
Metal-free photoinduced electron transfer–atom transfer radical polymerization (PET–ATRP) via a visible light organic photocatalyst†
Xiaodong
Liu
,
Lifen
Zhang
*,
Zhenping
Cheng
* and
Xiulin
Zhu
Suzhou key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: chengzhenping@suda.edu.cn; zhanglifen@suda.edu.cn; Fax: +86-512-65882787
First published on 26th November 2015
Abstract
The development of an atom transfer radical polymerization (ATRP) system without any transition metal catalyst for electronic and biomedical applications was considered to be in pressing need. Fluorescein (FL) was used as the organic photocatalyst for the polymerization of methyl methacrylate (MMA) via the proposed photoinduced electron transfer–atom transfer radical polymerization (PET–ATRP) mechanism. In the presence of electron donors provided by triethylamine (TEA), fluorescein can activate alkyl bromide and control radical polymerizations by a reductive quenching pathway. The polymerizations could be controlled by an efficient activation and deactivation equilibrium while maintaining the attractive features of “living” radical polymerization. The number-average molecular weight Mn,GPC increased with monomer conversion, and the controllability of molecular weight distributions for the obtained PMMA could be achieved in the polymerization processes. MALDI-TOF MS, 1H NMR spectroscopy and chain extension polymerizations show reserved chain-end functionality in the synthesized polymers and further confirm the “living” feature of the metal-free ATRP methodology. All these research results support the feasibility of the visible light mediated metal-free PET–ATRP platform for the synthesis of elegant macromolecular structures.
1. Introduction
In a wide range of possible controlling stimuli such as temperature,1 pH,2 light,3 electrochemistry4 or chemical changes,5 light-triggered reactions offer many attractive superiorities such as simple experimental setup, mild reaction conditions and minimal side reactions, tuneable and accessible light sources, and spatial and temporal control.6 Compared with costly and harmful UV irradiation with a relatively high energy (∼6 eV),7 visible light (400–700 nm, ∼2 eV) is more attractive and promising in synthetic applications. Polymerization mediated by visible light is considered to be an environment-friendly and green technology. Significant efforts have been dedicated to the development of photoinitiated and photoregulated radical polymerizations including photoinduced electron transfer reversible addition–fragmentation chain transfer (PET–RAFT) polymerization,8 photoredox-promoted atom transfer radical polymerization (ATRP),9 and organocatalytic,10 cobalt-mediated,11 Ir-catalysed6c,9c or tellurium-mediated12 methods and so on.13Transition metals (e.g., iridium and ruthenium) have been previously utilized as photoredox catalysts to mediate radical polymerization under light irradiation. Ruthenium(II)polypyridine complexes (e.g., RuII(bpy)3) can activate alkyl halides through the Ru(bpy)3+ photoredox cycle under visible light irradiation;14 while the excited fac-[Ir(ppy)3]* species15 could directly reduce an alkyl bromide initiator via a PET process to provide the desired alkyl radical and subsequently initiate polymerization. Besides, organo-cobalt porphyrins,16 both as a photoinitiator and a mediator, undergo photo-cleavage of the Co–C bond to give an organic radical and a cobalt(II) porphyrin metal-central radical for chain propagation with good control over the molecular weight and polydispersity. However, these obtained products and traditional ATRP polymers are inevitably contaminated with trace metal residues that cause distress for electronic and biomedical applications. The elimination of transition metals entirely eliminates the necessary step for purification of the synthesized polymers. Photocatalysis employed under visible light is undoubtedly one of the promising strategies to meet the increasing demand for more sustainable green chemical processes, and employing visible light photoredox catalysis for controlled/“living” radical polymerization is an emerging research focus area attracting significant interest in material and biological chemistry. However, it remains a great challenge to adopt metal-free catalytic systems to ATRP with an alkyl bromide commonly used as the initiator. Outstanding work reported by Hawker,10a Miyake10b and Matyjaszewski10c provided a good demonstration for further research, and the organic photoredox catalysts, 10-phenylphenothiazine (PTH) and perylene, have generated considerable interest in this research field.
The PET–RAFT technique employed by Boyer8b,17 was effectively conducted to activate/mediate controlled/“living” radical polymerization, and the PET mechanism is believed to be an efficient process to activate thiocarbonylthio compounds to generate radicals and thereby initiate controlled free radical polymerization. For example, Boyer et al. used a series of organo-dyes (e.g., eosin Y) as the catalysts to successfully conduct PET–RAFT for a variety of functional monomers, including N,N-dimethylaminoethyl methacrylate, hydroxyl ethyl methacrylate, pentafluorophenyl methacrylate, glycidyl methacrylate, oligoethylene glycol methyl ether methacrylate (OEGMA), and methacrylic acid.8f Various organic photocatalysts have been extensively exploited as green photoredox catalysts for a series of radical-mediated organic reactions because of their easy availability, low cost and toxicity.18 We envisaged that the unique properties of these catalysts would be promising for the development of photo-controlled living radical polymerization. Recently, fluorescein (FL) has been studied as a photoredox catalyst due to its visible region absorbance (∼450 nm), chemical stability, long excited-state lifetime, and favourable redox potential.18e Upon visible light photon absorption, FL is converted to its excited state FL*, and through a reductive quenching pathway by an electron donor (e.g., trialkylamines), a powerful single-electron reductant, FL˙− (E0 = −1.22 V), was generated, which can dechlorinate organic halides to provide carbon-central radical species. In this paper, based on this theoretical research background, we reported a novel methodology of metal-free photoinduced electron transfer–atom transfer radical polymerization (PET–ATRP), using methyl methacrylate (MMA) as the model monomer conducted by using FL as an effective organic visible-light photocatalyst. The controllability of the radical polymerization of MMA was achieved by the organic-catalyzed blue light mediated process. Through a reductive quenching pathway, the photocatalytic reductive dehalogenation of ethyl α-bromophenylacetate and subsequent initiation of the radical polymerization were realized by the aforementioned single-electron reductant FL˙− under blue light irradiation, and the polymerization process could be directly activated/deactivated by “on/off” light irradiation. The effects of acid–alkali polymerization conditions, initiator, light source, solvent, catalyst concentration, polymerization degree (DP) etc. on metal-free PET–ATRP of MMA were systematically investigated to optimize the polymerization conditions. Not only was the “living” feature of polymerization further clarified, but also the metal-free PET–ATRP mechanism was proposed by our research in detail.
2. Experimental section
2.1. Materials
Poly(ethylene glycol) methyl ether methacrylate (PEGMA, Mn = 475 g mol−1, mean degree of polymerization (DP) is 8–9; Aldrich) was passed through an alumina column to remove the inhibitor. Methyl methacrylate (MMA), acrylonitrile (AN), tert-butyl acrylate (tBA), glycidyl methacrylate (GMA) and benzyl methacrylate (BnMA) were all purchased from Sigma-Aldrich, and were purified by passing through a neutral alumina column before use. Ethyl α-bromophenylacetate (EBPA) (+97%), ethyl 2-bromoisobutyrate (EBiB, 98%, Aldrich), 4-methylbenzenesulfonyl chloride (p-TsCl), 2-bromopropanenitrile (BPN), methyl 2-bromopropanoate (MBP), methyl 2-chloropropanoate (MCP), 2-bromo-2-phenylacetonitrile (BPAN), 1-phenylethyl bromide (PEBr) were purchased from Alfa Aesar and used as received. Triethylamine (TEA), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA), methanol (MeOH), acetone, anisole and all other chemicals were obtained from Shanghai Chemical Reagents Co. Ltd and were used as received unless mentioned.2.2. Characterization
The number average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) values of the resulting polymers were determined using a TOSOH-HLC-8320 gel permeation chromatograph (GPC) equipped with a refractive index detector (Waters 2414), using TSK gel Super AWM-H columns (4.6 mm I.D. × 15 cm × 2) with measurable molecular weights ranging from 103 to 10 × 105 g mol−1. THF was used as the eluent at a flow rate of 0.35 mL min−1 at 40 °C. The GPC samples were injected by using a TOSOH plus autosampler, and were calibrated with PMMA standards obtained from TOSOH. The 1H NMR spectrum of the obtained polymer was recorded on a Bruker 300 MHz nuclear magnetic resonance (NMR) instrument using CDCl3 or DMSO-d6 as the solvent and tetramethylsilane (TMS) as the internal standard at ambient temperature. Matrix-assisted laser desorption ionization time-of-flight mass spectra (MALDI-TOF MS) were acquired on a Bruker Ultraflex-III TOF/TOF mass spectrometer (Bruker Daltonics, Inc., Billerica, MA) equipped with a Nd:YAG laser (355 nm). Fluorescence emission spectra were performed with a fused quartz cuvette (10 mm × 10 mm) on a Shimadzu RF-5301PC spectroscope with a R-928 photomultiplier at room temperature; the excitation and emission slit widths were all set at 3 nm.2.3. General procedure for metal-free PET–ATRP of MMA
Polymerizations were performed under blue light-emitting diode (LED) irradiation under an argon (Ar) atmosphere. A typical polymerization procedure with the molar ratio of [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 200/1/0.1/3 in the absence of air is shown as follows: a reaction mixture consisting of MMA (1.0 mL, 9.43 mmol), EBPA (8.25 μL, 0.047 mmol), fluorescein (1.56 mg, 0.0047 mmol), TEA (19.5 μL, 0.141 mmol) and 2.0 mL of DMSO was prepared in a 5 mL dried ampule. The reaction mixture was degassed by at least three freeze–pump–thaw cycles to provide an Ar atmosphere without any dissolved oxygen, and the ampule was flame-sealed. The mixture was then transferred to a magnetic drive stirring device surrounded with one double-density LED Flex strip (λmax = 458 nm, 0.85 mW cm−2) in a circle for LED irradiation. The polymerization was allowed to be conducted under stirring at room temperature (rt) for the irradiation duration specified in polymerization tables. After the desired polymerization time, the ampule was transferred into the dark. Then the reaction mixture was dissolved in THF (2.0 mL) followed by precipitation into 250 mL of methanol. The polymer was collected by filtration and dried under vacuum at 35 °C to a constant weight.2.4 Chain extension using PMMA as a macroinitiator
The macroinitiator-1 was synthesized under the molar ratio of [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 200/1/0.3/9 for 3 h (Mn,GPC = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1, Mw/Mn = 1.27), and the macroinitiator-2 was synthesized under the molar ratio of [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 200/1/0.4/12 for 2 h (Mn,GPC = 19400 g mol−1, Mw/Mn = 1.30). The polymerization and purification processes were performed under the general conditions as previously mentioned.
