
Bottom-up assembly of recyclable van der Waals-integrated photocatalysts towards efficient photoelectrocatalytic degradation†
Hua-Jun
Chen‡
a,
Zhao-Ming
Lu‡
a,
Yan-Ling
Yang‡
*b,
Xiao-Lei
Shi‡
c,
Jin-Geng
Chen
b,
Ze-Nan
Hu
a,
Bi-Ying
Zhang
a,
Yue-Feng
Chen
b,
Yu
Sun
b and
Zhi-Gang
Chen
 *c
*c
aSchool of Environment and Chemistry, Henan Provincial Engineering Research Centre for Catalytic Degradation and Energy Conservation, Luoyang Institute of Science and Technology, Luoyang, China
bSchool of Materials Science and Engineering, Shaanxi Key Laboratory of Green Preparation and Functionalization for Inorganic Materials, Shaanxi University of Science and Technology, Xi'an, China. E-mail: yangyanling@sust.edu.cn
cSchool of Chemistry and Physics, ARC Research Hub in Zero-emission Power Generation for Carbon Neutrality, and Centre for Materials Science, Queensland University of Technology, Brisbane, Queensland, Australia. E-mail: zhigang.chen@qut.edu.au
First published on 20th February 2025
Abstract
Large-scale synthesis of recyclable photocatalysts for pollutant degradation remains a challenge. Epitaxial growth of powdered photocatalysts on recyclable substrates is constrained by the dedicated materials with highly matched lattices and processing compatibility. Here, we propose a bond-free van der Waals-integrated (vdW-integrated) strategy for the seamless integration of materials with significantly different lattice structures and processing conditions. Amorphous carbon-coated photocatalysts with different dimensions can be physically integrated into recyclable carbon textiles (CTs) through vdW interactions for large-scale synthesis. The amorphous carbon coating effectively broadens the spectral response range and enhances the separation of photo-induced carriers. The recyclable van der Waals-integrated (vdW-integrated) photocatalyst can be employed as a recyclable anode to achieve synergistic degradation of 2,4-dinitrophenol by the combination of electrocatalysis and photocatalysis during the photoelectrocatalysis process. In contrast to conventional powdered photocatalysts, recyclable vdW-integrated catalysts demonstrate superior cycling stability and enhanced catalytic efficiency during both photocatalytic and photoelectrocatalytic processes. This straightforward bottom-up vdW-integrated strategy can be readily extended to assemble zero-dimensional (0D), one-dimensional (1D), or two-dimensional (2D) powdered photocatalysts with flexible CTs, enabling the assembly of recyclable vdW-integrated catalysts for various environment-related applications.
Introduction
In the last few decades, numerous photocatalysts, such as TiO2 microtubes,1,2 SnO2 nanoparticles,3 mesoporous Fe2O3 films,4 Cu2O solid microspheres,5 WO3 nanoparticles,6 Bi2O3 nanospheres,7 semimetal Bi,8 g-C3N4 nanosheets,9,10 and their combinations,11 have been developed to decontaminate pollutants in both liquid and gas phases.12–15 However, the practical application of photocatalytic technology is currently at a nascent stage.16–19 A general problem is that photocatalytic degradation is often hindered by the challenge of recycling powdered photocatalysts from aqueous solutions.8 The growth of photocatalysts on recyclable substrates would be effective in recycling powdered photocatalysts from aqueous solutions. The traditional chemical epitaxial growth method has provided many state-of-the-art photocatalytic devices to address the issue of recycling powdered photocatalysts from aqueous solutions after degradation. Nevertheless, the traditional chemical epitaxial growth of photocatalysts on recyclable substrates for their recycling is confined to materials that exhibit exceptional lattice matching and processing compatibility.18,20 Materials featuring significantly distinct lattice configurations from their substrates are unable to undergo a chemical epitaxial growth process to assemble recyclable photocatalysts. Therefore, it is imperative to seek a facile alternative strategy that is not limited by similar lattice structures and processing conditions to assemble recyclable photocatalytic devices for large-scale synthesis.Recently, it has been reported that the bottom-up van der Waals (vdW)-integrated strategy breaks through the limitations of lattice matching and processing compatibility, and diverse pre-fabricated two-dimensional (2D) materials featuring significantly distinct lattice structures and processing conditions are physically integrated through a bond-free vdW interaction.21–23 For example, mono- and bi-layer graphene devices were laminated onto hexagonal boron nitride (h-BN) substrates in a controlled way, which created a new complex graphene heterostructure.24 Bi2Te3 was deposited on the FeTe surface to fabricate a Bi2Te3/FeTe heterostructure through vdW interactions, which demonstrates superconductivity at the interface.25 The Bi2Te3/FeTe heterostructure provides a novel platform for realizing Majorana fermions. Although vdW integration has been well developed in 2D materials, the scalability of low-dimensional and three-dimensional (3D) materials has hardly been fully explored.21 In theory, beyond the limitation of lattice matching and processing compatibility, the bottom-up vdW-integrated strategy could be readily applied to assemble 0D/3D, 1D/3D, and 2D/3D vdW heterostructures. Therefore, such a bottom-up vdW-integrated strategy can afford an alternative approach to integrating photocatalysts on recyclable substrates to assemble recyclable photocatalytic devices.
In this study, we develop a facile bottom-up vdW-integrated strategy for assembling recyclable vdW-integrated catalysts to recycle powdered catalysts efficiently from aqueous solutions. First, 0D TiO2 nanospheres (P25, a commercial TiO2 nanosphere), 1D TiO2 fibers, and 2D g-C3N4 nanosheets are enveloped in amorphous carbon (AC) to fabricate P25@AC core/shell nanospheres, TiO2@AC core/shell fibers and g-C3N4@AC core/shell nanosheets, respectively. Then the prefabricated amorphous carbon-coated photocatalysts with different dimensions are physically integrated into recyclable vdW-integrated catalysts including P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs through the bottom-up vdW-integrated strategy. This strategy can break the limitations of lattice matching and processing compatibility to physically integrate two highly incompatible materials and create diverse vdW heterojunctions with unexpected functions and performance. Such a synthetic strategy can be easily scalable to integrate other 0D, 1D, or 2D powdered photocatalysts with CTs for assembling recyclable vdW-integrated catalysts that can be recycled from aqueous solutions. When evaluated as a recyclable photocatalytic anode for photoelectrocatalytic decomposition of 2,4-dinitrophenol, the as-prepared vdW-integrated catalysts have demonstrated a superior photoelectrocatalytic efficiency in comparison to the previously reported photoelectrocatalytic systems, which can be efficaciously employed for pollution oxidation processes, thereby significantly boosting the practical applications of photocatalytic and photoelectrocatalytic technologies in environmental fields.
Experimental
Synthesis of P25@AC core/shell nanospheres
A combination of polymerization of dopamine and carbonization was employed to fabricate P25@AC core/shell nanospheres. In a synthesis process, 0.20 g of P25 was ultrasonically dispersed into a prepared 200 mL of trihydroxy methyl-aminomethane solution at a concentration of 0.0100 mol L−1. Subsequently, the mixture underwent 4 hours of magnetic stirring to ensure thorough dissolution of 0.10 g of dopamine. Upon completion of this stage, the products were subjected to centrifugation, where they were sequentially washed with water and ethanol to eliminate impurities and residuals. After drying at 80 °C for 2 hours, the products were heated at 450 °C for 6 hours under an inert argon atmosphere, thereby yielding P25@AC core/shell nanospheres.Synthesis of TiO2@AC core/shell fibers
TiO2 fibers were synthesized using a combination of electrospinning and calcination. In a synthesis process, 1.2 g of polyvinyl pyrrolidone (PVP) was ultrasonically dissolved in 10 mL of ethanol to ensure thorough solvation, then 5 mL of ethanol/acetylacetone/tetrabutyl titanate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, volume ratio) mixed solution was added into the PVP solution with 48 hours of magnetic stirring to obtain a homogeneous solution. Subsequently, electrospinning was carried out on a commercial electrospinning apparatus (DP30, Yunfan Instruments Co., Ltd). The solution was transferred into a syringe with a 0.6 mm inner diameter capillary tip and injected using a micro-injection pump at a rate of 0.3 mL h−1. A high voltage of 18 kV was employed during electrospinning. The products were subjected to calcination in a muffle furnace at a temperature of 500 °C for 4 hours to ultimately yield TiO2 fibers.
