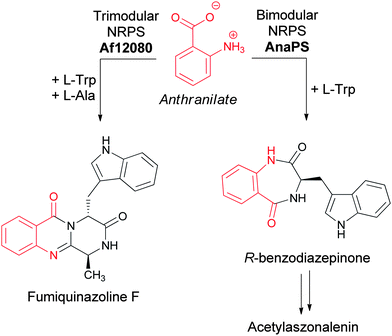 | ||
| Fig. 10 The assembly of acetylaszonalenin and fumiquinazoline F from anthranilate as chain initiating building block. | ||
These bimodular and trimodular NRPSs are thought to act similarly, using ATP to activate each amino acid monomer as aminoacyl-AMPs and then tethering these activated moieties as covalent pantetheinyl thioesters to 10 kDa carrier protein domains known as thiolation (T) domains. The Trp-activating module in each case has an embedded epimerization domain, presumed to convert an L-Trp-S-T domain thioester into a mixture of the D- and L-isomers, with only the D-Trp-thioester being carried forward. In the bimodular AnaPS, the dipeptidyl Ant-D-Trp-S-enzyme undergoes intramolecular attack of the anthranilyl-NH2 on the activated thioester carbonyl to release the cyclic benzodiazepinone (Fig. 11).
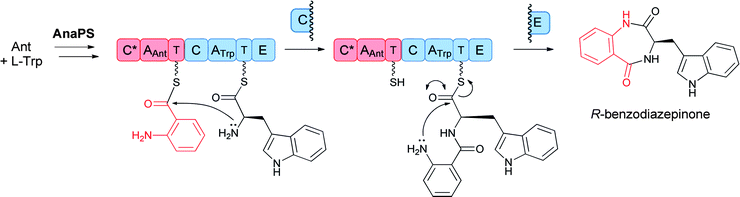 | ||
| Fig. 11 A two module nonribosomal peptide synthetase (NRPS) assembly line to convert anthranilate and L-Trp to a released 6,7-bicyclic benzodiazepinone. | ||
Two tailoring enzymes carry out subsequent prenylation at the β-carbon of the indole side chain with concomitant closure to the pyrroloindole group and subsequent N-acetylation of the indole nitrogen to yield acetylazonalenin (Fig. 12) with its 6-7-5-5-6 pentacyclic framework.25 Because the anthranilyl starter unit is a planar β- (rather than a typical α-aminoacyl moiety), the cyclisation yields a 6,7-bicyclic product with a fused diazepinone ring rather than a 6-6 system.
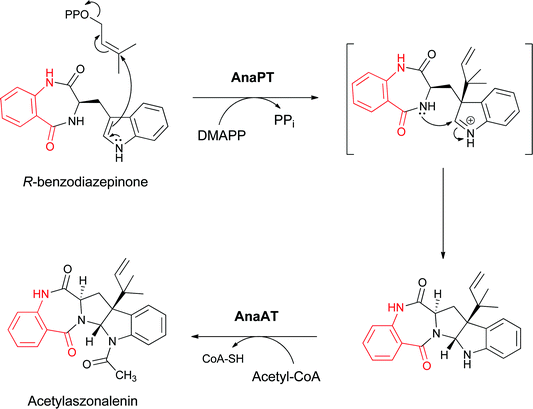 | ||
| Fig. 12 Conversion of the indolyl bezodiazepinone to the 6,7,5,5,6 pentacyclic framework of acetylaszonalenin by subsequent reverse prenylation and N-acetylation. | ||
Analogously for the FQF-generating trimodular NRPS assembly line, a linear anthranilyl-D-Trp-L-Ala-S-enzyme tripeptidyl intermediate is the likely precursor to the cyclising release step. Comparable attack of the anthranilyl-NH2 on the activated Ala-thioester would in this case give a bicyclic 6,10 ring system. That species undergoes dehydrative transannular ring closure to form the 6-6-6 tricyclic quinazalinone scaffold (Fig. 13, path a). Alternatively, formation of the tricyclic framework of FQF could proceed via initial diketopiperazine (DKP) formation by attack of the Trp derived amide nitrogen on the Ala-thioester followed by dehydrative cyclisation involving attack of the anthranilate-NH2 on the Ala-derived carbonyl of the DKP (Fig. 13, path b).
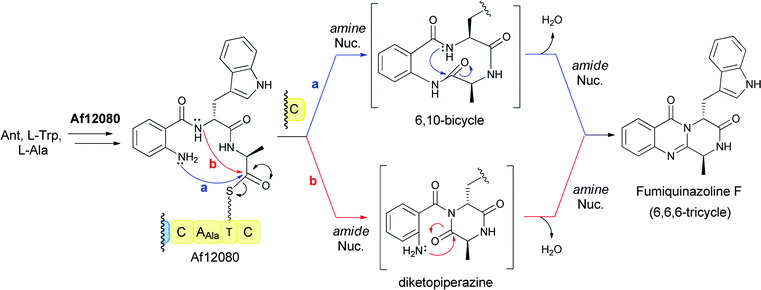 | ||
| Fig. 13 A three module NRPS assembly line for condensation of anthranilate, L-Trp, and L-Ala to form the 6,6,6-tricyclic fumiquinazoline F. | ||
The planar disposition of the carboxyl and amino groups on the anthranilate starter unit may be one of the driving forces for cyclising chain termination/release steps from the NRPS assembly lines. The corresponding 2-OH-benzoate (salicylate) is comparably activated as a starter unit by NRPS assembly lines but the ortho-hydroxyl appears to be too weak a nucleophile to initiate intramolecular cyclisation, in contrast to the ortho-NH2 in anthranilate rings.28 Instead the salicyl-OH is part of a ligand set for ferric iron chelation in bacterial siderophores,29e.g. in yersiniabactin and pyochelins from pseudomonads.30,31
The FQF metabolite is then subjected to a variety of additional enzymatic reactions that build in architectural complexity to this group of natural products. For example, FQF is sequentially processed to FQA and then FQC and FQD by FAD-dependent enzymes encoded in the FQ biosynthetic gene cluster.32,33 Of note is the annulation of the indole side chain to yield the observed tricyclic imidazoindolone ring system as FQF is converted to FQA by a two enzyme sequence. First the indole 2,3 double bond undergoes a formal epoxidation (the epoxide may be in equilibrium with a β-hydroxy-iminium species) and then is attacked by an L-alanine moiety in the form of an L-Ala-S-pantetheinyl generated by a monomodular NRPS. Intramolecular acylation by the indole nitrogen completes the ring annulation and yields FQA (Fig. 14).32,34
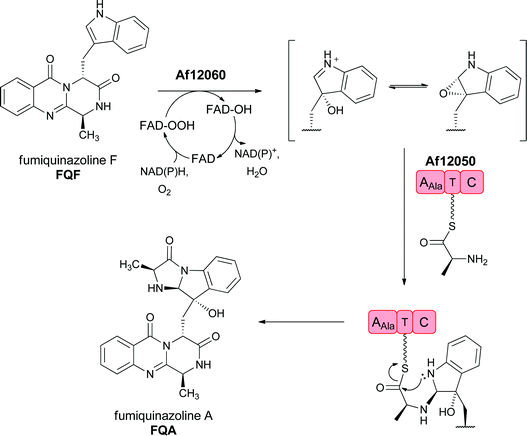 | ||
| Fig. 14 Enzymatic processing of fumiquinazoline F by sequential oxygenation at the pyrrolic double bond and annulation by an alanyl group carried on a single module NRPS generates an imidazoindolone scaffold in fumiquinazoline A. | ||
Subsequent processing by a second FAD enzyme Af12070, which appears to function as an amide oxidase on the pyrazinone ring of FQA, would set up a nascent imine that can be captured intramolecularly by an alcohol (FQC) or amine (FQD) to produce the highly structured aminal or hemiaminal products, respectively (Fig. 15).33In vitro reconstitution of Af12070 activity has shown that FQC is the initial kinetic product, and that FQC slowly converts non-enzymatically to the thermodynamic product FQD, presumably by interconversion to and capture of the imine intermediate. This transformation distorts the C ring out of plane in FQC and FQD, thus creating a distinct architectural scaffold (Fig. 15).
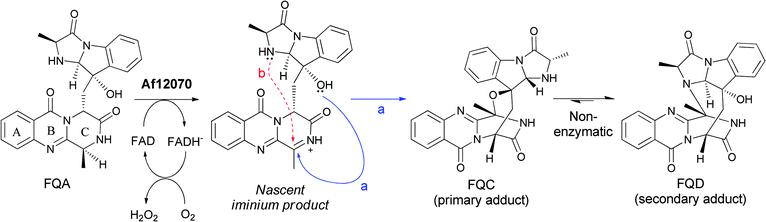 | ||
| Fig. 15 The maturation of fumiquinazoline A by an A. fumigatus tailoring oxidoreductase to FQC and FQD involves generation of a secondary imine that can be captured intramolecularly by an –OH to the hemiaminal (FQC) or by an –NH to the aminal (FQD). | ||
In Penicillium aethiopicum, the tremorgenic tryptoquialanine biosynthetic pathway also goes through the tricyclic FQF intermediate, undergoing annulation to an FQA analogue, 11′-dimethyl-2′-epi-FQA, and then being further processed by an apparent partial disassembly and opening of the quinazolinone core (Fig. 16).35 These anthranilate-initiated NRPS-mediated quinazolinone assemblies generate remarkable molecular architectures in short, efficient complexity-generating enzymatic pathways.