800 g mol−1, Mw/Mn = 1.27), and the macroinitiator-2 was synthesized under the molar ratio of [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 200/1/0.4/12 for 2 h (Mn,GPC = 19400 g mol−1, Mw/Mn = 1.30). The polymerization and purification processes were performed under the general conditions as previously mentioned.
In a typical experimental procedure for chain-extension polymerization of MMA, macroinitiator-1 (100 mg, 5.39 μmol, 0.19 equiv.), fluorescein (9.3 mg, 0.028 mmol, 1 equiv.), TEA (116 μL, 0.84 mmol, 30 equiv.) and MMA (1.00 mL, 9.43 mmol, 337 equiv.) were dissolved in 2.0 mL of DMSO, and the reaction was irradiated with a blue LED for 48 h before the polymer was isolated by precipitation from methanol. In chain-extension polymerization of St, macroinitiator-2 (100 mg, 5.22 μmol, 0.19 equiv.), fluorescein (9.3 mg, 0.028 mmol, 1 equiv.), TEA (116 μL, 0.84 mmol, 30 equiv.) and St (1.00 mL, 8.73 mmol, 312 equiv.) were dissolved in a mixture solution of DMSO (1.0 mL) and DMF (1.0 mL). The reaction mixture was irradiated with blue LED irradiation for 24 h before the polymer was purified as mentioned above.
3. Results and discussion
The polymerization operated through reductive and oxidative quenching pathways can be realized by visible light irradiated organic photocatalysts.19 An immediate challenge in developing a metal-free ATRP is to find out an organic catalyst with a high reducing ability to effectively catalyze controlled radical polymerization under visible light irradiation. The popular organic photocatalyst, 10-phenylphenothiazine (PTH), was adopted to catalyze ATRP under blue LED irradiation (λmax = 458 nm, 0.85 mW cm−2), yet favorable experiment results were not obtained (Table S1†). One possible reason is that PTH is typically not suitable for use as a photocatalyst under light in the visible region. After surveying several other possible candidates based on their maximum absorption wavelength, redox potential E0 and precedent studies on heterogeneous semiconductor-based (e.g., ZnO, CdS) photocatalysis, asymmetric organophotoredox catalysis and photosensitizers for solar cells,20 fluorescein (FL) was chosen as a photocatalyst for our subsequent studies owing to its favorable redox (E0 = −1.22 V) and photochemical characteristics. The experimental procedure for synthesis of poly(methyl methacrylate) (PMMA) by photoinduced metal-free PET–ATRP was conducted by using ethyl α-bromophenylacetate (EBPA) as the initiator, fluorescein (FL) as the visible-light photoredox catalyst and triethylamine (TEA) as the electronic donor in dimethyl sulfoxide (DMSO) under irradiation by a blue LED light (λmax = 458 nm, 0.85 mW cm−2) at room temperature. The afforded polymer was precipitated from methanol solution to provide PMMA as a white powder. Conversion of the monomer was determined gravimetrically, and the number-average molecular weight (Mn,GPC) and polydispersity (Mw/Mn) were detected by GPC using THF as the eluent.Control experiments proved that omission of any single component (FL, EBPA, TEA or light source) could result in either uncontrolled or no polymeric products (Table S1,† entries 1–4). These results clearly revealed that control over polymerization results from the photocatalyst mediating with light as an external stimulus. Of particular importance is to eliminate the disruptive effects from any possible iniferter agent. In the absence of triethylamine, trace amounts of uncontrolled polymers (2.6% of monomer conversion, Mn = 62100, Mw/Mn = 1.92) were obtained, indicating that fluorescein is proposed as an inefficient photoiniferter agent for photopolymerization reactions. While in the presence of triethylamine, no polymer was collected from the reaction system without any initiator, indicating that no obvious disadvantageous effects were generated from the photoiniferters on the polymerization. Specifically, in the synthesis of the block copolymer, this feature could prevent interference from homopolymers initiated from dye molecules. Besides, upon irradiation with blue LED light with an emission maximum at 458 nm, this xanthene-based dye was proved to be sufficiently stable under the reaction conditions.
3.1. Effects of acid and alkali on the metal-free PET–ATRP of MMA
Fluorescein derivatives have emerged as the scaffold for the detection of biologically relevant species (cysteine, pH, NO, ClO−, Zn2+, Fe3+, etc.).21 Worthy of concern is their optical properties of sensitivities to pH variation.22 Under acidic conditions, fluorescein is nonfluorescent and colorless in the spirocyclic form. In contrast, under basic conditions, fluorescein exists in the ring-opened form (Fig. 3a) and has strong fluorescence and longer absorption wavelength. The experimental results of the photomediated polymerizations catalyzed by FL in the alkali-induced lactone ring opening structure or lactone form under acidic conditions are shown in Table S2.† Encouragingly, the initial results showed that good catalytic efficiency and well-controlled polymerization could be obtained employing the photocatalyst in the ring-opened structure in the presence of TEA, while under acidic conditions, polymerization mediated by the spirocyclic structure is completely out of control. Furthermore, as triethylamine scales up, the controllability of molecular weight distribution of the obtained PMMA was improved to a certain extent within this system.Of particular note is that when the organic-based photoredox catalyst is employed under conditions without either TEA or acetic acid, the polymerization was absolutely out of control (entry 9 in Table S2,†Mn,GPC = 58500, Mw/Mn = 2.54). Meanwhile, the monomer conversion (Conv. 6.4%) and initiation efficiency (I* = 2.6%) were both rather low, indicating that the initiator was almost not consumed. This further verifies that an organic catalyst in a highly reducing state is needed to activate the alkyl bromide. Within the organic catalytic cycle, as the initial recyclable electron reservoir, the oxidation of TEA provides the electron for the reductive quenching of the dye's excited state FL*, and the formed FL˙− can reduce an alkyl halide under visible light irradiation to produce the electron-deficient alkyl radical for chain propagation (Table S2,† entries 1–8). Thus the important function of TEA is not only to ensure that the chemical structure of fluorescein is in ring-opened form, but also to act as an electron donor. The following sections mainly discuss the polymerization processes performed under the condition of TEA up to 30 times the content of photocatalyst.
3.2. Effects of initiator on metal-free PET–ATRP of MMA
The effect of the initiator type on the polymerization was then investigated, as shown in Table S3.† It can be seen that in most cases except EBPA and BPN, the molecular weights were higher than theoretical values, indicating the rather low initiation efficiencies and the poor control over the polymerization. Satisfactory optimization molecular weight distribution and initiation efficiency were maintained in the case of EBPA (64.5% Conv., Mw/Mn = 1.46, I* = 42.9%) and BPN (83.7% Conv., Mw/Mn = 1.54, I* = 47.0%), which was attributed to the higher activity of the halide component of the initiator. In consideration of significant effects by initiators on polymerization controllability and the facility of chain end characterization, the active EBPA, with a phenyl group as the internal standard, was selected as a more appropriate and efficient initiator for metal-free PET–ATRP.3.3. Effects of light source on metal-free PET–ATRP of MMA
Organic photocatalysts have certain maximum absorption and emission wavelengths, and a wide range of adjustment can be achieved by simple chemical modifications. Meanwhile, different covering wavelength areas can be displayed whenever the type of light is changed, and a lot of parameters such as the intensity and wavelength of light can be adjusted during the reaction time, which enables good control over the reaction. Therefore, it is necessary to investigate the effect of different light sources on the polymerization. As shown in Table S1,† transforming the light source to purple LED (λmax = 396 nm), green LED (λmax = 530 nm), yellow LED (λmax = 590 nm) or incandescent lamp (λ = 400–700 nm) resulted in higher measured molecular weight (Mn,GPC) and lower initiator efficiency (I*), which is probably due to the unmatched wavelength bands between light sources and the photocatalyst. For instance, the molecular weight given by GPC (Mn,GPC = 179000 g mol−1, Mw/Mn = 1.57) in the situation of yellow LED irradiation for 22 h deviated considerably from the theoretical value (Mn,th = 3970 g mol−1). While under blue LED irradiation, the polymerization was relatively well-controlled with high initiation efficiency (entries 5–7 in Table S1†). Thus in the following discussion, the polymerization was performed by exposure to blue LED irradiation.