:1:1, volume ratio) mixed solution was added into the PVP solution with 48 hours of magnetic stirring to obtain a homogeneous solution. Subsequently, electrospinning was carried out on a commercial electrospinning apparatus (DP30, Yunfan Instruments Co., Ltd). The solution was transferred into a syringe with a 0.6 mm inner diameter capillary tip and injected using a micro-injection pump at a rate of 0.3 mL h−1. A high voltage of 18 kV was employed during electrospinning. The products were subjected to calcination in a muffle furnace at a temperature of 500 °C for 4 hours to ultimately yield TiO2 fibers.
Subsequently, 0.20 g of TiO2 fibers was ultrasonically dispersed into 200 mL of trihydroxy methyl-aminomethane solution at a concentration of 10 mmol L−1. The mixture underwent 4 hours of magnetic stirring to ensure thorough dissolution of 0.10 g of dopamine. Then the products were subjected to centrifugation, where they were sequentially washed with water and ethanol to eliminate impurities and residuals. After drying at 80 °C for 2 hours, the products underwent carbonization at a temperature of 450 °C for 6 hours under an inert argon atmosphere, thereby successfully yielding TiO2@AC core/shell fibers.
Synthesis of g-C3N4@AC core/shell nanosheets
Polymerization of urea with the aid of water was employed to prepare g-C3N4@AC core/shell nanosheets. First, 6.00 g of urea and 30 mL of water were added to an alumina crucible. The crucible was sealed with foil and heated at a temperature of 550 °C for 2 hours, with the heating process being incrementally regulated at a consistent rate of 10 °C min−1. The polymerization products were ground into powder to successfully yield g-C3N4@ nanosheets.Afterward, 0.20 grams of g-C3N4 nanosheets were ultrasonically dispersed into a 200 mL solution containing trihydroxy methyl-aminomethane at a concentration of 10 mmol L−1, and then 0.10 g of dopamine was dissolved into the mixture with 4 hours of continuous magnetic stirring. Upon completion of this process, the resulting products were subjected to centrifugation for effective separation, where they were sequentially washed with water and ethanol to eliminate impurities and residuals. After drying at 80 °C for 2 hours, the products were carbonized at 450 °C for an extended duration of 6 hours under an inert argon atmosphere, thereby successfully yielding g-C3N4@AC core/shell nanosheets.
Assembly of vdW heterostructures
The procedures for the fabrication of P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs are illustrated in Fig. 1. 0.0100 g of freshly synthesized core/shell structural photocatalysts was ultrasonically dispersed into 100 mL of water. Subsequently, CTs measuring 2.5 cm in length, 5.0 cm in width, and 0.1 cm in thickness were immersed into the suspensions for 2 hours under magnetic stirring. During this stirring process, the above-mentioned core/shell structural photocatalysts were effectively assembled onto CTs through the vdW-integrated strategy. This strategic integration led to the assembly of recyclable P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs, respectively, each embodying a unique synergy between their components. | ||
| Fig. 1 Schematic illustration of a facile bottom-up vdW-integrated strategy of P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs. | ||
Morphology/microstructure characterization
An X-ray diffraction instrument (XRD, D8 Focus) was used to elucidate the crystalline architecture of photocatalysts. Utilizing state-of-the-art microscopy technology, a field-emission scanning electron microscope (FESEM, Sigma HD) and a transmission electron microscope (TEM, Tecnai G2 F20) were further employed to delve deeper into morphological and structural intricacies of photocatalysts. Moreover, to assess the critical optical absorption properties of photocatalysts, an advanced ultraviolet-visible diffuse reflection spectrometer (UV-vis DRS, UV-3600) was utilized to meticulously investigate and elucidate photocatalytic efficiency under the ultraviolet-visible spectrum. The photoluminescence spectrum (PL spectra, F-7000) was obtained to analyze the separation efficiency of photo-induced electron/hole.Catalytic experiments
The catalytic experiment of powdered photocatalysts was meticulously executed under the precise irradiation of a high-intensity xenon light source. In a typical experimental setup, precisely 0.0100 g of powdered photocatalysts was introduced into 100 mL of a 2,4-dinitrophenol solution at a concentration of 20 mg L−1. To ensure that thorough adsorption–desorption equilibrium was achieved, the mixture was dispersed for a period of 15 min. Subsequently, a precisely focused beam of xenon light was vertically oriented and masterfully directed from an elevated position onto the suspension below while it was being continuously stirred magnetically. The concentration of the 2,4-dinitrophenol solution (ct) was meticulously measured and utilized to calculate the oxidation rate using eqn (1). | (1) |
In the photocatalytic degradation process of 2,4-dinitrophenol, the first-order kinetic equation was judiciously employed to accurately quantify the rate constant (k). This pivotal step enables us to delve deeper into the dynamics and efficiency of catalytic oxidation.
 | (2) |
The photoelectrocatalytic process utilizing recyclable vdW-integrated catalysts was executed in a cylindrical reactor. Inside this vessel, a 100 mL solution containing 2,4-dinitrophenol and Na2SO4 at a concentration of 10 and 1.0 mg L−1, respectively, was prepared. A rectangular piece of the recyclable vdW-integrated photocatalyst (25 mm × 50 mm) served as the anode, while a stainless steel mesh electrode of equivalent dimensions acted as the cathode. Both were positioned horizontally within the reactor to ensure optimal interaction with the solution.
Following a 15 min period of dispersion to attain the adsorption–desorption equilibrium, recyclable vdW-integrated catalysts were subjected to vertical irradiation from a xenon light source. Simultaneously, an external bias voltage was applied to inhibit the recombination of photo-induced electron/hole. Throughout the photoelectrocatalytic procedure, the degradation rate and the kinetic behavior of 2,4-dinitrophenol were quantified using eqn (1) and (2), respectively, providing profound insights into the reaction mechanism.
