
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced charge separation and DNA self-repair depend on sequence directionality and stacking pattern†
Corinna L.
Kufner
 *a,
Sarah
Crucilla
ab,
Dian
Ding
cd,
Petr
Stadlbauer
ef,
Jiří
Šponer
ef,
Jack W.
Szostak
gh,
Dimitar D.
Sasselov
a and
Rafał
Szabla
*i
*a,
Sarah
Crucilla
ab,
Dian
Ding
cd,
Petr
Stadlbauer
ef,
Jiří
Šponer
ef,
Jack W.
Szostak
gh,
Dimitar D.
Sasselov
a and
Rafał
Szabla
*i
aDepartment of Astronomy, Harvard-Smithsonian Center for Astrophysics, 60 Garden Street, Cambridge, MA 02138, USA. E-mail: corinna.kufner@cfa.harvard.edu
bDepartment of Earth and Planetary Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
cHoward Hughes Medical Institute, Department of Molecular Biology and Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
dDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
eInstitute of Biophysics of the Czech Academy of Sciences, Královopolská 135, 61200 Brno, Czech Republic
fRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacky University Olomouc, Slechtitelu 241/27, 783 71, Olomouc – Holice, Czech Republic
gHoward Hughes Medical Institute, The University of Chicago, Chicago, IL 60637, USA
hDepartment of Chemistry, The University of Chicago, Chicago, Illinois 60637, USA
iInstitute of Advanced Materials, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, Wrocław, 50-370, Poland. E-mail: rafal.szabla@pwr.edu.pl
First published on 28th December 2023
Abstract
Charge separation is one of the most common consequences of the absorption of UV light by DNA. Recently, it has been shown that this process can enable efficient self-repair of cyclobutane pyrimidine dimers (CPDs) in specific short DNA oligomers such as the GAT![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) T sequence. The mechanism was characterized as sequential electron transfer through the nucleobase stack which is controlled by the redox potentials of nucleobases and their sequence. Here, we demonstrate that the inverse sequence TTAG promotes self-repair with higher quantum yields (0.58 ± 0.23%) than GATT (0.44 ± 0.18%) in a comparative study involving UV-irradiation experiments. After extended exposure to UV irradiation, a photostationary equilibrium between self-repair and damage formation is reached at 33 ± 13% for GATT and at 40 ± 16% for TTAG, which corresponds to the maximum total yield of self-repair. Molecular dynamics and quantum mechanics/molecular mechanics (QM/MM) simulations allowed us to assign this disparity to better stacking overlap between the G and A bases, which lowers the energies of the key A−˙G+˙ charge transfer state in the dominant conformers of the TTAG tetramer. These conformational differences also hinder alternative photorelaxation pathways of the TTAG tetranucleotide, which otherwise compete with the sequential electron transfer mechanism responsible for CPD self-repair. Overall, we demonstrate that photoinduced electron transfer is strongly dependent on conformation and the availability of alternative photodeactivation mechanisms. This knowledge can be used in the identification and prediction of canonical and modified DNA sequences exhibiting efficient electron transfer. It also further contributes to our understanding of DNA self-repair and its potential role in the photochemical selection of the most photostable sequences on the early Earth.
T sequence. The mechanism was characterized as sequential electron transfer through the nucleobase stack which is controlled by the redox potentials of nucleobases and their sequence. Here, we demonstrate that the inverse sequence TTAG promotes self-repair with higher quantum yields (0.58 ± 0.23%) than GATT (0.44 ± 0.18%) in a comparative study involving UV-irradiation experiments. After extended exposure to UV irradiation, a photostationary equilibrium between self-repair and damage formation is reached at 33 ± 13% for GATT and at 40 ± 16% for TTAG, which corresponds to the maximum total yield of self-repair. Molecular dynamics and quantum mechanics/molecular mechanics (QM/MM) simulations allowed us to assign this disparity to better stacking overlap between the G and A bases, which lowers the energies of the key A−˙G+˙ charge transfer state in the dominant conformers of the TTAG tetramer. These conformational differences also hinder alternative photorelaxation pathways of the TTAG tetranucleotide, which otherwise compete with the sequential electron transfer mechanism responsible for CPD self-repair. Overall, we demonstrate that photoinduced electron transfer is strongly dependent on conformation and the availability of alternative photodeactivation mechanisms. This knowledge can be used in the identification and prediction of canonical and modified DNA sequences exhibiting efficient electron transfer. It also further contributes to our understanding of DNA self-repair and its potential role in the photochemical selection of the most photostable sequences on the early Earth.
Introduction
Photoinduced charge separation is one of the main phenomena occurring in nucleic acids during the exposure to ultraviolet (UV) light.1 Despite its far reaching implications for biochemistry, biology and even materials sciences,2–5 the key experimental proof of photoinduced charge separation in native unmodified DNA was delivered less than 20 years ago. This experiment involving transient absorption spectroscopy demonstrated that UV excitation of polydeoxyadenosine could populate long-lived electronic states, which were assigned to charge transfer (CT) between the neighbouring stacked adenine nucleobases.6 While numerous examples of CT states have been corroborated in different modified and native forms of DNA since then,1,7–14 the key factors controlling the efficiency of this process still remain obscure. Consequently, prediction of DNA sequences capable of performing efficient UV-induced charge transfer is still a challenge.Among different processes that can be triggered by charge separation in DNA, self-repair of cyclobutane pyrimidine dimers (CPDs) attracted substantial attention, recently.15,16 CPDs are the most frequently formed photolesions during the exposure of DNA to ultraviolet light and their most characteristic structural element is the cyclobutane ring formed between two adjacent pyrimidine bases.17–21 Formation of this cyclobutane ring affects the structure of the sugar–phosphate backbone and precludes biochemical activity such as DNA replication and transcription.21,22 In biology, CPD repairing enzymes, such as photolyases, repair the lesions through the injection of an electron from the flavin adenine co-factor, after the absorption of visible light.23–27 Similarly, specific DNA sequences or alternative nucleobases were shown to trigger nonenzymatic DNA self-repair via photoinduced electron transfer.16,28–30 The most prominent examples of DNA self-repair were demonstrated for the damaged GATT sequence (“” representing the CPD) and for 2,6-diaminopurine (D) and 8-oxoguanine (O) nucleobases located in the vicinity of CPDs.31,32 In particular, the GATT sequence was described to undergo sequential electron transfer from guanine upon its photoexcitation.33–35 In other words, the yields of nonenzymatic DNA self-repair are a manifestation of how efficiently photoinduced charge separation can occur in specific DNA sequences and whether lifetimes of the CT states are sufficiently long to invoke a photochemical reaction.
It is worth emphasizing that highly efficient self-repair of CPDs greatly improves the photostability of specific sequences and was also suggested as a possible selection factor for primordial RNA and DNA oligomers from the rich pool of random sequences.1,15,36,37 More importantly, UV light has been proposed as one of the key sources of energy for selective prebiotic syntheses of nucleotides.38–46 This resulted in the consideration of the above mentioned D and O nucleobases as potential components of first informational polymers owing to their improved electron-donating and CPD-repairing properties when compared to canonical nucleobases.31,32,47 In particular, DNA trinucleotides containing a D nucleobase and a TT dimer were shown to repair the CPD with yields reaching up to 92% when irradiated at 280 nm and, thus, D could protect DNA oligomers from photodamage under prebiotic conditions.31
As shown by Bucher et al.1 the direction of charge transfer in stacked DNA is controlled by the oxidation potentials of the nucleobases. However, the efficiency with which excited CT states are populated seems to depend on much more subtle aspects related to the local environment of the sequence of stacked bases.2 This could be either governed by the spatial overlap of the neighbouring stacked bases as well as by the lifetimes of the different electronic states involved in photoinduced charge separation.31,48 More specifically, as suggested for DTT and TTD trimers, a possible factor which could enable funneling the excited-state population to a CT state is the inacessibility of efficient direct photorelaxation channels of the locally excited (LE) state on D via an S1/S0 conical intersection.31 Here, we present a surprising example of the damaged TTAG oligomer (5′-end denoted first), which exhibits self-repair quantum yields which are higher by ∼30% than in the case of the equivalent GATT oligomer with opposite sequence direction when exposed to UV irradiation at 285 nm. Since this difference cannot be explained by the ordering and oxidation potentials of nucleobases, we provide a mechanistic rationale for this process based on molecular dynamics simulations, quantum mechanics/molecular mechanics (QM/MM) calculations and explorations of excited-state potential energy surfaces using the algebraic diagrammatic construction to the second order method [ADC(2)].49,50 We show that in contrast to GATT, direct photorelaxation of the LE and CT intermediate states of the photoexcited TTAG tetramer is substantially hindered, which enhances sequential electron transfer between stacked bases and enables efficient and selective CPD self-repair.