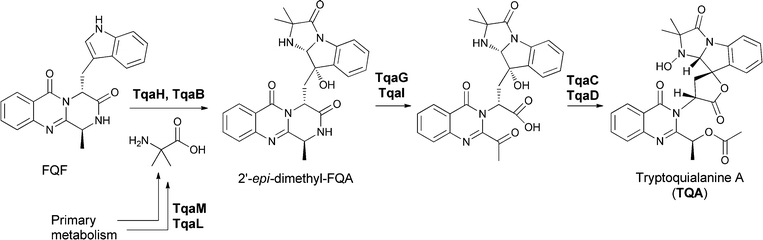 | ||
| Fig. 16 Enzymatic assembly of the tremorgenic tryptoquialanine proceeds through fumiquinazoline F on to 11′-dimethyl-2′-epi-fumiquinazoline A before directed fragmentation of the quinazoline and spirolactone formation. | ||
3.3 Substituted anthranilates as building blocks in the assembly of the benzodiazepinone scaffolds of the tomaymycin and sibiromycin antibiotic class
Pyrrolo-1,4-benzodiazepine antitumor antibiotics generated by Streptomyces are sequence-selective DNA alkylating agents.36–38 They include anthramycin, sibiromycin and tomaymycin (Fig. 17).![Assembly of the pyrrolo-[1,4]-benzodiazepine scaffolds of anthramycin, sibiromycin, tomaymycin antitumor antibiotics from different hydroxylated isomers of anthranilate.](/image/article/2012/NP/c1np00072a/c1np00072a-f17.gif) | ||
| Fig. 17 Assembly of the pyrrolo-[1,4]-benzodiazepine scaffolds of anthramycin, sibiromycin, tomaymycin antitumor antibiotics from different hydroxylated isomers of anthranilate. | ||
The framework of these antibiotics consists of an anthranilyl moiety condensed with a substituted dihydropyrrole moiety biosynthetically derived from tyrosine. The benzene ring derived from anthranilate undergoes distinct patterns of substitution in different producers. One pattern has a 5-hydroxy or a 4,5-dihydroxyanthranilate on the way to the 4-hydroxy-5-methoxy framework found in tomaymycin and congeners.39 These producers have an anthranilate synthase in the gene cluster and oxygenases suggesting the path progresses via direct hydroxylation of anthranilate to produce 4-OH-5-OMe-anthranilate for subsequent condensation.
The alternative substitution pattern, seen for example in anthramycin, involves 3-OH-anthranilate, which appears not to derive from de novo hydroxylation of anthranilate but instead from enzymatic degradation of tryptophan. Cleavage of Trp at the double bond in the 5-membered ring yields N-formyl kynurenine. Deformylation is followed by hydroxylation to give 3-OH-kynurenine, and a subsequent fragmentation that then yields 3-OH anthranilate.40 This can be methylated at C4 on the way to anthramycin or further hydroxylated at C5 to a 3-OH-4-Me-5-OH substitution pattern where the 5-hydroxyl is the site of glycosylation in sibiromycin.41
The mechanisms for condensation of the substituted anthranilates with the 4-substituted-2,3-dihydropyrrole-carboxylate are not yet established but there are NRPS modules in the tomaymycin biosynthetic cluster that, for example, may be involved in the activation of the anthranilyl and the dihydropyrrole-carboxylate building blocks as phosphopantetheinyl thioesters to generate a tethered anthranilyl-dihydropyrrole-thioester. The second NRPS module, encoded by tomB, appears to have a terminal reductase domain, suggesting that reductive release (of the corresponding aldehyde) mediates cleavage of the enzyme bound intermediate (Fig. 18). Subsequent (spontaneous) cyclisation generates the seven membered diazepine ring fused to the benzene moiety of the anthranilyl building block to create the fused pyrrolo-1,4-benzodiazepine scaffold. This cyclisation is comparable to that proposed for the bimodular acetylazonalenin synthetase, except that the linear dipeptidyl intermediate is tethered as an Ant-Trp-thioester in that case (Fig. 11).
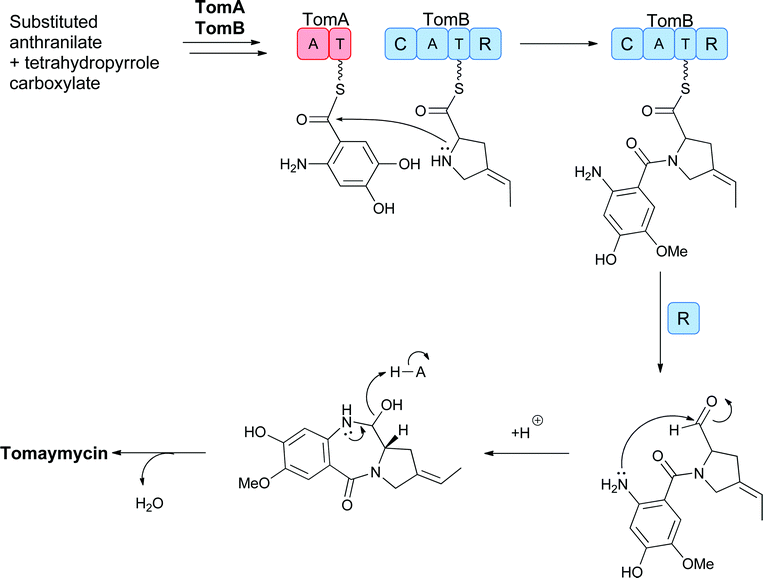 | ||
| Fig. 18 Proposed action of the two module NRPS assembly line to sequentially activate and condense 4-methyl-3-hydroxy-anthranilate and a dihydropyrrole carboxylate with cyclisation to release the 6,7,5-tricyclic scaffold of tomaymycin. | ||
The patterns of oxygenative and methyl substitution on the anthranilate moiety, the 4-substituent on the dihydropyrrole carboxylate and glycosylation on the 5-OH of the anthranilyl moiety with deoxysugars all build complexity and variation into the pyrrolo-1,4-benzodiazepine framework. One of these substituted anthranilates is also a building block of actinomycin, an antitumor antibiotic, the assembly of which is summarized in the next section.
Related systems proposed to build bicyclic heterocycles from 3-hydroxykynurenine are the quinoxaline-containing antibiotic dimers in the triostin and echinomycin series (Fig. 19).42 Tryptophan is proposed to be activated and tethered as a thioester to an NRPS module to allow for β-hydroxylation of part of the cellular pool of Trp in the producing streptomycete. Thiolytic release of 3-OH-Trp and then dioxygenative cleavage produces 3-OH-kynurenine. Rather than undergoing further cleavage to 3-OH-anthranilate (as seen in anthramycin biosynthesis), this substrate is suggested to undergo an oxygenative rearrangement to N-(2-aminophenyl)-3-OH-aspartate that undergoes further oxidation to the 3-keto species catalysed by Ecm3. Subsequent β-decarboxylation, cyclisation and aromatisation yields the quinoxaline carboxylate. This carboxylate then acts as the starter unit for a four module NRPS assembly line that creates the quinazolinyl tetrapeptide scaffold, which undergoes head to tail cyclisation in the release step en route to the mature triostin and echinomycin antibiotics. (Fig. 19).
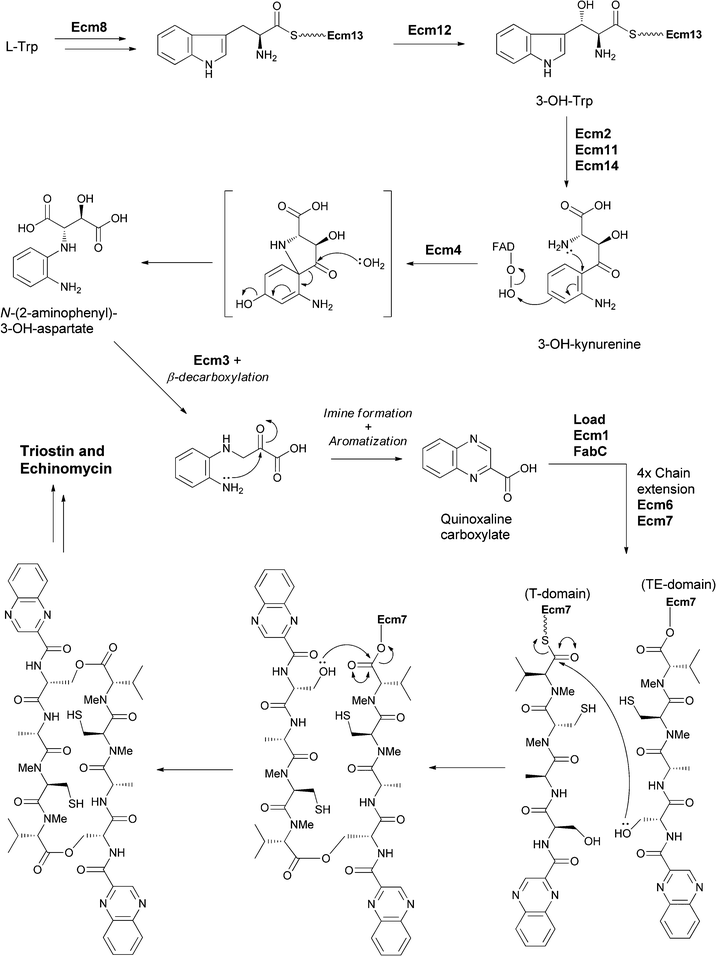 | ||
| Fig. 19 The enzymatic processing of tryptophan to quinoxaline building and subsequent assembly into antibiotics triostin and echinomycin. | ||
3.4 Substituted anthranilates as starter units for actinomycin biosynthesis
The chromopeptides of the actinomycin class have been studied for decades due to their antitumor activities.43,44 The central tricyclic phenoxazine ring system arises from the condensation of two peptidolactones that contain 4-methyl-3-hydroxy-anthranilate rings at their N-terminus (Fig. 20).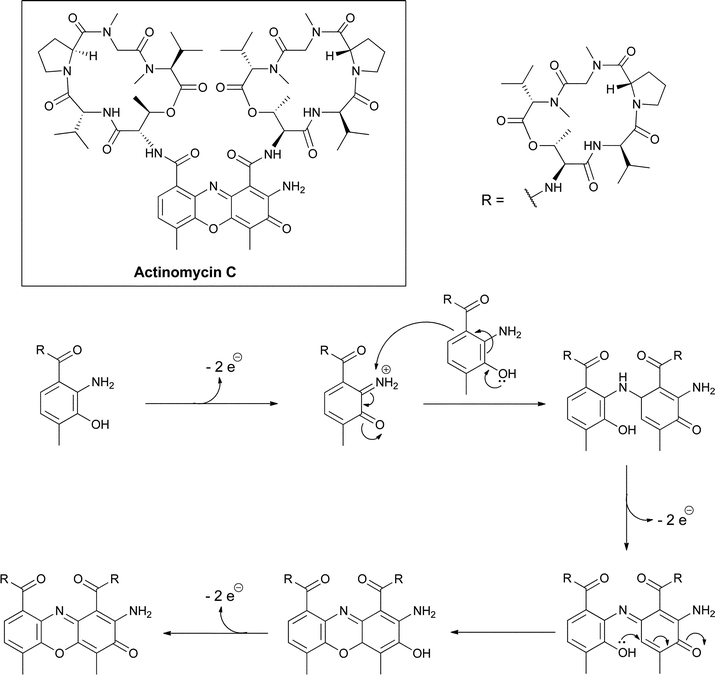 | ||