Results and discussion
During the process of synthesis, amorphous carbon was deposited onto commercial TiO2 nanospheres to create P25@AC core/shell nanospheres through an innovative combination of dopamine polymerization and subsequent carbonization. The X-ray diffraction (XRD) patterns, which illustrate the structural transformation, are elegantly presented in Fig. 2a. As evidenced by the XRD analysis, both samples essentially consist of anatase and rutile forms. Notably, the distinct diffraction peaks observed at 25.4°, 37.8°, 48.1°, 54.0°, and 55.2° can be confidently attributed to the (101), (004), (200), (105), and (211) crystal planes respectively, which are distinctive features of the anatase TiO2 phase.22 Meanwhile, the indexing process reveals that the diffraction patterns appearing at 27.4°, 36.1°, 41.2°, and 54.3° correspond precisely with the (110), (101), (111), and (211) crystal planes, respectively, which are exclusive signatures of the rutile TiO2 phase. In summary, the detailed diffraction analysis distinctly identifies the coexistence of both anatase and rutile phases in the TiO2 sample, each phase contributing its characteristic peaks that align perfectly with their corresponding well-known crystallographic planes. However, a notable observation is that no discernible carbon peak emerges in the XRD patterns associated with the P25@AC core/shell nanospheres. This intriguing absence serves as compelling evidence that the P25 nanospheres are effectively encapsulated by an amorphous carbon layer. | ||
| Fig. 2 (a and b) XRD patterns (a) and FESEM images (b) of P25@AC core/shell nanospheres. (c–e) Elemental mapping (c), TEM image (d), and HRTEM analysis (e) of P25@AC core/shell nanospheres. (f) UV-vis DRS spectra of P25 nanospheres and P25@AC core/shell nanospheres. (g) PL spectra of P25 nanospheres and P25@AC core/shell nanospheres. (h–j) The variance in electron density of P25@AC core/shell nanospheres (h), an adsorptive H2O molecule on P25@AC core/shell nanospheres (i), and an adsorptive O2 molecule on P25@AC core/shell nanospheres (j). The blue zone and red zone respectively symbolize the domains of electron depletion and enrichment. | ||
Fig. 2b shows the FESEM image, depicting the structure of P25@AC core/shell nanospheres. Numerous P25 nanospheres are wrapped together in an amorphous carbon shell. In Fig. 2c, an elemental mapping analysis of the P25@AC core/shell nanospheres is depicted, manifesting the coexistence of carbon, oxygen, and titanium elements within their composition. The TEM image elucidates the intricate structure of these P25@AC core/shell nanospheres. As evidenced by Fig. 2d, the P25 nanospheres are encapsulated by a layer of amorphous carbon, thereby demonstrating a core/shell architecture. Notably, the thickness of this amorphous carbon shell has been measured to be 3.1 nm. In Fig. 2e, the HRTEM image shows the P25@AC core/shell nanospheres, wherein an intricate lattice fringe of 3.5 Å corresponds to the (101) crystal plane of the anatase TiO2 phase. Concurrently, an additional lattice fringe, precisely measured at 3.2 Å, provides compelling evidence for the presence of the (110) crystal plane, which is an intrinsic feature of the rutile phase within the TiO2 structure.26 The HRTEM image thus confirms the coexistence and distinct crystalline attributes of both anatase and rutile phases in P25@AC core/shell nanospheres. Notably, no discernible lattice fringes indicative of a carbon layer are observed within the HRTEM image, which aligns harmoniously with the outcomes gleaned from the X-ray diffraction (XRD) patterns. This finding further substantiates the encapsulation of P25 by the amorphous carbon, thereby confirming the successful synthesis of the P25@AC core/shell nanostructure.
The carbon layer was further analyzed using Raman spectroscopy. As shown in Fig. S1,† the D-band and G-band confirm the formation of the carbon layer. The G-band at 1596 cm−1, characteristic of graphitic carbon, indicates the presence of sp2 carbon structures within the carbon layer of P25@AC core/shell nanospheres. The D-band at approximately 1341 cm−1 is associated with defective carbon in the layer. The intensity ratio of the D-band to the G-band reflects the degree of graphitization. The comparable intensities of the G-band and D-band suggest the presence of structural imperfections in the carbon layer of P25@AC core/shell nanospheres.
Fig. 2f compares the optical absorption efficiency of P25 nanospheres to that of P25@AC core/shell nanospheres, which demonstrate a superior capacity for absorbing UV-visible light in comparison to P25 nanospheres. This observation underscores the pivotal role of amorphous carbon in effectively broadening the spectral response scope of the P25 core, thereby augmenting its capability to harness a wider spectrum of UV-visible light.27 Additionally, the spectral absorption characteristics of both P25 nanospheres and P25@AC core/shell nanospheres were assessed by employing the intercept methodology. This approach allowed for a comprehensive evaluation of their respective absorption spectral ranges, thereby shedding light on the intrinsic optical properties of P25@AC core/shell nanospheres, as well as standalone P25 nanospheres. Fig. 2f vividly depicts that the λ values for P25 nanospheres and P25@AC core/shell nanospheres are measured at 391 nm and 771 nm, respectively. This compelling evidence reveals that the P25@AC core/shell nanospheres exhibit an enhanced spectral response range compared to P25. The P25@AC core/shell nanospheres exhibit an exceptional ability to be activated by visible light energy, a trait that significantly enhances their performance. The superior optical attributes of these nanospheres can be traced back to their distinctive formation of a heterostructure, which sets them apart in the realm of advanced materials. The creation of this P25@AC interface leads to a significant narrowing of the band gap, thereby augmenting its photoreactivity. In essence, the P25@AC core/shell nanospheres transcend the limitations of conventional P25, broadening the scope of light absorption and harnessing a more extensive spectrum of solar energy.
Mott–Schottky plots are useful for estimating the energy band structure of photocatalysts. As shown in Fig. S2,† the conduction band energy (Ec) of P25 and TiO2@AC core–shell nanospheres is approximately −0.56 and −0.23 eV, respectively. Since the conduction band potential of TiO2@AC core–shell nanospheres is more negative than the redox potential of ˙O2−/O2, adsorbed O2 molecules can be reduced to ˙O2− by photogenerated electrons in the conduction band of the TiO2@AC core–shell nanospheres.
To evaluate the separation efficiency of photo-induced electron/hole, the PL spectra of P25@AC core/shell nanospheres and P25 nanospheres excited by the irradiation of 223 nm UV light are shown in Fig. 2g. Compared to P25 nanospheres, the distinct dual peaks at 480 nm and 580 nm, characteristic of P25 nanospheres, are significantly suppressed in P25@AC core/shell nanospheres due to the encapsulation by the amorphous carbon shell. This quenching effect significantly amplifies the separation efficiency of photo-induced electron/hole within the P25@AC core/shell nanospheres. The conduction band (CB) of the P25 nanospheres exhibits a notably lower energy level than that of the Fermi level inherent to amorphous carbon.27 The lower energy level enables amorphous carbon to effectively act as a reservoir for photo-induced electrons originating from the CB of the P25 nanospheres. Consequently, amorphous carbon exhibits remarkable capability of encapsulating photo-induced electrons within the CB of P25 nanospheres. This unique electron trapping mechanism initiates a nonradiative decay process in the P25@AC core/shell nanospheres. This process effectively quenches the distinctive spectral peaks associated with the P25 nanospheres, thereby altering their photophysical properties.
Further insights into electron delivery in the P25@AC core/shell nanosphere were garnered through Density Functional Theory (DFT) calculations. Fig. 2h provides compelling evidence that electrons migrate from the CB of P25 towards amorphous carbon. Moreover, Fig. 2i reveals that the adsorption energy of an H2O molecule onto the surface of P25 reaches a substantial value of 0.91 eV. This high adsorption energy signifies that the bonding of the H2O molecule to P25 is stable chemisorption rather than weak physisorption, thereby underlining the potential reactivity of this interface. Furthermore, the adsorbed H2O molecule resides within the electron-depleted region, thereby suggesting that the adsorbed H2O molecule undergoes oxidation and transfers into ˙OH radicals. Fig. 2j depicts that the computed adsorption energy of an O2 molecule onto an amorphous carbon substrate reaches as low as 0.38 eV. This compelling evidence suggests that the interaction between the O2 molecule and the amorphous carbon surface is governed by a physical adsorption mechanism rather than chemical bond formation. The O2 molecule thus adheres to the substrate surface without undergoing any significant electron sharing or structural rearrangement characteristic of chemical bonding, affirming the predominantly physical nature of this adsorption process. Furthermore, the adsorbed O2 molecule is nestled within the electron-enriched domain, suggesting that there exists a profound inclination for the adsorbed O2 molecule to undergo reduction into ˙O2− on the surface of amorphous carbon, thereby shedding light on a critical mechanism. This mechanism divulges how photo-induced electrons are effectively transferred. It further illuminates the intricate formation pathways of both ˙OH and ˙O2− radicals.