Results and discussion
Experimental observation of higher self-repair quantum yields in T![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) TAG when compared to GATT
TAG when compared to GATT
In our previous works, we demonstrated the self-repair of the short single-stranded DNA sequence, GATT, via photoexcitation at 290 nm.33 After absorption of a UV photon, the GATT molecule sequentially populates minima on the potential energy surface of the lowest excited electronic state (S1 minima), which can be associated with the G+˙A−˙TT and G+˙ATT−˙ charge transfer states (Fig. 1 left).14,35 These states can be accessed right after the photoexcitation of the G base, which is responsible for the majority of the absorption of the oligomer at wavelengths longer than 280 nm.14,30 Here, we performed comparative UV irradiation experiments of the DNA oligonucleotides TTAG and GATT (Fig. 1). Exposure of the sequences to long-wavelength irradiation from an LED, centered around 285 nm (average power at the sample position 0.36 mW), allowed us to excite the Guanine (predominantly) and Adenine (partially) bases and minimized photoreversal from direct absorption of the TT dimer (Fig. 2A and S7†).51,52 As presented in Fig. 2B, continuous UV-irradiation of an aqueous TTAG solution (pH 6.9, ∼30 μM) in our system lead to an absorbance increase of several 10 mOD at 266 nm within 10 minutes. The irradiation induced difference spectra clearly correspond to typical absorption pattern exhibited by thymine bases in DNA, which is strongly indicative of the self-repair process (Fig. 2A). Complementary HPLC analysis corroborated this finding (Fig. 2C). The chromatogram of the damaged sequence TTAG as starting material (bottom) shows a peak at 8.4 min. Upon exposure to 285 nm light, the undamaged sequence TTAG gradually recovers, visible as an increase in absorption at 9.9 min. After 30 min of irradiation, the irradiated sample was spiked with the undamaged sequence TTAG for reference (top chromatogram). The emergence of one single peak at 9.9 min confirms the self-repair to TTAG. The corresponding chromatograms for the sequence GATT are shown in Fig. S1 in the ESI.†
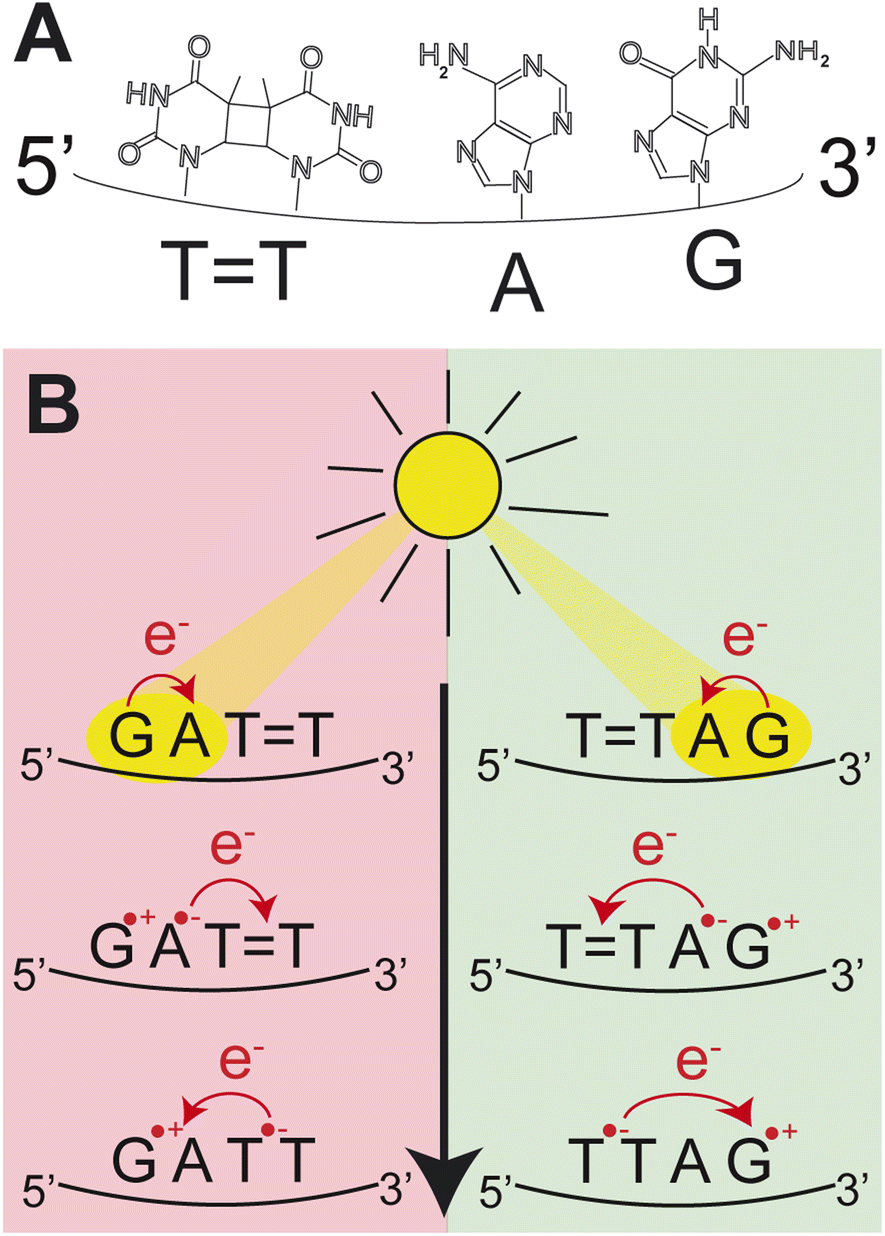 | ||
| Fig. 1 (A) Molecular structures of the nucleobases in the DNA sequence TTAG. (B) Schematic representation of the CPD self-repair mechanism promoted by photoinduced sequential electron transfer in the DNA sequences, GATT and TTAG. | ||
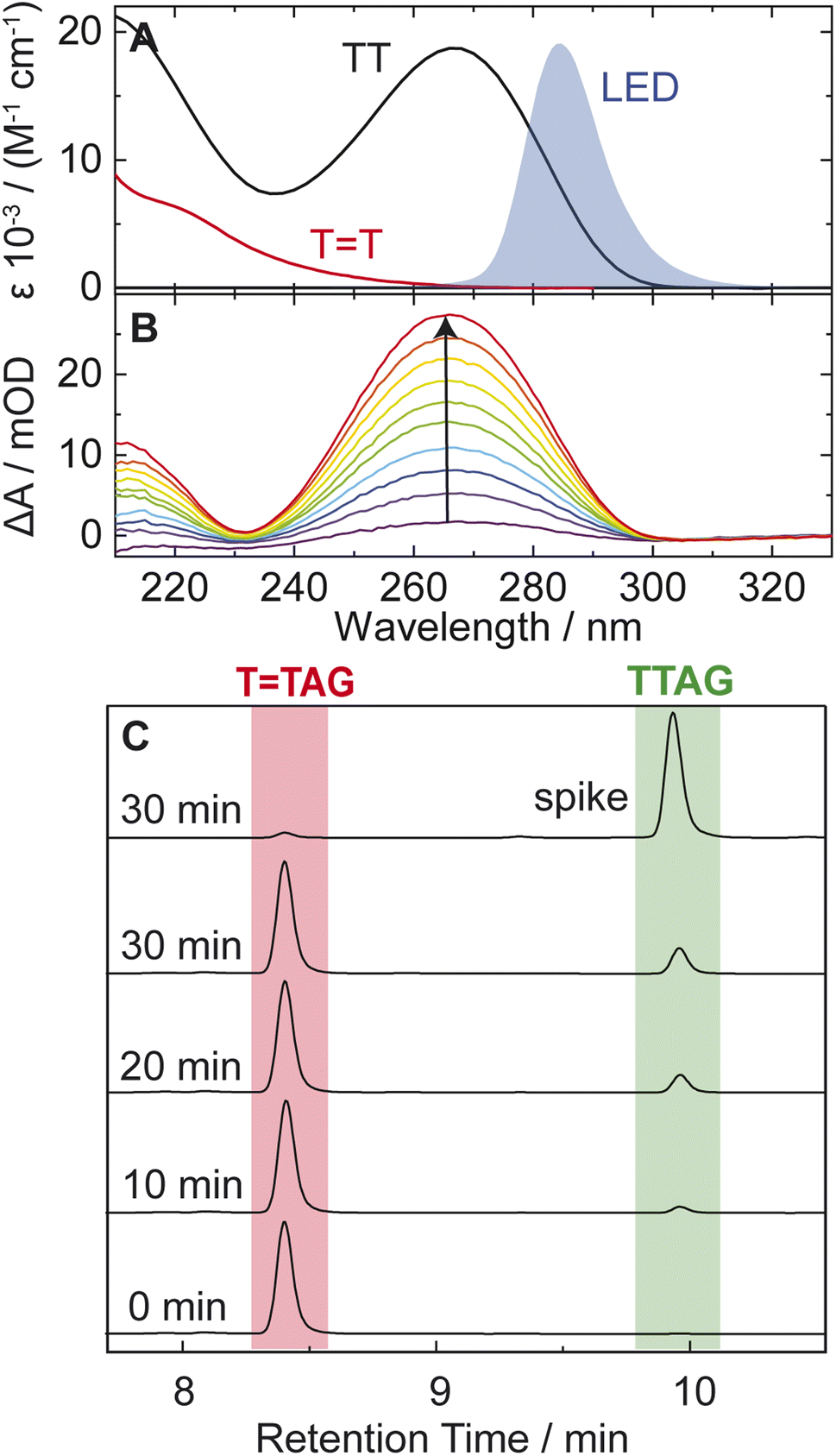 | ||
| Fig. 2 (A) Molar decadic absorption coefficients of the TT CPD lesion (red) and the undamaged TT dinucleotide (black). The emission spectrum of the LED centered around 285 nm, which was used for irradiation, is shown as blue shade for comparison. (B) UV absorption difference spectra of a 30 μM solution of the sequence TTAG after increasing times (1–10 min) of exposure to 285 nm irradiation with an average power of 0.36 mW. The recovery of the 266 nm absorption (arrow) is indicative of the self-repair to TTAG. (C) Analytical HPLC analysis of the sequence TTAG (8.4 min) after different irradiation times. Upon irradiation a recovery of the undamaged sequence TTAG (9.9 min) can be observed. The chromatogram on top is spiked with undamaged TTAG for reference. | ||