3.4. Effects of solvent type and volume on metal-free PET–ATRP of MMA
Investigation of the effects of different solvents on the polymerization provided the experimental observation that, satisfying the polymerization rate and high initiation efficiency can be acquired by photocatalytic polymerization in DMSO, whereas in the case of other solvents, such as acetone and anisole, the polymerization controllability was rather poor with a broad polydispersity and low initiation efficiency (Table S4†). This feature is ascribed to the better solubility of photocatalyst in the whole system and higher efficiencies provided by DMSO among commonly used solvents for acting in the polymerization. In particular, Fig. S1a and S1b† display the kinetic plots of photoinduced metal-free PET–ATRP at [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 100/1/0.1/3 in DMSO and DMF, respectively. The linear fitting curves (ln[M]0/[M] versus time) suggested the constant concentration of the propagating radicals during the course of polymerization. The apparent rate constant of the polymerization,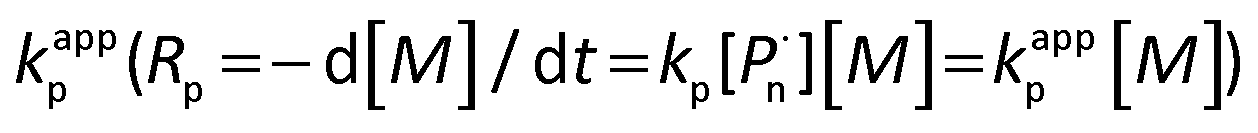 calculated from the slopes showed that the polymerization in DMSO (kappp = 6.78 × 10−5 s−1) was much faster than in DMF (kappp = 3.76 × 10−5 s−1), thus DMSO was selected as the most suitable solvent for this polymerization system.
calculated from the slopes showed that the polymerization in DMSO (kappp = 6.78 × 10−5 s−1) was much faster than in DMF (kappp = 3.76 × 10−5 s−1), thus DMSO was selected as the most suitable solvent for this polymerization system.
Subsequently, the effect of solvent volume of DMSO on polymerization was further studied, and the results are shown in Tables S5 and S6,† respectively. The polymerization rate decreased with the increase of solvent volume. In the molar ratio of [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 200/1/0.3/9, the monomer conversion reached up to 68.6% under 8 h irradiation in 1.0 mL of DMSO (entry 2 in Table S5†), whereas 38.9% and 77.2% of conversion for the cases with 2.0 mL of DMSO could be obtained after 7 h (entry 4 in Table S5†) and 35 h (entry 5 in Table S5†), respectively. However, as the volume of DMSO in the polymerization system was 4.0 mL, the reaction rate seemed to be higher than that in the case of 2.0 mL. This phenomenon was similar to the situation at the molar ratio of [MMA]0:[EBPA]0 = 200/1 with different photocatalyst contents as discussed in the following section, in which the polymerization rate would be faster in the system of lower photocatalyst concentration. Meanwhile, the growth trend of molecular weight seems to be modest. When 1.0 mL of DMSO was used as the solvent, the molecular weight at 68.6% conversion (Mn,GPC = 23500, Mw/Mn = 1.51) was lower than that at 34.4% conversion (Mn,GPC = 25100, Mw/Mn = 1.39). The possible reason is that under this condition the activation of bromide at polymer chain end was slightly less efficient, and continuous initiation activities were conducted throughout the whole polymerisation process. In the later polymerization period when most of monomers were consumed, the later growing polymers of low molecular weight took their share of the monomer conversion and the averaged molecular weight. Only in the case of 2.0 mL of solvent does the number-average molecular weight clearly increase, and initiation efficiency was also raised along with increasing conversion. Unfortunately, the molecular weights of the polymers determined by GPC deviated from the theoretical ones, and the molecular weight distributions become relatively broader with the increase of monomer conversion. At the molar ratio of [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 400:1:0.4:12, similar experimental results were obtained as well (entries 3–9 in Table S6†). Therefore, we focused our attention on the polymerization of MMA in 2.0 mL of DMSO in the following investigation.
3.5. Polymerization kinetics and effects of the photocatalyst concentration on metal-free PET–ATRP of MMA at [MMA]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :[EBPA]0 = 200:1
:[EBPA]0 = 200:1
In order to investigate the polymerization behaviour of MMA with the molar ratio of [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 200:1:x:y (x = 0.1–0.3, x:y = 1:30), the kinetic plots and the evolution of Mn,GPC and Mw/Mn with conversion at this polymerization degree were studied as illustrated in Fig. 1 and S1.† The molecular weights increased with conversion throughout the polymerization, and the experimentally measured molecular weights (Mn,GPC) were relatively greater than the theoretical ones, giving a slight low initiator efficiency (I*) probably caused by the low deactivator concentration and deactivation rate constants. When increasing the proportion of fluorescein ([MMA]0:[EBPA]0:[FL]0:[TEA]0) from 200/1/0.1/3 to 200/1/0.3/9, improved control over Mn,GPC could be achieved with molecular weight much closer to the theoretical values. In addition, as the first-order kinetic plot showed a downward curvature, the higher fluorescein proportion greatly decelerated the polymerization processes with a much lower polymerization rate than that of systems with lower dosage and ratio of the catalyst, and this was also accompanied by an increase in molecular weight distribution (Mw/Mn) to around 1.6. The calculated kappp values were 6.12 × 10−5 s−1, 1.77 × 10−5 s−1 and 1.12 × 10−5 s−1 corresponding to x = 0.1, 0.25 and 0.3, respectively, which further demonstrated that the polymerization rate decreased as the amount of fluorescein increased. One possible explanation would be related to the radical concentration; the high load of catalyst leads to higher concentration of polymer-end radicals which leads to more termination reaction.
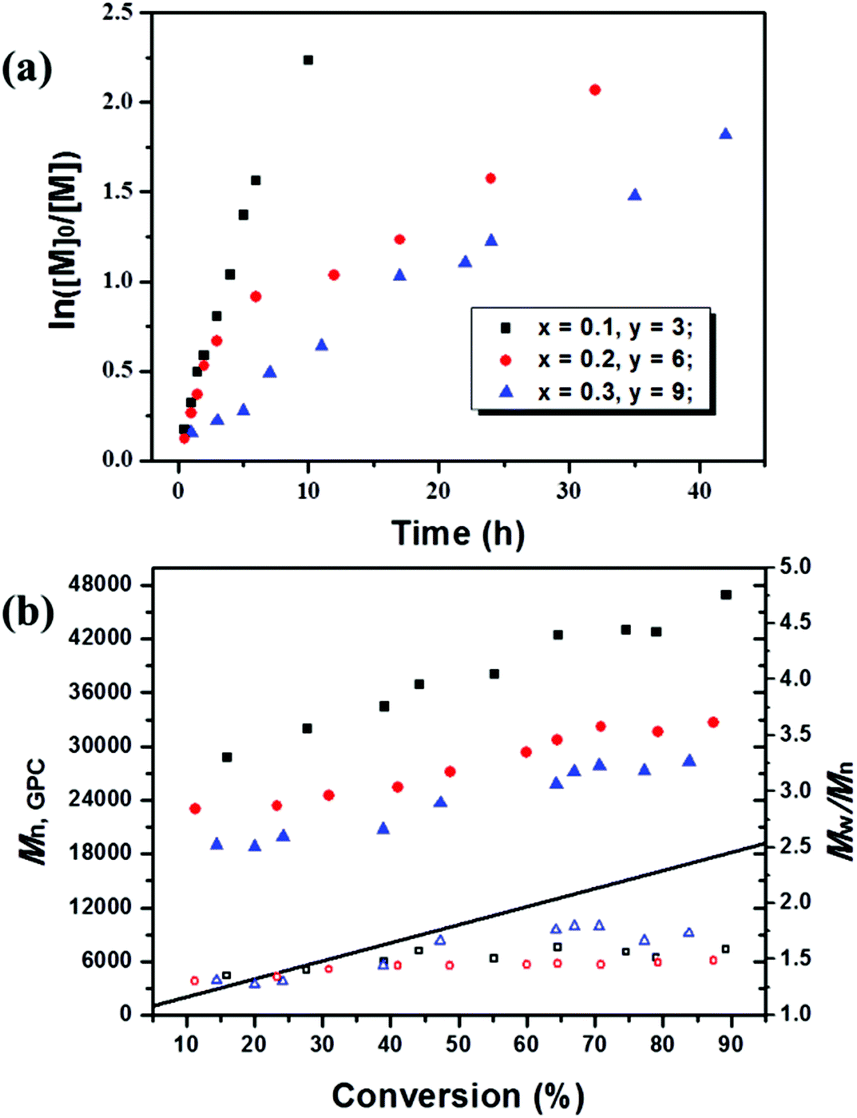 | ||
| Fig. 1 ln([M]0/[M]) as a function of time (a) and number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (b) for metal-free PET–ATRP of MMA. Polymerization conditions: [MMA]0/[EBPA]0/[FL]0/[TEA]0 = 200/1/x/y (x = 0.1, y = 3, square; x = 0.2, y = 6, circle; x = 0.3, y = 9, triangle), VMMA = 1.0 mL, VDMSO = 2.0 mL; samples were irradiated by a blue LED at rt. | ||
3.6. Polymerization kinetics and effects of the photocatalyst concentration on metal-free PET–ATRP of MMA at [MMA]0:[EBPA]0 = 400:1
Fig. 2 shows the kinetic plots of ln([M]0/[M]) versus time for metal-free PET–ATRP of MMA with molar ratios ([MMA]0:[EBPA]0:[FL]0:[TEA]0) of 400:1:0.3:9 and 400:1:0.4:12. The linearity of the kinetic plots in both cases indicated that the polymerization was approximately in a first order relationship with respect to the monomer concentration, and the propagation radicals kept constant during the polymerization process. When the conversion was less than 50%, the molecular weight (Mn,GPC) increased with monomer conversion and was higher than the theoretical values. The gap between molecular weights determined by GPC and theoretical ones was probably ascribed to the low initiation efficiency and lagging establishment of the dynamic equilibrium between active and dormant species. The photocatalytic reductive dehalogenation of initiator and polymer chain propagation always existed throughout the whole polymerization process, and molecular weights seem to approach theoretical values in more than about 50% conversion, suggesting the tendency of complete initiation with the increase of monomer conversion. Both at molar ratios ([MMA]0:[EBPA]0:[FL]0:[TEA]0) of 400:1:0.3:9 and 400:1:0.4:12 (entries 6–7 in Table S1†), the initiation efficiency was as high as 91% during the late period of polymerization (>85% Conv.). Yet, at the same time, the molecular weight distributions (Mw/Mn) become slightly broader than that in conversion less than 50%. By comparing the experimental results at 400:1:0.3:9 (kappp = 3.12 × 10−5 s−1) and at 400:1:0.4:12 (kappp = 1.98 × 10−5 s−1), polymerization regularity, that the reaction rate is reduced as the concentration of FL increases while bringing the molecular weights closer to theoretical values, was also discovered.