| Fig. 20 Oxidative dimerization of 4-methyl-3-hydroxy-anthranilyl tetrapeptide lactones creates the tricyclic phenoxazine scaffold in the actinomycin antitumor antibiotics. | ||
The peptide framework is assembled by a nonribosomal peptide synthetase where the substituted anthranilate acts as the chain-initiating building block and is typically activated as a phosphopantetheinyl thioester tethered to a carrier protein domain in the first NRPS module.45,46 The dimerization of two hydroxy-anthranilyl N-termini to create the peptidolactone-substituted dicarboxy phenoxazine platform for DNA intercalation is the last step in actinomycin maturation. This can occur nonenzymatically,47 reflecting the electron-rich autoxidizable aminophenol grouping in these substituted hydroxyanthranilates, but may also be enzyme-catalyzed (Fig. 20).48,49
Evidence for the biosynthetic origin of these substituted anthranilates comes from the gene cluster for actinomycin C, which has recently been cloned from S. chrysomallus. It was found to encode a dedicated tryptophan 2,3-dioxygenase, a kynurenase and a methyltransferase for the fragmentation of Trp across the pyrrole ring and hydrolysis of the resulting kynurenine in analogy to the pathways noted above for the pyrrolo-1,4-benzodiazepine scaffolds.45,50 A dedicated hydroxylase gene was not detected in the cluster but such a hydroxylase is presumed to act at the level of kynurenine. With the 3-OH installed, the 4-position of the anthranilyl moiety is sufficiently nucleophilic to attack the methyl group of S-adenosylmethionine, enabled by the methyltransferase, to complete the 4-Me-3-OH-2-NH2-benzoate building block. This is the same starter unit used in anthramycin assembly noted above.
3.5 Assembly of the benzoxazolinate ring in the enediyne antitumor agent C-1027
The family of enediyne natural products, containing a Z-1,5-diyne-3-ene array in a nine or ten membered ring system, are among the most cytotoxic agents known.51 They are produced by various strains of Streptomyces and other actinomycetes, often in combination with a chaperone protein to mitigate the reactivity that underlies their potent targeting of DNA by binding to and intercalating in the minor groove of target DNA prior to effecting DNA strand cleavage. The mechanism of enediyne activation had previously been established by Bergman and coworkers in an abiotic context, who showed that the enediyne unit can undergo cycloaromatisation (Fig. 21a) to a 1,4-benzenoid diradical intermediate.52 In the microenvironment of DNA this diradical can abstract hydrogen atoms and initiate DNA strand fragmentation.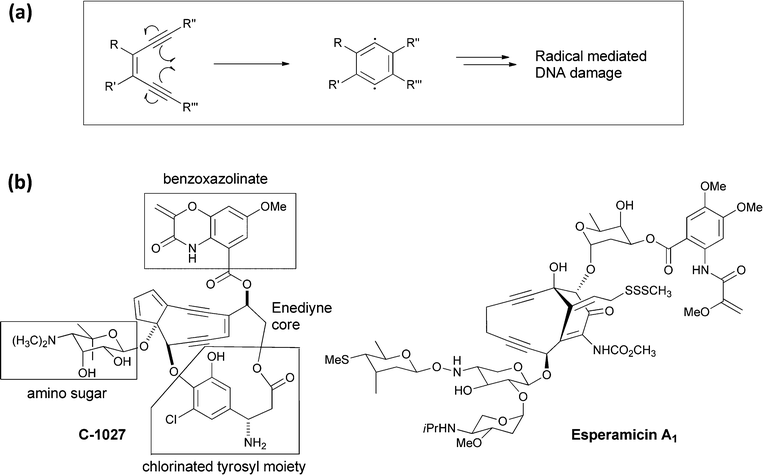 | ||
| Fig. 21 (a) A schematic for cyclisation of an enediyne to a benzenoid 1,4-diradical intermediate, followed by abstraction of two hydrogen atoms to yield a 1,2 disubstituted benzene. (b) The structure of the enediyne natural product C-1027 with its four component parts noted. Also shown is the structure of esperamycin A1. | ||
The framework of the enediyne natural products is typically multipartite with the enediyne unit flanked by deoxy (amino) sugars and by an organic monocyclic to tricyclic chromophore. For example, the C-1027 molecule (Fig. 21b) produced by Streptomyces globisporus has the enediyne core attached at three distinct points: to an amino sugar, to a chlorinated tyrosyl moiety,53 and a benzoxazolinate via an ester linkage.54 The benzoxazolinate bicyclic ring appears to be the binding determinant for the CagA chaperone protein during biosynthesis and secretion.
Examination of the biosynthesis of the benzoxazolinate moiety by Shen and colleagues has revealed a new enzymatic fate for anthranilate mediated by five enzymes encoded in the sgc gene cluster.55,56 The first step is the anticipated conversion of chorismate to aminodeoxyisochorismate by the SN2′′ amination noted above in section 2.1. Then, rather than the aromatizing elimination of the elements of pyruvate to yield anthranilate, an FMN and Fe/S-containing protein SgcG dehydrogenates the dihydroaromatic ring to the aromatic framework without loss of any of the substituents, resulting in release of enolpyruvyl anthranilate as a novel metabolite (Fig. 22).
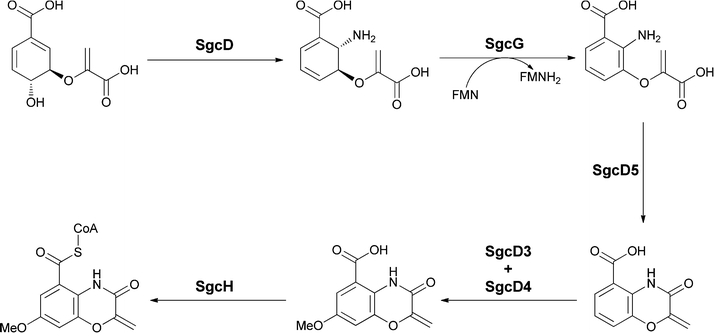 | ||
| Fig. 22 The enzymatic conversion of chorismate to the benzoxazolinyl carboxylate unit for incorporation onto C-1027. Aminodeoxyisochorismate is the first intermediate and undergoes aromatization to enolpyruvyl-anthranilate. A putative cyclisation followed by hydroxylase and O-methyltransferase action yields the heterobicyclic oxazoline-carboxylate. | ||
Subsequent intramolecular attack of the –NH2 on the carboxylate of the enolpyruvyl group would generate the bicyclic oxazolinate framework of the C-1027 chromophore. Further tailoring involves hydroxylation at the position para to the amide group and subsequent methylation of that –OH group to yield the free benzoxazolinate. Presumably, activation to the CoA thioester then sets up transfer to the alcohol group of the enediyne core. This is the first case of utilization of an enolpyruvyl moiety on an anthranilate scaffold to create a bisheterocyclic chromophore. In another enediyne antitumor antibiotic esperamycin A1 (Fig. 21b) there is a 4,5-dimethoxy-anthranilyl moiety esterified to a sugar, in turn connected to the enediyne core.57 Of note is the functionalization of the amino group of that substituted anthranilate with an enolpyruvyl ether moiety, perhaps arising from rearrangement of an enolpyruvyl anthranilate intermediate.