The increase in absorbance at 266 nm due to the recovery of undamaged TTAG from TTAG (black arrow in Fig. 1B) can be plotted as a function of the photon dose absorbed by the molecules (Fig. 3). At low irradiation doses, the increase in absorbance at 266 nm is linear. The data in this range can be fitted with a linear trendline (red). The slope of the initial absorbance increase is linearly proportional to the quantum yield of the CPD self-repair (see ESI† for details). The quantum yield of CPD self-repair was found to be 0.44 ± 0.18% for GATT (blue) and 0.58 ± 0.23% for TTAG (black), respectively. At higher absorbed doses, the slope of both plots decreases. This is largely the result of approaching the photostationary state of equilibrium, but can be also attributed to the formation of secondary products, which lower the yield of the net reaction. After absorption of high irradiation doses, a photostationary equilibrium between net damage formation and self-repair can be reached (Fig. 4). In case of the sequence GATT, the equilibrium is reached after the absorption of 3.5 J at a level of 33 ± 13% self-repair. This result is higher than the previously reported 25% at 290 nm irradiation as well as the corresponding quantum yields.33,53 The differences can be attributed to the 15 nm broad LED light source, in comparison to the previously used narrowband (3 nm broad) 290 nm excitation, as self-repair quantum yields and direct photoreversal may be higher at lower wavelengths. In case of TTAG, the equilibrium is reached only after absorption of 2.5 J at a higher level of 40 ± 16%. The error bars were estimated to 40% of the provided values according to the previous work by some of us were an analogous experimental setup was used.54 These results indicate that the ratio of the self-repair vs. the net rate of CPD formation is higher in case of TTAG when compared to GATT. The photostationary equilibrium indicates the maximum total yield of the repair in UV-rich environments, under persistent exposure to irradiation.
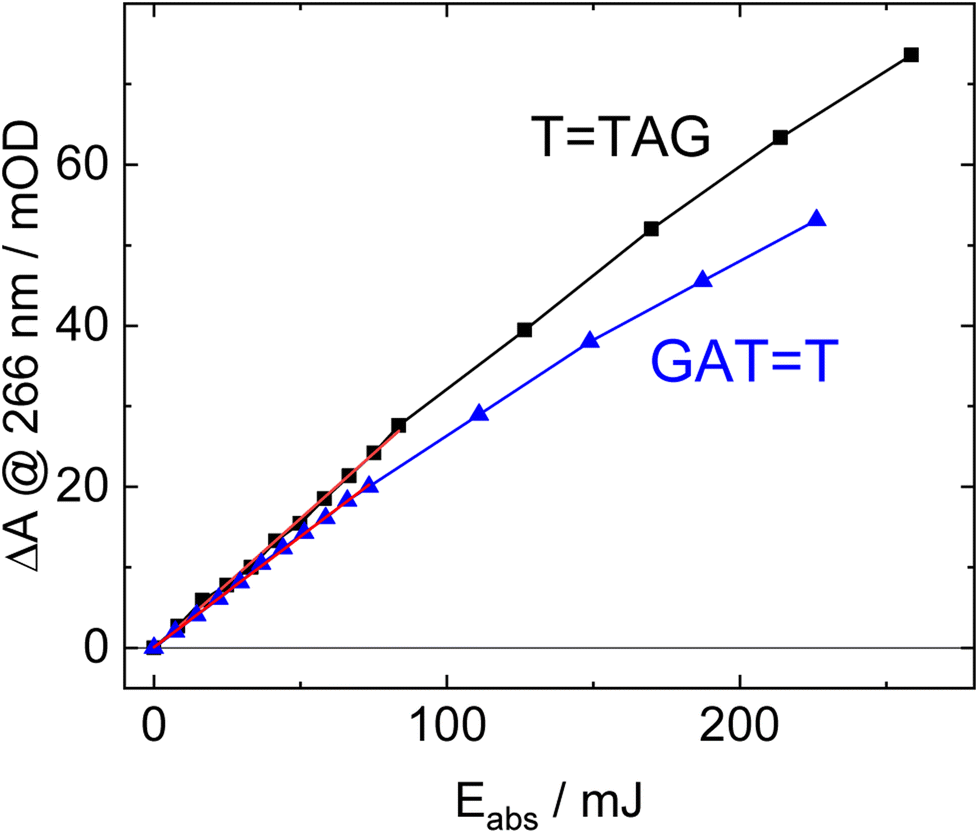 | ||
| Fig. 3 Absorbance difference spectra of TTAG (black) and GATT (blue) at 266 nm as a function of absorbed light dose at 285 nm. The data at low doses can be fitted with a linear trend line (red). The slope of the trend line is linearly proportional to the quantum yield of the self-repair. | ||
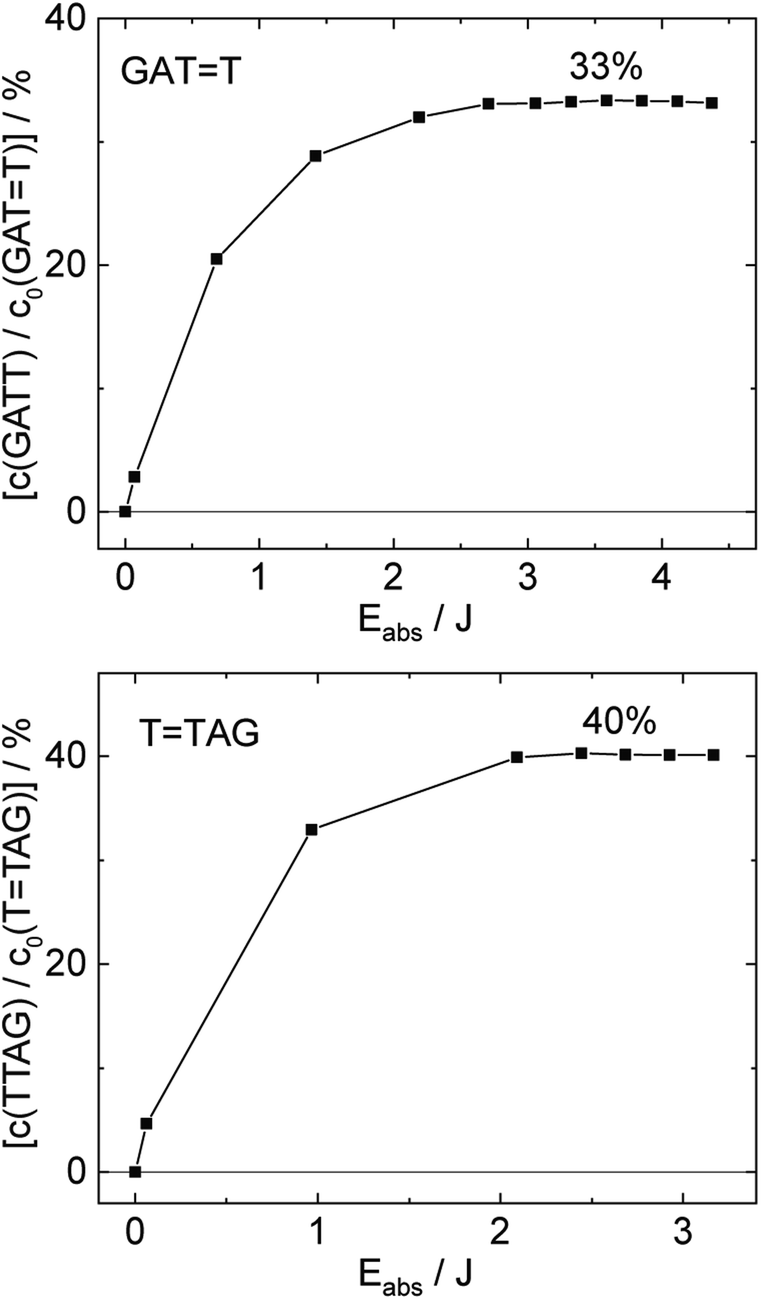 | ||
| Fig. 4 Concentration of the undamaged sequence GATT (top) and TTAG (bottom) divided by the initial concentration of the damaged sequence GATT (top) and TTAG (bottom) as starting material as a function of absorbed photon dose at 285 nm. In case of the GATT sequence a photostationary equilibrium between repair and damage formation is reached after absorption of 3.5 J at a level of ∼33%. The sequence TTAG reaches the equilibrium earlier after absorption of 2.5 J at a level ∼40%. The absorption coefficients at 266 nm were taken from Pan et al.30 | ||
Structural differences between the TTAG and GATT tetramers
We further performed classical molecular dynamics and quantum chemical simulations, to provide mechanistic rationale explaining this substantial difference in self-repairing activity between the TTAG and GATT tetramers. Since we previously performed such calculations for the GATT tetranucleotide, here, we applied an analogous computational protocol to the TTAG tetramer.35 The overall lengths of the trajectories of our force-field based molecular dynamics simulations for TTAG amounted to 10 μs per tetranucleotide. We selected two of the most representative stacked conformers, which have the highest contribution to the overall conformational space of TTAG (see the ESI† for more details). The AG-anti conformer of TTAG discussed in the main article is shown in Fig. 5. We used their averaged MD structures for subsequent QM/MM simulations in order to first optimize the ground-state geometry of the tetramer using density functional theory with dispersion correction (PBEh-3c functional) and next calculate their photophysical and photochemical properties.