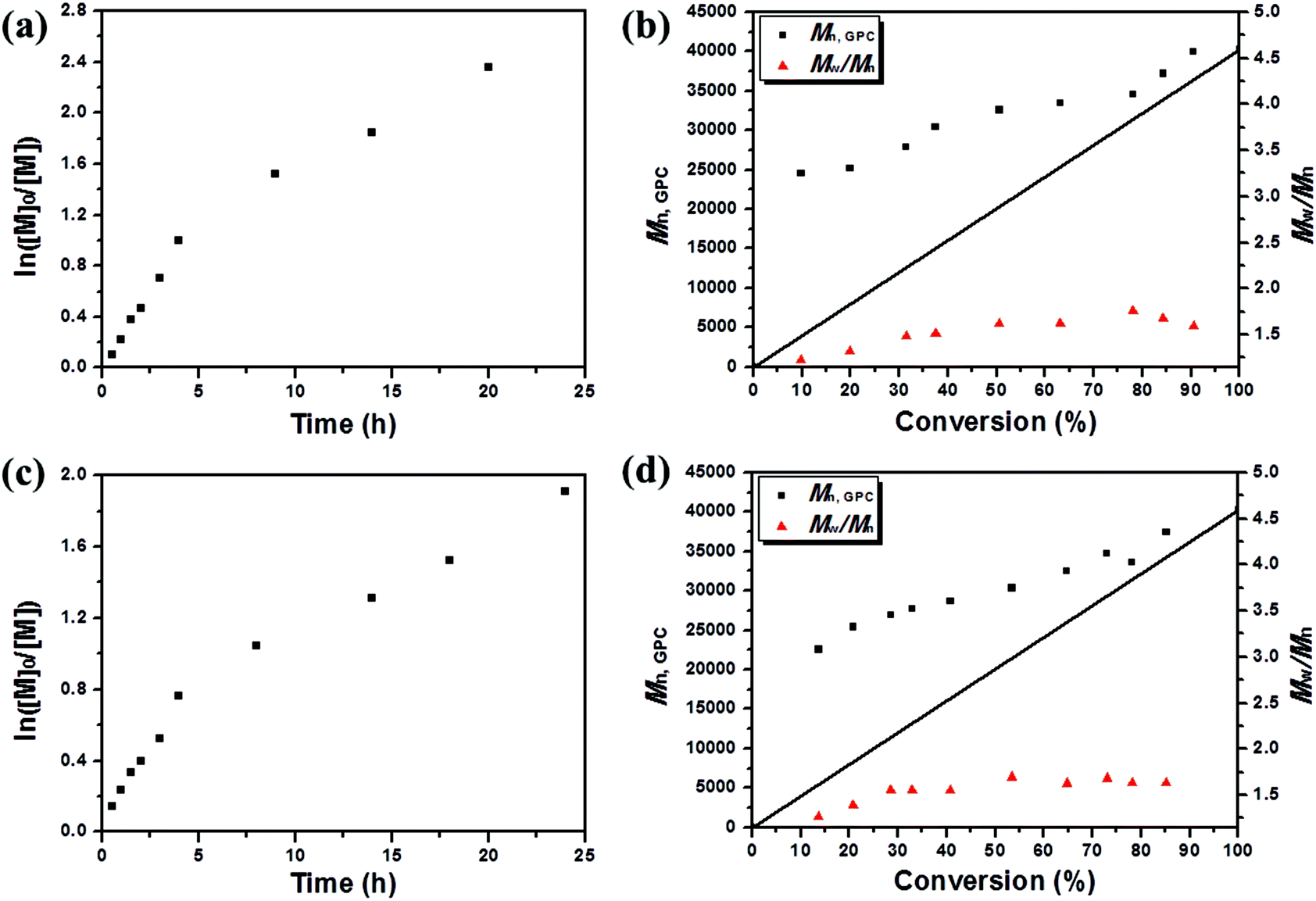 | ||
| Fig. 2 ln([M]0/[M]) as a function of time (a) and number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (b) for metal-free PET–ATRP of MMA. Polymerization conditions: [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 400:1:0.3:9, VMMA = 1.0 mL, VDMSO = 2.0 mL. ln([M]0/[M]) as a function of time (c) and number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (d) for metal-free PET–ATRP of MMA. Polymerization conditions: [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 400:1:0.4:12, VMMA = 1.0 mL, VDMSO = 2.0 mL; samples were irradiated by a blue LED at rt. | ||
Table S6† also shows the effect of the photocatalyst concentration on metal-free PET–ATRP of MMA at [MMA]0:[EBPA]0 = 400:1 in 2.0 mL or 4.0 mL of DMSO. In the polymerization system with 2.0 mL of solvent, the molecular weight Mn,GPC at the molar ratio ([MMA]0:[EBPA]0:[FL]0:[TEA]0) of 400:1:0.2:6 was higher than that of 400:1:0.4:12. For instance, the molecular weight was calculated to be 37500 g mol−1 at the monomer conversion of 85.2% (entry 7 in Table S6†) in the case of 400:1:0.4:12; while at the molar ratio of 400:1:0.2:6, the molecular weight reached up to 46500 g mol−1 when the monomer conversion was 86.3% (entry 2 in Table S6†). In the polymerization system containing 4.0 mL of solvent (entries 8–13 in Table S6†), the molecular weights provided under various conditions seem to show relatively few differences with each other, and by increasing the usage of catalyst, the polymerization rate would be slowed down, but the change of molecular weight along with monomer conversion was not distinct.
3.7. Effects of polymerization degree and monomer type on metal-free PET–ATRP of MMA
Higher molar ratios of [MMA]0/[EBPA]0 were also investigated in metal-free PET–ATRP under similar polymerization conditions. In the concentration of ∼1.0 mg mL−1 catalyst relative to monomer with a molar ratio of [MMA]0/[FL]0 = 100/0.2, the polymerization rate was decreased by increasing the polymerization degree (DP). As shown in Table S7,† monomer conversions of 57.8, 51.6 and 39.2% were achieved after 30 h in the cases of target DP of 500, 600 and 800, respectively. Compared with previous experimental data acquired under polymerization degrees of 200 and 400, it is easy to find that higher initiation efficiency was easier to access at high DP than at low DP. For example, at a DP of 500, the initiation efficiency was 77.5% when conversion reached 57.8%, while 86.2% of initiation efficiency could be obtained just as the monomer conversion was 39.25% at a DP of 800.In order to expand the scope and access of the applicability of this polymerization strategy to other functional monomers, the polymerization of styrene (St), poly(ethylene glycol) monomethyl ether methacrylate (PEGMA), acrylonitrile (AN), tert-butyl acrylate (tBA), glycidyl methacrylate (GMA) or benzyl methacrylate (BnMA) was also investigated as summarized in Table S8.† However, the extent of control over polymerization was inferior compared with that of PMMA. Further improvements for this metal-free method to enhance initiation efficiencies and provide polymers with controlled molecular weights, lower polydispersities as well as regular sequences and architectures are ongoing by our research group.