3.6 Anthranilate as starter unit for quinolones, including aurachins
A series of 2-alkyl-3-hydroxyquinolones function as quorum sensing molecules in the opportunistic pathogen Pseudomonas aeruginosa. The bicyclic quinolone framework, for example of 2-heptyl-4-quinolone, is encoded by the pqsABCDE operon. PqsA is an anthranilyl CoA ligase, which yields the activated anthranilyl fragment that is then condensed with 3-ketodecanoyl-S-ACP (acyl carrier protein) diverted from fatty acid biosynthesis.58 Imine formation, followed by Claisen condensation and then thioester hydrolysis and decarboxylation would yield 2-heptyl-4-quinolone. Subsequent oxygenation at C3 by PqsH generates the quorum sensing molecule PQS (Pseudomonas Quinolone Signal) (Fig. 23a).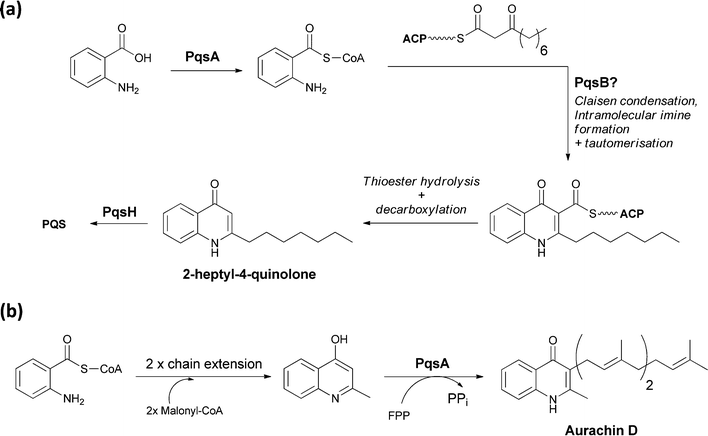 | ||
| Fig. 23 The enzymatic processing of anthranilate to quinolone scaffolds: (a) activation and condensation of anthranilate with fatty acid synthase derived intermediate en route to PQS (b) chain extension, cyclisation and prenylation of an anthranilate scaffold yields the electron transport inhibitor aurachin D. | ||
Aurachins are prenylated quinoline alkaloids from Stigmatella aurantiaca that act as electron transport inhibitors.59 The quinoline ring is also assembled from anthranilate and a C4 chain derived from two malonyl-CoA units. The isoprenyl unit derives from the C15 isoprenoid farnesyl-PP. Anthranilate is activated as anthranilyl CoA and then transthioesterified onto a carrier domain protein where it is used as the chain-initiating acyl group for two rounds of polyketide synthase chain elongation. The quinoline is then presumably formed by attack of the anthranilyl moiety –NH2 on the β carbonyl of the polyketide followed by thioester hydrolysis and decarboxylation.60 Prenylation presumably also occurs subsequent to release of the quinoline from the PKS assembly line (Fig. 23b).61
3.7 Phenazine biosynthesis from 3-hydroxy-2-aminodihydrobenzoate
Many variants of the tricyclic phenazine scaffold are seen in natural products derived from the rerouting of chorismate down amination pathways in bacteria. Pycocyanin (1-OH-N5-CH3-phenazine) was isolated 150 years ago as a blue compound from the bandages of soldiers that were infected with Pseudomonas aeruginosa.62 Other common phenazines include phenazine-1-COOH and the 1,6-dicarboxylic acid (Fig. 24).63–65 Because of their redox properties, including the ability to generate reactive, reduced oxygen species, phenazines are virulence factors in infections, e.g. cystic fibrosis, caused by producing microbes.66,67 They share some assembly analogy with the tricyclic phenoxazine scaffold of the actinomycins, in that they are assembled by an oxidative dimerisation of a 2-amino-3-hydroxy substituted metabolite. In this case though, the dimerizing unit is a dihydro form of 3-OH-anthranilate and there is no peptide chain attached to the (dihydro) anthranilyl carboxylate. A dicarboxy tricyclic phenazine framework results from this mode of head to tail dimerization.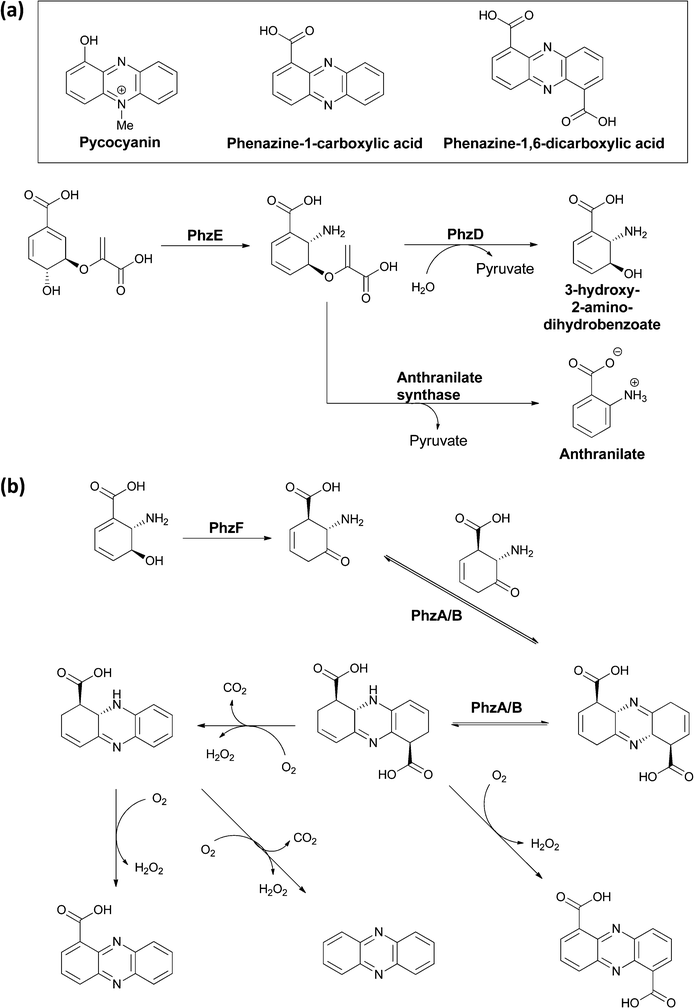 | ||
| Fig. 24 The microbial processing of chorismate to phenazines: (a) conversion of chorismate to aminodeoxychorismate is followed by PhzD-catalyzed hydrolysis of the enol ether kinkage to release 3-OH-2-NH2-2,3-dihydrobenzoate. (b) Isomerization by PhzF generates the 2-NH2-3-keto-5,6-ene tautomer that is a substrate for head to tail condensation by PhzA/B to a terahydrophenazine scaffold. Various routes of oxidation/decarboxylation can generate phenazine 1,6-dicarboxylate or phenazine-1-carboxylate. | ||
A five gene operon phzA–F encodes the pathway for their biosynthesis, with PhzE diverting chorismate to aminodeoxychorismate, as described in section 2.1.68 Then, in contrast to the aromatizing elimination of the elements of pyruvate (catalyzed by anthranilate synthase), which leads to anthranilate formation from that intermediate, an isochorismatase-like enzyme PhzD instead cleaves the vinyl ether, presumably by a net addition of water and pyruvate elimination, to yield 3-hydroxy-2-amino-dihydrobenzoate (Fig. 24a).69 The utility of this diverted dihydroaromatic molecule is evinced by its action as substrate for an isomerization catalyzed by PhzF to generate the 2-amino-3-keto-5,6-ene tautomer (Fig. 24b).70,71 This transient aminocyclohexenone can then undergo head to tail condensative dimerisation to yield a tetrahydro tricyclic-dicarboxy phenazine scaffold. This occurs by action of a PhzA/B complex, which accelerates a comparable uncatalyzed reaction.72,73 Oxidation (enzymatic and nonenzymatic) to the aromatized phenazine scaffold can then occur via single decarboxylation to phenazine-1-carboxylate, double decarboxylation to phenazine, or retention of both carboxylates in phenazine-1,6-carboxylate (Fig. 24b).
4 Meta-aminobenzoates as building blocks
4.1 3-Amino-5-hydroxybenzoate (AHBA) utilization as chain-initiating starter unit for ansa-bridged natural products
AHBA is the starting building block for the polyketide synthase assembly lines that build ansa-bridged scaffolds, including the rifamycins,20 ansamitocins,74 and geldanamycin75 molecules. AHBA has also been shown to be the nitrogen source for the terminal amide of the rifamycin related metabolites saliniketal A and B, in which a sequence of steps mediated by divergence at 34a-deoxyrifamycin W leave only the amino group derived from AHBA in the final product.23 The rif polyketide synthase assembly line has been intensively studied.20,21,76–79 During elongation of the tetraketidyl to the pentaketidyl tethered thioester the 3-amino-5-hydroxy benzoyl moiety is proposed to undergo a series of sequential oxidations of the aminophenol moiety to an aminobenzoquinone, which is attacked by an enolate (insert of Fig. 25), setting up for cyclisation to the hydroxy-amino-napthoquinone framework. This bicyclic aminonapthoquinone persists through to the full length undecaketidyl intermediate on the last thiolation domain of RifE (Fig. 25).78 A freestanding amide ligase, RifF, releases the chain by intramolecular cyclisation by using the amino group of the original AHBA starter unit (now the amino of the aminonapthoquinone nucleus) to attack the tethering pantetheinyl thioester of the undecaketidyl C-terminus.80 This step releases the first macrocyclic intermediate proansamycin X, which is then elaborated in a series of post assembly line modifications to the family of rifamycins. Thus, RifF makes a 23-membered macrolactam, which encompasses the functionalized napthoquinone ring system resulting from AHBA oxidation.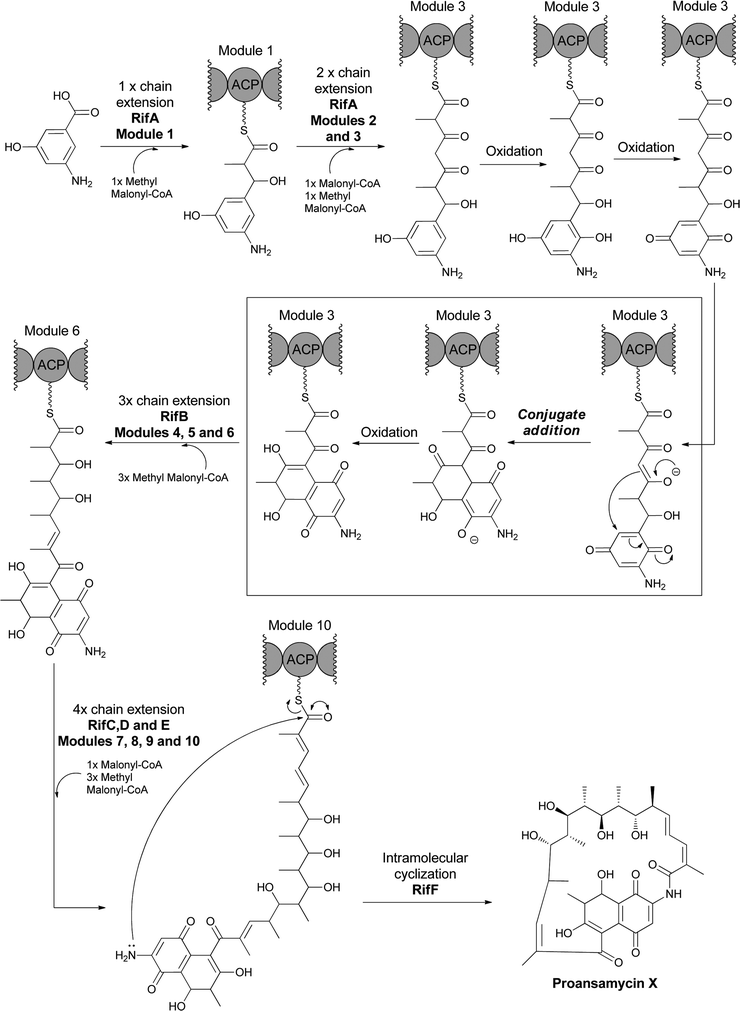 | ||
| Fig. 25 The multimodular polyketide synthase (PKS) assembly line for proansamycin in the rifamycin pathway. AHBA is the starter unit for the assembly line and intramolecular cyclisation to the aminonapthoquinone occurs during elongation of the tethered tetraketidyl to the pentaketidyl-S-enzyme species. Macrolactamizing release of the 23 membered macrocyclic proansamycin X is catalyzed by RifF acting as amide synthase with the aminonapthoquinone as an internal nitrogen nucleophile. | ||
In the related construction of ansamitocin scaffolds by PKS assembly lines the AHBA moiety is not hydroxylated during chain elongation and persists during elaboration of the heptaketidyl backbone.74 Analogous intramolecular lactamization to effect chain release of proansamitocin (Fig. 26) yields a 19-atom macrolactam. Hence, the proansamitocin assembly line manages not to oxidize the sensitive aminophenol moiety during all the elongation steps, in contrast to the rifamycin assembly line.