To better understand the charge transfer pathway, in situ X-ray photoelectron spectroscopy (XPS) was performed. As shown in Fig. S3,† the elements Ti, O, and C were detected in the survey XPS spectrum of P25@AC core–shell nanospheres, confirming the coexistence of P25 and carbon. For comparison, survey XPS spectra of amorphous carbon and commercial P25 were also analyzed. Under dark conditions, the characteristic peaks of P25 were observed at 458.8, 464.5, and 529.8 eV, corresponding to Ti 2p3/2, Ti 2p1/2, and the Ti–O bond, respectively (Fig. S4 and S5†). In Fig. S6,† the C 1s peak of amorphous carbon appeared at 284.68 eV. Interestingly, in the P25@AC core–shell nanospheres under dark conditions, the Ti 2p and O 1s peaks shifted to lower binding energies compared to those of P25, while the C 1s peak shifted positively relative to amorphous carbon. Since binding energy is inversely correlated with surface electron density, these results indicate that electrons from amorphous carbon accumulate in P25 after the formation of the P25@AC core–shell nanospheres. Under light irradiation, the binding energies of the typical peaks in the P25@AC core–shell nanospheres changed again. Compared to the dark conditions, the Ti 2p and O 1s peaks shifted positively, while the C 1s peak shifted negatively. These shifts confirm that photoexcited electrons migrate from P25 to amorphous carbon under light illumination, consistent with the results of DFT calculations.
Fig. 3a vividly portrays the structure of CTs. Evidently, the diameter of an individual fiber measures approximately 11 μm, which contributes to the unique three-dimensional architecture of CTs. This inherent design offers an expansive specific surface area that is highly conducive to fostering the efficient assembly of active photocatalysts, thereby enhancing their overall performance and functionality. To ensure the efficient and sustainable reuse of P25@AC core/shell nanospheres after use, they are physically bound to CTs for the fabrication of P25@AC/CTs, thereby integrating the benefits of both components into a synergistic whole. Fig. 3b illustrates the intricate structure of P25@AC/CTs, where a uniform dispersion and seamless adsorption of P25@AC core/shell nanospheres onto the surface of CTs can be observed. This SEM image clearly demonstrates the even distribution and strong interaction between the components. Fig. 3c–e portray a microcosmic perspective with SEM images of an individual carbon fiber. Evidently, P25@AC core/shell nanospheres form a robust bond with the carbon fiber surface through vdW forces. The designed nanostructures demonstrate an exceptional affinity to adhere to the carbon fiber surface, leveraging the inherent strength of the vdW interactions for a steadfast attachment. It is noteworthy that under neutral pH conditions, the zeta potential measurements reveal a positive charge for the P25@AC core/shell nanospheres.28 Consequently, P25@AC core/shell nanospheres exhibit a strong affinity towards negatively charged CTs, leading to their preferential adsorption. The photograph illustrates the P25@AC/CTs in bending and twisting states (Fig. 3f), demonstrating the extensive application of flexible P25@AC/CTs in a multitude of environmentally relevant fields.
 | ||
| Fig. 3 (a and b) FESEM images of CTs (a) and P25@AC/CTs (b). (c–e) FESEM images of the surface of the single carbon fiber. (f) The photograph of P25@AC/CTs in bending and twisting states. (g) FT-IR spectrum of P25@AC/CTs. (h and i) The comparison of Nyquist plots (h) and photocurrent spectra (i) between P25 nanospheres and P25@AC/CTs. | ||
Fig. 3g represents the FT-IR spectrum of the recyclable P25@AC/CT. Notably, the distinctive peak of stretching vibration corresponding to the Ti–O bond exhibits a resonant frequency of around 520 cm−1, signifying its unique molecular interaction. Moreover, a closer examination of the spectrum reveals three distinctive absorption peaks: one appearing at 1217 cm−1, which can be attributed to the stretching vibrations of C–O–C; an alternative peak at a wavenumber of 1726 cm−1, corresponding to –COOH; and finally, a peak at 2984 cm−1 that signifies the stretching vibrations of aliphatic C–H bonds.29 No discernible absorption peak can be ascribed to the Ti–O–C bond, which strongly indicates that the P25 nanospheres are interconnected with CTs through van der Waals interactions rather than covalent bonding.23 This absence of a characteristic spectral feature testifies to the non-chemical nature of the binding between these two components, thus underscoring the pivotal role of vdW forces in their interaction and assembly.
The intricate interplay between photoelectrochemical attributes is profoundly interconnected with the separation efficiency of photo-induced electron/hole.30,31 Consequently, to compare the efficiency in separating photo-induced electron/hole between P25 nanospheres and P25@AC/CTs, photocurrent spectra and Nyquist plots were meticulously obtained and analyzed. This comparative analysis serves as a powerful tool to discern and evaluate the disparities in their respective capabilities to separate and transport photo-induced electron/hole effectively. Fig. 3h presents the Nyquist plots for P25 nanospheres and P25@AC/CTs. These plots illustrate the radius of the Nyquist curve, which serves as an insightful indicator of the charge transfer resistance occurring at the interface of the electrode/electrolyte. Furthermore, it subtly unveils the separation efficiency of photo-induced electron/hole, thereby offering a comprehensive understanding of electrochemical performance of the materials. In essence, the smaller the radius of a material, the more facile the transfer of charge across the interface of the electrode/electrolyte becomes. In the case of P25@AC/CTs, its radius is notably diminutive compared to that of P25 nanospheres. This unique attribute serves as a cornerstone for enhanced performance. The charge transfer resistance (Rct) is a key indicator of charge transfer efficiency. For P25@AC/CTs, the Rct value was determined to be 8.17 Ω based on fitting to the Randles equivalent circuit model, which is significantly lower than that of P25 nanospheres. This reduced Rct value demonstrates the improved ability of P25@AC/CTs to enable rapid electron transfer, highlighting its superior photoelectrocatalytic activity. The assembly of P25@AC core/shell nanospheres onto CTs brings about a multitude of advantages. This synergistic fusion notably optimizes the charge transfer process across the electrode/electrolyte interface, thereby facilitating an efficient movement of photo-induced electron/hole. Moreover, this innovative combination effectively promotes the separation efficiency of photo-induced electron/hole, which is pivotal in augmenting photocatalytic efficiency. In essence, a relatively low Rct value of P25@AC/CTs coupled with its synergistic design not only lowers the resistance faced by transferring charges but also amplifies the photocatalytic efficiency through improved charge separation and transport dynamics. The photocurrent spectrum is vividly depicted in Fig. 3i. It is evident from the graph that the photocurrent density generated by the P25@AC/CTs surpasses that of the P25 nanospheres by nearly a factor of 1.6. This significant enhancement serves as compelling evidence that the innovative assembly constitutes an efficacious approach to inhibit the recombination efficiency of photo-induced electron/hole. Consequently, the innovative assembly yields a remarkable enhancement in the overall photocatalytic efficiency, underscoring its potential as a promising candidate for advanced photocatalytic applications.
