
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new Ru,Ru,Pt supramolecular architecture for photocatalytic H2 production†
Jessica Knoll
White‡
 * and
Karen J.
Brewer§
* and
Karen J.
Brewer§
Department of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: knoll.56@osu.edu
First published on 15th September 2015
Abstract
A new polyazine-bridged RuRuPt trimetallic supramolecular architecture emulates the photophysical properties of the previously reported Ru2RuPt tetrametallic architecture that exhibits photoinduced charge separation. The RuRuPt complexes are more robust H2O reduction photocatalysts with enhanced stability compared to the Ru2RuPt tetrametallic analogues.
Solar-to-chemical energy conversion is an important topic in the quest for clean and renewable energy.1–5 Converting H2O to H2 fuel by harvesting solar energy is a complicated multi-electron process that involves bond breaking and formation.1,6–8 Ru(II)–polyazine complexes, such as the prototypical [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), are attractive light absorbers (LA) for harnessing solar energy due to their broad UV and visible light absorption and long-lived, strongly reducing/oxidizing excited states.9–12 Molecular photocatalysts provide a means for analysing and understanding the complicated processes involved in H2O reduction.13
Supramolecular complexes,14 individual components each possessing their own function assembled into systems that perform a complex task, are an important class of molecular photocatalysts for H2 production from H2O. Several supramolecular photocatalysts featuring a Ru–polyazine LA coupled to a reactive metal (RM) such as Co,15 Rh,16–21 Pd,22–24 and Pt,25–29 are reported. The photocatalytic activity of a series of Ru,Pt bimetallic complexes, [(bpy)2Ru{phenNHCO(Rbpy)}PtCl2]2+ (phen = 1,10-phenanthroline, R = –COOH, –COOEt, or –CH3) depends on the nature of R. The charge separated (CS) excited state is most stabilized in the –COOH complex, providing the greatest activity in aqueous solution with 5 TON in 10 h.25,27 Dimerization of this architecture through the R-bpy unit doubles the efficiency by enhancing Pt–Pt dimerization for proton coupled electron transfer. A Ru,Pt bimetallic system [(tBu2bpy)2Ru(tpphz)PtX2]2+ (tBu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine; tpphz = tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine; X = Cl− or I−) produces H2 from H2O with 7 TON when X = Cl−, while a 40 fold increase in TON and enhanced stability is achieved when X = I−.30 It was recently discovered that the active catalyst of the PdIICl2 analogue, [(tBu2bpy)2Ru(tpphz)PdCl2]2+, is colloidal Pd,31 corroborating results of a previously reported Ru,Pd photocatalyst.32
The supramolecular architecture [{(TL)2Ru(dpp)}2Ru(BL)PtCl2]6+ (Ru2RuPt; TL = phen or Ph2phen = 4,7-diphenyl-1,10-phenanthroline; BL = dpp = 2,3-bis(2-pyridyl)pyrazine or dpq = 2,3-bis(2-pyridyl)quinoxaline) is active in photocatalytic H2 production from H2O by virtue of photoinduced charge separation in which the HOMO is localized on the terminal Ru and the LUMO is localized on BL coordinated to the Pt RM.33,34 The catalytic efficiency of this architecture is strongly influenced by the nature of BL; the complexes with BL = dpq display greatly enhanced catalysis compared to their BL = dpp counterparts owing to the stabilized LUMO and enhanced driving force for intramolecular electron transfer toward the reactive Pt centre. Spectroscopic analysis of these complexes is difficult due to the presence of multiple overlapping, strongly absorbing intraligand (IL) π → π* and metal-to-ligand charge transfer (MLCT) transitions in the UV and visible regions, respectively.