3.8. Polymerization mechanism and controlled “on–off” light switching regulation
The possible mechanism of this metal-free photoinduced electron transfer–atom transfer radical polymerization (PET–ATRP) mediated by fluorescein under visible light irradiation is shown in Fig. 3b. Fluorescein can absorb visible light to afford its excited species FL* which can transport electrons by the photo-induced electron transfer (PET) process;23 TEA is a reducing agent,24 capable of serving as a recyclable electron donor. As shown in Fig. 3b, under visible light irradiation, fluorescein in the excited state (FL*) was provided with an electron and reductively quenched by the electron donor TEA to generate an FL radical anion (FL˙−) in a reductive pathway rather than in an oxidative pathway, and an amine radical cation Et3N˙+ derived from the oxidation of TEA was also formed in this process, which was then involved in the single electron oxidation of bromine anion to generate the bromine radical. Then FL˙−, as a reductant processing the desired reduction potential, would transfer an electron to the alkyl bromide of the initiator or subsequent polymer chain ends to generate an electron-deficient alkyl radical or a propagating radical, and at the same time the initial ground state photocatalyst was regenerated. Addition of the alkyl radical to the olefin of the monomer would provide the propagating polymer chains. Meanwhile, the deactivation of the propagating radical should formally occur by the reaction with the bromine radical which was generated by a single electron oxidation of the bromine anion with the radical cation Et3N˙+. The reversed cycle can be established by the processes of further reduction of the dormant polymer, addition of monomers to alkyl radical chain ends, and formation of dormant chain-terminated species. This feature of the organic photocatalyst enables the establishment of an efficient activation and deactivation of polymerization equilibrium for control over molecular weights and polydispersities.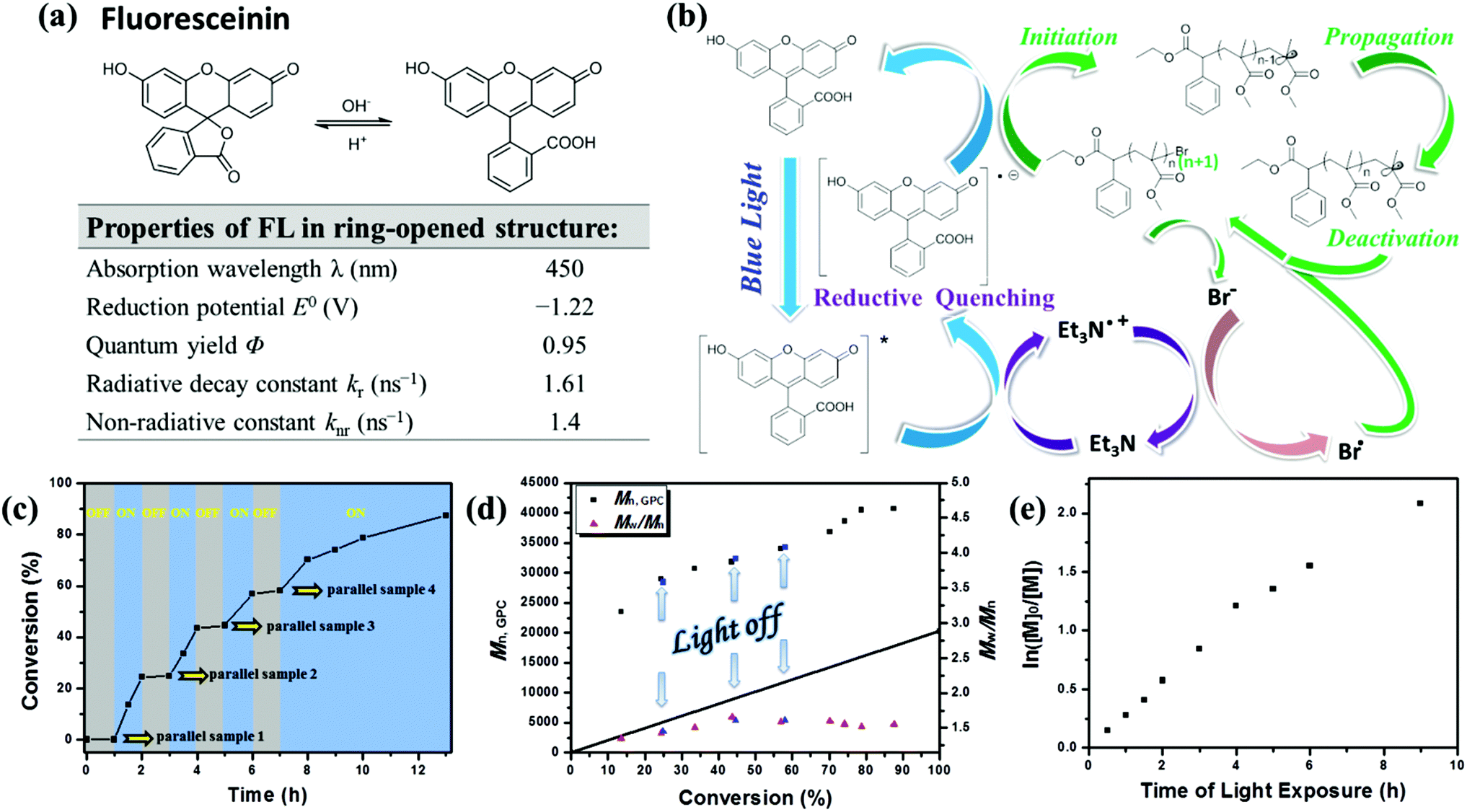 | ||
| Fig. 3 (a) pH-dependent equilibrium between the spirolactone form and the ring-opened form of fluorescein (FL) and optical properties of fluorescein in ring-opened structure.25 (b) Proposed mechanism of metal-free PET–ATRP photo-mediated with fluorescein as an organic photocatalyst. (c) Plot of monomer conversion versus time demonstrating the effect of blue light on the control over polymerization propagation through repeated “on-off” cycling of irradiation (blue regions) and light source removal (shaded regions); number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) versus conversion (d) and ln([M]0/[M]) as a function of exposure time (e) for metal-free PET–ATRP of MMA in the “on/off” light irradiation experiment. Polymerization conditions: [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 200:1:0.15:4.5, VMMA = 1.0 mL, VDMSO = 2.0 mL; samples were irradiated by a blue LED at rt. | ||
Fluorescence quenching studies demonstrated the reductive quenching effect. Fluorescence intensity of FL in DMSO solution decreased over time under blue light irradiation (Fig. S3a†), then the fluorescence – provided instantly with treatment of light avoidance in the dark environment – would recover within 30 min (Fig. S3b†); while for the solution of FL (0.1 mM) and TEA (30 equiv.) in DMSO, the quenched fluorescence caused by blue light irradiation (Fig. S3c†) was not recoverable even after being kept out of light for an adequate time (Fig. S3d†). This illustrates that an effective and irreversible fluorescence quenching effect had taken place in the presence of TEA. Meanwhile, the fluorescence of the polymerization system still had not been quenched throughout the whole reaction period (Fig. S2†), indicating that the initial ground state photocatalyst was regenerated after an electron transferred from FL˙− to the alkyl bromide of the initiator, and these results further confirmed the reversed catalysed cycle as illustrated in Scheme 1. Furthermore, the fluorescence intensity of FL with TEA (30 equiv.) in DMSO solution was much stronger than FL at the same concentration, illustrating that the chemical structure of fluorescein treated with TEA was maintained in the ring-opened form.
To confirm the necessity of light and the efficient nature of photocatalyst for reversible activation and deactivation of the polymerization process, an intermittent light irradiation sequence was introduced to investigate the temporal control. A repeated circulation of 1 h of irradiation followed by the same period of “dark” conditions was performed by using a parallel-group comparison trial method. The polymerization system was at first subject to the dark environment for 1 h, and the parallel sample 1 indicated that no polymer was obtained. The reaction mixture was then exposed to blue LED light, reaching 13.5% and 24.4% conversion after 0.5 h and 1 h light exposure, respectively (Fig. 3c). After the subsequent removal of irradiation from the system during the course of 1 h, the polymerization was suspended as demonstrated by parallel sample 2 with almost no monomer consumption and molecular weight Mn,GPC change observed during the “dark” periods. Upon re-exposure to light, as displayed in Fig. 3d, the polymerization process was reactivated, and the molecular weight reached up to 31800 g mol−1 after 4 h, along with the monomer conversion changing to 43.6%, proving that polymer chain termination did not occur when the light was turned off. This “on–off” light switching could be recycled multiple times, indicating that in the absence of light irradiation, the polymerization paused with the chain end resting as the dormant alkyl bromide, and the dormant species were protected from any irrelevant radical reaction but were available to be reactivated upon re-exposure to light. These experimental achievements confirmed that the polymerization processes were strictly driven by the light stimulus and only occurred during irradiation time, and stagnant conversion and molecular weight could be detected in the absence of light. Particularly, a proportionality relationship between molecular weight and monomer conversion was expressed up to a high conversion (>85%) even with three on/off cycles (Fig. 3d). Furthermore, a linear relationship between ln([M]0/[M]) and time of light exposure indicated a constant propagation radical concentration throughout the whole polymerization process (Fig. 3e) and illustrated the light-stimulus responsibility and “living” feature of the photocatalyst mediated metal-free polymerization.
3.9. Chain end analysis by 1H NMR and MALDI-TOF MS
Under the polymerization condition of [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 200:1:0.3:9 in 2.0 mL of DMSO for 5 h, PMMA obtained via the metal-free PET–ATRP mechanism was analyzed by 1H NMR spectroscopy in CDCl3 (Fig. 4a) and in DMSO-d6 (Fig. 4b). The ethyl phenylacetate and bromine end groups are observable in the 1H NMR spectrum scanned in CDCl3 solution. The aromatic protons He (around δ 7.35 ppm) and ethyl groups Hf (δ 4.09 ppm) were assigned to the initiator EBPA segment, as indicated in the chemical structure. The chemical shift Hd at δ 3.72 ppm is attributed to the methyl ester group at the end of the polymer chain, deviated from other methyl ester groups in PMMA due to the electron-attracting impact of the ω-bromide atom. The 1H NMR spectrum obtained in DMSO-d6 was used to calculate the molecular weight Mn,1H NMR by comparison of the integral area of the phenyl unit from EBPA (He) with the methyl groups in the polymer backbone (Ha) and methyl ester groups (Hc). However, the chemical shift of the methyl ester group at the polymer chain end observed in this situation is overlapped by other methyl ester groups (Fig. 4b, peak c). The polymerization degree (DP) and the molecular weight Mn,1H NMR were calculated as follows:| DP = [Area (Ha) − Area (Ha,EBPA) + Area (Hc)]/6 = 195; |
| Mn,1H NMR = MEBPA + DP × MMMA = 19800 g mol−1. |
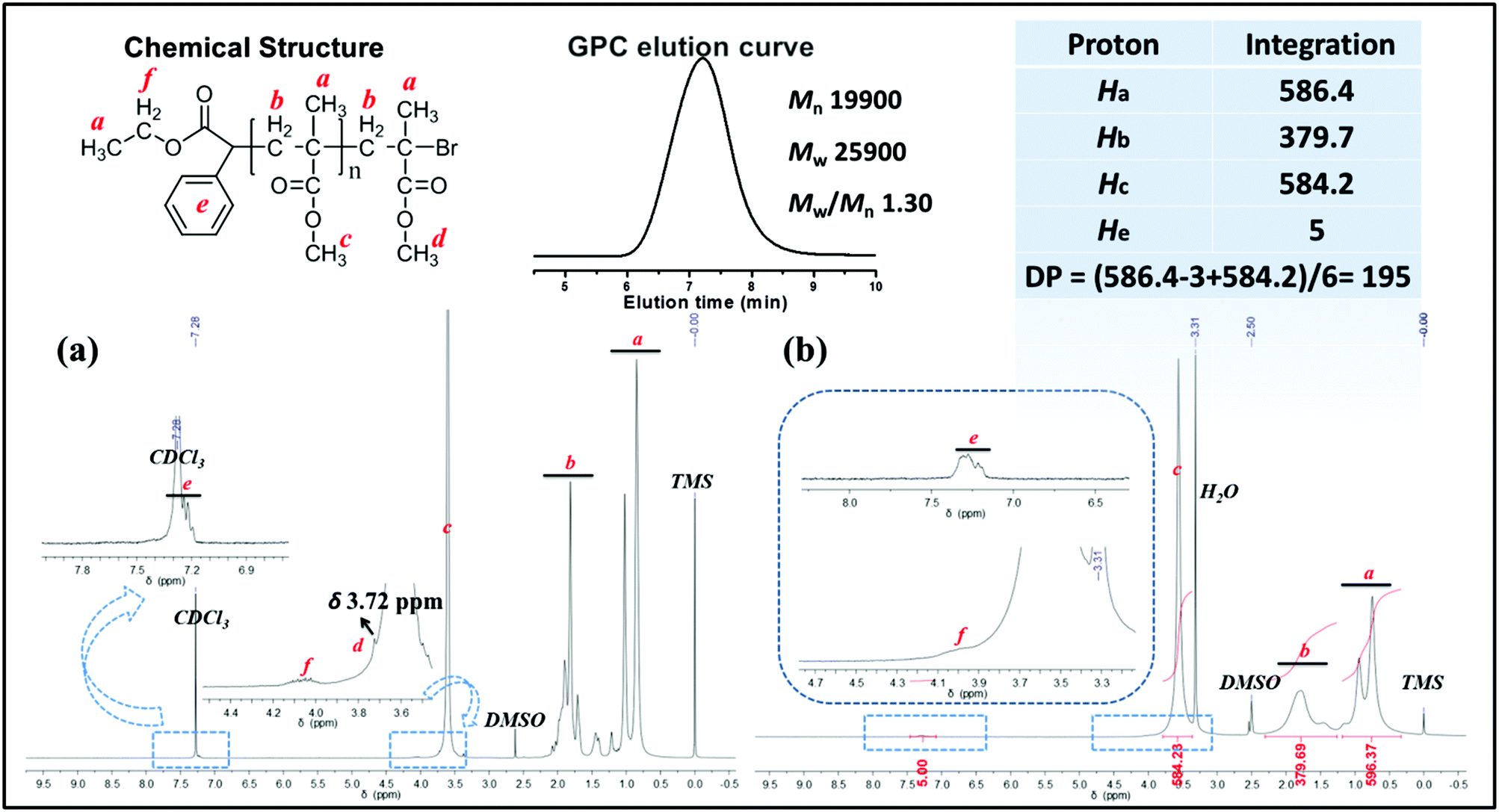 | ||
| Fig. 4
1H NMR spectra of PMMA in CDCl3 (a) and DMSO-d6 (b), obtained by photoinduced metal-free PET–ATRP of MMA under conditions of [MMA]0:[EBPA]0:[FL]0:[TEA]0 = 200:1:0.3:9 in 2.0 mL of DMSO for 5 h. Mn,GPC = 19900 g mol−1, Mw/Mn = 1.30, Mn,1H NMR = 19800 g mol−1. | ||
The molecular weight (Mn,1H NMR = 19800 g mol−1) calculated by the 1H NMR spectrum was very close to the molecular weight obtained by GPC analysis (Mn,GPC = 19900 g mol−1, Mw/Mn = 1.30), indicating that the obtained PMMA was end-capped by EBPA segments with high fidelity. To further confirm the initiation mechanism of the polymerization process, a polystyrene (PS) (entry 6 in Table S8,†Mn,GPC = 6200 g mol−1, Mw/Mn = 1.35) was analyzed by MALDI-TOF MS (Fig. 5). A series of main peaks is separated by an interval corresponding to a St repeating unit (104 mass units). This major set of peaks in the spectrometry indicated the existence of ethyl 2-phenyl acetate and bromide groups at the end of the polymer chains, suggesting that reversible activation and deactivation occurred effectively within this system. A PMMA sample (entry 2 in Table S4,†Mn,GPC = 13800 g mol−1, Mw/Mn = 1.53) was also analyzed by MALDI-TOF MS as shown in Fig. 6. The interval of the main series of peaks by a m/z ratio of 100.04–100.06 was assigned to a MMA repeating unit. This set of peaks belonging to PMMA demonstrated the existence of an ethyl 2-phenyl acetate group at the chain end but missing the bromide functional groups (Fig. 6), the absence of bromide chain-end groups was probably due to the fact that they could be obliterated from polymers during MALDI-TOF MS analysis as usually observed in polymers produced by traditional ATRP.26
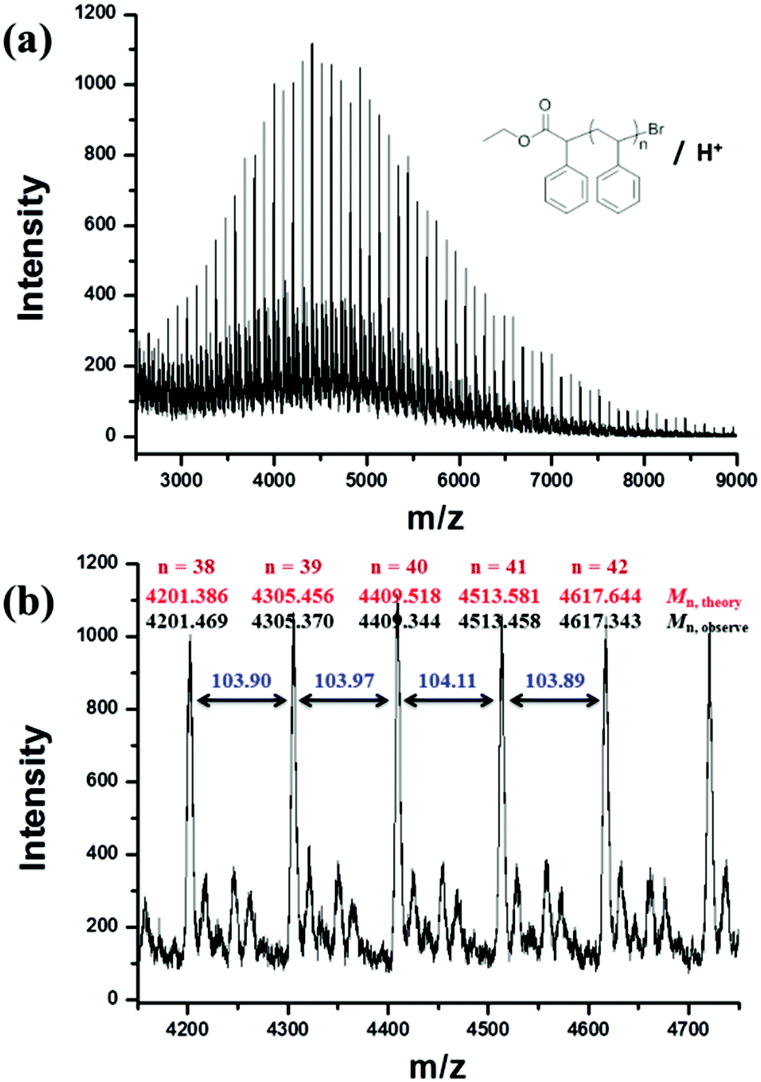 | ||
| Fig. 5 (a) MALDI-TOF mass spectrometry results in the linear mode of PS (Mn,GPC = 6200 g mol−1, Mw/Mn = 1.35) obtained by metal-free ATRP of MMA with fluorescein (FL) as an organic photocatalyst. (b) Enlargement of the MALDI-TOF-MS of PS from m/z 4150 to 4750. | ||
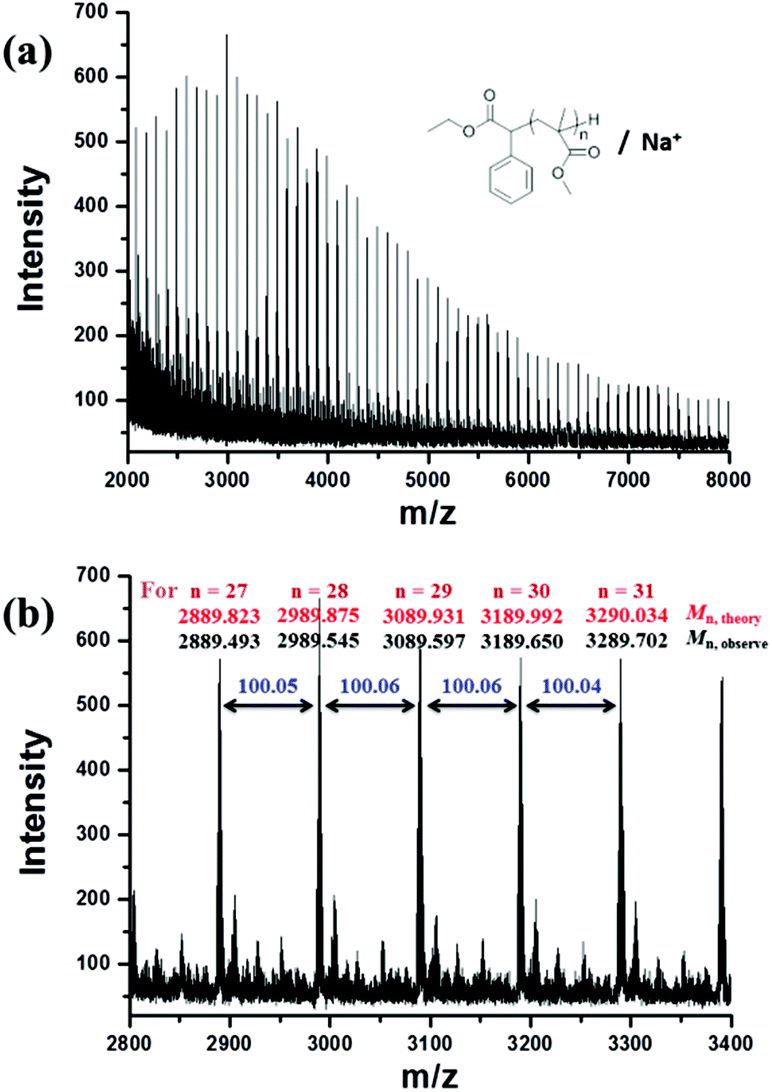 | ||
| Fig. 6 (a) MALDI-TOF mass spectrometry results in the round mode of PMMA (Mn,GPC = 13800 g mol−1, Mw/Mn = 1.53) obtained by metal-free ATRP of MMA with fluorescein (FL) as an organic photocatalyst. (b) Enlargement of the MALDI-TOF-MS of PMMA from m/z 2800 to 3400. | ||
3.10. Chain extension polymerization
To further confirm the “living” feature of metal-free PET–ATRP with fluorescein as an organic photocatalyst as well as provide additional evidence for the existence of active bromide in the chain ends, the obtained PMMA homopolymers, purified by precipitation from methanol at least twice to remove any possible residual EBPA initiator, was utilized as a macroinitiator agent in the chain-extension experiment with MMA or St as the monomer. The chain extension reactions carried out via the metal-free PET–ATRP mechanism were conducted with various fluorescein contents and were implemented using the macroinitiator in the form of similar molecular weight (approximately 19000 g mol−1) and polydispersity (Mw/Mn ∼ 1.30). Initially, as the employed content of FL was 1.73 equiv., there was an obvious increase of Mn,GPC of PMMA with a relatively wide distribution (Mn,GPC = 37400 g mol−1, Mw/Mn = 1.62) after the 48 h chain extension reaction (Fig. S4a†). When the chain extension photocatalyzed reaction was conducted using 3.46 equiv. fluorescein, GPC traces illustrated a peak shift from the macroinitiator (Mn,GPC = 19400 g mol−1, Mw/Mn = 1.30) to the obtained PMMA (Mn,GPC = 31000 g mol−1, Mw/Mn = 1.60) as observed in Fig. S4b.† The GPC elution curve exhibited that some starting macroinitiators remained in the extended polymers, which may be perhaps caused by the difficulty in activating the bromide groups in the long polymer chain ends.
Chain extension of macroinitiator-1 (Mn,GPC = 18800 g mol−1, Mw/Mn = 1.27) with MMA was carried out with 5.2 equiv. fluorescein at rt under blue LED irradiation for 48 h (Fig. 7a), providing a well-defined polymer (Mn,GPC = 29000, Mw/Mn = 1.44). The GPC elution trace clearly shifted to the higher molecular species, suggesting the sufficient alkyl bromine chain-end functionality and high propagation efficiency of the macroinitiator. The effects of fluorescein content found in these experiments were quite significant, which suggests that in order to gain control over polymers in chain extension polymerization, it is necessary to increase the dosage of photocatalyst in the polymerization system to establish the equilibrium between the activation of the dormant bromide chain terminated polymer and the deactivation of the propagating radical chain.
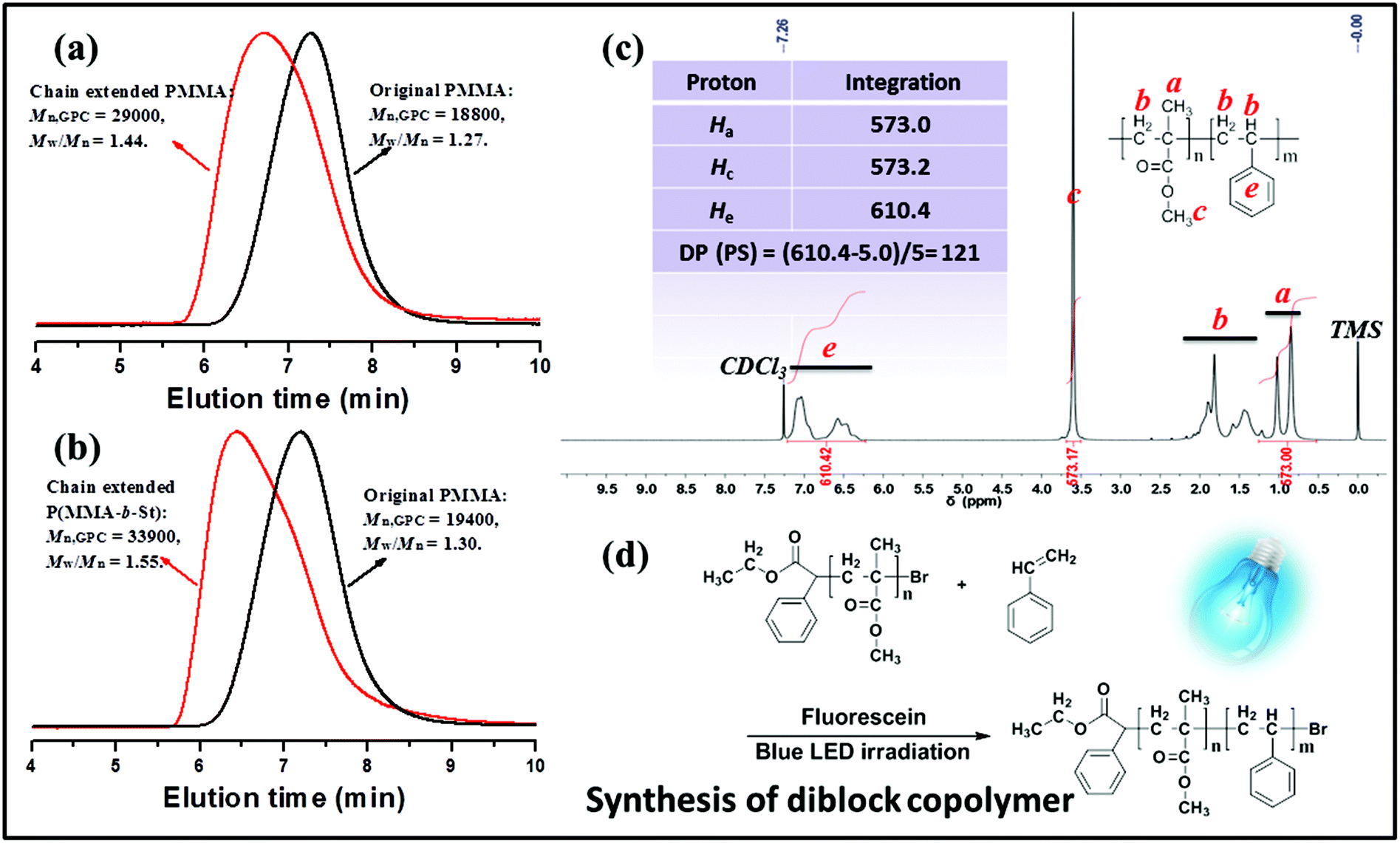 | ||
| Fig. 7 (a) GPC traces of PMMA before and after chain extension using PMMA (Mn,GPC = 18800 g mol−1, Mw/Mn = 1.27) prepared by metal-free PET–ATRP of MMA as the macroinitiator. (b) GPC traces of a poly(methyl methacrylate)-b-(styrene) diblock copolymer P(MMA-b-St) before and after chain extension using PMMA (Mn,GPC = 19400 g mol−1, Mw/Mn = 1.30) prepared by metal-free PET–ATRP of MMA as the macroinitiator. (c) 1H NMR spectrum of P(MMA-b-St) in CDCl3. (d) Synthesis of P(MMA-b-St) by the photoinduced metal-free PET–ATRP mechanism. | ||
In Fig. 7b, the chain extension employing styrene as the fresh monomer resulted in a block copolymer of macroinitiator-2 (Mn,GPC = 19400 g mol−1, Mw/Mn = 1.30) P(MMA-b-St) (Mn,GPC = 33900 g mol−1, Mw/Mn = 1.55) with an obvious GPC peak shift. Moreover, as shown in Fig. 7, the chemical shifts at δ 6.37–7.08 ppm assigned to the aromatic protons in PS segments indicated the successful synthesis of the block copolymer. The molecular weight of such a block copolymer measured by 1H NMR is 32000 g mol−1, and the calculated DP of the PS segment was 121. The difference between Mn,GPC and 1H NMR data was probably attributed to the use of linear PMMA as the GPC standard. All these results exemplify the photocatalytic nature of FL via the metal-free PET–ATRP mechanism, and indicate that the macroinitiator with bromide chain-end groups can be used to reinitiate polymerization under external visible light stimulation to construct block copolymers.
4. Conclusions
In this work, the photoinduced living radical polymerization technique named photoinduced electron transfer–atom transfer radical polymerization (PET–ATRP) using a reductive pathway was developed for the first time, which provides new opportunities for the synthesis of well-controlled polymer architectures through a photochemical approach. Serving as an organic photocatalyst for the reduction of alkyl bromides to generate carbon-central radicals in the presence of TEA as an electronic reservoir under visible light irradiation, fluorescein has been successfully evaluated as an efficient catalyst for photoinduced metal-free PET–ATRP of methyl methacrylate (MMA) via a reductive quenching cycle. Temporal control over the polymerization in “on/off” switch experiment was easily demonstrated by activation and deactivation processes manipulated by light-on and off periods, respectively. MALDI-TOF MS and 1H NMR spectroscopy demonstrated that ethyl phenylacetate and bromine end groups corresponding to the initiator EBPA were directly introduced at the polymer chain end. Chain-extension polymerizations further confirmed the “living” feature of metal-free PET–ATRP using fluorescein as an organic photoredox catalyst. The utility of photoinduced metal-free PET–ATRP for the synthesis of block copolymers was achieved by the chain extension of PMMA homopolymers with styrene. However, the photocatalyzed polymerization has been extended to the synthesis of polystyrene (PS) and other methacrylate functional monomers with predictable molecular weights and polydispersities which needs to be further improved.Acknowledgements
The financial support from the National Natural Science Foundation of China (no. 21174096, 21274100, 21234005), the Specialized Research Fund for the Doctoral Program of Higher Education (no. 20123201130001), the Project of Science and Technology Development Planning of Suzhou (no. ZXG201413, SYG201430), the Project of Science and Technology Development Planning of Jiangsu Province (no. BK20141192) and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) are gratefully acknowledged.Notes and references
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Chem. Commun., 2012, 48, 10951–10953 RSC.
- (a) M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS PubMed; (b) X. Liu, Q. Chen, G. Yang, L. Zhang, Z. Liu, Z. Cheng and X. Zhu, J. Mater. Chem. B, 2015, 3, 2786–2800 RSC; (c) X. Liu, B. Chen, X. Li, L. Zhang, Y. Xu, Z. Liu, Z. Cheng and X. Zhu, Nanoscale, 2015, 7, 16399–16416 RSC.
- A. Ohtsuki, L. Lei, M. Tanishima, A. Goto and H. Kaji, J. Am. Chem. Soc., 2015, 137, 5610–5617 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- M. E. Honigfort, W. J. Brittain, T. Bosanac and C. S. Wilcox, Macromolecules, 2002, 35, 4849–4851 CrossRef CAS.
- (a) M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS; (b) A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580–7583 CrossRef CAS PubMed; (c) N. J. Treat, B. P. Fors, J. W. Kramer, M. Christianson, C.-Y. Chiu, J. R. d. Alaniz and C. J. Hawker, ACS Macro Lett., 2014, 3, 580–584 CrossRef CAS.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- (a) J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed; (b) J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS; (c) S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed; (d) T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS; (e) S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed; (f) J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- (a) K. Ishizu and H. Kakinuma, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 63–70 CrossRef CAS; (b) D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS; (c) B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed; (d) M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036–2042 CrossRef CAS; (e) B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC; (f) X. Jiang, J. Wu, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2014, 35, 1879–1885 CrossRef CAS PubMed; (g) V. Nikolaou, A. Anastasaki, F. Alsubaie, A. Simula, D. J. Fox and D. M. Haddleton, Polym. Chem., 2015, 6, 3581–3585 RSC; (h) A. Anastasaki, V. Nikolaou, F. Brandford-Adams, G. Nurumbetov, Q. Zhang, G. J. Clarkson, D. J. Fox, P. Wilson, K. Kempe and D. M. Haddleton, Chem. Commun., 2015, 51, 5626–5629 RSC; (i) Q. Yang, F. Dumur, F. Morlet-Savary, J. Poly and J. Lalevée, Macromolecules, 2015, 48, 1972–1980 CrossRef CAS.
- (a) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed; (b) G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS; (c) X. Pan, M. Lamson, J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS.
- (a) A. Debuigne, M. Schoumacher, N. Willet, R. Riva, X. Zhu, S. Rutten, C. Jerome and C. Detrembleur, Chem. Commun., 2011, 47, 12703–12705 RSC; (b) Y. Zhao, M. Yu and X. Fu, Chem. Commun., 2013, 49, 5186–5188 RSC.
- (a) S. Yamago, Y. Ukai, A. Matsumoto and Y. Nakamura, J. Am. Chem. Soc., 2009, 131, 2100–2101 CrossRef CAS PubMed; (b) Y. Nakamura and S. Yamago, Macromolecules, 2015, 48, 6450–6456 CrossRef CAS.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- (a) H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600–13603 CrossRef CAS PubMed; (b) J. T. Xu, A. Atme, A. F. Martins, K. Jung and C. Boyer, Polym. Chem., 2014, 5, 3321–3325 RSC.
- C.-H. Peng, T.-Y. Yang, Y. Zhao and X. Fu, Org. Biomol. Chem., 2014, 12, 8580–8587 CAS.
- (a) S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS; (b) J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC; (c) S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- (a) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed; (d) D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC; (e) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed; (f) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169–171 CrossRef CAS; (g) J. T. Xu and C. Boyer, Macromolecules, 2014, 48, 520–529 CrossRef.
- J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS.
- (a) M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed; (b) J. Moser and M. Graetzel, J. Am. Chem. Soc., 1984, 106, 6557–6564 CrossRef CAS; (c) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS; (d) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- (a) S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831–7841 CrossRef CAS PubMed; (b) E. Azuma, N. Nakamura, K. Kuramochi, T. Sasamori, N. Tokitoh, I. Sagami and K. Tsubaki, J. Org. Chem., 2012, 77, 3492–3500 CrossRef CAS PubMed; (c) M. D. Pluth, M. R. Chan, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2011, 9385–9392 CrossRef CAS PubMed; (d) X. Xiong, F. Song, G. Chen, W. Sun, J. Wang, P. Gao, Y. Zhang, B. Qiao, W. Li, S. Sun, J. Fan and X. Peng, Chem. – Eur. J., 2013, 19, 6538–6545 CrossRef CAS PubMed; (e) H. Zheng, X.-Q. Zhan, Q.-N. Bian and X.-J. Zhang, Chem. Commun., 2013, 49, 429–447 RSC.
- L. Yuan, W. Lin and Y. Feng, Org. Biomol. Chem., 2011, 9, 1723–1726 CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35–41 CrossRef CAS.
- H. D. Burrows, Photochem. Photobiol., 1974, 19, 241–243 CrossRef CAS PubMed.
- (a) G. R. Fleming, A. W. E. Knight, J. M. Morris, R. J. S. Morrison and G. W. Robinson, J. Am. Chem. Soc., 1977, 99, 4306–4311 CrossRef CAS; (b) D. Magde, R. Wong and P. G. Seybold, Photochem. Photobiol., 2002, 75, 327–334 CrossRef CAS PubMed.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 7791–7805 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01765c |
| This journal is © The Royal Society of Chemistry 2016 |