To enhance the functionality and properties of TiO2 fibers, a combination of polymerization of dopamine and carbonization was employed to envelop TiO2 fibers with an amorphous carbon layer, thereby meticulously crafting TiO2@AC core/shell fibers. Fig. 4a presents the XRD patterns that depict the crystalline structures of both TiO2 fibers and TiO2@AC core/shell fibers. The results of XRD patterns indicate that both TiO2 fibers and TiO2@AC core/shell fibers comprise the anatase and rutile phases. XRD patterns indicate that the diffraction peaks corresponding to carbon are hardly found owing to the amorphous nature of the carbon coating.32Fig. 4b showcases a representative FESEM image of TiO2@AC core/shell fibers. The image thus provides compelling evidence of the successful synthesis of these fibers, which uniformly encapsulate a TiO2 core within an amorphous carbon shell, thereby endowing them with remarkable homogeneity and dispersibility throughout the material. Fig. 4c presents a typical TEM image of TiO2@AC core/shell fibers, in which the products are pictured as being made up of many small particles. TiO2 fibers are encapsulated within an amorphous carbon layer, thus TiO2@AC core/shell fibers are successfully synthesized. The thickness of the amorphous carbon coating is measured around 2 nm. In Fig. 4d, the HRTEM image vividly exhibits the structural details of TiO2@AC core/shell fibers. This image reveals a lattice fringe measuring 3.5 Å, which can be confidently attributed to the (101) crystalline plane of the anatase TiO2 phase. Concurrently, a lattice fringe with a dimension of 3.2 Å is discernible and aligns with the (110) crystal plane of the rutile TiO2 phase. Notably, the HRTEM image does not exhibit any discernible lattice fringes representative of the carbon coating layer. This observation aligns seamlessly with the outcomes gleaned from the XRD patterns, thereby substantiating the amorphous nature of the carbon coating on the TiO2@AC core/shell fibers. In Fig. 4e, a compelling UV-visible DRS comparison is depicted between TiO2 fibers and TiO2@AC core/shell fibers. Fig. 4e reveals that the TiO2@AC core/shell fibers manifest an enhanced capability in absorbing UV-visible light compared to TiO2 fibers. This observation serves as compelling evidence that the incorporation of amorphous carbon significantly boosts the efficiency of UV-visible light harvesting in TiO2 fibers, thereby underlining its pivotal role in enhancing the overall photocatalytic efficiency.33 Additionally, the λ values for TiO2 fibers and TiO2@AC core/shell fibers have been estimated using the intercept method, revealing a noteworthy disparity. Specifically, the calculated λ for TiO2 fibers is 401 nm, while TiO2@AC core/shell fibers exhibit an enhanced λ value of 940 nm, respectively. The outcomes clearly demonstrate that TiO2@AC core/shell fibers can be excited using visible, ultraviolet, and near-infrared light.
 | ||
| Fig. 4 (a) XRD patterns of TiO2 fibers and TiO2@AC core/shell fibers. (b–d) SEM, TEM, and HRTEM images of TiO2@AC core/shell fibers. (e and f) UV-vis DRS and PL spectraof TiO2 fibers and TiO2@AC core/shell fibers. (g) SEM image of TiO2@AC/CTs. (h and i) The comparison of photocurrent spectra (h) and Nyquist plots (i) between TiO2 fibers and TiO2@AC/CTs. | ||
In Fig. 4f, the two distinct peaks at 480 and 580 nm, characteristic of TiO2 fibers, are significantly suppressed in the TiO2@AC core/shell fibers. This significant quenching effect can be attributed to the encapsulation by amorphous carbon, which effectively suppresses the PL properties when compared with the standalone TiO2 fibers. Therefore, TiO2@AC core/shell fibers exhibit a significantly improved capability in separating photo-induced electron/hole. The CB of the TiO2 fibers is significantly beneath the Fermi level of the amorphous carbon substrate.27 This sophisticated design thus propels amorphous carbon to trap photo-induced electrons within the CB of TiO2 fibers, thereby enhancing the overall separation efficiency. The process of electron trapping leads to a non-radiative decay within TiO2@AC core/shell fibers, effectively diminishing the distinctive peaks of TiO2 fibers. Leveraging a bottom-up vdW-integrated approach, these TiO2@AC core/shell fibers are meticulously adsorbed onto CTs through physical interaction, thereby assembling recyclable TiO2@AC/CTs, which can be efficiently retrieved and recycled from aqueous solutions. Fig. 4g presents an SEM image of TiO2@AC/CTs. Herein, TiO2@AC core/shell fibers are observed to be uniformly dispersed and firmly anchored onto the outer surface of the carbon fiber through vdW interaction. This results in an intimate interfacial bonding that ensures the effective integration and stability of TiO2@AC/CTs.
In Fig. 4h, a compelling photocurrent spectrum comparison is depicted between TiO2 fibers and TiO2@AC/CTs. As shown in Fig. 4h, the photocurrent density demonstrated by TiO2@AC/CTs exhibits a notably superior performance, approximately 3.5 times more substantial than that of TiO2 fibers, thereby signifying that our bottom-up vdW-integrated strategy serves as an efficacious approach to enhance the rate of separation performance of photo-induced electron/hole. This, in turn, significantly boosts the overall photocatalytic efficiency, validating the effectiveness of our approach. In Fig. 4i, a compelling Nyquist plot comparison is depicted between TiO2 fibers and TiO2@AC/CTs. Fig. 4i shows that the Rct values for TiO2@AC/CTs and TiO2 fibers were determined to be 4.43 and 8.31 Ω, respectively, based on fitting to the Randles equivalent circuit model. The significantly lower Rct value of TiO2@AC/CTs compared to TiO2 fibers indicates more efficient charge transfer and separation, demonstrating the superior electrochemical performance of TiO2@AC/CTs.
A combination of polymerization of dopamine and carbonization was employed to envelop g-C3N4 nanosheets with an amorphous carbon layer, thereby meticulously crafting g-C3N4@AC core/shell nanosheets. Fig. 5a depicts a comparison of the photocurrent spectra between g-C3N4 nanosheets and g-C3N4@AC core/shell nanosheets. The diffraction patterns manifested at 13.6° and 27.6° are meticulously indexed, revealing the presence of tri-s-triazine units (100) and conjugated aromatic systems (002), thereby through a process of thermal polymerization, g-C3N4 nanosheets were fabricated through the utilization of urea as the primary precursor, with water serving as an essential auxiliary agent in this innovative synthesis approach. The diffraction peaks of the carbon coating are notably elusive in XRD patterns of g-C3N4@AC core/shell nanosheets owing to the non-crystalline structure of the carbon coating. Fig. 5b and c present typical FESEM images of g-C3N4 and g-C3N4@AC core/shell nanosheets with wrinkles, showing a nanosheet similar to pristine g-C3N4 nanosheets. The TEM image indicates that g-C3N4@AC core/shell nanosheets are made up of thick and dense nanosheets (Fig. 5d). Combination of XRD, FESEM, and TEM results shows that amorphous carbon is enveloped on g-C3N4 nanosheets, thereby confirming that the g-C3N4@AC core/shell nanosheets are successfully synthesized.
 | ||
| Fig. 5 (a) An in-depth comparison of XRD patterns between g-C3N4 nanosheets and g-C3N4@AC core/shell nanosheets. (b and c) The vivid comparison of SEM images between g-C3N4 nanosheets (b) and g-C3N4@AC core/shell nanosheets (c). (d) TEM image of g-C3N4@AC core/shell nanosheets. (e and f) The comparison of UV-vis DRS (e) and PL spectra (f) between g-C3N4 nanosheets and g-C3N4@AC core/shell nanosheets. (g) SEM image of g-C3N4@AC/CTs. (h and i) An in-depth comparison of Nyquist plots (h) and photocurrent spectra (i) between g-C3N4 and g-C3N4@AC/CTs. | ||
Fig. 5e depicts a comparison of UV-vis DRS between g-C3N4 nanosheets and g-C3N4@AC core/shell nanosheets. The unique architecture of the g-C3N4@AC core/shell nanosheets plays a pivotal role in enhancing their light-harvesting properties. The synergy between g-C3N4 and amorphous carbon creates efficient absorption of UV-vis light.33 This results in a superior light absorption capacity in comparison with g-C3N4 nanosheets alone. The estimated λ values for individual g-C3N4 nanosheets and innovative g-C3N4@AC core/shell nanosheets, as derived through the intercept method, are respectively 461 nm and 473 nm. These findings indicate that the optical bandgap of g-C3N4 nanosheets is slightly narrower than that of g-C3N4@AC core/shell nanosheets, manifesting a wider spectral response range of g-C3N4@AC core/shell nanosheets.