Reported herein is the new trimetallic supramolecular architecture [(Ph2phen)2Ru(dpp)Ru(bpy)(BL)PtCl2]4+ (labelled RuRuPt; BL = dpp or dpq) designed to provide analogous redox, spectroscopic, photophysical, and photocatalytic properties compared to the Ru2RuPt tetrametallic complexes. Reducing the number of (Ph2phen)2RuII(dpp) LA units from two to one simplifies the molecular architecture and provides more active photocatalysts for H2O reduction compared to the Ru2RuPt analogues. Fig. 1 depicts the structures of the new RuRuPt trimetallic complexes, [(Ph2phen)2Ru(dpp)Ru(bpy)(dpp)PtCl2]4+ (RuRudppPt) and [(Ph2phen)2Ru(dpp)Ru(bpy)(dpq)PtCl2]4+ (RuRudpqPt), and the previously reported Ru2RuPt tetrametallic complexes (Ru2RudppPt and Ru2RudpqPt). The synthesis, redox and spectroscopic properties, and photocatalytic activity towards H2O reduction of this new architecture are discussed below.
 | ||
| Fig. 1 Structural representations of the RuRuPt and Ru2RuPt complexes. | ||
The RuRuPt complexes were assembled using a six step building block process. Synthetic details are provided in ESI.† The synthetic approach to these structurally complicated supramolecules required initial assembly of the bimetallic complex [(Ph2phen)2Ru(dpp)RuCl2(bpy)](PF6)2 by coupling [(Ph2phen)2Ru(dpp)](PF6)235 and [(bpy)RuCl2(DMSO)2]36 monometallic precursors. This complex was reacted with either dpp or dpq to provide an open coordination site to extend the molecular architecture, thereby generating bimetallics of the design [(Ph2phen)2Ru(dpp)Ru(bpy)(BL)](PF6)4. The RuRuPt trimetallics, [(Ph2phen)2Ru(dpp)Ru(bpy)(BL)PtCl2](PF6)4 (RuRudppPt and RuRudpqPt), were prepared upon reacting the BL-containing bimetallic with cis-[PtCl2(DMSO)2]. ESI-MS data is consistent with the desired product for each complex.
Electrochemical analysis of the RuRuPt complexes and their bimetallic precursors indicates orbital energetics that are similar to the analogous Ru2RuPt tetrametallic complexes and their trimetallic precursors. Square wave voltammograms are provided in Fig. S1 and the data and assignments are given in Table S1 (ESI†). Fig. 2a highlights the similarities between the cyclic voltammograms of RuRudpqPt and Ru2RudpqPt. The new RuRuPt complexes possess the same spatially separated HOMO and LUMO that was reported in the Ru2RuPt complexes and is necessary for photoinduced charge separation.33,34,37 The first oxidation process at 1.54–1.57 V vs. Ag/AgCl is assigned the terminal RuII/III oxidation in all four Pt-containing complexes. This couple for Ru2RudppPt and Ru2RudpqPt has approximately twice the peak current of RuRudppPt and RuRudpqPt, consistent with the number of terminal Ru centres (two and one, respectively). The first reduction is assigned as BL0/−; this reduction occurs at −0.39 V and −0.08 V for RuRudppPt and RuRudpqPt, respectively, and this trend is consistent with dpq's stabilized π* orbitals compared to those of dpp. The BL0/− potentials are quite similar to those of the corresponding tetrametallic complexes (−0.33 V and −0.02 V for Ru2RudppPt and Ru2RudpqPt, respectively). This supports the validity of the RuRuPt complexes as analogues for the Ru2RuPt complexes.