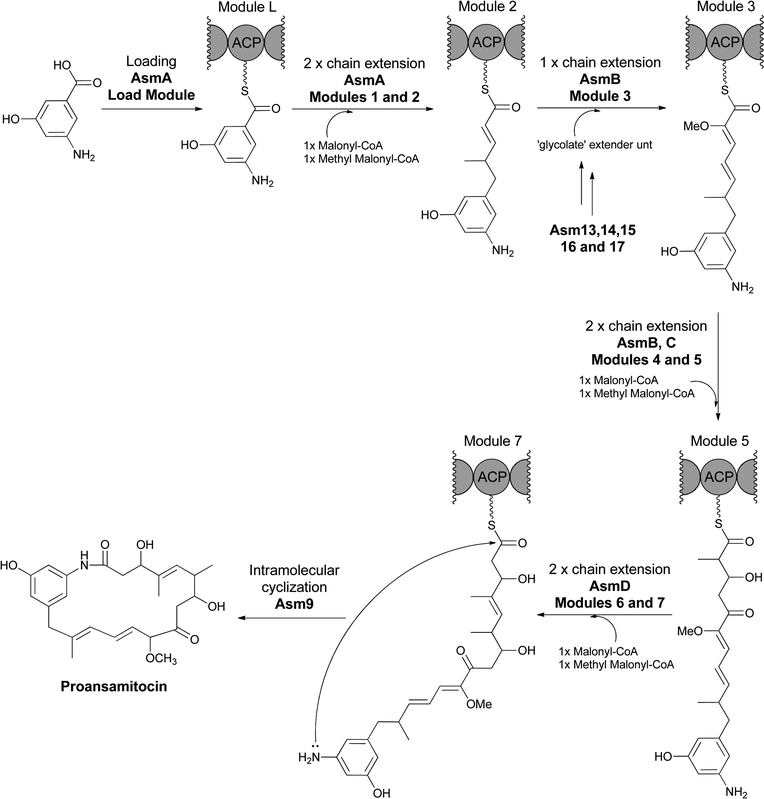 | ||
| Fig. 26 AHBA as starter unit for the proansamitocin PKS assembly line. Release of the full length chain by Asm9 creates the 19 membered macrolactam via internal attack of the AHBA amino group on the thioester carbonyl. | ||
The size of these 19 and 23 membered ansa-bridged rings, built ultimately from a meta-aminobenzoate starter unit, is drastically different from the cyclisation of the corresponding ortho-aminobenzoate starter unit that yields a 6,6- or 6,7-bicyclic framework as noted earlier. Notably, the carbonyl oxidation state persists in the anthranilate-derived quinazoline natural products during the action of bi- and trimodular NRPS assembly lines while the processing of AHBA as a starter unit by polyketide synthase machinery results in a net reduction of the AHBA carbonyl to the methylene group seen in the ansa-bridged natural product scaffolds. The presence of the 5-OH in the meta-benzoate scaffolds represents an easily oxidizable framework that is clearly consequential for napthoquinone formation in the rifamycins, aminobenzoquinone formation in the geldanamycins, and adjacent chlorination in ansamitocin-3 and congeners.
4.2 3-Amino-2-hydroxybenzoate (aminosalicylate) as a building block for antimycins
The antimycins, inhibitors of mitochondrial electron transport chains, are produced by Streptomyces antibioticus.81–83 They have a 3-aminosalicyl ring amide linked to a threonyl residue that in turn is part of a nine membered-hexyl-bilactone moiety (Fig. 27). The 3-amino group of the salicyl ring is N-formylated.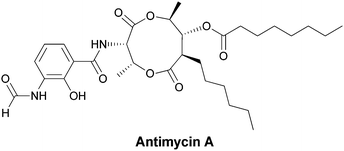 | ||
| Fig. 27 The mitochondrial respiratory poison antimycin A has an N-formyl-aminosalicylamide attached to a threonyl-bis-nonalactone. Assembly is presumed to go through the N-formyl-aminosalicyl-Thr dipeptidyl precursor as a tethered thioester. | ||
This 3-amino-2-OH-benzoate is a distinct regioisomer from the 3-amino-5-OH-benzoate (AHBA) noted above as the key starter subunit for ansa-bridged natural products. No biosynthetic gene cluster has yet been described for antimycin family members so it is not yet clear if the aminosalicyl starter unit arises from amination of salicylate, by an as yet unknown mechanism, or by 2-hydroxylation of a 3-aminobenzoate precursor via action of an oxygenase akin to those that hydroxylate the anthranilate ring noted in section 3 above. The timing of N-formylation is likewise unknown, as is the immediate source of the formyl group. The bilactone substructure suggests hybrid NRPS–PKS assembly machinery building off the N-formyl-aminosalicyl starter unit to an aminosalicyl-threonyl-S-enzyme intermediate that then switches to PKS assembly logic for chain extension and bis-lactonization. Formylation of the amino group may be acting as a mechanism to suppress intramolecular lactam formation and thus allow the observed macrolactonisation instead.
4.3 3-Amino-4-hydroxybenzoate (3,4-AHBA) for grixazone biosynthesis
A third hydroxylated meta-aminobenzoate regioisomer, 3,4-AHBA, has also been found as a streptomycete metabolite where it serves as a building block for the chromophoric phenoxazinones grixazone A and B from Streptomyces griseus.84 Grixazone A differs from grixazone B (Fig. 28) by reduction of a carboxylate to the corresponding aldehyde, which appears to be mediated by the action of GriCD from the grixazone biosynthetic cluster.85 The substitution pattern in 3,4-AHBA, like that of 3,5-AHBA above, is not readily derivable from a shikimate intermediate and studies by Horinuchi and colleagues indeed indicated diversion of two primary metabolites, aspartate semialdehyde and dihydroxyacetone-P, by a nonshikimate pathway in the producer S. griseus. GriI catalyses an initial aldol condensation to give the acyclic 2-amino-4,5-dihydroxy-6-one-heptanoate-7-P, after which GriH was required for cyclodehydrative aromatization to 3,4-AHBA.86 The phenoxazinone arises from an oxidative dimerization process with analogies to the formation of the phenoxazine chromophore in the actinomycins noted above in section 3.4. GriF has been shown to favour phenoxazine formation via the oxidation of 3-amino-4-hydroxybenzaldehyde (3,4-AHBAL) and 3,4-AHBA to the corresponding quinone imines (albeit at very different rates), which then undergo a seemingly spontaneous dimerization.87 GriF catalysed oxidation to the quinone imine intermediates not only favours dimerization but also appeared to be required to mediate the non-enzymatic conjugate addition of N-acetylcysteine seen during grixazone biosynthesis.87 3,4-AHBA is also thought to be incorporated into other phenoxazine metabolites including michigazone,88 exfoliazone89 and N-acetylquestiomycin,90 and has been shown to be a building block in the assembly of the manumycin family of antibiotics, such as asukamycin.91,92 This pathway may reflect an alternate route to the shikimate pathway for microbial benzene ring constructions.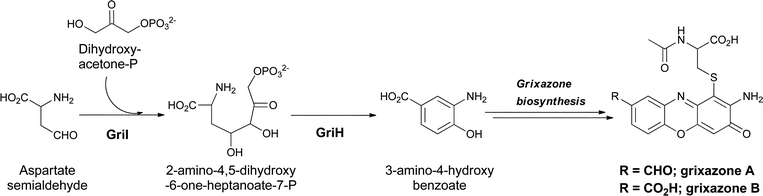 | ||
| Fig. 28 The enzymatic shuttling of primary metabolites towards 3-amino-4-hydroxybenzoate biosynthesis, and subsequent dimerization to give grixazone in Streptomyces griseus. | ||
4.4 3-Amino-2,4-dihydroxybenzoate in platencin and platensimycin
The 3,4-AHBA building block can undergo further hydroxylation at C2 to yield the 3-amino-2,4-dihydroxybenzoate (2,3,4-ADHBA) unit found in an amide linkage in the antibiotics platencin and platensimycin (Fig. 29). These antibiotics are generated by Streptomyces platensis and bind with nanomolar potency to the acyl enzyme intermediate of the FabF β-ketoacyl synthases in bacterial fatty acid biosynthesis.93,94 They are bacteriocidal and curative in infections in animal models.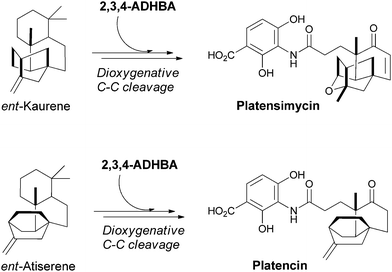 | ||
| Fig. 29 The biosynthesis of the potent bacteriocidal metabolites platensimycin and platencin. Dioxygenative cleavage of terpene derived multicyclic scaffolds, ent-kaurene and ent-antiserene, and subsequent condensation with 2,3,4-ADHBA yields the final antibiotic scaffolds. | ||
The multicyclic isoprenoid framework in platensimycin arises from geranylgeranyl diphosphate, which undergoes a series of cation-mediated cyclisations via copalyl diphosphate to produce ent-kaurene, the enantiomer of the plant terpene kaurene.95 The ent-kaurene scaffold must then undergo dioxygenative cleavage to the keto acid, and the carboxylate is subsequently ligated to the aminodihydroxybenzoate. The isoprenoid scaffold in platencin is similar and is thought to derive from ent-atiserene rather than ent-kaurene.