 | ||
| Fig. 5 Averaged structures of the major stacked conformers obtained from the classical MD simulations of TTAG (left) and GATT (right)35 tetranucleotides. The stacking of the G and A bases is presented at the bottom. | ||
Notably, as shown in Fig. 5, the highest populated stacked conformers of TTAG and GATT differ by the relative spatial orientation of the nucleobases and the stacking patterns. The positioning of the G and A bases at the 3′-end of the TTAG tetramer results in the two nucleobases predominantly populating the anti orientations with respect to the sugar scaffold. This entails a very good overlap of the six-membered counterparts of the purine rings and partial overlap of the five-membered subunits for most of the populated conformers. While the TTAG conformer presented in Fig. 5 was populated in 9% of the simulation time, we also observed this AG-anti arrangement for conformers with partially unstacked or overhanging TT dimer. Overall, the AG-anti arrangement was present in >50% of sampled conformations of TTAG (see Fig. S4 in the ESI† to this article). In contrast, the G base of the GATT tetramer prefers to form a hydrogen bond between its N3 atom and the free 5′-OH group of the sugar, which is accompanied by the syn orientation of the nucleobase with respect to the sugar ring.35 As observed previously, the neighbouring adenine of GATT also prefers the syn orientation to maintain better stacking and the resulting GA-syn stacked conformer was determined to be dominant (populated in over >30% of conformations).35 It is worth noting though that the G and A bases of this conformer are somewhat displaced with respect to one another and less favorably stacked, having only the five- and six-membered counterparts of G and A bases stacked. As previously indicated for long-range electron transport in DNA, the degree of stacking and conformational arrangement could strongly affect the rate and yield of CT along the stack.2 Therefore, these structural differences are the first indication that disparities between the photochemistry of the two tetranucleotides should be expected.
Photophysical properties of TTAG
According to the vertical excitation energies computed for the TTAG tetramer (see the ADC(2)/MM results Table 1), the charge transfer states can be found in the higher energy range of the UV absorption spectrum, whereas the lower energy range of the spectrum is dominated by local ππ* excitations of the purine bases and nπ* excitations of the T bases constituting the TT dimer. The lowest-energy CT state in the Frank–Condon region involves an electron transferred from G to A and can be identified as the S6 state with the  molecular orbital character. The analogous
molecular orbital character. The analogous 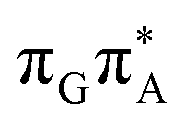 CT state of the GA-syn stacked conformer of GATT has the vertical excitation energy higher by 0.2 eV and was identified as the S10 state. This demonstrates that the stacking overlap between the G and A bases can strongly affect the ordering of the electronic states and the energies of CT excitations.
TAG tetranucleotides obtained at the ADC(2)/TZVP level of theory, assuming the ground-state minimum-energy geometry optimized with the PBEh-3c method. The results for the GATT tetranucleotide were taken from ref. 33 and obtained with the same approach
CT state of the GA-syn stacked conformer of GATT has the vertical excitation energy higher by 0.2 eV and was identified as the S10 state. This demonstrates that the stacking overlap between the G and A bases can strongly affect the ordering of the electronic states and the energies of CT excitations.
TAG tetranucleotides obtained at the ADC(2)/TZVP level of theory, assuming the ground-state minimum-energy geometry optimized with the PBEh-3c method. The results for the GATT tetranucleotide were taken from ref. 33 and obtained with the same approach
| State/transition | E exc [eV] | f OSC | λ [nm] | |
|---|---|---|---|---|
AG-anti conformer of T![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) TAG TAG
|
||||
| S1(LE) |

|
4.95 | 0.097 | 250.7 |
| S2(LE) |

|
5.07 | 1.04 × 10−3 | 244.5 |
| S3(LE) |

|
5.12 | 1.65 × 10−3 | 242.0 |
| S4(LE) |
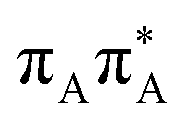
|
5.15 | 0.070 | 240.7 |
| S5(LE) |

|
5.28 | 8.06 × 10−3 | 234.7 |
| S6(CT) |

|
5.40 | 0.388 | 229.4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
|
GA-syn conformer of GATT
|
||||
| S4(LE) |

|
5.00 | 0.056 | 248.0 |
| S7(CT) |

|
5.32 | 4.25 × 10−4 | 233.1 |
| S10(CT) |

|
5.60 | 0.015 | 221.4 |
We previously identified another CT excitation in the Franck–Condon region of the GATT tetramer, namely the  CT state associated with an electron transferred between the A base and the TT dimer. In fact, this excitation is the lowest energy CT state found for the ground-state geometry of the GA-anti conformer of GATT, and was identified as the S10 state with the excitation energy of 5.81 eV. In the case of the GA-syn conformer of the GATT tetranucleotide, the
CT state associated with an electron transferred between the A base and the TT dimer. In fact, this excitation is the lowest energy CT state found for the ground-state geometry of the GA-anti conformer of GATT, and was identified as the S10 state with the excitation energy of 5.81 eV. In the case of the GA-syn conformer of the GATT tetranucleotide, the 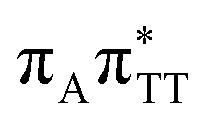 CT state was found to be the S7 state with the vertical excitation energy of 5.32 eV. Nevertheless, we did not find this electronic state among the ten lowest vertical excitations of the TTAG tetranucleotide. Furthermore, low self-repair yields of canonical trinucleotides containing CPDs was ascribed to the limited accessibility of the
CT state was found to be the S7 state with the vertical excitation energy of 5.32 eV. Nevertheless, we did not find this electronic state among the ten lowest vertical excitations of the TTAG tetranucleotide. Furthermore, low self-repair yields of canonical trinucleotides containing CPDs was ascribed to the limited accessibility of the 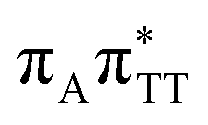 CT state.30,31 Therefore, as proposed for the GATT tetranucleotide, we postulate that the initial photoinduced charge separation event in TTAG is initiated with the population of the
CT state.30,31 Therefore, as proposed for the GATT tetranucleotide, we postulate that the initial photoinduced charge separation event in TTAG is initiated with the population of the 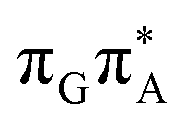 state outside of the Franck–Condon region. The details of this mechanism are discussed in the following section.
state outside of the Franck–Condon region. The details of this mechanism are discussed in the following section.