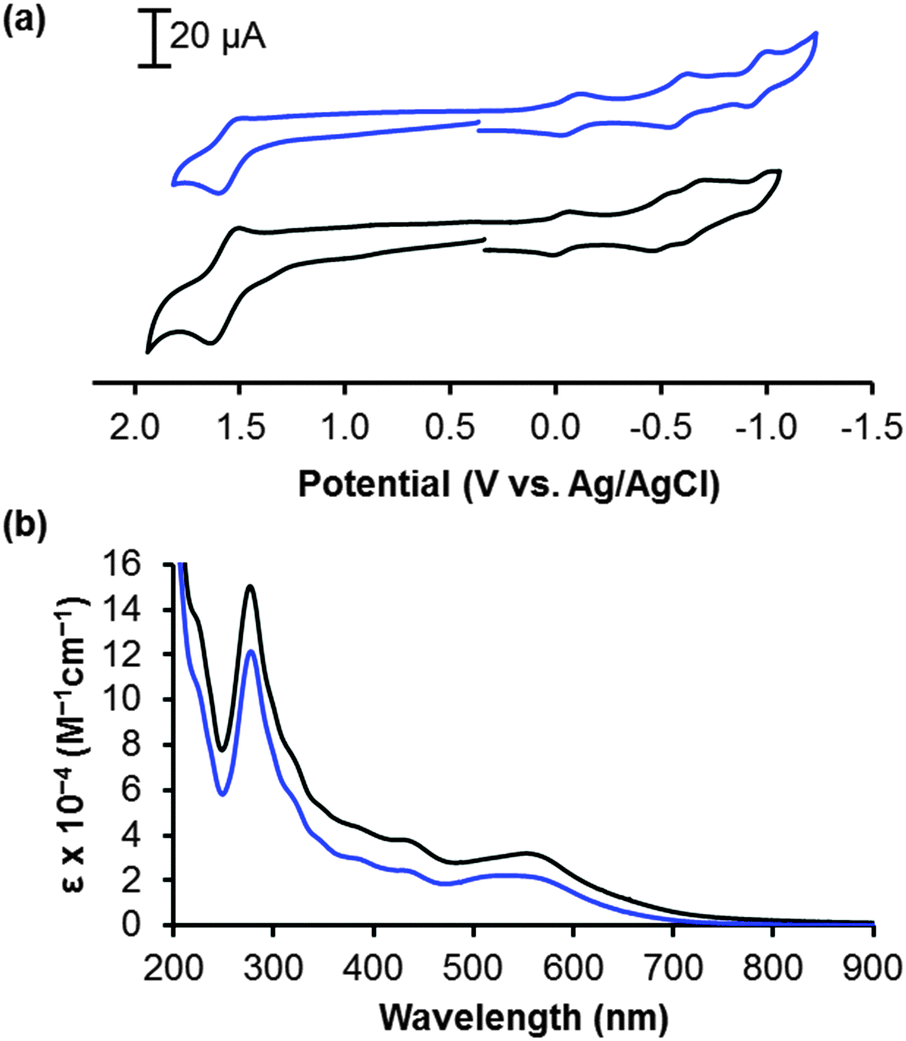 | ||
| Fig. 2 Cyclic voltammograms measured in CH3CN with 0.1 M Bu4NPF6 (a) and electronic absorption spectra in CH3CN (b) of Ru2RudpqPt (black) and RuRudpqPt (blue). | ||
The electronic absorption spectrum for each of the RuRuPt complexes is similar to the Ru2RuPt analogue while exhibiting a decrease in the molar absorptivity throughout the UV and visible regions due to the absence of a strongly absorbing (Ph2phen)2RuII(dpp) unit. This is shown in Fig. 2b and Fig. S2 and Table S2 (ESI†). Similar to related Ru(II)–polyazine complexes, the UV and visible regions are dominated by IL and MLCT transitions, respectively, with transitions involving dpp or dpq red-shifted compared to Ph2phen transitions. The broad, lowest energy absorption band for RuRudpqPt is centred at 543 nm (ε = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) with contributions from Ru → dpp and Ru → dpq 1MLCT transitions. The lowest energy band for Ru2RudpqPt is centred at 550 nm with ε = 31000 M−1 cm−1 due to additional central Ru → dpp and terminal Ru → dpp 1MLCT transitions. Similar trends are observed in comparing RuRudppPt with Ru2RudppPt (Table S2, ESI†). The RuRuPt architecture maintains the broad light absorption that covers the entire visible region which is desirable in solar energy conversion, while decreasing the number of possible transitions to allow for further studies such as transient absorption spectroscopy.
000 M−1 cm−1) with contributions from Ru → dpp and Ru → dpq 1MLCT transitions. The lowest energy band for Ru2RudpqPt is centred at 550 nm with ε = 31000 M−1 cm−1 due to additional central Ru → dpp and terminal Ru → dpp 1MLCT transitions. Similar trends are observed in comparing RuRudppPt with Ru2RudppPt (Table S2, ESI†). The RuRuPt architecture maintains the broad light absorption that covers the entire visible region which is desirable in solar energy conversion, while decreasing the number of possible transitions to allow for further studies such as transient absorption spectroscopy.
The emissive nature of the RuRuPt complexes provides a convenient probe into the excited state dynamics that are similar to those observed for the Ru2RuPt complexes. A simplified state diagram for RuRudpqPt is pictured in Fig. S3 and emission spectroscopy data is presented in Table S3 and Fig. S4 and S5 (ESI†). Emission is observed from the terminal Ru → dpp 3MLCT excited state in each case. The two [(Ph2phen)2Ru(dpp)Ru(bpy)(BL)](PF6)4 bimetallic complexes emit at 762 nm with τ = 120 ns and Φem = 1.5 × 10−3. These homobimetallics serve as photophysical models to study charge separation in the Pt-containing complexes. Coordination of cis-PtCl2 results in quenched emission (Φem = 1.1 × 10−3 and 5.2 × 10−4 for BL = dpp and dpq, respectively) and shortened excited state lifetimes (τ = 90 and 100 ns when BL = dpp and dpq, respectively) as the Pt unit stabilizes the BL(π*) orbitals and enables intramolecular electron transfer to populate a low-lying, non-emissive charge separated (3CS) state. As observed in the Ru2RuPt complexes, the degree of emission quenching and charge separation is strongly dependent on the nature of BL.34,37 The emissive 3MLCT state is populated with 98% and 43% efficiency for RuRudppPt and RuRudpqPt with λexc = 540 nm (equations in ESI†). These efficiencies are close to the analogous Ru2RuPt complexes (99% and 51% for Ru2RudppPt and Ru2RudpqPt, respectively).34 Less efficient emissive state population results from more efficient population of the 3CS state. The degree by which Φem and τ of the 3MLCT state are quenched varies substantially due to population of the 3CS from both the emissive 3MLCT state and a higher-energy 3MLCT state, as discussed previously.34,37 The agreement in photophysical properties of the Ru2RuPt and RuRuPt complexes further demonstrates the suitability of the new architecture as analogues to the tetrametallic complexes.
The RuRuPt complexes exhibit remarkably enhanced photocatalytic activity and stability towards H2 production from H2O compared to their Ru2RuPt analogues. Table 1 features the amount of H2 produced and turnover number (TON = moles of H2 produced/moles of catalyst) for RuRudppPt and RuRudpqPt as well as for the previously reported Ru2RudppPt and Ru2RudpqPt following 470 nm irradiation (flux = 2.3 × 1019 photons per min) for 20 hours in RT CH3CN with 50 μM metal complex, 0.62 M H2O, 1.5 M DMA sacrificial electron donor, and 110 μM [DMAH+][SO3CF3−]. The volume of the solution and the headspace are 4.5 mL and 15.3 mL, respectively. The reported values are the average of three experiments. To highlight the greater activity of the new architecture, a plot of H2 production vs. time for RuRudpqPt and Ru2RudpqPt is provided in Fig. 3, and the plot for the dpp analogues are provided in Fig. S6 (ESI†). RuRudpqPt is the most active catalyst in this series, producing 52 ± 4 μmol of H2 in 20 hours and undergoing 230 ± 20 TON (quantum yield of H2 production, Φ = 1.1 × 10−3). The enhancement in photocatalytic activity within the trimetallic series of RuRudpqPt and RuRudppPt (29 ± 2 μmol of H2, 130 ± 9 TON, Φ = 6.2 × 10−4) is related to the enhanced 3CS state population in the BL = dpq complex and agrees with the trend observed in the analogous Ru2RuPt complexes.34 For each BL, the H2 production of the RuRuPt complex is greater than the Ru2RuPt analogue (shown in Fig. 3 for the BL = dpq analogues) despite the smaller molecule's less efficient absorption at 470 nm. The RuRuPt architecture is expected to provide less demanding steric bulk near the cis-PtCl2 site, whereas the presence of two bulky (Ph2phen)2RuII(dpp) units in the Ru2RuPt architecture may hinder interactions between the substrate and the catalytic site. Three dimensional models highlighting the difference in sterics for different geometric isomers of the Ru2RudpqPt and RuRudpqPt architectures are provided in Fig. S7 (ESI†). The RuRuPt and Ru2RuPt complexes are expected to exist in up to 16 and 32 isomers, respectively, as a result of Λ and Δ optical isomers as well as the AB chelating nature of the bridging ligands. The distribution of isomers in each sample is unknown; however, steric effects on reactivity, charge separation distance, and orbital overlap are expected to result from structural variations among isomers.
| Complex | μmol H2 | TONb | Φ × 104 |
|---|---|---|---|
| a 50 μM catalyst in spectral grade CH3CN, 0.62 M H2O, 1.5 M DMA, 110 μM [DMAH+][SO3CF3−], λirr = 470 ± 10 nm (flux = 2.3 × 1019 photons per min). Values represent H2 production after 20 h photolysis. b TON = moles of H2 produced/moles of catalyst. c Quantum yield of H2 production. | |||
| Ru2RudppPt | 7.1 ± 3.2 | 32 ± 14 | 1.6 ± 0.7 |
| Ru2RudpqPt | 25 ± 1 | 110 ± 6 | 5.5 ± 0.3 |
| RuRudppPt | 29 ± 2 | 130 ± 9 | 6.2 ± 0.4 |
| RuRudpqPt | 52 ± 4 | 230 ± 20 | 11 ± 1 |
 | ||
| Fig. 3 Photocatalytic H2 production with RuRudpqPt (black circles) and Ru2RudpqPt (white circles) with 50 μM catalyst in spectral grade CH3CN, 0.62 M H2O, 1.5 M DMA, and 110 μM [DMAH+][SO3CF3−]. Solutions were irradiated with λ = 470 ± 10 nm. | ||
The new RuRuPt trimetallic supramolecular architecture features a Ru(II)–polyazine LA and a cis-PtCl2 RM site with a spatially separated HOMO and LUMO that imparts unusual photophysical properties. This architecture provides redox and photophysical properties analogous to the previously reported Ru2RuPt architecture, demonstrating the ability to design supramolecular complexes with desired properties through knowledge of previously studied systems. The architecture features a terminal Ru-based HOMO and a remote BL-based LUMO which enables photoinduced charge separation. The nature of BL dictates the energy of the LUMO and largely impacts the population of the 3CS state in competition with population of the emissive 3MLCT state, with BL = dpq affording a complex with a more efficiently populated 3CS state compared to the BL = dpp analogue. The enhanced charge separation in RuRudpqPt is important in providing an active and stable catalyst for H2 production from H2O (52 μmol of H2 and 230 TON in 20 h) while RuRudppPt is less active (29 μmol of H2 and 130 TON in 20 h). Both of these complexes are superior to their Ru2RuPt analogues, possibly due to steric factors provided by the bulky (Ph2phen)2RuII(dpp) units or a varied distribution of geometric isomers. Future studies include further probing the unusual excited state dynamics using transient absorption spectroscopy enabled by the less complicated spectroscopy by virtue of the absence of a strongly absorbing LA unit.
Acknowledgement is made to the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Sciences, U.S. Department of Energy (DE FG02-05ER15751) for their generous support of the development of new LAs used in this research.
References
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- D. Gust and T. A. Moore, Science, 1989, 244, 35–41 CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- N. D. McDaniel and S. Bernhard, Dalton Trans., 2010, 39, 10021–10030 RSC.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CrossRef CAS.
- R. J. Watts, J. Chem. Educ., 1983, 60, 834–842 CrossRef CAS.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- V. Balzani, L. Moggi and F. Scandola, in Supramolecular Photochemistry, ed. V. Balzani, NATO ASI Series 214, Reidel, Dordrecht, The Netherlands, 1987, pp. 1–28 Search PubMed.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS PubMed.
- M. Elvington, J. Brown, S. M. Arachchige and K. J. Brewer, J. Am. Chem. Soc., 2007, 129, 10644–10645 CrossRef CAS PubMed.
- G. F. Manbeck and K. J. Brewer, Coord. Chem. Rev., 2013, 257, 1660–1675 CrossRef CAS PubMed.
- T. A. White, S. L. H. Higgins, S. M. Arachchige and K. J. Brewer, Angew. Chem., 2011, 123, 12417–12421 CrossRef PubMed.
- T. A. White, B. N. Whitaker and K. J. Brewer, J. Am. Chem. Soc., 2011, 133, 15332–15334 CrossRef CAS PubMed.
- R. Zhou, G. F. Manbeck, D. G. Wimer and K. J. Brewer, Chem. Commun., 2015, 51, 12966–12969 RSC.
- T. Stoll, M. Gennari, J. Fortage, C. E. Castillo, M. Rebarz, M. Sliwa, O. Poizat, F. Odobel, A. Deronzier and M.-N. Collomb, Angew. Chem., Int. Ed., 2014, 53, 1654–1658 CrossRef CAS PubMed.
- M. Karnahl, C. Kuhnt, F. W. Heinemann, M. Schmitt, S. Rau, J. Popp and B. Dietzek, Chem. Phys., 2012, 393, 65–73 CrossRef CAS PubMed.
- M. Karnahl, C. Kuhnt, F. Ma, A. Yartsev, M. Schmitt, B. Dietzek, S. Rau and J. Popp, ChemPhysChem, 2011, 12, 2101–2109 CrossRef CAS PubMed.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215–6218 CrossRef CAS PubMed.
- H. Ozawa, M.-A. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926–4927 CrossRef CAS PubMed.
- H. Ozawa and K. Sakai, Chem. Lett., 2007, 36, 920–921 CrossRef CAS.
- H. Ozawa, Y. Yokoyama, M.-A. Haga and K. Sakai, Dalton Trans., 2007, 1197–1206 RSC.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS PubMed.
- K. Sakai, H. Ozawa, H. Yamada, T. Tsubomura, M. Hara, A. Higuchi and M.-A. Haga, Dalton Trans., 2006, 3300–3305 RSC.
- M. G. Pfeffer, T. Kowacs, M. Wächtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 6627–6631 CrossRef CAS PubMed.
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044–5048 CrossRef CAS PubMed.
- P. Lei, M. Hedlund, R. Lomoth, H. Rensmo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2007, 130, 26–27 CrossRef PubMed.
- J. D. Knoll, S. M. Arachchige and K. J. Brewer, ChemSusChem, 2011, 4, 252–261 CAS.
- J. D. Knoll, S. L. H. Higgins, T. A. White and K. J. Brewer, Inorg. Chem., 2013, 52, 9749–9760 CrossRef CAS PubMed.
- M. T. Mongelli and K. J. Brewer, Inorg. Chem. Commun., 2006, 9, 877–881 CrossRef CAS PubMed.
- M. Toyama, K.-I. Inoue, S. Iwamatsu and N. Nagao, Bull. Chem. Soc. Jpn., 2006, 79, 1525–1534 CrossRef CAS.
- J. D. Knoll, S. M. Arachchige, G. Wang, K. Rangan, R. Miao, S. L. H. Higgins, B. Okyere, M. Zhao, P. Croasdale, K. Magruder, B. Sinclair, C. Wall and K. J. Brewer, Inorg. Chem., 2011, 50, 8850–8860 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, electrochemical, spectroscopic, and photocatalytic data, three dimensional models. See DOI: 10.1039/c5cc06463e |
| ‡ Current address: Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH, 43210, USA. |
| § Karen J. Brewer is deceased (October 24, 2014). |
| This journal is © The Royal Society of Chemistry 2015 |