4.5 Condensation of 3-hydroxy-anthranilate and 3,4-hydroxy-aminobenzamide in farinamycin biosynthesis
A convergence of utilization of two of the aminobenzoate isomers, ortho-aminobenzoate and meta-aminobenzoate, occurs in the recently reported quinazoline metabolite farinamycin, isolated from S. griseus HKI0545 when cultured in a farina-based fermentation.96 This strain was known to produce phenoxazine metabolites, such as grixazone and pitucamycin, indicating it makes 3,4-AHBA (and the corresponding aldehyde, see section 4.3 above).97,98 Nett and Hertweck postulate condensation of the 3-OH-anthranilate and 3,4-AHBA building blocks in the assembly of farinamycin. Presumably, the 3-amino-4-hydroxybenzamide is condensed with 3-OH-anthranilate or its CoA thioester to give the 8-hydroxy-quinazoline-4-one bicyclic scaffold. That intermediate is then proposed to couple with the known epoxyquinone antibiotic enaminomycin C99 to generate farinamycin (Fig. 30). Use of both types of aminobenzoate building blocks increases the range of heterocyclic frameworks that can be assembled.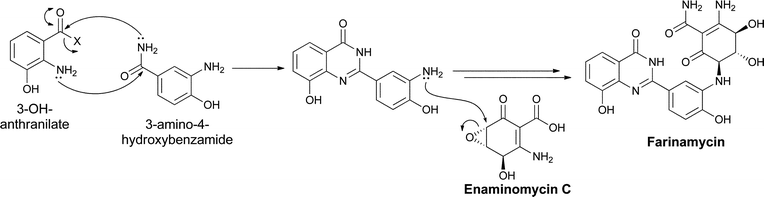 | ||
| Fig. 30 The biosynthesis of farinamycin in Streptomyces griseus. The cyclic condensation of two different aminobenzoate building blocks is followed by a coupling with the known epoxyquinone natural product enaminomycin C. | ||
5 PABA, dihydropteroate synthase and target of the sulfanilamides
While PABA is rarely used in construction of secondary metabolites, it does serve as a starter unit for candicidin family members and related aromatic polyenes with antifungal activities (Fig. 31). The para-aminoacetophenone moiety derives from PABA incorporated at the front end of a 20 module Type I polyketide synthase assembly line spread over five proteins. Adjacent to the pks genes in the candicidin biosynthetic gene cluster are genes encoding ADC synthase and ADC lyase to generate the PABA utilized in chain initiation.100–102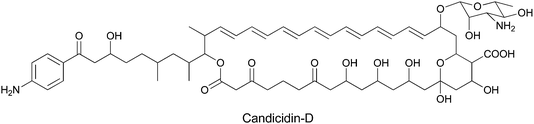 | ||
| Fig. 31 The structure of the aromatic polyene antibiotic candicidin-D. PABA is incorporated as the starter unit for a multimodular PKS that performs 22 rounds of chain extension to install C2 or C3 units at varying oxidation states, before macrolactonization and other decorations yield the final product. | ||
In plants such as Arabidopsis PABA can be enzymatically esterified to glucose and accumulated in vacuoles, presumably as a storage reservoir.103 PABA is also found to be amide linked to a Δ2-6-OH-heptanoic acid unit that serves as a linker between PABA and ascarylose, which is appended via a β-glycoside linkage to the terminal hydroxy group.104 This results in one of a related mixture of ascarosides secreted by adult forms of Caenorhbditis elegans that act to delay development of stage II larvae (Fig. 32). The ascarosides send the larvae into stages known as dauer diapause, a non-aging life stage, when food supplies are scarce.
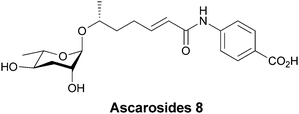 | ||
| Fig. 32 Ascaroside 8 is one of a related mixture of developmental signalling molecules containing a β-glycoside sugar, which is in this case linked to PABA. | ||
PABA has a primary role in the construction of the skeleton of folate coenzymes, necessary for provision of thymidylate for DNA biosynthesis in microbes and plants. Humans must obtain folates through dietary means, mainly from plants. In plants the pterin ring moiety is made in the cytoplasm, PABA in the chloroplasts, and the glutamylation that yields the folate coenzyme occurs in mitochondria.105–107 Folate, in its tetrahydro oxidation state, is also the coenzyme that mediates most of C1 metabolism at the level of itinerant methanol and formaldehyde oxidation states in cells. PABA is a substrate for the enzyme dihydropteroate synthase where the para-amino group is the nucleophile for reaction with cosubstrate 6-hydroxymethyl-7,8-dihydropteridine-PP.108 The C–N bond is formed between the exocyclic CH2 group of the pteridine as the C–O pyrophosphate bond is broken. The pyrophosphate linkage was installed in the previous step by a pyrophosphokinase to create the leaving group for C–O bond cleavage in the dihydropteroate synthase step (Fig. 33). The dihydropteroate is then subjected to one or more amide-forming steps, forging glutamyl linkages on the way to the family of dihydrofolates (pteroyl-oligoglutamates). Subsequent reduction to the 5,6,7,8-tetrahydro-pteridine oxidation state (= H4-folates) yields the active form of the coenzyme for carrying C1 units at either N5 or N10.
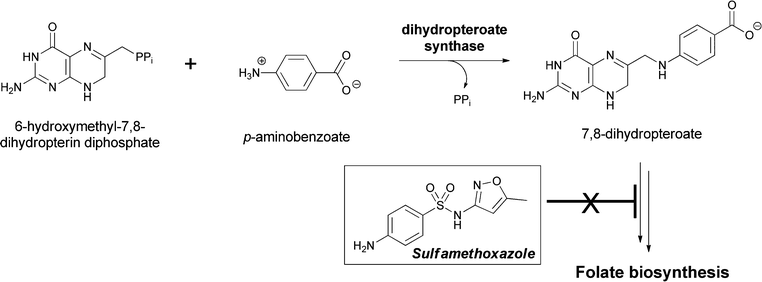 | ||
| Fig. 33 The dihydropteroate synthase reaction involves condensation of PABA with 7,8-dihydropterin-6-hydroxymethyl-PP. Release of the good leaving group PPi occurs concomitant with amide bond formation in dihydropteroate. Subsequent glutamylation yields the folate coenzyme scaffold. Sulfonamide antibiotics act as PABA analogs on this enzymatic step in folate biosynthesis. | ||
Dihydropteroate synthase has been a famous enzyme in the antibacterial community for decades because p-amino-benzenesulfonamides are potent competitive inhibitors of this crucial step in folate biosynthesis.109,110 Dozens of variant sulfonamides, including a current drug sulfamethoxazole, have been produced by medicinal chemistry efforts and this class of antibiotics has been in continuous use since the 1930s.111,112 Blockage of the de novo generation of tetrahydrofolate leaves organisms unable to add the CH3 group to dUMP to make dTMP, and the resulting lack of dTMP and subsequently dTTP abrogates bacterial DNA synthesis. Despite its lack of incorporation in secondary metabolites, PABA is by far the most famous of the aminobenzoate isomers in this arena of metabolism and biosynthesis.
6 Summary
All three regioisomers of aminobenzoate are generated as building blocks for microbial metabolism. The 2-amino and 4-aminobenzoates (anthranilate and PABA) are generated by a homologous pair of aminating enzymes that divert some of the chorismate pool from within producer cells. 3-aminobenzoate occurs notably as the 3-amino-5-hydroxy-benzoate scaffold in microbes that produce ansa-bridged polyketide natural products. The 3-amino-regioisomer is not available from chorismate and instead arises from the action of a set of enzymes that install the amino group at C3 of fructose and then carry out transketolase-mediated fragmentation to generate an erythrose-4-P-1-imine that is trapped by C–C bond formation with PEP, resulting in the formation of a stable 3-amino group. A subsequent set of transformations parallel to the classical shikimate pathway disgorge 3-amino-5-hydroxy-benzoate (AHBA) as the building block for rifamycins, ansamitocins, geldanamicins, and other related scaffolds.Anthranilate can also be hydroxylated at C3, C4, or C5 (actinomycins and benzo-1,4-pyrrolone frameworks, respectively). All these aminophenol regioisomers (including AHBA) are subject to facile oxidation (by one and/or two electron pathways); this can be a liability or can be used advantageously to enable oxidative condensations, as in phenoxazine ring formation in actinomycins and the amino-napthoquinone nucleus of rifamycins. The C-methylations found adjacent to particular hydroxy groups, for example, in the anthranilate scaffolds of actinomycins and benzo-pyrrole natural products, are also enabled by the carbanionic character of the carbons ortho to such hydroxyl groups.