As shown in Fig. 5f, g-C3N4 nanosheets exhibit higher PL intensity than g-C3N4@AC core/shell nanosheets. The characteristic peaks at 464 nm is significantly weakened in the presence of amorphous carbon, indicating that amorphous carbon with its lower Fermi level can capture photo-induced electrons and enhance the separation of photo-induced electron/hole.27 Therefore, the lower PL intensity of g-C3N4@AC core/shell nanosheets is ascribed to the migration of photo-induced electrons within the CB of g-C3N4 nanosheets towards a lower Fermi level of amorphous carbon rather than nonradiative recombination. Subsequently, g-C3N4@AC core/shell nanosheets are physically assembled onto CTs through a bottom-up vdW-integrated strategy to assemble recyclable g-C3N4@AC/CTs. Fig. 5g shows an SEM image of g-C3N4@AC/CTs, from which it can be observed that g-C3N4@AC core/shell fibers are distributed evenly and tightly on the outer surface of CTs. Fig. 5h shows that the Rct values for g-C3N4@AC/CTs and g-C3N4 nanosheets were determined to be 8.25 and 11.31 Ω, respectively, based on fitting to the Randles equivalent circuit model. The lower Rct value of g-C3N4@AC/CTs indicates more efficient transfer and separation of photoinduced electron–hole pairs compared to that of g-C3N4 nanosheets. Fig. 5i shows an in-depth comparison of photocurrent spectra between g-C3N4 and g-C3N4@AC/CTs; the photocurrent density demonstrated by g-C3N4@AC/CTs exhibits a notably superior performance, approximately 5 times more substantial than that of g-C3N4 nanosheets, indicating that the innovative assembly can effectively inhibit the recombination efficiency of photo-induced electron/hole within g-C3N4 nanosheets.
The catalytic performance of P25@AC/CTs is first examined through the oxidative degradation of 2,4-dinitrophenol. An astute observation is depicted in Fig. 6a, where the concentration of 2,4-dinitrophenol exhibits a subtle yet significant decrease during the preliminary adsorption (−15 to 0 min). However, P25@AC/CTs exhibit remarkable efficiency during photoelectrocatalytic degradation, successfully eradicating 100% of 2,4-dinitrophenol within a mere 60 min. In contrast, when subjected to standalone photocatalysis and electrocatalysis processes using the same P25@AC/CTs, only 35.0% and 24.4% of 2,4-dinitrophenol were decomposed respectively. This significant disparity underscores the synergistic interplay between photocatalysis and electrocatalysis mechanisms during the process of photoelectrocatalysis, thereby amplifying its effectiveness. Kinetic studies give further insight into cooperative interaction. As shown in Fig. 6d, the catalytic oxidation process of 2,4-dinitrophenol adheres to the principles of the first-order kinetic equation. Remarkably, the kinetic constant in the context of photoelectrocatalysis exhibits an impressive augmentation, estimated to be approximately 13.2 and 20.1 times higher than its counterparts in photocatalysis and electrocatalysis respectively. This significant escalation can be attributed to the innovative electrochemical-assisted technique that incorporates a positive bias, which effectively boosts the separation of photo-induced electron/hole, thereby enhancing the overall efficiency and performance of the system.
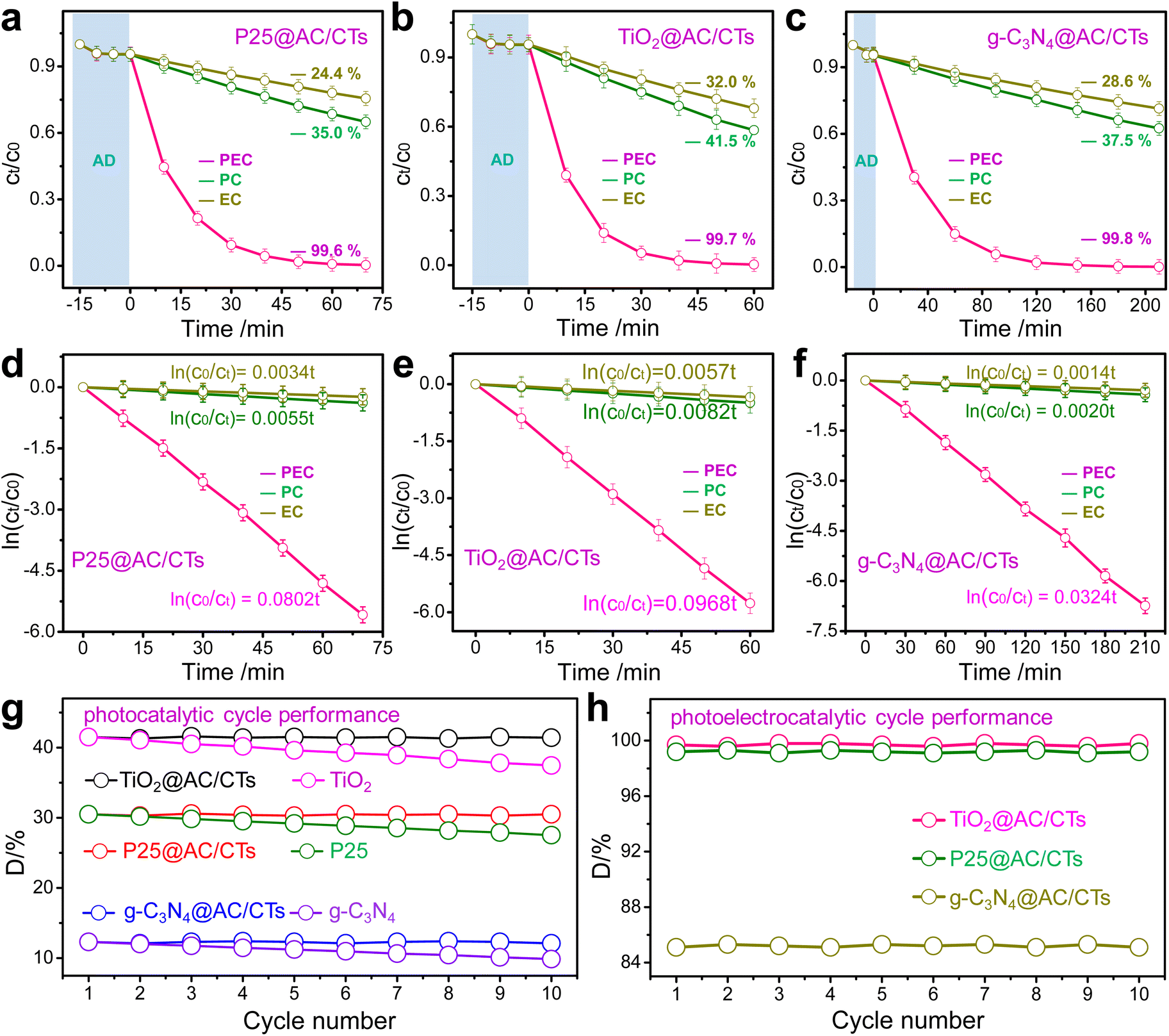 | ||
| Fig. 6 (a–f) The catalytic efficiency and kinetics of P25@AC/CTs (a and d), TiO2@AC/CTs (b and e), and g-C3N4@AC/CTs (c and f). (g) The photocatalytic cycling performance of conventional powdered photocatalysts and vdW-integrated catalysts, and the time for a single cycle is 60 min. (h) The cycling performance of P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs in the process of photoelectrocatalysis. | ||
In P25@AC core/shell nanospheres, the transfer phenomenon of photo-induced electrons occurs from the P25 nanospheres to the amorphous carbon layer because of the lower Fermi level of amorphous carbon.27 Moreover, the electrochemical-assisted technique holds the potential to significantly augment the process of segregating photo-induced electron/hole, thereby achieving an even higher degree of efficiency in the separation process of photo-induced electron/hole. Therefore, photo-induced electrons undergo a seamless transfer from P25@AC/CTs to stainless steel mesh through an external circuit in the presence of positive bias, leaving holes in the VB of P25. The reported DFT calculations show that H2O molecules can be chemically adsorbed on the (101) crystal faces of P25 nanospheres,23 and then oxidized to ˙OH by holes in the VB of P25 nanospheres. Simultaneously, photo-induced electrons transfer to the stainless steel mesh and reduce dissolved oxygen to ˙O2−. ˙OH and ˙O2− serve as reactive oxygen species (ROS) during photoelectrocatalysis of 2,4-dinitrophenol.34 The cooperative interaction between electrocatalysis and photocatalysis indicates that the electrochemical-assisted technique plays an indispensable role in the separation of photo-induced electron/hole than electron capture by CTs with excellent electrical conductivity.
Fig. 6b, c, e and f show that TiO2@AC/CTs and g-C3N4@AC/CTs exhibit similar synergy between electrocatalysis and photocatalysis to that of P25@AC/CTs during photoelectrocatalysis. However, TiO2@AC/CTs exhibit higher catalytic efficiency than P25@AC/CTs and g-C3N4@AC/CTs. Especially, the photoelectrocatalytic kinetic constant of TiO2@AC/CTs exhibits a remarkable enhancement, being approximately 1.2 to 3.0 times more efficient than that of P25@AC/CTs and g-C3N4@AC/CTs, which can be attributed to the lower recombination efficiency of photo-induced electron/hole within TiO2@AC/CTs. The strategic assembly of photocatalysts featuring amorphous carbon coatings, each with their unique dimensions and CTs through a universal bottom-up vdW-integrated strategy can serve as a foundational guideline for crafting recyclable vdW-integrated catalysts for the practical application of photocatalytic and photoelectrocatalytic technologies, which can significantly reduce the recycling cost as well as enhance the catalytic efficiency through electrochemical-assisted techniques.
To substantiate the efficacy of TiO2@AC/CTs for authentic wastewater, both municipal effluent and dyeing wastewater were selected as representative test subjects in our study. The municipal secondary effluent under investigation is known for its intricate composition featuring significant amounts of tryptophan-like and fulvic acid-like matters.35 The TOC removal efficiency of TiO2@AC/CTs, P25@AC/CTs, and g-C3N4@AC/CTs was 98.6, 97.2 and 96.3% (Fig. 7a). Similarly, in the dyeing wastewater, tryptophan-like and aromatic protein-like matters were detected, indicating the presence of proteins and tryptophan.35 In the TiO2@AC/CTs, P25@AC/CTs, and g-C3N4@AC/CTs systems, the TOC removal was 97.7, 96.2, and 95.1% during photoelectrocatalysis, respectively (Fig. 7b). These findings further substantiate the photoelectrocatalytic properties of vdW-integrated catalysts when applied to authentic wastewater, demonstrating a remarkable non-selective capability in degrading authentic wastewater. Consequently, the implementation of vdW-integrated catalysts presents itself as a highly promising and innovative alternative for the treatment of authentic wastewater.
 | ||
| Fig. 7 TOC removal efficiency of municipal effluent (a) and dyeing wastewater (b) in different photoelectrocatalytic systems. | ||
Furthermore, an in-depth comparison was conducted on the photoelectrocatalytic efficiency of P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs against a range of previously reported photoelectrocatalysts. The comparative photoelectrocatalysts used in this study fall under the category of recyclable photoelectrocatalysts, ensuring a high degree of comparability. This is essential for conducting a fair and meaningful evaluation of their performance and properties under similar conditions. As evidenced by Table 1, it is noteworthy that our vdW-integrated catalysts exhibit a significantly superior photoelectrocatalytic performance compared to the existing state-of-the-art photoelectrocatalytic systems.
| Photoelectrocatalysts | Wastewater | Light source | Photoelectrocatalytic efficiency | Ref. |
|---|---|---|---|---|
| TiO2@AC/CTs, 2 × 2.5 cm2 | 10 mg L−1, 2,4-dinitrophenol, 100 mL | Xenon light | T 90% = 25 min k = 0.0973 min−1 | Our sample 1 |
| P25@AC/CTs, 2 × 2.5 cm2 | 10 mg L−1, 2,4-dinitrophenol, 100 mL | Xenon light | T 90% = 40 min k = 0.0803 min−1 | Our sample 2 |
| g-C3N4@AC/CTs, 2 × 2.5 cm2 | 10 mg L−1, 2,4-dinitrophenol, 100 mL | Xenon light | T 90% = 100 min k = 0.0326 min−1 | Our sample 3 |
| Dy2O3/graphite/TiO2/Ti nanocomposite | 10 mg L−1, Maxilon blue | Visible light lamps | T 90% = 180 min | 36 |
| Highly ordered TiO2 nanotube arrays, 1.9 cm2 | 20 mg L−1, phenol, 70 mL | Mercury–xenon lamp | T 85% = 400 min k = 0.00358 min−1 | 37 |
| Ag/AgCl/TiO2 nanotube arrays, 3 × 5 cm2 | Microcystin, 1.00 mg L−1, 40 mL | Incandescent lamps | T 100% = 300 min k = 0.0082 min−1 | 38 |
To delve deeper into cycling stability, a series of cycle experiments were designed and executed to shed light on the superior cycling stability of recyclable vdW-integrated catalysts compared to their conventional powdered counterparts. The primary objective was to assess and highlight the enhanced cycling stability that vdW-integrated catalysts exhibit during repetitive usage, thereby setting them apart as a promising advancement in the field of photocatalysis technology. Fig. 6g plots the cycling performance of vdW-integrated catalysts and conventional powdered photocatalysts during the photocatalytic process. Compared with vdW-integrated catalysts, conventional powdered photocatalysts including P25, TiO2 fibers, and g-C3N4 nanosheets, suffer from an inherent limitation in their ability to maintain consistent performance during repetitive photocatalytic cycles. This decline in photocatalytic efficiency is exacerbated by the cumulative effect of catalyst attrition; they are inherently difficult to recycle from the aqueous medium after degradation, thereby causing a steady decline in their overall photocatalytic efficiency with each successive cycle.
Fig. 6h plots the photocatalytic and photoelectrocatalytic cycling performance of vdW-integrated catalysts. In Fig. 6h, it is vividly illustrated that the photocatalytic and photoelectrocatalytic efficiencies of P25@AC/CTs, TiO2@AC/CTs, as well as g-C3N4@AC/CTs remain almost unaltered over the course of 10 consecutive cycles. This steadfast efficiency serves as compelling evidence that the vdW-integrated catalysts exhibit exceptional cycling stability throughout both photocatalytic and photoelectrocatalytic processes. The remarkable cycling stability demonstrated by these vdW-integrated catalysts including P25@AC/CTs, TiO2@AC/CTs, and g-C3N4@AC/CTs, highlights their stability and durability under repetitive cycles. Their photocatalytic and photoelectrocatalytic efficiencies show negligible variation across a full sequence of 10 cycles, affirming their stability during extended periods of photocatalytic and photoelectrocatalytic processes. This steadfast efficiency signifies that these vdW-integrated catalysts have achieved a high level of cycling stability, which is a critical attribute for practical applications in authentic wastewater treatment. The high cycling stability of vdW-integrated catalysts is attributed to strong vdW interactions between amorphous carbon-coated photocatalysts with different dimensions and CTs. The exceptional cycling stability observed in vdW-integrated catalysts can be ascribed to the robust vdW interactions occurring between amorphous carbon-coated photocatalysts with different dimensions and CTs. These interactions play a pivotal role in maintaining structural integrity and functionality, thereby enhancing the overall performance and stability of the photocatalytic and photoelectrocatalytic systems. The synergistic effect of the amorphous carbon coating and the vdW forces ensures the remarkable cycling stability of vdW-integrated catalysts.
Conclusions
In summary, amorphous carbon-coated photocatalysts, including 0D P25@AC core/shell nanospheres, 1D TiO2@AC core/shell fibers, and 2D g-C3N4@AC core/shell nanosheets have been meticulously adhered to CTs through a bottom-up vdW-integrated strategy to assemble recyclable vdW-integrated catalysts for recycling them from aqueous solutions. The coating of amorphous carbon can amplify the spectral response range and inhibit the recombination efficiency of photo-induced electron/hole in pristine photocatalysts, thereby enhancing their overall performance. The vdW-integrated catalysts achieve a remarkable synergy between electrocatalysis and photocatalysis in degrading 2,4-dinitrophenol during the photoelectrocatalytic process. Compared with conventional powdered photocatalysts, vdW-integrated catalysts achieve high cycling stability in the process of photocatalysis and photoelectrocatalysis, which can significantly reduce the recycling cost of conventional powdered photocatalysts as well as enhance catalytic efficiency through electrochemical-assisted techniques. Our low-energy vdW interactions can be a general strategy to integrate other 0D, 1D, or 2D powdered photocatalysts with CTs to craft recyclable vdW-integrated catalysts for effectively recycling them from aqueous solutions after degradation.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the Key R&D Plan of Shaanxi Province (Grant No. 2023-YBGY-492) and the Xi'an International Science and Technology Cooperation Base of Semiconductor Thermoelectric Materials and Devices. ZGC acknowledges financial support from the Australian Research Council, the QUT capacity building professor program, and the HBIS-UQ Innovation Centre for Sustainable Steel (ICSS) project. This work was enabled by the use of the Central Analytical Research Facility hosted by the Institute for Future Environments at QUT.References
- X. Zou, Y. Yang, H. Chen, X.-L. Shi, G. Suo, X. Ye, L. Zhang, X. Hou, L. Feng and Z.-G. Chen, J. Colloid Interface Sci., 2020, 579, 463–469 CrossRef CAS PubMed.
- F. Cheng, L. Liang, G. Lin and S. Xi, J. Mater. Chem. A, 2024, 12, 3879–3885 RSC.
- P. V. Viet, C. M. Thi and L. V. Hieu, J. Nanomater., 2016, 2016, 22 Search PubMed.
- K. Jiang, B. Sun, M. Yao, N. Wang, W. Hu and S. Komarneni, Microporous Mesoporous Mater., 2018, 265, 189–194 CrossRef CAS.
- H. Gao, J. Zhang and M. Wang, Mater. Res. Bull., 2013, 48, 3431–3437 CrossRef CAS.
- A. A. Ashkarran, A. Iraji zad, M. M. Ahadian and S. A. Mahdavi Ardakani, Nanotechnology, 2008, 19, 195709 CrossRef CAS PubMed.
- H. L. Chen, X. Dong, Y. Dong and C. Gangbin, Ceram. Soc., 2012, 40, 1483–1488 CAS.
- Y. Yang, H. Chen, X. Zou, X.-L. Shi, W. D. Liu, L. Feng, G. Suo, X. Hou, X. Ye, L. Zhang, C. Sun, H. Li, C. Wang and Z.-G. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 24845–24854 CrossRef CAS PubMed.
- F. Guo, L. Wang, H. Sun, M. Li and W. Shi, Inorg. Chem. Front., 2020, 7, 1770–1779 RSC.
- B. Jing, Z. Ao, Z. Teng, C. Wang, J. Yi and T. An, Sustainable Mater. Technol., 2018, 16, 12–22 CrossRef CAS.
- X. Zou, Y. Yang, H. Chen, X. L. Shi, S. Song and Z. G. Chen, Mater. Des., 2021, 202, 109542 CrossRef CAS.
- M. Saranya, R. Ramachandran, P. Kollu, S. K. Jeong and A. N. Grace, RSC Adv., 2015, 5, 15831–15840 RSC.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. M. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef PubMed.
- H. J. Chen, R. Wang, Y. L. Yang, X. L. Shi, S. Lu and Z. G. Chen, Prog. Nat. Sci., 2021, 31, 858–864 CrossRef CAS.
- B. Tang, S. C. Zhu, H. Liang, S. Li, B. J. Liu and F. X. Xiao, J. Mater. Chem. A, 2022, 10, 4032–4042 RSC.
- X. Y. Dao and W. Y. Sun, Inorg. Chem. Front., 2021, 8, 3178–3204 RSC.
- H. J. Chen, Y. L. Yang, M. Hong, J. G. Chen, G. Q. Suo, X. J. Hou, L. Feng and Z. G. Chen, Sustainable Mater. Technol., 2019, 21, e00105 CrossRef CAS.
- H. J. Chen, J. Z. Zhang, X. J. Xi and W. J. Tian, Environ. Sci. Nano, 2023, 10, 1468–1481 RSC.
- B. Ma, R. Lin, H. He, Q. Wu and S. Chen, Composites, Part B, 2021, 185, 107782 CrossRef.
- K. S. Varma, R. J. Tayade, K. J. Shah, P. A. Joshi, A. D. Shukla and V. G. Gandhi, Water-Energy Nexus, 2020, 3, 46–61 CrossRef.
- Y. Liu, Y. Huang and X. Duan, Nature, 2019, 567, 323–333 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- H. J. Chen, Y. L. Yang, X. X. Zou, X. L. Shi and Z. G. Chen, J. Mater. Sci. Technol., 2022, 98, 143–150 CrossRef CAS.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed.
- Q. L. He, H. Liu, M. He, Y. H. Lai, H. He, G. Wang, K. T. Law, R. Lortz, J. Wang and I. K. Sou, Nat. Commun., 2014, 5, 4247 CrossRef CAS PubMed.
- W. J. Tian, H. J. Chen, L. Zhao, L. L. Zheng, Z. N. Hu and Y. L. Yang, Colloids Surf., A, 2023, 679, 132530 CrossRef CAS.
- M. Olek, T. Büsgen, M. Hilgendorff and G. Michael, J. Phys. Chem. B, 2006, 110, 12901–12904 CrossRef CAS PubMed.
- D. L. Liao, G. S. Wu and B. Q. Liao, Colloids Surf., A, 2009, 348, 270–275 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- I. Mintsouli, N. Philippidis, I. Poulios and S. Sotiropoulos, J. Appl. Electrochem., 2006, 36, 463–474 CrossRef CAS.
- J. Georgieva, S. Sotiropoulos, S. Armyanov, N. Philippidis and I. Poulios, J. Appl. Electrochem., 2011, 41, 173–181 CrossRef CAS.
- Y. Sun, Y. L. Yang, H. J. Chen, J. Liu, X. L. Shi, G. Suo, X. Hou, X. Ye, L. Zhang, S. Lu and Z. G. Chen, J. Colloid Interface Sci., 2023, 651, 284–295 CrossRef CAS PubMed.
- H. Liu, L. Li, C. Guo, J. Ning, Y. Zhong and Y. Hu, Chem. Eng. J., 2020, 385, 123929 CrossRef CAS.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- X. Jin, R. Wang, P. Jin, X. Shi, Y. Wang, L. Xu, X. Wang and H. Xu, Chem. Eng. J., 2021, 413, 127528 CrossRef CAS.
- B. Ayoubi-Feiz, D. Soleimani and M. Sheydaei, J. Electroanal. Chem., 2019, 849, 113377 CrossRef CAS.
- Z. Liu, X. Zhang, S. Nishimoto, M. Jin, D. A. Tryk, T. Murakami and A. Fujishima, J. Phys. Chem. C, 2008, 112, 253–259 CrossRef CAS.
- W. Liao, Y. Zhang, M. Zhang, M. Murugananthan and S. Yoshihara, Chem. Eng. J., 2013, 231, 455–463 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07813f |
| ‡ The first four authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |