
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous determination of Cd, Pb, Cu and Zn as total and labile fractions in soil using a small-sized electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometer after diffusive gradients in thin-film passive accumulation†
Simion Bogdan
Angyus
abc,
Marin
Senila
a,
Eniko
Covaci
 bc,
Michaela
Ponta
bc,
Maria
Frentiu
a and
Tiberiu
Frentiu
*bc
bc,
Michaela
Ponta
bc,
Maria
Frentiu
a and
Tiberiu
Frentiu
*bc
aNational Institute for Research and Development of Optoelectronics INOE 2000 INCD Bucharest, Research Institute for Analytical Instrumentation, Donath 67, 400293 Cluj-Napoca, Romania
bBabes-Bolyai University, Faculty of Chemistry and Chemical Engineering, Arany Janos 11, 400028 Cluj-Napoca, Romania. E-mail: tiberiu.frentiu@ubbcluj.ro
cBabes-Bolyai University, Research Center for Advanced Analysis, Instrumentation and Chemometrics, Arany Janos 11, 400028 Cluj-Napoca, Romania
First published on 6th November 2023
Abstract
The study presents for the first time the figures of merit of a completely miniaturized instrumentation based on a capacitively coupled plasma microtorch as the core element interfaced with a small-sized electrothermal vaporization device and a low-resolution microspectrometer for the simultaneous determination of Cd, Pb, Cu and Zn by optical emission spectrometry in soil as total and labile fractions after diffusive gradients in thin-film (DGT) accumulation. The coupling of the low-power and low-argon consumption plasma (15 W; 150 mL min−1) with the DGT passive accumulation technique, although requiring a too-long time for sample preparation, allowed a considerable improvement of the detection limits and avoidance of the non-spectral matrix effects, otherwise a recognized process in low power microplasmas, when a complex matrix is analysed, like environmental samples. The detection limits for the total content in soil were (mg kg−1), 0.10(Cd), 0.40(Pb), 0.15(Cu), and 0.03(Zn), one order of magnitude better than in the procedure without DGT accumulation and 10–3300-times lower than the guide values in soil. In the DGT-based labile fraction exhibiting the highest bioavailability the detection limits were (μg kg−1) 0.01(Cd, Cu, and Zn) and 0.03(Pb), which allowed the determination of Cd, Pb, Cu and Zn in the concentration range (μg kg−1) of 0.3–2.0, 0.8–18.4, 2.4–56.3 and 9.4–60.6, respectively. Validation through the analysis of certified reference materials (CRMs) showed a recovery of 85–123% with a relative expanded uncertainty of 19–35% (k = 2) for the total content of analytes. The analysis of the certified reference materials highlighted that the DGT accumulation was not affected by the multielemental matrix, since the experimental diffusion coefficients of the analytes were similar in the four analyzed CRMs and to those provided by the manufacturer, respectively. Precision for the measurements of the total content and DGT-labile fraction in real samples evaluated from the combined uncertainty was 10–19% and 10–15%, respectively. The Bland–Altman plot applied to the results of real samples indicated the lack of statistical differences versus line-source graphite furnace atomic absorption spectrometry for both the total content and DGT-based labile fraction.
Introduction
Because of the exposure risk of the population and environment to toxic elements (Cd, Pb, Hg, As, etc.) and potentially toxic, such as Cu, Zn and Cr, the development of sample preparation procedures and advanced analytical techniques based on atomic and mass spectrometry for the determination and monitoring of these elements in the environment, food, beverages and biological materials continues to be a challenge for academic and routine analysis laboratories.1,2 High-sensitivity multielement analytical techniques, such as high-resolution continuum source atomic absorption spectrometry (HR-CS-AAS), inductively coupled plasma mass spectrometry (ICP-MS), inductively coupled plasma optical emission spectrometry (ICP-OES), laser-induced breakdown spectrometry (LIBS), total-reflection X-ray fluorescence spectrometry (TXRF) and energy dispersive X-ray fluorescence spectrometry (EDXRF) are still the most commonly used for the determination of element traces.3 To achieve the required sensitivity, these spectrometric techniques are often coupled with preconcentration/accumulation procedures by solid phase extraction, liquid–liquid extraction, direct liquid or solid micro-sampling by electrothermal vaporization (ETV) and laser ablation (LA), or derivatization by generating chemical vapor using the classical method or the UV photoinduced procedure.4–15 Although the ICP-OES and ICP-MS techniques are high-performing and provide excellent sensitivity, the laboratory-scale equipment involves high operating costs related to energy consumption and Ar as a support gas for plasma generation and is still not easily accessible for many laboratories. On the other hand, line-source atomic absorption is by its very nature a sequential, slow method with high sample consumption and less competitive with the simultaneous methods in the current state of the art. The methods based on derivatization, providing a highly sensitive and selective determination of several elements through the avoidance of the non-spectral interferences, although easily accessible in laboratories, remain complicated by the large number of parameters that need to be optimized, and the use of expensive and sometimes unstable reagents. The alternative is the unconventional technology using completely miniaturized instrumentation, in which the core element is a microplasma-based excitation/ionization source interfaced with a low-resolution microspectrometer or mass spectrometer.16,17 This led to the development of very attractive analytical applications obeying the principles of green analytical chemistry (GAC) in spectrometry and, more recently, of white analytical chemistry (WAC).18–22 Since introducing a gaseous sample into a microplasma was found to be a relatively simple task, the microplasma technology was mainly developed as a specific high-sensitivity detector by optical emission spectrometry (OES) and mass spectrometry (MS) in gas chromatography (GC) or as a device for introducing derivatized species of elements generated under UV irradiation by plasma, known as plasma-induced vapor generation technology.23–28 However, there are several limitations in the development of microplasma analytical technologies, the most important of which are the lack of stability and poor excitation capability in the case of samples with a complex matrix, because of the low operating power.16 Thus, the discharge stability is disturbed in the case of direct introduction of liquid samples because of low tolerance for water-loaded aerosols. It was shown that this shortcoming could be overcome by introducing liquid microsamples by ETV using the ideal miniaturized electrothermal vaporized-microplasma coupling, which provided the simultaneous determination of several elements with limits of detection (LODs) at the ng mL−1 or pg level, similar to ICP-OES.29,30 In our laboratory it was demonstrated that a fully miniaturized analytical system consisting of a small-sized electrothermal vaporization device (SSETV), a low-power (15 W) and low argon consumption (150 mL min−1) capacitively coupled plasma microtorch (μCCP) interfaced with a low-resolution microspectrometer with a full width at half maximum (FWHM) of 0.35 nm accomplished instantaneous and efficient vaporization of a liquid microsample and high flow of analytes into plasma. Under these conditions the simultaneous determination of several elements was achieved, including the priority hazardous ones (Hg, Cd and Pb) in environmental samples and foods with LODs similar to those of ICP-OES.31–34 Unfortunately, elemental determination using a small-sized electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometer (SSETV-μCCP-OES) proved to be affected by non-spectral interferences when the Rh filament was heated around 1500 °C, a temperature necessary for the vaporization of less volatile elements (such as Cu), so it was necessary to use standard addition instead of external calibration for measurements. However, the non-spectral effects could be overcome by the selective vaporization of analytes at 1300 °C and was successfully applied to the speciation of Hg as Hg2+ and CH3Hg+ in food, and determination of As, Sb, Bi, Se, Te, Hg and Sn in environmental samples.35,36The diffusive gradients in thin-film (DGT) technique proposed by Davison and Zhang37,38 has been extensively used in the analysis of surface water, soil, water sediment, food, etc. as a passive sampling technique.39,40 The DGT technique is based on the diffusion of labile species through a diffusive gel and passive accumulation in a suitable binding gel over a deployment period of the order of hours or days followed by the elution of analytes in solution, e.g. 1 mol L−1 HNO3 for metals. Next, it follows their quantification in the eluent and calculation of the time-averaged mean concentration in the uptake solution (cDGT) based on an equation derived from Fick's first law, which takes into account the characteristics of the DGT gel.40
The DGT passive accumulation technique mimics the retention of metal ions in soil solution by plant roots at the root–soil interface, which involves first the transfer of ions from the solid phase of soil into solution, and then absorption by the roots. Studies regarding the bioavailability of toxic metals have demonstrated that the labile fraction in the soil solution (cDGT) obtained after the DGT passive sampling was a very good indicator to measure the bioavailability of metals through roots, given the good correlation coefficients between metal concentrations in plants and cDGT.41–44
In two inter-laboratory studies, the DGT procedure using the standard DGT Chelex-100 devices (DGT Research Ltd., Lancaster, UK) coupled exclusively with simultaneous multielement analysis techniques such as ICP-MS and ICP-OES was validated as in situ sampling for surface water monitoring.45,46 Until now, there have been no studies on the validation of the passive DGT sampling for the determination of the total and labile fraction in soil using miniaturized instrumentation with microplasma sources equipped with low-resolution microspectrometers. The first study published by us highlights that the coupling between the DGT passive sampling with SSETV-μCCP-OES (DGT-SSETV-μCCP-OES) instrumentation is suitable for surface water monitoring related to the determination of elements with excitation energy below 7 eV.47 Consequently, a highly sensitive method providing improved LODs was developed meeting the principles of GAC (AGREEprep) and WAC (Red/Green/Blue-RGB-12 concept) and providing high both green and white scores as a result of cost-effective SSETV-μCCP-OES instrumentation and in situ DGT passive sampling.47 Increasing the technological maturity of the microplasma sources from the laboratory scale prototype to mature commercial instrumentation is a challenge and involves the development and validation of cost-effective and easy-to-run applications for the determination of priority hazardous elements and other traces providing analytical performance similar to the much more sophisticated laboratory equipment.
In line with the state of the art, the present study is focused for the first time on the development and validation of an analytical method free of non-spectral interference for the determination of the total metal content and labile fraction in soil, after DGT passive accumulation coupled with SSETV-μCCP-OES equipped with a low-resolution microspectrometer. The study was carried out for the simultaneous determination of Cd, Pb, Cu and Zn, four elements of interest for the environmental assessment. The working hypothesis in avoiding the non-spectral effects in the determination of these elements is sustained by the high selectivity of the Chelex-100 commercial resin towards transition elements, above that for alkaline and alkaline-earth elements, which would allow separation of analytes from the multielemental matrix of soil.48 The simultaneous determination by SSETV-μCCP-OES coupled with DGT is ensured by the fact that a certain binding resin (e.g., Chelex-100) can be used for the sampling/accumulation of several transition metals. The DGT passive accumulation was conducted in ex situ batch experiments. The characteristics of the emission spectrum recorded by SSETV-μCCP-OES in the eluate after the accumulation by DGT samplers and a study about the analytical performance are presented. The DGT-SSETV-μCCP-OES method was validated in terms of LODs in comparison with those without DGT accumulation, accuracy for the total content in certified reference materials (CRMs) of soil and precision from measurements of total content and DGT-based labile fractions in several real soil samples. The validation of the DGT-SSETV-μCCP-OES method was also conducted against the line-source graphite furnace atomic absorption spectrometry (GFAAS). The results were compared using the statistical Tukey's and Dunnett's tests and Bland–Altman plot (p > 0.05).49–51
Experimental
SSETV-μCCP-OES instrumentation and DGT passive accumulation
Determinations were performed on a completely miniaturized SSETV-μCCP-OES set-up previously described.47,52,53 The instrumentation consists of a μCCP (Babes-Bolyai University, Cluj-Napoca, Romania) with a Mo tip microelectrode (1.25 mm diameter, Goodfellow, Cambridge, UK) powered by an r.f. generator (10–30 W, 13.56 MHz, 15 × 17 × 24 cm3, Technical University, Cluj-Napoca, Romania) for plasma generation at 50–200 mL min−1 Ar in a quartz tube (5 mm i.d., 25 mm length, 160 nm cut-off, H. Baumbach & Co. Ltd., Ipswich, UK). A home-made SSETV (Babes-Bolyai University, Cluj-Napoca, Romania) consisting of 4 turns with 1.5 mm diameter of Rh filament (250 μm diameter, 99.9% purity, tempered and annealed Goodfellow, Cambridge, UK), mounted in a vaporization quartz chamber was used for liquid microsample evaporation. A programmable DC Power Supply Tenma, model 72-13360 (Premier Farnel, Leeds, UK) with adjustable heating ramp up to 60 V CC, output current up to 15 A, 1 mV/1 mA resolution and an USB interface, was used to heat the Rh filament. A Maya2000 Pro microspectrometer equipped with a charge-coupled detector (CCD), 165–309 nm spectral range, 0.35 nm FWHM, Ar purged CCD chamber (Ocean Optics, Dunedin USA) was used for recording the episodic emission spectra. A two-way valve SMCEVT307-5 DZ-01 F-Q supplied by the DC power source DC HY3003 (Mastech, Premier Farnell, Leeds, UK) was used to direct the Ar flow into the vaporization chamber or bypass it. The basic assembly is presented in ref. 47 The capacitively coupled plasma operated at 15 W appears as a diffuse bluish discharge with a length of 8–10 mm and a width of 2–3 mm developed at the tip of the Mo microelectrode.31 In the superficial luminescence zone, over the 0–1 mm viewing heights, appears the element excitation that allows the simultaneous multielemental emission spectra recording at 0.8 mm observation height through a lens, without fiber optics.53 Although its operating power is low, the plasma energy is efficiently used only for atomization and excitation processes, because the sample is introduced into the plasma as dry vapor. It has been shown that the μCCP operated at 15 W power has an excitation capability similar to other microplasma sources so that the emission spectrum consists of few resonant and non-resonant spectral lines with excitation energies (Eex) below 7 eV.47 Due to the simple spectrum, a low-resolution microspectrometer such as Maya2000 Pro (FWHM 0.35 nm) could be used for episodic spectra recording. As a reference, the argon-hydrogen microplasma, studied by Karanassios' group and developed on a hybrid chip for elemental analysis of microsamples, operated at a lower power (4 W) compared to our plasma, has a length/width of 20/0.75 mm and allows the excitation of elements with Eex below 7 eV.30The optimum working conditions for the determination of total and DGT-based labile fraction of Cd, Pb, Cu and Zn in soil using SSETV-μCCP-OES equipment are summarized in the ESI (ESI, Section 1 and Table S1).†
Commercial DGT devices (DGT Research Ltd., Lancaster, UK) equipped with agarose cross-linked polyacrylamide (APA) diffusive gel, membrane filters (polyethersulphone) and Chelex-100 binding resin of iminodiacetate with high selectivity for transition metals were used for the accumulation of the target ions.48 All assembly/disassembly operations of the DGT devices, handling and sample processing were performed in a clean room.
For the determination of Cd, Pb, Cu and Zn in eluates by atomic absorption spectrometry, the PerkinElmer PinAAcle 900T GFAAS spectrometer (Norwalk, USA) was used under the operating conditions previously presented.54 The spectrometer is equipped with electrodeless discharge lamps (EDLs) for Cd and Pb, and hollow cathode lamps (HCLs) for Cu and Zn, a transversally heated graphite furnace atomizer and allows the background correction by the longitudinal Zeeman effect. Chemical modifiers were used according to the recommendation of the manufacturer, using 5 μL of chemical modifier for a sample aliquot of 20 μL. The PinAAcle 900T spectrometer can be used for sequential multielemental analysis of any type of sample, including soil analysis. Compared to this common sequential atomic absorption technique of high sensitivity, the (DGT)-SSETV-μCCP-OES technique is simultaneous. It requires validation to check if it is an appropriate alternative for the determination of Cd, Pb, Cu and Zn in total and DGT-based labile fraction in soil.
The ICP-OES 5300 Optima DV spectrometer from PerkinElmer (Waltham, Massachusetts, USA) was used for the determination of Na, K, Ca, Mg, Sr, Fe, Ni, Cr, Co and Mn in a multielemental matrix. The 761 Compact IC Metrohm ion chromatograph, Metrohm (Herisau, Switzerland) was used for the determination of anions (F−, Cl−, NO3− and SO42−) in soil solution in which the DGT-based labile fraction of Cd, Pb, Cu and Zn was determined. The concentration of dissolved organic carbon (DOC) and dissolved inorganic carbon (DIC) fractions was quantified in soil solution using the 2100S Multi N/C analyzer, Analytik Jena (Jena, Germany). The Multi 350i, Geotech (Denver, USA) was used for pH measurement of soil solution and for pH adjustment of solutions in which the DGT devices were immersed for the determination of total content of Cd, Pb, Cu and Zn in soil. All these procedures were previously described.47
The Milli-Q water purification system Millipore (Bedford, USA) and the microwave digester Berghof MWS3+ (Berghof, Germany) were employed for the preparation of ultrapure water and microwave-assisted digestion of the samples, respectively.
Reagents, solutions and certified reference materials (CRMs)
Hydrochloric acid ultrapure (30%), nitric acid ultrapure (60%), ammonia solution (25%) suprapur, and ICP multielement standard solution IV 1000 mg L−1 were purchased from Merck (Darmstadt, Germany). Multielement (n = 7 point calibration curves for Cd, Pb, Cu and Zn) solutions containing 0–100 μg L−1 Cd and 0–1000 μg L−1 Pb, Cu and Zn in 2% (v/v) HNO3 were prepared to be used for the calibration of the SSETV-μCCP-OES equipment. Multielement solutions of Cd, Pb, Cu and Zn in the range of 0–10 μg L−1 in 2% (v/v) HNO3 were used for the calibration of the GFAAS instrument. Arsenic calibration standards in the range of 0–100 μg L−1 in 2% (v/v) HNO3 were also used for the determination of As by GFAAS and 0–1000 μg L−1 by SSETV-μCCP-OES at 189.042 nm, respectively. The appropriate chemical modifiers (PerkinElmer Pure, Shelton, USA) were used to compensate for the matrix effects in GFAAS (Cd and Pb: 1% NH4H2PO4 + 0.06% Mg(NO3)2, Cu: 0.1% Pd(NO3)2 + 0.06% Mg(NO3)2, and Zn: 0.1% Mg(NO3)2). A solution of 0.1 mol L−1 NH3 was used to adjust the pH in the uptake solution to 4.0 ± 0.1 prior to immersion of the DGT devices for the passive accumulation of analytes for the determination of their total concentration in soil. A solution of 1 mol L−1 HNO3 was used for the elution of the target elements from the binding gel.The accuracy of the (DGT)-SSETV-μCCP-OES and (DGT)-GFAAS methods for total concentration of Cd, Pb, Cu and Zn was checked by analyzing four CRMs: SQC001 Metals in soil and CRM048 Trace metals – Sand 1 from Sigma Aldrich Chemie GmbH (Taufkirchen, Germany), Metranal-34 Loam from Analytika Spol (Vysocany, Czech Republic) and CRM025-050 Metals in soil from Resource Technology Corporation (Laramie, USA), after ex situ DGT passive accumulation at pH 4.0 ± 0.1 in 100 mL solution obtained by 10 and 20-fold dilution of aliquot volumes of digest. The CRMs were used also to study the uptake kinetics of the target elements and determination of the diffusion coefficients and elution factors.
Glassware, the DGT supports, storage boxes, elution tubes and digestion vessels were cleaned by soaking in 10% (v/v) HNO3 for 12 h and rinsing with ultrapure water, dried and stored in clean plastic bags.46
Sample preparation
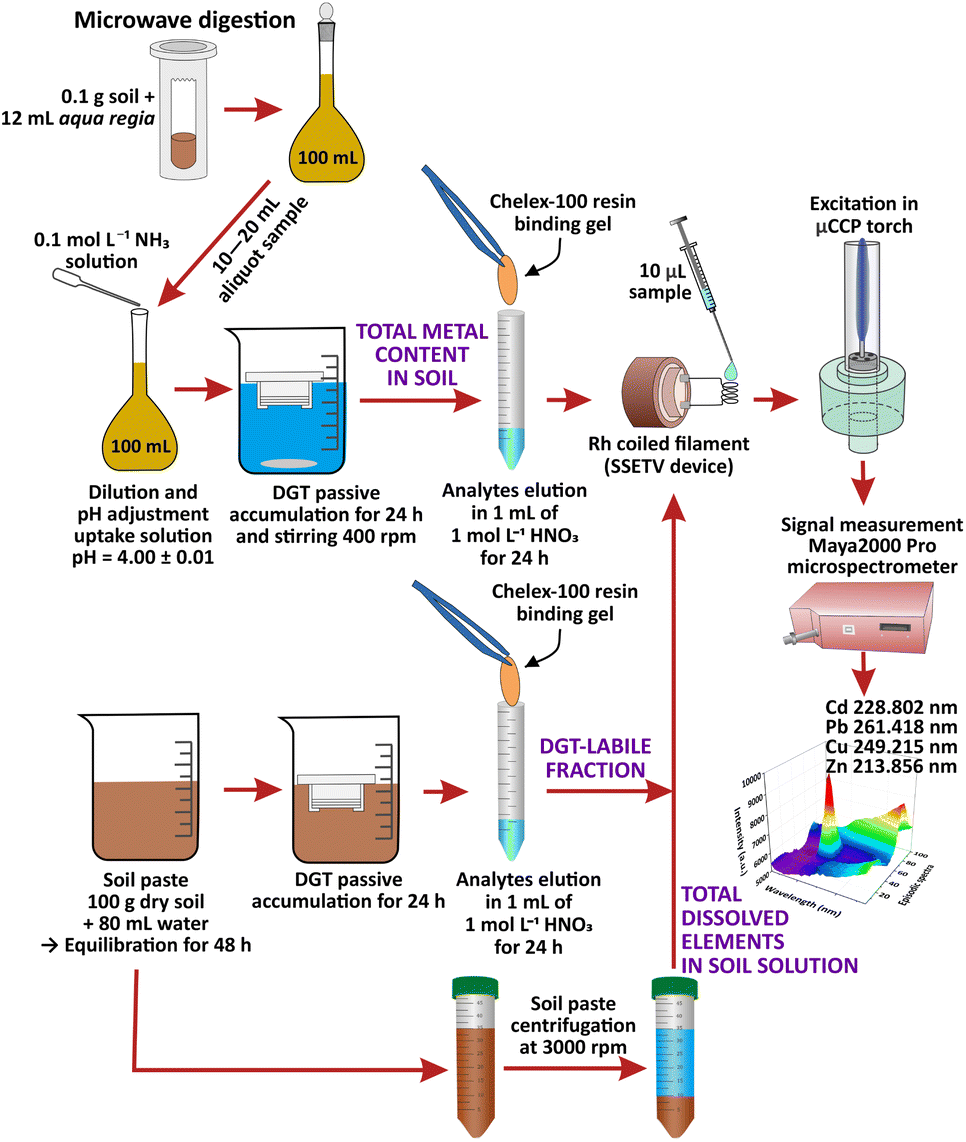 | ||
| Fig. 1 Schematic diagram of soil sample preparation for the determination of total content and labile fraction of Cd, Pb, Cu and Zn in soil by SSETV-μCCP-OES after DGT passive accumulation on Chelex-100 resin. | ||
Amounts of 2.5 kg agricultural soil of luvisol type with clay texture were collected from the topsoil layer (20 cm depth), from three locations, north-western Romania (city of Baia Mare), in the vicinity of former tailing ponds, where the tailings and wastewater resulted from Pb and Cu mining and ores processing, were stored. The samples were air-dried at room temperature, then crushed, homogenized and sieved to <2 mm to remove stones and roots. Next, 1 kg sample was oven-dried at 105 ± 5 °C, ground in a ball mill and sieved to <100 μm. Amounts of 0.1 g soil sample were subjected to microwave-assisted digestion with 12 mL aqua regia using a 4-step protocol previously presented.31 The digest was diluted to 100 mL with ultrapure water and filtered (0.45 μm). The uptake solutions for DGT accumulation were prepared from aliquot volumes of 10 to 20 mL diluted with water and the pH adjustment to 4.0 ± 0.1 by potentiometric titration with 0.1 mol L−1 NH3 and finally made up to 100 mL. The working pH value of 4.0 ± 0.1 was selected based on a previous study showing that in the 4–7 pH range the accuracy of the ratio of the concentration determined using the DGT sampling to the analytical concentration of the uptake solution (cDGT/csol) was in the range of 0.83–1.13 with a relative extended uncertainty of ±20% (k = 2).47 After assembling the DGT devices (filter membrane of polyethersulphone, the APA diffusive gel and the Chelex-100 binding gel) were immersed for 24 h in the uptake solution in triplicate (3 × 100 mL solution) avoiding touching the diffusion surface with the vessel walls. The solutions were gently stirred at 400 (× g). The solution temperature measured at least 3 times during accumulation was 21 ± 1 °C. After exposure, the DGT devices were extracted from the uptake solutions, rinsed with ultrapure water jets and disassembled. The Chelex-100 gel was separated with tweezers, washed with ultrapure water and subjected to elution of analytes by immersion in 1 mL of 1 mol L−1 HNO3 for 24 h at 21 ± 1 °C in a clean tube. The eluate was analyzed for the determination of the total content of Cd, Pb, Cu and Zn in soil by SSETV-μCCP-OES and GFAAS using external calibration.
The CRM soils analyzed for the assessment of the accuracy of the DGT-SSETV-μCCP-OES method were processed using the same protocol and 24 h accumulation time.
Calculation of the total concentration of metals and DGT-based labile fractions is presented in the ESI (Section 2).†
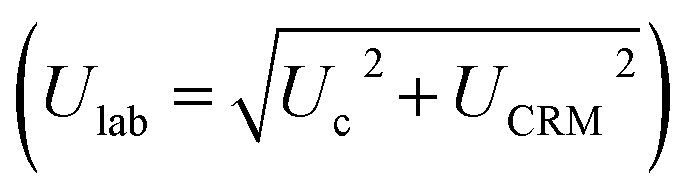 . The average concentrations were calculated using both the experimental diffusion coefficients and elution factors (Dexp, fexp), and those provided by the manufacturer. The existence of significant differences (p > 0.05) between the found mean and certified value was investigated by verifying the fulfillment of relation Δm < Ulab, where Δm is the difference between the found mean and certified value. The bias between the results obtained by (DGT)-SSETV-μCCP-OES and (DGT)-GFAAS methods was statistically checked using Tukey's test, while the bias of the two methods against the certified values by Dunnett's test (p > 0.05).49,50
. The average concentrations were calculated using both the experimental diffusion coefficients and elution factors (Dexp, fexp), and those provided by the manufacturer. The existence of significant differences (p > 0.05) between the found mean and certified value was investigated by verifying the fulfillment of relation Δm < Ulab, where Δm is the difference between the found mean and certified value. The bias between the results obtained by (DGT)-SSETV-μCCP-OES and (DGT)-GFAAS methods was statistically checked using Tukey's test, while the bias of the two methods against the certified values by Dunnett's test (p > 0.05).49,50
The acceptance criterion for accuracy was that the interval (R ± Ulab%) contained the 100% theoretical recovery value with a confidence limit of ± 30%, considered for the coupling of a spectrometric method with DGT passive accumulation.45,46
The precision of the DGT-SSETV-μCCP-OES method for the determination of the total content and DGT-based labile fraction of analytes was evaluated from the percentage relative standard deviation (RSD%) resulting from ulab for DGT accumulation in triplicate for each real sample. For the validation of the method, a threshold of maximum 30% RSD was considered.45,46 The precision of the DGT-SSETV-μCCP-OES method was compared to that of DGT-GFAAS.
Results and discussion
Matrix characterization of CRMs and soil samples
The agricultural Luvisols analyzed in this study contained 25–33% sand, 40–43% silt and 27–32% clay (ESI, Section 3 and Table S2†). The concentration of the multielemental matrix in the uptake solution of the analyzed CRMs in which the DGT devices were immersed for the determination of the total content of analytes is presented in the ESI (Section 3 and Table S3).† Tables S4 and S5† (ESI, Section 3) exhibit the matrix characterization in the uptake solution resulting after digestion in aqua regia and soil solution obtained by centrifugation of soil paste. The multielemental matrix is dominated by Ca, Mg, Fe, K and Na as cations, and SO42− and NO3− as anions. The carbon speciation in the soil solution revealed that the DOC fraction was dominant (32–170 mg L−1) compared to DIC (6.5–55 mg L−1).The emission spectrum of a solution obtained by CRM048-50G sample digestion, that contains concentrations of (μg L−1) 90(Cd), 320(Pb), 80(Cu), 420(Zn) and 100(As) are presented in the ESI (Section 3, Fig. S1).† Emission of As at 189.042 nm and the spectral interference of the Cd line at 228.802 nm with As at 228.812 nm can be observed in the emission spectrum, although the As emission in the microplasma was found to be 25 times lower than that of Cd. Therefore, As was considered as part of the matrix composition. According to the 3σ criterion, the LOD for As at 189.042 nm in SSETV-μCCP-OES was 0.014 mg L−1, while in GFAAS it was 0.001 mg L−1. Consequently, As concentration in uptake and soil solution was determined by GFAAS. However, the experimental determination of total Cd, Pb, Cu and Zn by SSETV-μCCP-OES with DGT passive accumulation was achieved in solutions with a supplementary dilution of 10–20 times after digestion. The dilution was performed to diminish the concentration of concomitants such as Fe, Mn, As, etc. and thus to minimize the competing process of their retention in the binding gel. Otherwise, there would be saturation of the binding gel with concomitants, the decrease in the accumulation of analytes and, thereby, the decrease of sensitivity of the SSETV-μCCP-OES method. In other words, the objective was to achieve a quantitative separation of the analytes from concomitants through retention by the Chelex-100 gel and an adequate preconcentration factor, to improve sensitivity, correlated with the deployment period of DGT devices.
The capability of the DGT device with Chelex-100 gel to separate Cd from As in the accumulation step of analytes was also checked in the eluate. The emission spectrum of the eluate, obtained from CRM048-50G at the determination of total analyte content, presented in the ESI (Section 3 and Fig. S2),† demonstrates the lack of As lines in the spectrum. Consequently, the determination of total/DGT-based labile fraction of Cd at the most sensitive line of 228.802 nm in soil by SSETV-μCCP-OES was addressed in CRM analysis with concentration of Cd in the range of 1.44–369 mg kg−1 and As in the range of 42.4–339 mg kg−1, as concomitant. Arsenic concentration in uptake and soil solution determined by GFAAS is shown in the ESI (Section 3 and Tables S1–S3).†
Working conditions for the DGT passive accumulation. Determination of experimental diffusion coefficients and elution factors (Dexp and fexp)
The DGT passive accumulation technique was validated in terms of Dexp and fexp. The experimental value of the diffusion coefficient was calculated from the slope of the curve for the linear accumulation of analytes in time, in which the analyte accumulated mass/analyte mass in the uptake solution ratio was plotted against the deployment time (s). The accumulated mass of analyte (M) in the binding gel was calculated as the difference between the initial and final mass in the uptake solution, taking into account the solution volume and the concentration determined before and after DGT accumulation. The accuracy of the diffusion coefficient (Dexp ± Ulab, k = 2) was assessed based on the uncertainty of the uptake kinetics slope for the four CRMs analyzed and that of the element concentration in solutions used in the study of the uptake kinetics, and the influence of temperature on diffusion for ±1 °C. The studies were conducted in batch mode by immersing the DGT devices for 8–48 h in 100 mL uptake solutions with pH 4.00 ± 0.01 at 21 ± 1 °C prepared from the digests of the four CRMs in aqua regia. The expression for Dexp (cm2 s−1) was: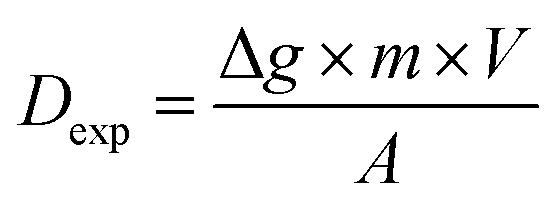 | (1) |
The kinetic curves presented in Fig. 2 show a linear increase of the analyte accumulation over the immersion period, with determination coefficients in the range of 0.9965–0.9977 and analyte retention (%) of 10–38(Cd), 12–55(Pb), 8–37(Cu) and 13–42(Zn). The slope of the uptake kinetics curve was statistically independent of the nature of the CRM sample, which means that reproducibility was better than 5% for the diffusion coefficients through the APA gel and retention of analytes by the Chelex-100 binding resin from one sample to another. In other words, the multimineral matrix in the uptake solution (ESI, Section 3 and Table S3†) has no significant effect on the diffusion and retention of analytes.
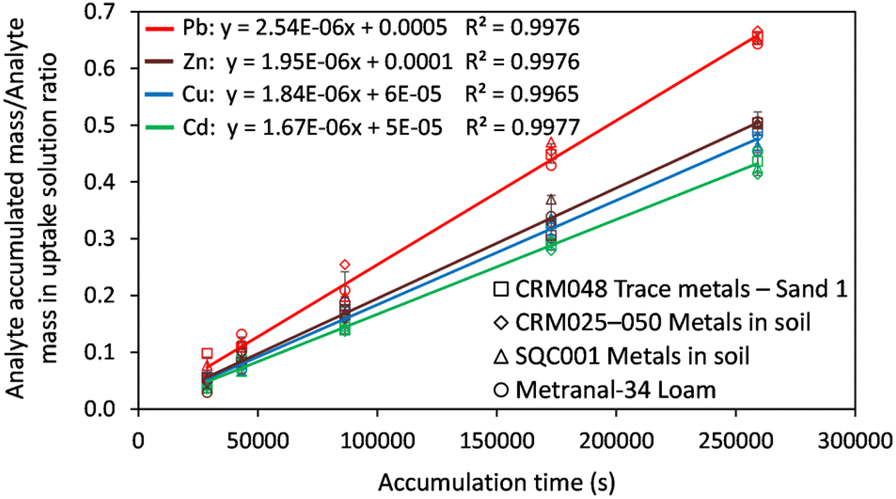 | ||
| Fig. 2 Uptake kinetics of target elements by Chelex-100 DGT for the uptake solution prepared from four CRMs. Conditions: volume of the uptake solution: 100 mL prepared by 10-fold dilution of the digested CRMs; pH: 4.0 ± 0.1; temperature: 21 ± 1 °C; elution in 1 mL solution of 1 mol L−1 HNO3. Error bars correspond to the standard deviation obtained for the four CRMs. | ||
The fexp was calculated as the ratio between the analyte mass in the eluate and the mass difference of the analyte in the uptake solution before and after removing the DGT device. The expanded uncertainty of fexp was evaluated from a study on the elution repeatability (n = 3) and uncertainty in the mass balance in the analysis of CRMs (fexp ± Ulab, k = 2). The found values for fexp and Dexp were compared with those provided by the manufacturer and reported in the literature (Table 1).
| Parameter | Experimental value (mean ± Ulab)a,b | Values recommended by the manufacturer (mean ± Ulab)a,b | ||||||
|---|---|---|---|---|---|---|---|---|
| Cd | Pb | Cu | Zn | Cd | Pb | Cu | Zn | |
| a Diffusion coefficient determined for accumulation by Chelex-100 resin at pH 4.0 ± 0.1 and 21 ± 1 °C; Ulab is the absolute expanded uncertainty (k = 2) assessed using the uncertainty of the uptake kinetics slope and influence of the temperature for a variation of ±1 °C. b Elution factor in 1 mL of 1 mol L−1 HNO3 solution for 24 h; Ulab is the expanded uncertainty (k = 2) for 3 parallel measurements for each CRM. | ||||||||
| D (cm2 s−1)10−6 | 5.00 ± 0.38 | 7.60 ± 0.50 | 5.50 ± 0.40 | 5.82 ± 0.38 | 5.46 ± 0.32 | 7.19 ± 0.40 | 5.58 ± 0.32 | 5.44 ± 0.30 |
| f e | 0.89 ± 0.06 | 0.91 ± 0.08 | 0.76 ± 0.10 | 0.81 ± 0.08 | 0.80 ± 0.06 | 0.80 ± 0.06 | 0.80 ± 0.06 | 0.80 ± 0.06 |
The data in Table 1 show a good agreement between the experimental values (Dexp and fexp), and the standard values recommended by the manufacturer of the DGT devices. Therefore, the values found (Dexp and fexp) and those recommended by the manufacturer were used to calculate the total content and DGT-based labile fraction of Cd, Cu, Pb and Zn in soil CRMs and all test samples. The results were statistically compared according to the validation section of the DGT-SSETV-μCCP-OES method.
Limits of detection
In a previous study, the instrumental LODs for the SSETV-μCCP-OES method without DGT passive accumulation (3σ criterion) were (μg L−1): 0.12(Cd), 0.20(Cu), 0.80(Pb) and 0.05(Zn).47 By comparison, the corresponding values in GFAAS were (μg L−1): 0.12(Cd), 0.60(Cu), 0.60(Pb) and 0.36(Zn).47 Accumulation of analytes by Chelex-100 DGT for 24 h led to one order of magnitude improvement in LODs expressed in μg L−1, namely 0.01(Cd and Zn), 0.02(Cu), and 0.07(Pb), and allowed quantification of elements in river water by DGT-SSETV-μCCP-OES at concentrations below the mentioned instrumental LODs.47 In comparison, the LODs reported in the literature for ICP-MS, one of the most sensitive methods, coupled with DGT passive accumulation were (μg L−1): 0.006 and 0.0032(Cd), 0.005 and 0.019(Pb), 0.051 and 0.047(Cu), and 0.58(Zn).56,57 Poorer LODs in DGT-SSETV-μCCP-OES, compared to DGT-ICP-MS should be judged in terms of lower power for plasma generation (15 W) versus (1000 W), and lower sensitivity of the detection system based on a low-resolution microspectrometer equipped with a CCD. However, the SSETV-μCCP-OES instrumentation is cost-effective and easy-to-run, compared to lab-scale ICP-MS instrumentation.Table 2 presents the LODs for the total content and DGT-based labile fraction of target analytes by SSETV-μCCP-OES and GFAAS methods, for 24 h under the optimal working conditions. According to data in Table 2, the LODs of the SSETV-μCCP-OES method for the determination of total content of analytes after the DGT passive accumulation for 24 h were in the range (mg kg−1) 0.03(Zn)–0.40(Pb), while the LOD range was 0.10(Cd)–0.40(Cu) for GFAAS. One can observe the improvement of the LODs by one order of magnitude as a result of the preconcentration using the DGT technique, similar to the results obtained in the monitoring of Cd, Cu, Pb and Zn in river water.47 The LODs for total content determination were 10 to 3300-times lower than the guide values in soil showing that Cd, Pb, Cu, and Zn could be quantified below these concentrations. In contrast, SSETV-μCCP-OES without DGT preconcentration provided quantification for contents above (mg kg−1) 3.6(Cd), 24(Pb), 6(Cu), and 1.5(Zn), which would be useful only for Cu and Zn determination. The LODs of the SSETV-μCCP-OES method for the DGT-based labile fraction of analytes were (μg L−1): 0.008(Cd, Cu and Zn) and 0.024(Pb), corresponding to (μg kg−1) in dry soil: 0.01(Cd, Cu and Zn) and 0.03(Pb). Compared to GFAAS, the LODs obtained by SSETV-μCCP-OES coupled with DGT passive sampling were similar for Cd and Pb, and better for Cu and Zn.
| Method | LODs for total contenta (mg kg−1) | LODs for the DGT-based labile fractionb (μg kg−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| Cd | Pb | Cu | Zn | Cd | Pb | Cu | Zn | |
| a LOD for 0.1 g sample made up to 100 mL followed by 10-times dilution. b LOD for 100 g sample and 80 mL ultrapure water. c Guide values in soil according to Romanian legislation (order no. 756/1997 for the approval of the regulation regarding the assessment of environmental pollution, published in Official Monitor of Romania, no. 303 bis from 6 November 1997, in Romanian). https://legislatie.just.ro/Public/DetaliiDocument/13572 (accessed 14 June 2022). | ||||||||
| SSETV-μCCP-OES without DGT accumulation | 1.2 | 8.0 | 2.0 | 0.5 | — | — | — | — |
| SSETV-μCCP-OES with DGT accumulation | 0.10 | 0.40 | 0.15 | 0.03 | 0.01 | 0.03 | 0.01 | 0.01 |
| GFAAS without DGT accumulation | 1.2 | 6.0 | 6.0 | 3.6 | — | — | — | — |
| GFAAS with DGT accumulation | 0.10 | 0.30 | 0.40 | 0.25 | 0.01 | 0.03 | 0.03 | 0.02 |
| Guide valuesc | 1 | 20 | 20 | 100 | — | — | — | — |
Accuracy of the DGT-SSETV-μCCP-OES method for the determination of the total content of analytes
The found values by SSETV-μCCP-OES and external calibration without DGT passive accumulation indicated negative systematic errors compared to the certified values with recovery in the range of 58–83% and relative expanded uncertainty of 12–37% (k = 2) (ESI, Section 4 and Table S6†). Dunnett's test revealed a statistical difference (p > 0.05) between found and certified results. The negative bias was caused by the presence of depressive non-spectral effects coming from the multielemental matrix on the emission signal of Cd, Cu, Pb and Zn in SSETV-μCCP-OES, generating significant systematic errors compared to certified values. Previous results indicated that in the case of soil analysis by SSETV-μCCP-OES the depressive effects of the multielemental matrix could be compensated by using the standard addition method, which is tedious compared to external calibration.31–34Table 3 presents the results for the analysis of soil CRMs after DGT passive accumulation in an uptake solution with 4.0 ± 0.1 pH for 24 h at 21 ± 1 °C using experimental and recommended diffusion coefficients and elution factors. All quantifications were based on external calibration.
| CRM/analyte | Certified concentration mean ± UCRMa (mg kg−1) | Experimental diffusion coefficients and elution factors | Recommended diffusion coefficients and elution factors | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Found concentration mean ± Ulabb (mg kg−1) | Accuracy recovery ± Ulabc (%) | Found concentration mean ± Ulabb (mg kg−1) | Accuracy recovery ± Ulabc (%) | ||||||
| DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | ||
| a – UCRM is the absolute expanded uncertainty for certified concentration (k = 2; 95% confidence level). b – Ulab is the absolute expanded uncertainty in the laboratory for found concentration (k = 2, n = 3 parallel measurements and 95% confidence level). c – Ulab% is the relative expanded uncertainty in the laboratory for found concentration (k = 2, n = 3 parallel measurements and 95% confidence level). | |||||||||
| SQC001 metals in soil | |||||||||
| Cd | 118 ± 2 | 123 ± 28 | 113 ± 28 | 105 ± 23 | 95 ± 25 | 126 ± 25 | 115 ± 25 | 107 ± 19 | 98 ± 22 |
| Pb | 144 ± 2 | 147 ± 49 | 145 ± 38 | 102 ± 33 | 101 ± 26 | 176 ± 56 | 175 ± 40 | 123 ± 32 | 121 ± 23 |
| Cu | 330 ± 4 | 363 ± 94 | 359 ± 102 | 110 ± 26 | 109 ± 28 | 341 ± 69 | 337 ± 79 | 103 ± 20 | 102 ± 23 |
| Zn | 874 ± 11 | 827 ± 278 | 921 ± 260 | 95 ± 34 | 105 ± 28 | 902 ± 280 | 1004 ± 252 | 103 ± 31 | 115 ± 25 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| CRM048 trace metals – sand 1 | |||||||||
| Cd | 92.8 ± 1.55 | 93.7 ± 32.00 | 102.8 ± 27.18 | 101 ± 34 | 111 ± 26 | 96.0 ± 30.81 | 105.2 ± 25.17 | 103 ± 32 | 113 ± 24 |
| Pb | 320 ± 6.27 | 327 ± 103.38 | 324 ± 87.04 | 102 ± 32 | 101 ± 27 | 394 ± 113.62 | 390 ± 89.40 | 123 ± 29 | 122 ± 23 |
| Cu | 84.3 ± 1.45 | 78.4 ± 19.33 | 80.5 ± 19.61 | 93 ± 25 | 95 ± 24 | 73.7 ± 13.92 | 75.6 ± 14.00 | 87 ± 19 | 90 ± 19 |
| Zn | 425 ± 9.14 | 428 ± 148.41 | 414 ± 103.64 | 101 ± 35 | 97 ± 25 | 466 ± 147.94 | 451 ± 93.54 | 110 ± 32 | 106 ± 21 |
|
|||||||||
| Metranal-34 loam | |||||||||
| Cd | 1.44 ± 0.07 | 1.33 ± 0.38 | 1.38 ± 0.36 | 93 ± 28 | 96 ± 26 | 1.37 ± 0.34 | 1.41 ± 0.32 | 95 ± 25 | 98 ± 22 |
| Pb | 83.1 ± 2.3 | 75.9 ± 24.4 | 71.9 ± 17.6 | 91 ± 32 | 87 ± 24 | 91.3 ± 27.4 | 86.5 ± 17.9 | 110 ± 30 | 104 ± 21 |
| Cu | 167 ± 1 | 151 ± 49 | 153 ± 41 | 91 ± 33 | 92 ± 27 | 142 ± 40 | 144 ± 31 | 85 ± 28 | 86 ± 22 |
| Zn | 198 ± 6 | 168 ± 56 | 170 ± 38 | 85 ± 33 | 86 ± 22 | 180 ± 55 | 182 ± 32 | 91 ± 31 | 92 ± 18 |
|
|||||||||
| CRM025–050 metals in soil | |||||||||
| Cd | 369 ± 19.0 | 372 ± 107.1 | 375 ± 94.0 | 101 ± 29 | 101 ± 25 | 381 ± 100.4 | 383 ± 85.5 | 103 ± 26 | 104 ± 22 |
| Pb | 1447 ± 88 | 1234 ± 372 | 1216 ± 315 | 85 ± 30 | 84 ± 26 | 1487 ± 415 | 1465 ± 319 | 103 ± 28 | 101 ± 22 |
| Cu | 7.76 ± 0.73 | 7.99 ± 2.32 | 8.38 ± 2.01 | 103 ± 29 | 108 ± 24 | 7.53 ± 1.96 | 7.92 ± 1.82 | 97 ± 26 | 102 ± 23 |
| Zn | 51.8 ± 3.35 | 50.1 ± 13.41 | 46.1 ± 13.00 | 97 ± 27 | 89 ± 28 | 54.6 ± 12.63 | 50.3 ± 12.45 | 105 ± 23 | 97 ± 25 |
The weight of the individual sources in the relative combined uncertainty of the mean concentration of analytes in the CRM soil samples found by DGT-SSETV-μCCP-OES and DGT-GFAAS is presented in the ESI (Section 5, Fig. S3 and S4).†
In the case of analysis performed after DGT accumulation, the differences (Δm) for all four elements were lower than Ulab (k = 2) of found results, evaluated based on the combined uncertainty of sample preparation and analysis steps in the laboratory and certified uncertainty. In addition, Dunnett's test (p > 0.05) indicated no significant difference between the found results in both methods and reference values, when DGT accumulation was used (p-values 0.052–0.999). Tukey's test (p > 0.05) performed using the average results and combined uncertainty values found by DGT-SSETV-μCCP-OES and DGT-GFAAS, using both experimental and recommended values for diffusion coefficient and elution factor indicated the absence of significant differences (p values 0.202–0.985). The negative bias between the results caused by the differences between the experimental diffusion coefficients and elution factors and those recommended by the manufacturer was only 2%(Cd), 4%(Cu), 7%(Zn) and 17%(Pb), which fit in any case in the analysis errors expressed as combined uncertainty. Therefore, in the case of coupling the analysis method with DGT accumulation, the recovery of the certified concentrations was in the range of 85–123% with a relative expanded uncertainty of 19–35% (k = 2) for SSETV-μCCP-OES and 84–122% with a relative expanded uncertainty of 18–28% (k = 2) for GFAAS, respectively. The good agreement between found results and certified values indicated the lack of non-spectral interference from the multielemental matrix, including As, in the analysis of CRMs for the total content of analytes by SSETV-μCCP-OES when DGT passive accumulation was used. Arsenic was determined by GFAAS, with a LOD of 0.001 mg L−1, and not by SSETV-μCCP-OES or ICP-OES, as its concentration in the uptake solution or soil solution was usually below the LOD of 0.014 mg L−1 of these methods.36 The lack of matrix effects can be observed even in the case of Cd, which demonstrates its separation from As in the uptake solution using the DGT devices with Chelex-100 at concentration levels of As in the range of 0.004–0.036 mg L−1, presented in the ESI (Section 3 and Table S3).† The coupling of the DGT selective, passive accumulation with detection by optical emission in a low-power microplasma, prone to non-spectral effects caused by the multielemental matrix, is a suitable approach. Besides, analyte accumulation in the DGT sampling led to substantial improvement of sensitivity and LODs.
As shown in Fig. S3 and S4 (Section 5),† the combined uncertainty in the laboratory (ulab) for the found concentration by DGT-SSETV-μCCP-OES and DGT-GFAAS was higher than the uncertainty provided for certified concentration (uCRM), because the main contribution came from aliquot analysis, followed by the DGT accumulation, including the uncertainty of diffusion coefficient dependent on temperature and pH. However, the uncertainty of recovery in the analysis of CRMs was generally below ±30% recommended in interlaboratory studies conducted for the validation of analysis methods involving the DGT passive sampling or accumulation.45,46
Determination of total and labile fractions of Cd, Pb, Cu, and Zn found in the soil samples using DGT-SSETV-μCCP-OES
Tables 4 and 5 present the results for the total content and labile fraction of the analytes in soil obtained by DGT-SSETV-μCCP-OES and DGT-GFAAS calculated using the experimental parameters related to accumulation and elution. Quantification was made using external calibration with multielement synthetic standards. The total contents (Table 4) referred to the DGT passive accumulation for 24 h in 100 mL of 1:10 diluted digest (n = 3) at 4.0 ± 0.1 pH. The pH values for soil and soil solution were 6.5–7.3 and 6.3–7.4, respectively, also suitable for DGT accumulation.47 The DGT-based labile fraction (Table 5) was determined using DGT accumulation from soil paste prepared according to the procedure presented earlier. Comparatively, the total dissolved concentration of analytes found in soil solution after separation by centrifugation without acid digestion is also presented.
| Sample | Total Cd content mean ± Ulaba (mg kg−1) | Total Pb content mean ± Ulaba (mg kg−1) | Total Cu content mean ± Ulaba (mg kg−1) | Total Zn content mean ± Ulaba (mg kg−1) | ||||
|---|---|---|---|---|---|---|---|---|
| DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | |
| a – Ulab is the absolute expanded uncertainty in the laboratory for found concentration (k = 2, n = 3 parallel measurements and 95% confidence level). b – RSD is the relative standard deviation evaluated from the combined uncertainty (n = 3 parallel measurements and 95% confidence level). | ||||||||
| S1 | 4.1 ± 1.2 | 4.8 ± 1.2 | 95.2 ± 29.4 | 104 ± 33 | 142 ± 33 | 131 ± 33 | 321 ± 95 | 306 ± 93 |
| S2 | 4.8 ± 1.5 | 6.0 ± 1.5 | 71.0 ± 21.6 | 69.5 ± 14.8 | 59.0 ± 15.7 | 63.4 ± 18.9 | 135 ± 34 | 111 ± 31 |
| S3 | 14.5 ± 3.3 | 14.1 ± 3.9 | 171 ± 37 | 160 ± 37 | 432 ± 137 | 420 ± 128 | 359 ± 90 | 372 ± 89 |
| S4 | 4.4 ± 0.9 | 5.6 ± 1.5 | 35.6 ± 9.3 | 37.0 ± 8.0 | 75.2 ± 28.0 | 83.6 ± 23.4 | 86.4 ± 32.2 | 64.5 ± 20.2 |
| S5 | 7.3 ± 1.8 | 7.2 ± 2.2 | 74.2 ± 20.3 | 71.3 ± 15.0 | 78.2 ± 22.3 | 79.4 ± 19.8 | 246 ± 68 | 255 ± 52 |
| S6 | 8.9 ± 2.3 | 8.6 ± 2.3 | 71.0 ± 17.0 | 77.1 ± 18.2 | 85.8 ± 29.1 | 78.0 ± 22.9 | 202 ± 62 | 179 ± 44 |
| S7 | 11.1 ± 2.8 | 11.9 ± 3.7 | 91.4 ± 28.5 | 92.4 ± 23.4 | 49.7 ± 13.1 | 50.6 ± 18.4 | 114 ± 32 | 125 ± 35 |
| S8 | 4.9 ± 1.5 | 5.6 ± 1.4 | 228 ± 57 | 215 ± 69 | 74.8 ± 18.7 | 70.6 ± 20.0 | 152 ± 46 | 162 ± 50 |
| S9 | 13.5 ± 3.7 | 13.2 ± 4.7 | 146 ± 31 | 146 ± 31 | 79.3 ± 25.1 | 79.8 ± 22.2 | 227 ± 57 | 207 ± 64 |
| S10 | 8.1 ± 1.9 | 9.5 ± 2.3 | 128 ± 30 | 135 ± 38 | 135 ± 40 | 145 ± 36 | 223 ± 54 | 204 ± 55 |
| RSDb (%) | 12–19 | 12–18 | 10–16 | 12–18 | 11–16 | 11–16 | 12–19 | 10–16 |
:8 compared to the total dissolved concentration in soil solution separated by centrifugation
| Sample | Labile Cd content mean ± Ulaba (μg kg−1) | Dissolved Cd content mean ± Ulaba (μg kg−1) | Labile Pb content mean ± Ulaba (μg kg−1) | Dissolved Pb content mean ± Ulaba (μg kg−1) | ||||
|---|---|---|---|---|---|---|---|---|
| DGT-SSETV-μCCP-OES | DGT-GFAAS | SSETV-μCCP-OESb | GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | SSETV-μCCP-OESb | GFAAS | |
| a – Ulab is the absolute expanded uncertainty in the laboratory for found concentration (k = 2, n = 3 parallel measurements and 95% confidence level). b – Concentration determined by the standard addition method for matrix effect compensation. c – RSD is the relative standard deviation evaluated from the combined uncertainty (n = 3 parallel measurements and 95% confidence level). | ||||||||
| S1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 1.0 ± 0.1 | 0.9 ± 0.1 | 2.7 ± 0.6 | 2.3 ± 0.6 | 75.6 ± 9.8 | 88.0 ± 12.2 |
| S2 | 0.7 ± 0.2 | 0.8 ± 0.1 | 1.4 ± 0.2 | 1.0 ± 0.2 | 2.8 ± 0.8 | 3.2 ± 0.7 | 17.9 ± 3.9 | 14.2 ± 3.8 |
| S3 | 1.5 ± 0.4 | 1.9 ± 0.4 | 3.4 ± 0.5 | 2.9 ± 0.6 | 9.5 ± 2.4 | 7.9 ± 1.7 | 43.3 ± 8.1 | 45.1 ± 8.5 |
| S4 | 0.5 ± 0.1 | 0.5 ± 0.1 | 1.0 ± 0.2 | 0.8 ± 0.1 | 1.8 ± 0.5 | 1.7 ± 0.4 | 8.5 ± 1.6 | 6.3 ± 0.9 |
| S5 | 0.8 ± 0.2 | 1.0 ± 0.2 | 4.9 ± 0.7 | 1.5 ± 0.2 | 1.0 ± 0.2 | 0.9 ± 0.2 | 20.2 ± 2.6 | 21.0 ± 6.1 |
| S6 | 0.8 ± 0.2 | 0.8 ± 0.2 | 4.6 ± 0.8 | 4.1 ± 0.5 | 2.5 ± 0.6 | 2.7 ± 0.9 | 39.2 ± 6.6 | 31.0 ± 4.1 |
| S7 | 0.7 ± 0.1 | 0.7 ± 0.1 | 4.4 ± 0.8 | 4.0 ± 0.7 | 0.8 ± 0.2 | 0.9 ± 0.2 | 28.3 ± 3.8 | 21.2 ± 3.0 |
| S8 | 1.3 ± 0.4 | 1.6 ± 0.3 | 10.4 ± 2.1 | 7.2 ± 1.0 | 18.4 ± 4.4 | 18.1 ± 4.0 | 157 ± 26 | 150 ± 36 |
| S9 | 2.0 ± 0.4 | 1.7 ± 0.3 | 6.8 ± 1.0 | 5.4 ± 0.8 | 15.0 ± 3.5 | 15.4 ± 4.6 | 114 ± 16 | 128 ± 25 |
| S10 | 1.6 ± 0.4 | 1.6 ± 0.3 | 5.2 ± 0.9 | 6.4 ± 1.5 | 3.5 ± 0.7 | 4.1 ± 1.0 | 42.7 ± 6.5 | 46.9 ± 6.9 |
| RSDc (%) | 10–14 | 8–12 | 7–10 | 7–12 | 10–15 | 11–16 | 6–11 | 7–14 |
| Sample | Labile Cu content mean ± Ulaba (μg kg−1) | Dissolved Cu content mean ± Ulaba (μg kg−1) | Labile Zn content mean ± Ulaba (μg kg−1) | Dissolved Zn content mean ± Ulaba (μg kg−1) | ||||
|---|---|---|---|---|---|---|---|---|
| DGT-SSETV-μCCP-OES | DGT-GFAAS | SSETV-μCCP-OESb | GFAAS | DGT-SSETV-μCCP-OES | DGT-GFAAS | SSETV-μCCP-OESb | GFAAS | |
| S1 | 2.4 ± 0.6 | 1.9 ± 0.4 | 70.0 ± 14 | 51.4 ± 10.8 | 12.0 ± 2.6 | 14.9 ± 3.1 | 81.7 ± 10.5 | 51.4 ± 15.2 |
| S2 | 5.4 ± 1.5 | 5.0 ± 1.2 | 75.3 ± 16.2 | 78.2 ± 16.9 | 24.3 ± 5.7 | 21.3 ± 4.9 | 25.2 ± 5.2 | 28.2 ± 9.2 |
| S3 | 56.3 ± 13.5 | 81.7 ± 18.3 | 295 ± 49 | 345 ± 63 | 15.9 ± 4.5 | 19.5 ± 4.5 | 116 ± 18 | 120 ± 25 |
| S4 | 7.4 ± 1.6 | 8.6 ± 2.1 | 76.4 ± 10.1 | 78.2 ± 15.8 | 21.9 ± 5.5 | 21.8 ± 4.1 | 27.3 ± 5.4 | 28.2 ± 5.3 |
| S5 | 8.7 ± 2.4 | 7.9 ± 2.0 | 100 ± 22 | 67.2 ± 13.0 | 60.6 ± 12.2 | 70.2 ± 11.9 | 70.8 ± 10.1 | 67.2 ± 8.6 |
| S6 | 15.5 ± 4.3 | 12.2 ± 2.7 | 85.8 ± 12.6 | 120 ± 23 | 31.1 ± 7.9 | 24.3 ± 5.4 | 38.2 ± 4.6 | 40.3 ± 4.1 |
| S7 | 7.5 ± 2.1 | 6.4 ± 1.4 | 60.7 ± 10.7 | 67.6 ± 13.3 | 26.7 ± 6.9 | 30.4 ± 5.1 | 33.6 ± 4.1 | 37.6 ± 4.5 |
| S8 | 20.1 ± 4.9 | 23.6 ± 6.2 | 166 ± 22 | 175 ± 25 | 9.4 ± 2.5 | 11.9 ± 2.8 | 282 ± 64 | 282 ± 57 |
| S9 | 13.8 ± 3.1 | 12.1 ± 3.7 | 105 ± 23 | 107 ± 25 | 10.0 ± 2.6 | 13.2 ± 2.5 | 198 ± 24 | 197 ± 54 |
| S10 | 22.3 ± 5.2 | 26.1 ± 6.8 | 84.6 ± 17.7 | 90.6 ± 21 | 19.7 ± 4.8 | 15.9 ± 4.0 | 92.6 ± 12.1 | 90.6 ± 11.5 |
| RSDc (%) | 11–14 | 11–15 | 7–11 | 7–12 | 10–15 | 11–16 | 6–11 | 5–16 |
The results indicated the lack of multielemental matrix effects, including As at concentrations in the range of 0.004–0.075 mg L−1, for the determination of the total concentration and DGT-based labile fraction of Cd, Pb, Cu and Zn in soil, attributed to the selective retention of analytes against the dominant metals and separation of Cd from As found by GFAAS in the matrix.
The total contents (mg kg−1) of 4.1–13.5(Cd), 35.6–228(Pb), 49.7–432(Cu) and 86.4–359(Zn) in soil were determined with precision in the range of 10–19% calculated from combined uncertainty. Precision in the DGT-SSETV-μCCP-OES measurement was found to be similar to that in DGT-GFAAS. The concentrations of the DGT-based labile fractions (μg kg−1) were 0.3–2.0(Cd), 0.8–18.4(Pb), 2.4–56.3(Cu) and 9.4–60.6(Zn) and measurement precision derived from combined uncertainty was in the range of 10–15%, similar to that in DGT-GFAAS.
Fig. S5 and S6† (ESI, Section 6) depict the Bland–Altman plots for the comparison of the results obtained by DGT-SSETV-μCCP-OES and DGT-GFAAS for the total content and labile fraction in soil. Graphics show no significant difference (p > 0.05) for both sets of determination, since the bias between methods is much lower than the determined concentration, its confidence interval includes the zero value and the differences between the pair results fall within the limits of agreement. In short, the DGT-SSETV-μCCP-OES method provides reliable results both for total content and labile fraction of Cd, Pb, Cu and Zn in soil samples after DGT passive sampling used for preconcentration of analytes and separation from the matrix.
Unfortunately, several data presented in Table 5, show statistically significant differences between the SSETV-μCCP-OES and GFAAS methods in the determination of total dissolved metals in soil solution, although quantification in microplasma was performed using the standard addition method. These differences most probably are due to the presence of organic matter in relatively high concentration in the range of 32–170 mg L−1 in soil solution, which affects the evaporation process of analytes from the Rh microfilament. Periodically, a carbon-like residue was observed on the filament, which required its periodic cleaning by evaporating a mixture of HNO3 and H2O2. These secondary phenomena, caused by the presence of organic matter, could have been avoided if the soil solution had been acid digested when organic matter is destroyed.
The time-integrated mean concentration of elements (cDGT) reported to the total dissolved concentration determined in the bulk soil solution (csol) provides the ratio (R) as an indicator of the ability of the solid phase of soil to resupply elements into soil solution.58,59
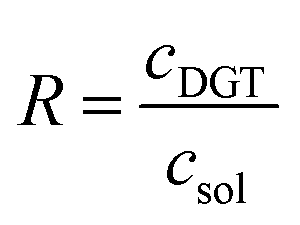 | (2) |
R > 0.8 stands for a fast and sustained supply capacity from the solid phase; R < 0.1 means low resupply capacity in soil solution and limited mobilization of elements from the solid phase; 0.1 < R < 0.8 resupply is intermediary.
The status of these indices for the target analytes in our study is presented in the ESI (Section 7 and Fig. S7).† In the case of Cd, the plot indicated an intermediary capacity of soil to resupply the soil solution, with R values in the range of 0.13–0.50. In the case of Pb and Cu, the resupply indices were rather weak, with intermediary values of 0.03–0.22 and 0.03–0.26, respectively. Only for Zn resupply was intermediary or fast and sustained, with R values in the range of 0.03–0.96. The results were in agreement with the low percentage of total dissolved in soil solution, below 0.2% for Cd, Cu and Zn and 0.08% for Pb, compared to the total content in soil.
Conclusions
The study highlighted for the first time the unique capability of cost-effective instrumentation based on optical emission spectrometry with a low-power and low-argon consumption plasma microtorch interfaced with a low-resolution microspectrometer compared to GFAAS analytical instrumentation to determine the total and the labile fraction of some elements of interest in soil solution after DGT passive sampling/accumulation. The proposed method allowed the quantification of priority hazardous elements, such as Cd and Pb. First of all, the selective accumulation of analytes provided by the DGT passive sampling led to overcoming the non-spectral interference in the low-power plasma caused by the dominant elements in soil and allowed the use of external calibration instead of the standard addition. Furthermore, it was possible to determine Cd by DGT-SSETV-μCCP-OES at the most sensitive line, without spectral interference from As, as the Chelex-100 binding gel ensures the selective retention of Cd versus As. Another advantage when using DGT accumulation was the improvement of LODs, enabling the determination of the labile fraction of the elements with the highest toxicity. However, significant differences have been observed between SSETV-μCCP-OES and GFAAS in the determination of total dissolved metals in soil solution caused by the presence of organic matter that affects the evaporation process of metals from the Rh filament. Although the use of the passive DGT accumulation as a time-integrated technique would be an impediment to developing fast methods, the present study exhibits a new idea by implementing DGT devices in a simultaneous multielement method on a completely miniaturized spectral instrumentation. However, the advantages of the new easy-to-run set-up that does not require a skilled analyst, make it a viable alternative to the classic laboratory methods broadly used, such as ICP-MS and GFAAS, the latter considered as a reference for validation of the proposed method. Anyway, the versatility of the DGT technique is a challenge for the development of analytical technologies incorporating microtorch/microplasma with high green and white scores, and capable of widening the area of applicability. The study put in evidence the opportunity to convert the miniaturized spectrometric instrumentation used so far in academic laboratories into mature analytical systems, to be used for analytical applications where their figures of merit are suitable for fit-of-purpose.Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
This work was supported by a grant from the Romanian Ministry of Research and Innovation, CNCS/CCCDI-UEFISCDI, contract nr. 733PED/2022, project number PN-III-P2-2.1-PED2021-0151, within PNCDI III.References
- J. R. Bacon, O. T. Butler, W. R. L. Cairns, O. Cavoura, J. M. Cook, C. M. Davidson and R. Mertz-Kraus, J. Anal. At. Spectrom., 2023, 38, 10–56 RSC.
- M. Patriarca, N. Barlow, A. Cross, S. Hill, A. Robson and J. Tyson, J. Anal. At. Spectrom., 2023, 38, 496–577 RSC.
- E. H. Evans, J. Pisonero, C. M. M. Smith and R. N. Taylor, J. Anal. At. Spectrom., 2022, 37, 942–965 RSC.
- S. Cerutti, P. H. Pacheco, R. Gil and L. D. Martinez, Curr. Opin. Green Sustainable Chem., 2019, 19, 76–86 CrossRef.
- I. Lavilla, V. Romero, I. Costas and C. Bendicho, TrAC, Trends Anal. Chem., 2014, 61, 1–10 CrossRef CAS.
- R. E. Sturgeon, J. Anal. At. Spectrom., 2017, 32, 2319–2340 RSC.
- M. Slachcinski, J. Anal. At. Spectrom., 2019, 34, 257–273 RSC.
- K. A. Romanovskiy, M. A. Bolshov, A. V. Munz, Z. A. Temerdashev, M. Y. Burylin and K. A. Sirota, Talanta, 2018, 187, 370–378 CrossRef CAS PubMed.
- Z. H. Fernandez, L. A. V. Rojas, A. M. Alvarez, J. R. E. Alvarez, J. A. dos Santos Junior, I. P. Gonzalez, M. R. Gonzalez, N. A. Macias, D. I. Sanchez and D. H. Torres, Food Control, 2015, 48, 37–42 CrossRef CAS.
- C. M. M. Santos, M. A. G. Nunes, A. B. Costa, D. Pozebon, F. A. Duarte and V. L. Dressler, Anal. Methods, 2017, 9, 3497–3504 RSC.
- R. A. Althobiti and D. Beauchemin, J. Anal. At. Spectrom., 2021, 36, 535–539 RSC.
- A. S. Henn, E. M. M. Flores, V. L. Dressler, M. F. Mesko, J. Feldmann and P. A. Mello, J. Anal. At. Spectrom., 2018, 33, 1384–1393 RSC.
- L. Chirita, E. Covaci, A. Mot, M. Ponta, A. Gandea and T. Frentiu, J. Anal. At. Spectrom., 2021, 36, 267–272 RSC.
- L. Chirita, E. Covaci, M. Ponta and T. Frentiu, Anal. Methods, 2023, 15, 1734–1746 RSC.
- I. S. Denmark, E. Begu, Z. Arslan, F. X. Han, J. M. Seiter-Moser and E. M. Pierce, Anal. Chim. Acta, 2018, 1041, 68–77 CrossRef CAS PubMed.
- M. Wang, B. Qian, Q. X. Tian, J. L. Lin, J. Hao, Y. Zhang and B. J. Han, Appl. Spectrosc. Rev., 2023, 58, 443–488 CrossRef CAS.
- S. Liu, Y. L. Yu and J. H. Wang, J. Anal. At. Spectrom., 2017, 32, 2118–2126 RSC.
- C. Bendicho, I. Lavilla, F. Pena-Pereira and V. Romero, J. Anal. At. Spectrom., 2012, 27, 1831–1857 RSC.
- W. Wojnowski, S. Tobiszewski, F. Pena-Pereira and E. Psillakis, TrAC, Trends Anal. Chem., 2022, 149, 116553 CrossRef CAS.
- P. M. Nowak, R. Wietecha-Posluszny and J. Pawliszyn, TrAC, Trends Anal. Chem., 2021, 138, 116223 CrossRef CAS.
- C. M. Hussain, C. G. Hussain and R. Kecili, TrAC, Trends Anal. Chem., 2023, 159, 116905 CrossRef CAS.
- E. Covaci and T. Frentiu, Stud. Univ. Babes-Bolyai, Chem., 2022, 67, 7–25 CAS.
- B. J. Han, X. M. Jiang, X. D Hou and C. B. Zheng, Anal. Chem., 2014, 86, 936–942 CrossRef CAS PubMed.
- M. Li, Y. Deng, C. B. Zheng, X. M. Jiang and X. D. Hou, J. Anal. At. Spectrom., 2016, 31, 2427–2433 RSC.
- M. F. Mirabelli, J. C. Wolf and R. Zenobi, Analyst, 2017, 142, 1909–1915 RSC.
- M. Zhang, Q. S. Tang, P. X. Li, L. He, X. D. Hou and X. M. Jiang, Anal. Chem., 2023, 95, 5151–5158 CrossRef CAS PubMed.
- E. Covaci, M. Senila, C. Tanaselia, S. B. Angyus, M. Ponta, E. Darvasi, M. Frentiu and T. Frentiu, J. Anal. At. Spectrom., 2018, 33, 799–808 RSC.
- K. Greda, M. Welna, A. Szymczycha-Madeja and P. Pohl, Talanta, 2022, 249, 123694 CrossRef CAS PubMed.
- X. M. Jiang, Y. Chen, C. B. Zheng and X. D. Hou, Anal. Chem., 2014, 86, 5220–5224 CrossRef CAS PubMed.
- S. Weagant, G. Dulai, L. Li and V. Karanassios, Spectrochim. Acta, Part B, 2015, 106, 75–80 CrossRef CAS.
- T. Frentiu, E. Darvasi, S. Butaciu, M. Ponta, D. Petreus, A. I. Mihaltan and M. Frentiu, Talanta, 2014, 129, 72–78 CrossRef CAS PubMed.
- T. Frentiu, E. Darvasi, S. Butaciu, M. Ponta, D. Petreus, R. Etz and M. Frentiu, Microchem. J., 2015, 121, 192–198 CrossRef CAS.
- S. Butaciu, T. Frentiu, M. Senila, E. Darvasi, S. Cadar, M. Ponta, D. Petreus, R. Etz and M. Frentiu, Food Control, 2016, 61, 227–234 CrossRef CAS.
- T. Frentiu, S. Butaciu, E. Darvasi, M. Ponta, M. Frentiu and D. Petreus, Chem. Pap., 2017, 71, 91–102 CrossRef CAS.
- S. B. Angyus, E. Darvasi, M. Ponta, D. Petreus, R. Ratz, M. Senila, M. Frentiu and T. Frentiu, Talanta, 2020, 217, 121067 CrossRef CAS.
- S. B. Angyus, E. Levei, D. Petreus, R. Etz, E. Covaci, O. T. Moldovan, M. Ponta, E. Darvasi and T. Frentiu, Molecules, 2021, 26, 2642 CrossRef CAS PubMed.
- W. Davison and H. Zhang, Nature, 1994, 367, 546–548 CrossRef CAS.
- W. Davison, Diffusive Gradients in Thin-Films for Environmental Measurements, Cambridge University Press, UK, 2016 Search PubMed.
- C. Li, S. M. Ding, I. Y. Yang, Y. Wang, M. Y. Ren, M. S. Chen, X. F. Fan and E. Lichtfouse, Environ. Chem. Lett., 2019, 17, 801–831 CrossRef CAS.
- M. Reichstadter, P. Divis, E. Abdulbur-Alfakhoury and Y. Gao, Talanta, 2020, 217, 121059 CrossRef CAS PubMed.
- S. Wagner, J. Santer, J. Irrgeher, M. Puschenreiter, S. Happel and T. Prohaska, Anal. Chem., 2022, 94, 6338–6346 CrossRef CAS PubMed.
- S. Zarrouk, A. Bermond, N. K. Benzina, V. Sappin-Didier and L. Denaix, Environ. Chem. Lett., 2014, 12, 191–199 CrossRef CAS.
- S. Marrugo-Madrid, M. Turull, H. Zhang and S. Diez, Environ. Chem. Lett., 2021, 19, 3761–3788 CrossRef CAS.
- R. Chen, X. L. Mu, J. X. Liu, N. Cheng, R. G. Shi, M. M. Hu, Z. O. Chen and H. Wang, Sci. Total Environ., 2023, 860, 160523 CrossRef CAS PubMed.
- A. Dabrin, J. P. Ghestem, E. Uher, J. L. Gonzalez, I. J. Allan, M. Schintu, N. Montero, J. Balaam, E. Peinerud, C. Miege and M. Coquery, Environ. Pollut., 2016, 208, 299–308 CrossRef CAS PubMed.
- J. L. Gonzalez, I. Amouroux, S. Guesdon, F. Menet-Nedelec, E. Ponzevera, N. Montero, B. Marras, M. Schintu, M. Caetano, M. C. Dos Santos, M. R. Sanz, V. M. Gabet, G. R. Jose, M. J. Belzunce-Segarra, J. Larreta, I. Menchaca, P. Bersuder, T. Bolam, F. Regan, B. White and H. Zhang, Sci. Total Environ., 2022, 847, 157499 CrossRef CAS PubMed.
- S. B. Angyus, M. Senila, T. Frentiu, M. Ponta, M. Frentiu and E. Covaci, Talanta, 2023, 259, 124551 CrossRef CAS PubMed.
- H. Zhang and W. Davison, Anal. Chem., 1995, 67, 3391–3400 CrossRef CAS.
- J. M. Tukey, Biometrics, 1949, 5, 99–114 CrossRef CAS.
- C. W. Dunnett, J. Am. Stat. Assoc., 1955, 50, 1096–1121 CrossRef.
- J. M. Bland and D. G. Altman, Stat. Methods Med. Res., 1999, 8, 135–160 CrossRef CAS PubMed.
- T. Frentiu, M. Ponta, E. Darvasi, S. Butaciu, S. Cadar, M. Senila, A. Mathe, M. Frentiu, D. Petreus, R. Etz, F. Puskas and D. Sulea, RO Patent 131066 B1, 2020.
- T. Frentiu, D. Petreus, M. Senila, A. I. Mihaltan, E. Darvasi, M. Ponta, E. Plaian and E. A. Cordos, Microchem. J., 2011, 97, 188–195 CrossRef CAS.
- V. Ivanova-Petropulos, B. Balabanova, E. Bogeva, T. Frentiu, M. Ponta, M. Senila, R. Gulaboski and F. D. Irimie, Food Anal. Methods, 2017, 10, 459–468 CrossRef.
- B. Babalola and H. Zhang, Groundw. Sustain. Dev., 2021, 12, 100493 CrossRef.
- O. A. Garmo, O. Royset, E. Steinnes and T. P. Flaten, Anal. Chem., 2003, 75, 3573–3580 CrossRef CAS PubMed.
- C. Gaulier, C. Zhou, Y. Gao, W. Guo, M. Reichstadter, T. Ma, W. Baeyens and G. Billon, Sci. Total Environ., 2021, 757, 143827 CrossRef CAS PubMed.
- M. P. Harper, W. Davison, H. Zhang and W. Tych, Geochim. Cosmochim. Acta, 1998, 62, 2757–2770 CrossRef CAS.
- H. M. Conesa, R. Schulin and B. Nowack, Environ. Sci. Pollut. Res., 2010, 17, 657–664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00258f |
| This journal is © The Royal Society of Chemistry 2024 |