Sequential electron transfer in the TTAG tetranucleotide
According to the calculations of vertical excitation energies, irradiation of the TTAG tetranucleotide at 285 nm primarily results in the population of the S1(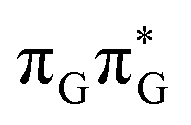 ) state of the G base. We argue that subsequent electron transfer events will occur on the hypersurface of the lowest excited singlet state involving changes of molecular orbital character until the key S1/S0 state crossing is reached. Such a mechanism leading to partial splitting of the cyclobutane ring is presented in Fig. 6 and involves three intermediate S1 minima. Therefore, the presented self-repair mechanism of TTAG is analogous to the sequential electron transfer (SET) process described for the GATT tetranucleotide.35 However, we identified the key differences in the SET mechanism for the GATT and TTAG tetranucleotides which could further explain the higher self-repair yields found for TTAG.
) state of the G base. We argue that subsequent electron transfer events will occur on the hypersurface of the lowest excited singlet state involving changes of molecular orbital character until the key S1/S0 state crossing is reached. Such a mechanism leading to partial splitting of the cyclobutane ring is presented in Fig. 6 and involves three intermediate S1 minima. Therefore, the presented self-repair mechanism of TTAG is analogous to the sequential electron transfer (SET) process described for the GATT tetranucleotide.35 However, we identified the key differences in the SET mechanism for the GATT and TTAG tetranucleotides which could further explain the higher self-repair yields found for TTAG.
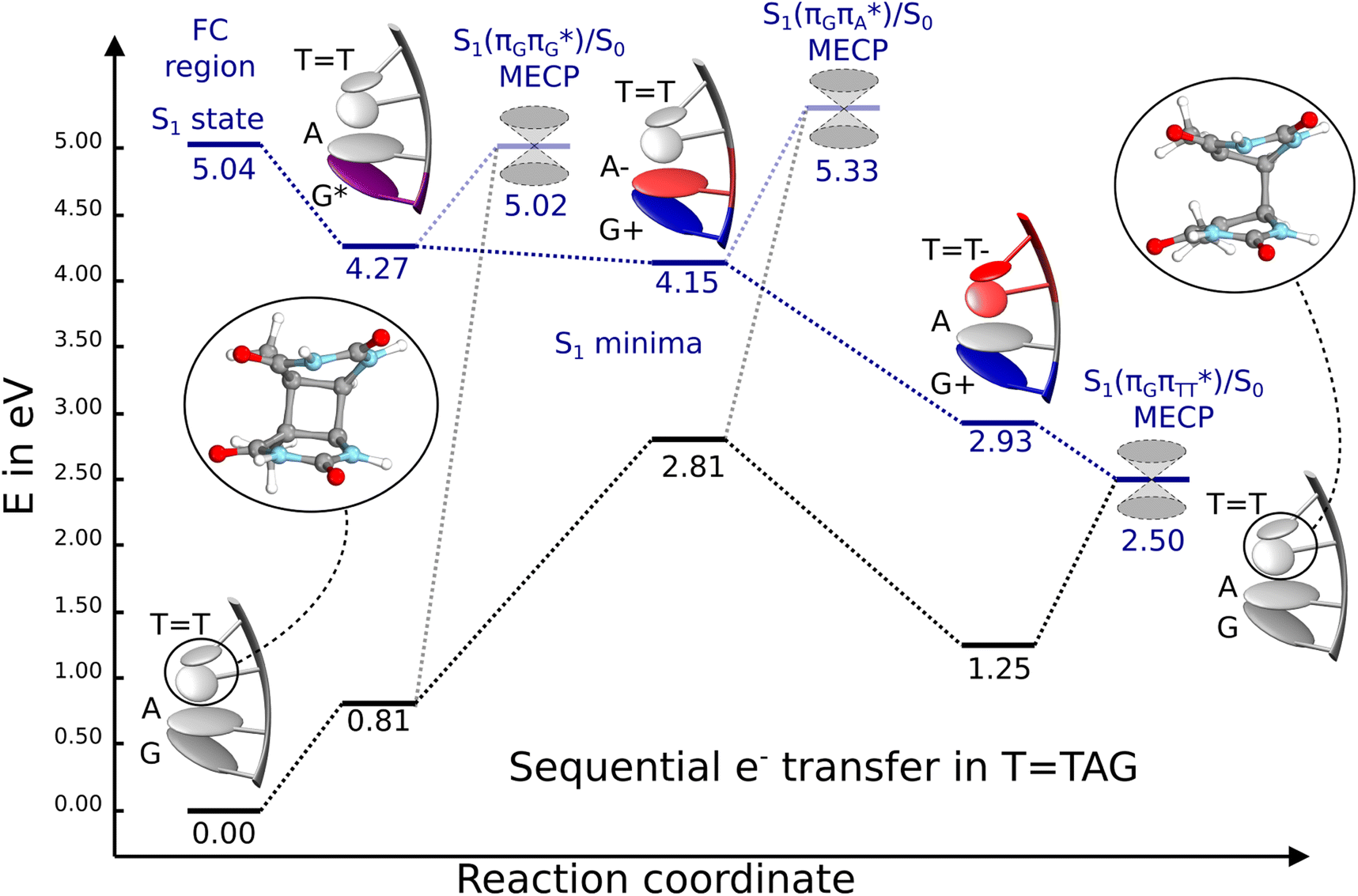 | ||
Fig. 6 Energy diagram showing the sequential electron transfer mechanism initiated with the photoexcitation of the guanine base of the TTAG tetranucleotide to the S1(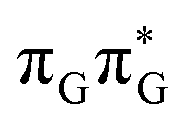 ) state in the Franck–Condon (FC) region (left-hand side of the diagram). The leftmost structure corresponds to the ground-state geometry of the AG-anti conformer of the TTAG tetramer. The three middle energy levels are associated with the key S1 minima. The partly transparent pathways leading to S1/S0 MECPs demonstrate the possible competing direct photorelaxation mechanism from the G* and A−˙G+˙ intermediate states. The rightmost structure is associated with the key S1/S0 MECP responsible for C5–C5 bond breaking. The energies were obtained with the QMbases/MM setup at the ADC(2)/def2-SVP level of theory (see the Computational Methods section in the ESI† for more details). ) state in the Franck–Condon (FC) region (left-hand side of the diagram). The leftmost structure corresponds to the ground-state geometry of the AG-anti conformer of the TTAG tetramer. The three middle energy levels are associated with the key S1 minima. The partly transparent pathways leading to S1/S0 MECPs demonstrate the possible competing direct photorelaxation mechanism from the G* and A−˙G+˙ intermediate states. The rightmost structure is associated with the key S1/S0 MECP responsible for C5–C5 bond breaking. The energies were obtained with the QMbases/MM setup at the ADC(2)/def2-SVP level of theory (see the Computational Methods section in the ESI† for more details). | ||
Initial vibrational relaxation of the  state results in modest puckering of the aromatic ring of guanine and reaching the vicinity of the first S1 minimum that participates in the SET mechanism. This minimum is denoted as G* in Fig. 6 and lies 4.27 eV above the ground-state structure of the AG-anti conformer of TTAG. Similarly as in the case of GATT, ring puckering is most pronounced at the C4 and C5 atoms and greater out of plane distortion of the C4 and N3 atoms and rotation about the C4N3 bond leads to the S1(
state results in modest puckering of the aromatic ring of guanine and reaching the vicinity of the first S1 minimum that participates in the SET mechanism. This minimum is denoted as G* in Fig. 6 and lies 4.27 eV above the ground-state structure of the AG-anti conformer of TTAG. Similarly as in the case of GATT, ring puckering is most pronounced at the C4 and C5 atoms and greater out of plane distortion of the C4 and N3 atoms and rotation about the C4N3 bond leads to the S1(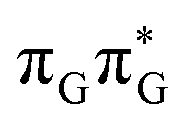 )/S0 minimum-energy crossing point (MECP) lying ∼0.75 eV above the G* S1 minimum (see Fig. 6 and 7). Considerably sloped topography of this state crossing and the associated energy barrier hinder the direct photorelaxation of the G base. We anticipate that this will increase the importance of the competitive forward electron transfer process from the G base to the neighbouring A base. It is worth noting that the analogous S1(
)/S0 minimum-energy crossing point (MECP) lying ∼0.75 eV above the G* S1 minimum (see Fig. 6 and 7). Considerably sloped topography of this state crossing and the associated energy barrier hinder the direct photorelaxation of the G base. We anticipate that this will increase the importance of the competitive forward electron transfer process from the G base to the neighbouring A base. It is worth noting that the analogous S1(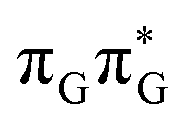 )/S0 MECP of the of GATT tetranucleotide was reported to lie only 0.15 eV and 0.50 eV above the S1(G*) minima for the GA-syn and GA-anti conformers, respectively. This indicates that direct photorelaxation of the G base should be more efficient in the GATT tetranucleotide, whereas UV-excited TTAG tetramer should more easily undergo charge separation.
)/S0 MECP of the of GATT tetranucleotide was reported to lie only 0.15 eV and 0.50 eV above the S1(G*) minima for the GA-syn and GA-anti conformers, respectively. This indicates that direct photorelaxation of the G base should be more efficient in the GATT tetranucleotide, whereas UV-excited TTAG tetramer should more easily undergo charge separation.
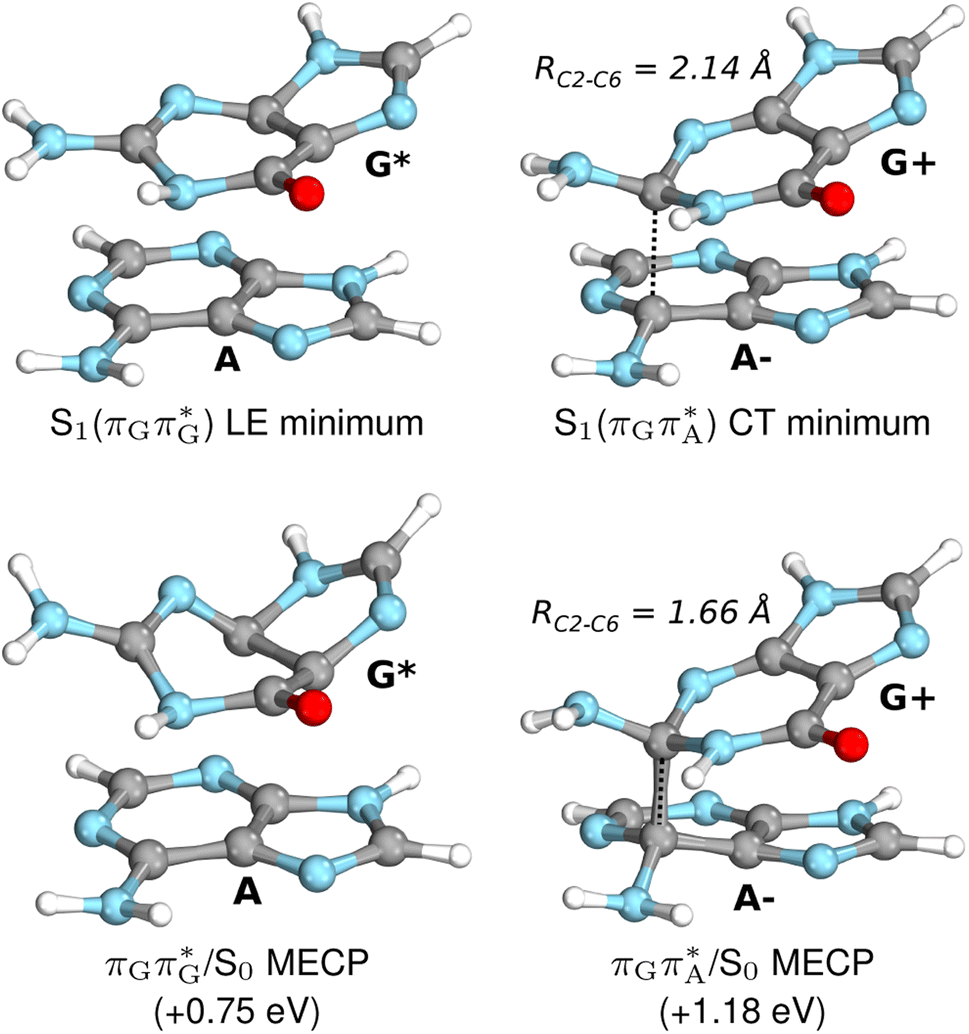 | ||
| Fig. 7 Geometries of the G and A bases located for the intermediate excited-state (S1) G* and A−˙G+˙ minima (top) and the geometries of the corresponding S1/S0 MECPs. The presented geometries and energies were obtained with the QMbases/MM setup at the ADC(2)/def2-SVP level of theory (see the Computational Methods section in the ESI† for more details). | ||
Subsequent excited-state electron transfer from the G base may allow to reach the vicinity of the A−˙G+˙ S1 minimum. This entails structural changes in the ring-puckering pattern of the G base, with most pronounced pyramidalization of the C2 atom and additional pyramidalization of the C6 atom of A. Consequently, the two pyramidalized C atoms create the main contact between these purine bases in the A−˙G+˙ CT minimum. This S1 minimum lies merely 0.12 eV below the G* minimum of TTAG AG-anti conformer, which implies a weaker driving force for e− transfer than in the case of the GATT tetramer (ΔE = −0.9 eV for the GA-anti conformer). While driving force is an important component of the electron rate within the Marcus model, the efficiency of photoinduced CT between stacked nucleobases is also dependent on the excited-state lifetime of the donor state. Given that, the direct photorelaxation of the G base is hindered in major conformers of TTAG, we expect the G to A electron transfer process to be an important contributor to the photochemistry of this tetranucleotide.
Similarly as in the case of the 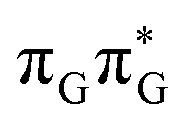 state, the direct photorelaxation from the A−˙G+˙ minimum is hindered owing to very high energy of the S1(
state, the direct photorelaxation from the A−˙G+˙ minimum is hindered owing to very high energy of the S1(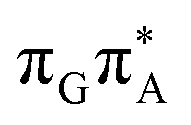 )/S0 state crossing, which lies 1.18 eV above the corresponding S1 minimum. The energy of this state crossing is even higher than the vertical excitation energy of lowest optically bright state of the G base. This state crossing involves formation of a (transient) covalent bond between the C2 atom of G and C6 atom of A. Consequently, the second electron transfer event from the radical anion of the A base to the TT dimer should be the preferred event occurring after the population of the
)/S0 state crossing, which lies 1.18 eV above the corresponding S1 minimum. The energy of this state crossing is even higher than the vertical excitation energy of lowest optically bright state of the G base. This state crossing involves formation of a (transient) covalent bond between the C2 atom of G and C6 atom of A. Consequently, the second electron transfer event from the radical anion of the A base to the TT dimer should be the preferred event occurring after the population of the  of the TTAG tetranucleotide. In contrast, direct photorelaxation of GATT from its
of the TTAG tetranucleotide. In contrast, direct photorelaxation of GATT from its 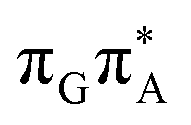 state is again more efficient than for TTAG, since the analogous S1(
state is again more efficient than for TTAG, since the analogous S1(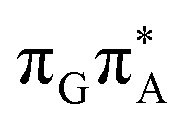 )/S0 state crossing lies 0.42 eV above and 0.25 eV below the G+˙A−˙ minimum located for the GA-syn and GA-anti conformers, respectively.
)/S0 state crossing lies 0.42 eV above and 0.25 eV below the G+˙A−˙ minimum located for the GA-syn and GA-anti conformers, respectively.
The above interpretation is further supported by the recent investigation of the photodynamics of the GA and AG dinucleotides with transient absorption spectroscopy, which showed that the yield of excited-state interbase charge transfer is higher by ∼75% for GA than for AG. Similarly the GA dinucleotide was reported to exhibit a longer lifetime of the CT state (170 ± 10 ps) than the AG dinucleotide (112 ± 12 ps).55 While in the case of these dinucleotides the trend is clearly opposite than for the damaged GATT and TTAG tetranucleotides, dinucleotides are characterized by very different conformational spaces than longer oligomers owing to both purine bases being located at the termini of the mini-strands. Consequently, the results of Petropoulos et al.55 are consistent with the picture emerging from our QM/MM calculations, that is, that the population of DNA excited CT states is strongly affected by the interbase stacking pattern.
The second excited-state e− transfer allows the tetranucleotide to reach the TT−˙AG+˙ electronic configuration that enables direct photoreversal of the CPD. This CT event is associated with much stronger driving force for electron transfer as the TT−˙AG+˙ S1 minimum lies 1.22 eV below the A−˙G+˙ minimum. Similarly as in the case of the GATT tetranucleotide as well as damaged trimers containing 2,6-diaminopurine, a very modest barrier (<0.1 eV) separates the final S1 CT minimum of TTAG from the S1( )/S0 state crossing responsible for CPD repair (see the PES presented in Fig. S10 in the ESI†). Beyond this barrier, the C5–C5 bond breaking process can occur spontaneously and the S1(
)/S0 state crossing responsible for CPD repair (see the PES presented in Fig. S10 in the ESI†). Beyond this barrier, the C5–C5 bond breaking process can occur spontaneously and the S1(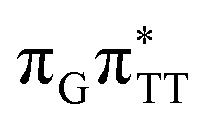 )/S0 conical intersection has a peaked topography (see Fig. S10 in the ESI†). This geometry of the TT dimer in this MECP is characterized by the C5⋯C5 distance (between constituent T bases) equal to 2.55 Å, which means that this covalent bond of the CPD is completely broken at the point of conical intersection. The remaining C6–C6 bond of the CPD maintains its length of 1.56 Å at the S1/S0 state crossing, but can be subsequently broken in the hot electronic ground state, which completes the CPD self-repair process.35,56,57 In other words, CPD reversal is a stepwise process and as reported previously for enzymatic repair of thymine dimers, the barrier associated with C6–C6 bond breaking does not exceed 0.15 eV (3.1 kcal mol−1).56,57
)/S0 conical intersection has a peaked topography (see Fig. S10 in the ESI†). This geometry of the TT dimer in this MECP is characterized by the C5⋯C5 distance (between constituent T bases) equal to 2.55 Å, which means that this covalent bond of the CPD is completely broken at the point of conical intersection. The remaining C6–C6 bond of the CPD maintains its length of 1.56 Å at the S1/S0 state crossing, but can be subsequently broken in the hot electronic ground state, which completes the CPD self-repair process.35,56,57 In other words, CPD reversal is a stepwise process and as reported previously for enzymatic repair of thymine dimers, the barrier associated with C6–C6 bond breaking does not exceed 0.15 eV (3.1 kcal mol−1).56,57
We emphasize, that reaching this S1(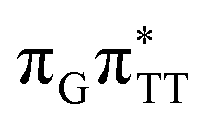 )/S0 state crossing does not ensure CPD self-repair, since the C5–C5 bond may still be reformed after non-radiative transition to the S0 state. Therefore, the experimentally measured self-repair quantum yield cannot be directly associated with the quantum yield of the SET process in TTAG. However, the peaked topography of this state crossing indicates that C5–C5 bond rupture involves relatively high momentum, which should generally drive the C5 atoms of the T bases towards greater separation, which can be subsequently followed by C6–C6 bond breaking.31,35
)/S0 state crossing does not ensure CPD self-repair, since the C5–C5 bond may still be reformed after non-radiative transition to the S0 state. Therefore, the experimentally measured self-repair quantum yield cannot be directly associated with the quantum yield of the SET process in TTAG. However, the peaked topography of this state crossing indicates that C5–C5 bond rupture involves relatively high momentum, which should generally drive the C5 atoms of the T bases towards greater separation, which can be subsequently followed by C6–C6 bond breaking.31,35
Conclusions
In summary, we demonstrated that the directionality of DNA sequences can substantially influence the efficiency of photoinduced electron transfer through the base stack. We present this based on the example of photoinduced self-repair of cyclobutane pyrimidine dimers in the GATT and TTAG sequences, which is controlled by the sequential electron transfer mechanism.35 In particular, the TTAG sequence is characterized by higher quantum yields of CPD self-repair when compared to the GATT tetramer (0.58% vs. 0.44%). Overall, the TTAG tetranucleotide can repair up to 40% of the formed photodimers before reaching photostationary equilibrium when irradiated at 285 nm. In comparison, we managed to achieve only up to 33% of self-repair for the GATT sequences under equivalent conditions.
We ascribe this phenomenon to the differences in the conformational ensembles between the two tetranucleotides. Firstly, for the major conformers of the TTAG tetramer (AG-anti), we observe much higher degree of stacking between the G and A bases than in the case of the GATT tetranucleotide. Better stacking overlap lowers the energy of  CT state in the Franck–Condon region. This electronic CT state is responsible for the first electron transfer event in the CPD self-repair process. Secondly, the S1/S0 minimum-energy crossing points (MECPs) responsible for the direct photorelaxation of the intermediate
CT state in the Franck–Condon region. This electronic CT state is responsible for the first electron transfer event in the CPD self-repair process. Secondly, the S1/S0 minimum-energy crossing points (MECPs) responsible for the direct photorelaxation of the intermediate  LE and
LE and  CT states are practically energetically inaccessible in the TTAG tetramer. As a result, excited-state electron transfer process is a much more competitive and favorable photorelaxation mechanism for TTAG than in the case of the GATT tetranucleotide. Even though many more photorelaxation mechanisms are usually available in DNA strands, involving e.g. locally-excited nπ* or πσ* states, our calculations demonstrate that the well-stacked conformation of the TTAG tetramer can effectively restrain some of these channels and promote interbase electron transfer.
CT states are practically energetically inaccessible in the TTAG tetramer. As a result, excited-state electron transfer process is a much more competitive and favorable photorelaxation mechanism for TTAG than in the case of the GATT tetranucleotide. Even though many more photorelaxation mechanisms are usually available in DNA strands, involving e.g. locally-excited nπ* or πσ* states, our calculations demonstrate that the well-stacked conformation of the TTAG tetramer can effectively restrain some of these channels and promote interbase electron transfer.
We anticipate that these alternative photorelaxation mechanisms are the main reason for the modest self-repair quantum yields resulting from our measurements. Here, we were able to identify two other photorelaxation pathways of the intermediate 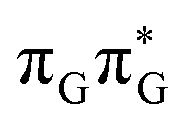 LE and
LE and 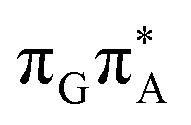 CT electronic states with our static excited-state PES explorations. However, in a dynamic picture higher electronic states can easily interchange their order with the S1 state and lead to other S1/S0 state crossings, which are more challenging to grasp with the static QM/MM approach. These state crossings can potentially enable back-electron transfer which was observed for polyadenosine sequences.58 Furthermore, as discussed above, reaching the key S1(
CT electronic states with our static excited-state PES explorations. However, in a dynamic picture higher electronic states can easily interchange their order with the S1 state and lead to other S1/S0 state crossings, which are more challenging to grasp with the static QM/MM approach. These state crossings can potentially enable back-electron transfer which was observed for polyadenosine sequences.58 Furthermore, as discussed above, reaching the key S1(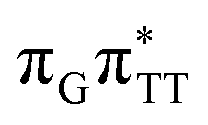 )/S0 state crossing which entails C5–C5 bond breaking may still be followed by CPD reformation. Nevertheless, the quantum yields are sufficiently high to enable the accumulation of high quantities of repaired material during continuous irradiation (∼33% for GATT and ∼40% for TTAG).
)/S0 state crossing which entails C5–C5 bond breaking may still be followed by CPD reformation. Nevertheless, the quantum yields are sufficiently high to enable the accumulation of high quantities of repaired material during continuous irradiation (∼33% for GATT and ∼40% for TTAG).
Overall, we show that photoinduced electron transfer in DNA and the associated CPD self-repair process are strongly dependent on conformation and the availability of alternative (direct) photorelaxation channels of the intermediate states. This demonstrates that the efficiency of electron transfer cannot be simply predicted based on sequence. However, prior computational exploration of the conformational spaces of DNA sequences and their associated photochemical properties (including the energetics of CT states and S1/S0 state crossings) can offer valuable predictive capacity for the identification of DNA oligomers that can undergo efficient charge separation upon UV absorption.
Data availability
Computational data, including geometries and results of QM/MM calculations can be found under: https://doi.org/10.6084/m9.figshare.24711825.Author contributions
Conceptualization: C. L. K. and R. S.; investigation and formal analysis: C. L. K., S. C., D. D., P. S. and R. S.; funding acquisition: D. D. S., J. W. S., J. S. and R. S.; resources: J. S. and J. W. S.; writing – original draft: R. S.; visualisation: C. L. K. and R. S.; writing – review and editing: C. L. K. and R. S.; supervision: D. D. S., C. L. K., J. W. S., J. S. and R. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Fanny Ng for assistance during conduction of the experiments, and Prof. Carlos E. Crespo-Hernández for helpful discussion. P. S. and J. S. acknowledge support by the Czech Science Foundation (21-23718S). This work was supported in part by a grant from the Simons Foundation (SCOL 290360 to D. D. S., and SCOL 290363 to J. W. S.), the National Science Foundation (Grant No. 1933505 to D. D. S., DGE 2140743 to S. J. C.), and by a grant from the National Science Centre, Poland (2020/37/B/ST4/04092 to R. S.).Notes and references
- D. B. Bucher, B. M. Pilles, T. Carell and W. Zinth, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4369–4374 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS PubMed.
- M. A. Grodick, N. B. Muren and J. K. Barton, Biochemistry, 2015, 54, 962–973 CrossRef CAS PubMed.
- C. Guo, K. Wang, E. Zerah-Harush, J. Hamill, B. Wang, Y. Dubi and B. Xu, Nat. Chem., 2016, 8, 484–490 CrossRef CAS PubMed.
- S. M. Hart, J. L. Banal, M. A. Castellanos, L. Markova, Y. Vyborna, J. Gorman, R. Häner, A. P. Willard, M. Bathe and G. S. Schlau-Cohen, Chem. Sci., 2022, 13, 13020–13031 RSC.
- C. E. Crespo-Hernández, B. Cohen and B. Kohler, Nature, 2005, 436, 1141–1144 CrossRef PubMed.
- I. Buchvarov, Q. Wang, M. Raytchev, A. Trifonov and T. Fiebig, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4794–4797 CrossRef CAS PubMed.
- C. T. Middleton, K. d. L. Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernández and B. Kohler, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS PubMed.
- C. Su, C. T. Middleton and B. Kohler, J. Phys. Chem. B, 2012, 116, 10266–10274 CrossRef CAS PubMed.
- Y. Zhang, J. Dood, A. A. Beckstead, X.-B. Li, K. V. Nguyen, C. J. Burrows, R. Improta and B. Kohler, J. Phys. Chem. B, 2015, 119, 7491–7502 CrossRef CAS PubMed.
- V. A. Spata, W. Lee and S. Matsika, J. Phys. Chem. Lett., 2016, 7, 976–984 CrossRef CAS PubMed.
- F. D. Lewis, R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2018, 51, 1746–1754 CrossRef CAS PubMed.
- M. Duchi, M. P. O'Hagan, R. Kumar, S. J. Bennie, M. C. Galan, B. F. E. Curchod and T. A. A. Oliver, Phys. Chem. Chem. Phys., 2019, 21, 14407–14417 RSC.
- C. L. Kufner, W. Zinth and D. B. Bucher, ChemBioChem, 2020, 21, 2306–2310 CrossRef CAS PubMed.
- C. L. Kufner, D. B. Bucher and D. D. Sasselov, ChemSystemsChem, 2023, 5, e202200019 CrossRef CAS.
- W. Lee and S. Matsika, J. Phys. Chem. B, 2023, 127, 18–25 CrossRef CAS PubMed.
- T. Douki, M. Court, S. Sauvaigo, F. Odin and J. Cadet, J. Biol. Chem., 2000, 275, 11678–11685 CrossRef CAS PubMed.
- W. J. Schreier, T. E. Schrader, F. O. Koller, P. Gilch, C. E. Crespo-Hernández, V. N. Swaminathan, T. Carell, W. Zinth and B. Kohler, Science, 2007, 315, 625–629 CrossRef CAS PubMed.
- W. J. Schreier, J. Kubon, N. Regner, K. Haiser, T. E. Schrader, W. Zinth, P. Clivio and P. Gilch, J. Am. Chem. Soc., 2009, 131, 5038–5039 CrossRef CAS PubMed.
- C. Rauer, J. J. Nogueira, P. Marquetand and L. González, J. Am. Chem. Soc., 2016, 138, 15911–15916 CrossRef CAS PubMed.
- V. Pagès and R. P. Fuchs, Oncogene, 2002, 21, 8957–8966 CrossRef PubMed.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CrossRef CAS PubMed.
- A. Mees, T. Klar, P. Gnau, U. Hennecke, A. P. M. Eker, T. Carell and L.-O. Essen, Science, 2004, 306, 1789–1793 CrossRef CAS PubMed.
- W. Lee, G. Kodali, R. J. Stanley and S. Matsika, Chem.–Eur. J., 2016, 22, 11371–11381 CrossRef CAS PubMed.
- M. Zhang, L. Wang, S. Shu, A. Sancar and D. Zhong, Science, 2016, 354, 209–213 CrossRef CAS PubMed.
- M. Zhang, L. Wang and D. Zhong, Arch. Biochem. Biophys., 2017, 632, 158–174 CrossRef CAS PubMed.
- C. Tan, Z. Liu, J. Li, X. Guo, L. Wang, A. Sancar and D. Zhong, Nat. Commun., 2015, 6, 7302 CrossRef CAS PubMed.
- D. J.-F. Chinnapen and D. Sen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 65–69 CrossRef CAS PubMed.
- M. R. Holman, T. Ito and S. E. Rokita, J. Am. Chem. Soc., 2007, 129, 6–7 CrossRef CAS PubMed.
- Z. Pan, J. Chen, W. J. Schreier, B. Kohler and F. D. Lewis, J. Phys. Chem. B, 2012, 116, 698–704 CrossRef CAS PubMed.
- R. Szabla, M. Zdrowowicz, P. Spisz, N. J. Green, P. Stadlbauer, H. Kruse, J. Šponer and J. Rak, Nat. Commun., 2021, 12, 3018 CrossRef PubMed.
- K. V. Nguyen and C. J. Burrows, J. Am. Chem. Soc., 2011, 133, 14586–14589 CrossRef CAS PubMed.
- D. B. Bucher, C. L. Kufner, A. Schlueter, T. Carell and W. Zinth, J. Am. Chem. Soc., 2016, 138, 186–190 CrossRef CAS PubMed.
- V. Piccinni, S. Reiter, D. Keefer and R. de Vivie-Riedle, J. Phys. Chem. A, 2020, 124, 9133–9140 CrossRef CAS PubMed.
- R. Szabla, H. Kruse, P. Stadlbauer, J. Šponer and A. L. Sobolewski, Chem. Sci., 2018, 9, 3131–3140 RSC.
- A. A. Beckstead, Y. Zhang, M. S. d. Vries and B. Kohler, Phys. Chem. Chem. Phys., 2016, 18, 24228–24238 RSC.
- C. L. Kufner, S. Krebs, M. Fischaleck, J. Philippou-Massier, H. Blum, D. B. Bucher, D. Braun, W. Zinth and C. B. Mast, Sci. Rep., 2023, 13, 2638 CrossRef CAS PubMed.
- S. Ranjan and D. D. Sasselov, Astrobiology, 2016, 16, 68–88 CrossRef CAS PubMed.
- S. Ranjan and D. D. Sasselov, Astrobiology, 2017, 17, 169–204 CrossRef CAS PubMed.
- J. Xu, M. Tsanakopoulou, C. J. Magnani, R. Szabla, J. E. Šponer, J. Šponer, R. W. Góra and J. D. Sutherland, Nat. Chem., 2017, 9, 303–309 CrossRef CAS PubMed.
- S. J. Roberts, R. Szabla, Z. R. Todd, S. Stairs, D.-K. Bučar, J. Šponer, D. D. Sasselov and M. W. Powner, Nat. Commun., 2018, 9, 4073 CrossRef PubMed.
- J. Xu, V. Chmela, N. J. Green, D. A. Russell, M. J. Janicki, R. W. Góra, R. Szabla, A. D. Bond and J. D. Sutherland, Nature, 2020, 582, 60–66 CrossRef CAS PubMed.
- B. W. F. Colville and M. W. Powner, Angew. Chem., 2021, 133, 10620–10624 CrossRef.
- J. Xu, N. J. Green, D. A. Russell, Z. Liu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 14482–14486 CrossRef CAS PubMed.
- N. J. Green, J. Xu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 7219–7236 CrossRef CAS PubMed.
- R. Szabla, in Prebiotic Photochemistry: From Urey–Miller-like Experiments to Recent Findings, ed. F. Saija and G. Cassone, The Royal Society of Chemistry, 2021, ch. 5, pp. 79–106 Search PubMed.
- I. Anusiewicz, I. Świerszcz, P. Skurski and J. Simons, J. Phys. Chem. A, 2013, 117, 1240–1253 CrossRef CAS PubMed.
- W. Lee and S. Matsika, Faraday Discuss., 2019, 216, 507–519 RSC.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CAS.
- C. Hättig, Advances in Quantum Chemistry, Academic Press, 2005, vol. 50, pp. 37–60 Search PubMed.
- H. E. Johns, S. A. Rapaport and M. Delbrück, J. Mol. Biol., 1962, 4, 104–114 CrossRef CAS PubMed.
- W. J. Schreier, P. Gilch and W. Zinth, Annu. Rev. Phys. Chem., 2015, 66, 497–519 CrossRef CAS PubMed.
- C. L. Kufner, PhD thesis, Ludwig-Maximilians-Universität München, 2018.
- S. J. Crucilla, D. Ding, G. G. Lozano, J. W. Szostak, D. D. Sasselov and C. L. Kufner, Chem. Commun., 2023, 59, 13603–13606 RSC.
- V. Petropoulos, L. Uboldi, M. Maiuri, G. Cerullo, L. Martinez-Fernandez, E. Balanikas and D. Markovitsi, J. Phys. Chem. Lett., 2023, 14, 10219–10224 CrossRef CAS PubMed.
- F. Masson, T. Laino, I. Tavernelli, U. Rothlisberger and J. Hutter, J. Am. Chem. Soc., 2008, 130, 3443–3450 CrossRef CAS PubMed.
- D. Huang, S. Chen, J. Pu, X. Tan and Y. Zhou, J. Phys. Chem. A, 2019, 123, 2025–2039 CrossRef CAS PubMed.
- B. Bauer, R. Sharma, M. Chergui and M. Oppermann, Chem. Sci., 2022, 13, 5230–5242 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, theoretical methods and additional results. Computational data, including geometries and results can be found under https://doi.org/10.6084/m9.figshare.24711825. See DOI: https://doi.org/10.1039/d3sc04971j |
| This journal is © The Royal Society of Chemistry 2024 |