Two further variations of enzymatic processing of anthranilate moieties also set up for ensuing heterocyclisations. In the phenazine pathway the aminodeoxyisochorismate (ADIC) generated in the first step of anthranilate synthase type enzymes is not subsequently aromatized but instead undergoes a directed vinyl ether hydrolysis to the 2-amino-3-hydroxy-dihydrobenzoate. A tautomer of this dihydro amino alcohol can undergo head to tail dimerization to create the tricyclic phenazine ring frameworks found in various pseudomonads. Another example of the versatility of the 2-aminobenzoate for directed heterocyclisation is illustrated by the recent discovery a third enzymatic fate for ADIC in C-1027 biosynthesis: dehydrogenation to 3-enolpyruvyl anthranilate that can then undergo cyclisation to give the bicyclic benzoxazolinate chromophore. This moiety provides a crucial role by directing interaction of C-1027 with the protein chaperone that protects the producer organism from self-inflicted DNA damage during production of this highly reactive enediyne metabolite.
We have noted that anthranilate and AHBA can be starter units for NRPS or PKS assembly lines. With anthranilyl starter units this β-amino acid can yield 6.7-bicyclic benzodiazepinone or 6,6,6-tricyclic quinazolinone natural product scaffolds. By contrast, the AHBA starter unit can be elongated with or without oxidation along the way, before the amino group acts as the internal nucleophile in chain-releasing events that give rise to the large ansa bridge macrolactams in various AHBA-initiating assembly lines. Finally, biological chemistry open to PABA seems more restricted, but in acting as a central substrate in folate coenzyme formation it plays a key role in providing the dTTP building block for DNA synthesis. Given the role of anthranilate in tryptophan biosynthesis and PABA in the folate pathway these two aminobenzoates qualify as primary microbial metabolites. AHBA is more properly viewed as a conditional or secondary metabolite. Metabolic installation of an amino group at C2, C3, or C4 of benzoate generates a set of building blocks that open up a wide range of condensation chemistries to producing microbes.
7 References
- P. R. Andrews, G. D. Smith and I. G. Young, Biochemistry, 1973, 12, 3492–3498 CrossRef CAS.
- S. D. Copley and J. R. Knowles, J. Am. Chem. Soc., 1987, 109, 5008–5013 CrossRef CAS.
- P. Kast, C. Grisostomi, I. A. Chen, S. Li, U. Krengel, Y. Xue and D. Hilvert, J. Biol. Chem., 2000, 275, 36832–36838 CrossRef CAS.
- S. G. Sogo, T. S. Widlanski, J. H. Hoare, C. E. Grimshae, G. A. Berchtold and J. R. Knowles, J. Am. Chem. Soc., 1984, 106, 2701–2703 CrossRef CAS.
- R. M. Romero, M. F. Roberts and J. D. Phillipson, Phytochemistry, 1995, 39, 263–276 CrossRef CAS.
- V. Viswanathan, J. Green and B. Nichols, J. Bacteriol., 1995, 177, 5918–5923 CAS.
- J. Liu, N. Quinn, G. A. Berchtold and C. T. Walsh, Biochemistry, 1990, 29, 1417–1425 CrossRef CAS.
- K. T. Ziebart and M. D. Toney, Biochemistry, 2010, 49, 2851–2859 CrossRef CAS.
- O. Kerbarh, E. M. Bulloch, R. J. Payne, T. Sahr, F. Rebeille and C. Abell, Biochem. Soc. Trans., 2005, 33, 763–766 CrossRef CAS.
- D. A. Miller, L. Luo, N. Hillson, T. A. Keating and C. T. Walsh, Chem. Biol., 2002, 9, 333–344 CrossRef CAS.
- B. P. Nichols and J. M. Green, J. Bacteriol., 1992, 174, 5309–5316 CAS.
- M. Siebert, K. Severin and L. Heide, Microbiology, 1994, 140, 897–904 CrossRef CAS.
- M. Siebert, A. Bechthold, M. Melzer, U. May, U. Berger, G. Schroder, J. Schroder, K. Severin and L. Heide, FEBS Lett., 1992, 307, 347–350 CrossRef CAS.
- M. S. DeClue, K. K. Baldridge, D. E. Kunzler, P. Kast and D. Hilvert, J. Am. Chem. Soc., 2005, 127, 15002–15003 CrossRef CAS.
- Z. He, K. D. S. Lavoie, P. A. Bartlett and M. Toney, J. Am. Chem. Soc., 2004, 126, 2378–2385 CrossRef CAS.
- Q. Z. Ye, J. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9391–9395 CrossRef CAS.
- H. G. Floss, J. Nat. Prod., 1996, 69, 158–169 CrossRef.
- H. G. Floss, Nat. Prod. Rep., 1997, 14, 433–452 RSC.
- P. R. August, L. Tang, Y. J. Yoon, S. Ning, R. Muller, T. W. Yu, M. Taylor, D. Hoffmann, C. G. Kim, X. Zhang, C. R. Hutchinson and H. G. Floss, Chem. Biol., 1998, 5, 69–79 CrossRef CAS.
- T.-W. Yu, R. Müller, M. Müller, X. Zhang, G. Draeger, C.-G. Kim, E. Leistner and H. G. Floss, J. Biol. Chem., 2001, 276, 12546–12555 CrossRef CAS.
- H. G. Floss and T.-W. Yu, Chem. Rev., 2005, 105, 621–632 CrossRef CAS.
- J. Guo and J. W. Frost, J. Am. Chem. Soc., 2002, 124, 528–529 CrossRef CAS.
- M. C. Wilson, T. A. M. Gulder, T. Mahmud and B. S. Moore, J. Am. Chem. Soc., 2010, 132, 12757–12765 CrossRef CAS.
- J. McMurry and T. Begley, The Organic Chemistry of Biological Pathways, Roberts & Company, 1995 Search PubMed.
- W. B. Yin, A. Grundmann, J. Cheng and S. M. Li, J. Biol. Chem., 2009, 284, 100–109 CrossRef CAS.
- J. C. Frisvad, C. Rank, K. F. Nielsen and T. O. Larsen, Med. Mycol., 2009, 47, S53–S71 CrossRef CAS.
- B. D. Ames and C. T. Walsh, Biochemistry, 2010, 49, 3351–3365 CrossRef CAS.
- B. D. Ames, X. Gao, S. W. Haynes, Y. Tang and C. T. Walsh, Unpublished observations, 2011 Search PubMed.
- C. Ratledge and L. G. Dover, Annu. Rev. Microbiol., 2000, 54, 881–941 CrossRef CAS.
- R. D. Perry, P. B. Balbo, H. A. Jones, J. D. Fetherston and E. DeMoll, Microbiology, 1999, 145, 1181–1190 CrossRef CAS.
- C. D. Cox, K. L. Rinehart, M. L. Moore and J. C. Cook, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4256–4260 CrossRef CAS.
- B. D. Ames, X. Liu and C. T. Walsh, Biochemistry, 2010, 49, 8564–8576 CrossRef CAS.
- B. D. Ames, S. W. Haynes, X. Gao, B. S. Evans, N. L. Kelleher, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 8756–8769 CrossRef CAS.
- S. W. Haynes, B. D. Ames, X. Gao, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 5668–5679 CrossRef CAS.
- X. Gao, Y. H. Chooi, B. D. Ames, P. Wang, C. T. Walsh and Y. Tang, J. Am. Chem. Soc., 2011, 133, 2729–2741 CrossRef CAS.
- M. L. Kopka, D. S. Goodsell, I. Baikalov, K. Grzeskowiak, D. Cascio and R. E. Dickerson, Biochemistry, 1994, 33, 13593–13610 CrossRef CAS.
- L. A. Masterson, S. J. Croker, T. C. Jenkins, P. W. Howard and D. E. Thurston, Bioorg. Med. Chem. Lett., 2004, 14, 901–904 CrossRef CAS.
- P. G. Baraldi, A. Bovero, F. Fruttarolo, D. Preti, M. A. Tabrizi, M. G. Pavani and R. Romagnoli, Med. Res. Rev., 2004, 24, 475–528 CrossRef CAS.
- W. Li, S. Chou, A. Khullar and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2958–2963 CrossRef CAS.
- L. H. Hurley, Acc. Chem. Res., 1980, 13, 263–269 CrossRef CAS.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869–2878 CrossRef CAS.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS.
- E. Frei, Cancer Chemother. Rep., 1974, 58, 49–54 Search PubMed.
- U. Hollstein, Chem. Rev., 1974, 74, 625–652 CrossRef CAS.
- F. Schauwecker, F. Pfennig, W. Schroder and U. Keller, J. Bacteriol., 1998, 180, 2468–2474 CAS.
- U. Keller and F. Schauwecker, Prog. Nucleic Acid Res. Mol. Biol., 2001, 70, 233–289 CrossRef CAS.
- G. H. Jones, Antimicrob. Agents Chemother., 2000, 44, 1322–1327 CrossRef CAS.
- E. Katz and H. Weissbach, J. Biol. Chem., 1962, 237, 882–886 CAS.
- C. E. Barry, P. G. Nayar and T. P. Begley, Biochemistry, 1989, 28, 6323–6333 CrossRef CAS.
- U. Keller, M. Lang, I. Crnovcic, F. Pfennig and F. Schauwecker, J. Bacteriol., 2010, 192, 2583–2595 CrossRef CAS.
- K. C. Nicolaou, A. L. Smith and E. W. Yue, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5881–5888 CrossRef CAS.
- R. G. Bergman, Acc. Chem. Res., 1973, 6, 25–31 CrossRef CAS.
- S. Lin, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2008, 130, 6616–6623 CrossRef CAS.
- J. L. Hu, Y. C. Xue, M. Y. Xie, R. Zhang, T. Otani, Y. Minami, Y. Yamada and T. Marunaka, J. Antibiot., 1988, 41, 1575–1579 CAS.
- W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef CAS.
- S. G. Van Lanen, S. Lin and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 494–499 CrossRef CAS.
- J. Golik, G. Dubay, G. Groenewold, H. Kawaguchi, M. Konishi, B. Krishnan, H. Ohkuma, K. Saitoh and T. W. Doyle, J. Am. Chem. Soc., 1987, 109, 3462–3464 CrossRef CAS.
- J. P. Coleman, L. L. Hudson, S. L. McKnight, J. M. Farrow, III, M. W. Calfee, C. A. Lindsey and E. C. Pesci, J. Bacteriol., 2008, 190, 1247–1255 CrossRef CAS.
- B. Kunze, G. Hofle and H. Reichenbach, J. Antibiot., 1987, 40, 258–265 CAS.
- G. Höfle and B. Kunze, J. Nat. Prod., 2008, 71, 1843–1849 CrossRef.
- E. Stec, D. Pistorius, R. Muller and S. M. Li, ChemBioChem, 2011, 12, 1724–1730 CrossRef CAS.
- J. Fordos, Recl. Trav. Soc. d'Emulation Sci. Pharm., 1859, 3, 30 Search PubMed.
- J. M. Turner and A. J. Messenger, in Adv. Microb. Physiol., ed. A. H. Rose and D. W. Tempest, Academic Press, 1986, Vol. 27, pp. 211–275 Search PubMed.
- J. B. Laursen and J. Nielsen, Chem. Rev., 2004, 104, 1663–1686 CrossRef CAS.
- D. V. Mavrodi, W. Blankenfeldt and L. S. Thomashow, Annu. Rev. Phytopathol., 2006, 44, 417–445 CrossRef CAS.
- L. Allen, D. H. Dockrell, T. Pattery, D. G. Lee, P. Cornelis, P. G. Hellewell and M. K. B. Whyte, J. Immunol., 2005, 174, 3643–3649 CAS.
- H. Heijerman, J. Cystic Fibrosis, 2005, 4, 3–5 CrossRef CAS.
- M. McDonald, D. V. Mavrodi, L. S. Thomashow and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 9459–9460 CrossRef CAS.
- J. F. Parsons, K. Calabrese, E. Eisenstein and J. E. Ladner, Biochemistry, 2003, 42, 5684–5693 CrossRef CAS.
- W. Blankenfeldt, A. P. Kuzin, T. Skarina, Y. Korniyenko, L. Tong, P. Bayer, P. Janning, L. S. Thomashow and D. V. Mavrodi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16431–16436 CrossRef CAS.
- J. F. Parsons, F. Song, L. Parsons, K. Calabrese, E. Eisenstein and J. E. Ladner, Biochemistry, 2004, 43, 12427–12435 CrossRef CAS.
- E. G. Ahuja, D. V. Mavrodi, L. S. Thomashow and W. Blankenfeldt, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1129–1131 CrossRef.
- E. G. Ahuja, P. Janning, M. Mentel, A. Graebsch, R. Breinbauer, W. Hiller, B. Costisella, L. S. Thomashow, D. V. Mavrodi and W. Blankenfeldt, J. Am. Chem. Soc., 2008, 130, 17053–17061 CrossRef CAS.
- T.-W. Yu, L. Bai, D. Clade, D. Hoffmann, S. Toelzer, K. Q. Trinh, J. Xu, S. J. Moss, E. Leistner and H. G. Floss, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7968–7973 CrossRef CAS.
- W. He, L. Wu, Q. Gao, Y. Du and Y. Wang, Curr. Microbiol., 2006, 52, 197–203 CrossRef CAS.
- T. Schupp, C. Toupet, N. Engel and S. Goff, FEMS Microbiol. Lett., 1998, 159, 201–207 CrossRef CAS.
- H. G. Floss and T. W. Yu, Curr. Opin. Chem. Biol., 1999, 3, 592–597 CrossRef CAS.
- T. W. Yu, Y. Shen, Y. Doi-Katayama, L. Tang, C. Park, B. S. Moore, C. R. Hutchinson and H. G. Floss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9051–9056 CrossRef CAS.
- A. Stratmann, T. Schupp, C. Toupet, W. Schilling, L. Oberer and R. Traber, J. Antibiot., 2002, 55, 396–406 CAS.
- A. Stratmann, C. Toupet, W. Schilling, R. Traber, L. Oberer and T. Schupp, Microbiology, 1999, 145, 3365–3375 CAS.
- D. Kluepfel, S. N. Sehgal and C. Vezina, J. Antibiot., 1970, 23, 75–80 CAS.
- N. Neft and T. M. Farley, Antimicrob. Agents Chemother., 1972, 1, 274–276 CAS.
- K. Shiomi, K. Hatae, H. Hatano, A. Matsumoto, Y. Takahashi, C. L. Jiang, H. Tomoda, S. Kobayashi, H. Tanaka and S. Omura, J. Antibiot., 2005, 58, 74–78 CrossRef CAS.
- Y. Ohnishi, Y. Furusho, T. Higashi, H. K. Chun, K. Furihata, S. Sakuda and S. Horinouchi, J. Antibiot., 2004, 57, 218–223 CAS.
- Y. Ohnishi and S. Horinouchi, Biofilms, 2004, 1, 319–328 CrossRef.
- H. Suzuki, Y. Ohnishi, Y. Furusho, S. Sakuda and S. Horinouchi, J. Biol. Chem., 2006, 281, 36944–36951 CrossRef CAS.
- H. Suzuki, Y. Furusho, T. Higashi, Y. Ohnishi and S. Horinouchi, J. Biol. Chem., 2006, 281, 824–833 CrossRef CAS.
- G. Wolf, J. Worth and H. Achenbach, Arch. Microbiol., 1975, 106, 245–249 CrossRef CAS.
- S. Imai, A. Shimazu, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1990, 43, 1606–1607 CAS.
- N. N. Gerber and M. P. Lechevalier, Biochemistry, 1964, 3, 598–602 CrossRef CAS.
- S. J. Gould, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1996, 118, 9228–9232 CrossRef CAS.
- Z. Rui, K. Petříčková, F. Škanta, S. Pospíšil, Y. Yang, C.-Y. Chen, S.-F. Tsai, H. G. Floss, M. Petříček and T.-W. Yu, J. Biol. Chem., 2010, 285, 24915–24924 CrossRef CAS.
- J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, A. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully and S. B. Singh, Nature, 2006, 441, 358–361 CrossRef CAS.
- J. Wang, S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. L. Colletti, K. Herath, R. Cummings, O. Salazar, I. Gonzalez, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. F. Cully and S. B. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7612–7616 CrossRef CAS.
- M. J. Smanski, Z. Yu, J. Casper, S. Lin, R. M. Peterson, Y. Chen, E. Wendt-Pienkowski, S. R. Rajski and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13498–13503 CrossRef CAS.
- M. Nett and C. Hertweck, J. Nat. Prod., 2011, 74, 2265–2268 CrossRef CAS.
- P. B. Gomes, M. Nett, H.-M. Dahse and C. Hertweck, J. Nat. Prod., 2010, 73, 1461–1464 CrossRef CAS.
- P. B. Gomes, M. Nett, H.-M. Dahse, I. Sattler, K. Martin and C. Hertweck, Eur. J. Org. Chem., 2010, 2010, 231–235 CrossRef.
- Y. Itoh, T. Haneishi, M. Arai, T. Hata, K. Aiba and C. Tamura, J. Antibiot., 1978, 31, 838–846 CAS.
- A. B. Campelo and J. A. Gil, Microbiology, 2002, 148, 51–59 CAS.
- S. Chen, X. Huang, X. Zhou, L. Bai, J. He, K. J. Jeong, S. Y. Lee and Z. Deng, Chem. Biol., 2003, 10, 1065–1076 CrossRef CAS.
- J. F. Martin and J. F. Aparicio, Methods Enzymol., 2009, 459, 215–242 CAS.
- A. Eudes, G. G. Bozzo, J. C. Waller, V. Naponelli, E. K. Lim, D. J. Bowles, J. F. Gregory, 3rd and A. D. Hanson, J. Biol. Chem., 2008, 283, 15451–15459 CrossRef CAS.
- C. Pungaliya, J. Srinivasan, B. W. Fox, R. U. Malik, A. H. Ludewig, P. W. Sternberg and F. C. Schroeder, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7708–7713 CrossRef CAS.
- A. D. Hanson and J. F. Gregory III, Annu. Rev. Plant Biol., 2011, 62, 105–125 CrossRef CAS.
- A. D. Hanson and J. F. Gregory III, Curr. Opin. Plant Biol., 2002, 5, 244–249 CrossRef CAS.
- F. Rebeille, D. Macherel, J.-M. Mouillon, J. Garin and R. Douce, EMBO J., 1997, 16, 947–957 CrossRef CAS.
- W. S. Dallas, J. E. Gowen, P. H. Ray, M. J. Cox and I. K. Dev, J. Bacteriol., 1992, 174, 5961–5970 CAS.
- G. M. Brown, J. Biol. Chem., 1962, 237, 536–540 CAS.
- H. G. Vinnicombe and J. P. Derrick, Biochem. Soc. Trans., 1999, 27, 53–58 CAS.
- W. T. Hughes, S. Kuhn, S. Chaudhary, S. Feldman, M. Verzosa, R. J. A. Aur, C. Pratt and S. L. George, N. Engl. J. Med., 1977, 297, 1419–1426 CrossRef CAS.
- S. Hawser, S. Lociuro and K. Islam, Biochem. Pharmacol., 2006, 71, 941–948 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |