
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An aqueous one-pot route to gold/quantum rod heterostructured nanoparticles functionalized with DNA†
C.
Hamon
a,
C.
Martini
b,
P.
Even-Hernandez
a,
B.
Boichard
a,
H.
Voisin
b,
L.
Largeau
c,
C.
Gosse
c,
T.
Coradin
b,
C.
Aimé
*b and
V.
Marchi
*a
aUMR 6226 Institut des Sciences Chimiques de Rennes, Université de Rennes 1, CNRS, Avenue du Général Leclerc, 35042 Rennes, France. E-mail: valerie.marchi@univ-rennes1.fr
bSorbonne Universités, UPMC Univ Paris 06, CNRS, Collège de France, Laboratoire de Chimie de la Matière Condensée de Paris, 11 place Marcelin Berthelot, 75231 Paris, France. E-mail: carole.aime@upmc.fr
cLaboratoire de Photonique et de Nanostructures, LPN-CNRS, Route de Nozay, 91460 Marcoussis, France
First published on 10th September 2015
Abstract
We report an original approach exploiting the photoelectrochemical properties of quantum rods and the versatility of Au(I) organometallic chemistry to control DNA surface grafting. This one-pot aqueous approach provides Janus biofunctionalized nanoparticles, the assembly of which should results in the emergence of synergistic properties.
Tailoring the properties of heterostructured nanoparticles composed of metals and semi-conductors at the scale of a few to tens of nanometers remains a main challenge in nanosciences. In most cases, noble metals (Au and Pt) have been reduced on semi-conductor nanocrystals leading to new devices with synergistic properties for photocatalysis and nanoelectronics.1 A common strategy for deriving such heterostructured nanoparticles (HNPs) is to nucleate the metal by reduction of the corresponding metal ions onto the semi-conductor surface. This process can be induced either by temperature2 or by light irradiation3 in an organic solvent.4 Based on their charge separation ability, the resulting metal/semiconductor HNPs are very attractive for light-harvesting applications as they provide at the single particle scale both tunable absorption of the solar light and a rapid charge transfer.5 They also offer new input for further development of photocatalytic systems.6 Yet, these developments are highly dependent on the possibility of controlling the nanoparticle organization at larger scales.
HNP assembly in a colloidal suspension can be directed by selective functionalization of the metal domain. This requires the simultaneous control of the location of this domain and its regioselective functionalization. In this context, anisotropic semi-conductor quantum rods (QRs), for instance made of CdSe/CdS, are particularly attractive to specifically reduce metal salts at their tips thanks to light-induced anisotropic charge separation.2,3,7,8 Moreover, in terms of HNP organization, such anisotropic rods should favor a defined packing in condensed phases.9 Among possible strategies based on non-specific10 or specific interactions (such as dithiol2 or biotin/streptavidin11), self-assembled organic linkers are used to spontaneously direct nanoparticle organization by weak molecular interactions and to build ordered macroscopic structures.9 However, the extension of these approaches to biological linkers of interest, such as DNA,12 is rendered difficult by the poor colloidal stability of HNPs in aqueous media.
Here we present an original strategy combining the unique photoelectrochemical properties of CdSe-seeded CdS quantum rods (CdSe@CdS QRs) and the versatility of Au(I) organometallic chemistry. It not only provides a new method for the synthesis of Janus HNPs in water but also opens the door for their biofunctionalization by a one-pot reaction.
We focused on CdSe@CdS QRs that are key platforms for applications in photocatalysis13 and imaging,14 due to the possibility of tuning their optical properties by adjusting the size of the seed.15 The nanoparticles were dispersed in water thanks to a robust peptide exchange strategy, as reported for CdSe@ZnS quantum dots16 and CdSe@CdS QRs.17 More precisely, we relied on a PEGylated polycysteine (C3E6D, Scheme 1) developed in our laboratory to replace the ligand used for the nanoparticle synthesis. We selected such peptides because of their strong affinity for the semiconducting surface, their chemical versatility, and their easy accessibility. In the same way, bidentate ligands such as dihydrolipoic acid–PEG derivatives could also be good candidates to control the photoreduction of the QRs.18 In our case, after QR purification from the free ligands, this cysteine-based multi-thiol anchor group successfully limits aggregation19 contrary to that observed with other transfer ligands, such as mono-thiol mercaptoundecanoic acid20 or D–L cysteine.21 CdSe@CdS QRs with different aspect ratios were synthesized and their ability to photoreduce resazurin, a model acceptor dye, was tested.22 Comparing photoreduction kinetics indeed permits the selection of an efficient aspect ratio for charge separation in aqueous medium. As expected the photoreduction rate increases when the aspect ratio varies between 10 and 30, before decreasing with higher values due to the presence of crystalline defects in long rods (Fig. S1, ESI†). Two batches of CdSe@CdS QRs with an optimized aspect ratio were prepared for further investigations, with an aspect ratio of 14 and 15 (Fig. S2 and S3, ESI†).7
 | ||
| Scheme 1 Schematic representation of the photo-induced synthesis of HNPs in water, including the chemical structure of the C3E6D peptide and the chloro-Au(I) salt. | ||
We next synthesized HNPs in water by photoreduction on the QR–peptide surface of the monovalent gold complex chloro-Au(I), i.e. chloro[diphenyl(3-sulfonatophenyl) phosphine]gold(I) sodium salt (Scheme 1).6c,23 Using Au(I) salts in the synthesis of HNPs presents several advantages in comparison to using Au(III) salts. The redox potential of Au(I)/Au (1.692 V versus the standard hydrogen electrode (SHE)) is higher than that of Au(III)/Au (1.498 V versus SHE). As only one electron is consumed for reduction instead of three electrons for Au(III) reduction, the photooxidation process altering the CdSe@CdS QRs is limited.24 The photoreduction was realized at room temperature using a LED working at 405 nm. Importantly, to prevent nanoparticle oxidation under light irradiation, the QR–peptide suspension was reacted with chloro-Au(I) in the presence of ascorbic acid (AA) as a classical reducing agent and triethanolamine (TEA)25 as a photohole scavenger (see sample 1 in Table S1 for the concentrations, ESI†). Because of its high oxidation potential (i.e. weak bound electron) at −3.4 eV vs. vacuum at pH 8, TEA is known to recombine with the photohole trapped at the surface of the nanoparticle on a time scale faster than the photooxidation of the CdS semiconductor material. The reaction was monitored by UV-visible and fluorescence spectroscopies and by transmission electron microscopy (TEM).
After 20 minutes, the increase of the plasmon band (490–700 nm) indicates the growth of the gold domain on the QR and the formation of HNPs (Fig. 1a). This is further supported by the quenching of QR fluorescence, as a consequence of the formation of the metal/semiconductor heterojunction (Fig. 1b). Indeed, the gold domain acts as an electron sink and drags the photo-induced electron, leading to an increase of the charge separation.1 In parallel the decrease in absorbance between 400 and 490 nm (Fig. 1a) is associated with the photocorrosion of the QRs.20 Meanwhile, TEM was used to confirm the formation of HNPs that appear to be highly monodisperse in terms of rod size, as well as gold domain size and position (Fig. 1c). Importantly, these observations show the presence of one unique gold domain per HNP, identified by the higher electron density of the metal. This domain was found to be located approximately between 1/4 and 1/5 of the total rod length. This preferential location of the metal reduction reaction on the QR has been previously reported for reduction performed in organic solvents and it is attributed to the tendency of photogenerated electrons to localize near the CdSe seed. A phenomenon called electrochemical Ostwald ripening may also be at work.1,25 Surface energy stabilizes larger metal crystals versus the smaller ones. The standard reduction electrochemical potential of the metal nanoparticles is negatively shifted when the size decreases. These driving forces facilitate the electron transfer from small to large nanoparticles on the quantum rods acting as conductive substrates.26 In addition, in the absence of light irradiation, no HNP is formed (Fig. 1d). The latter observation confirms that the metal growth is photochemically controlled.
 | ||
| Fig. 1 Kinetics of HNP formation: (a) UV-visible and (b) fluorescence spectra over 2 hour photo-reduction (spectra recorded every 15 min, starting spectrum with open circles). Corresponding TEM images: (c) after 20 min irradiation and (d) without irradiation. | ||
In a second phase, TEM was used more systematically to investigate HNP morphology. Well-defined objects were obtained after an irradiation time of 20 minutes, with a slight decrease in length from L = 62.8 ± 5.2 nm for the starting QRs to L = 58.5 ± 5.0 nm for the processed ones. In contrast, for longer irradiation times a more pronounced shortening of the QR is observed: L = 49.7 ± 13.1 nm after 1 h and L = 19.1 ± 8.5 nm after 2 h (Fig. S4, ESI†). This degradation of the QRs is attributed to the CdS photo-induced oxidation. We secondly monitored the position of the gold domain over the total HNP length by plotting the l = f(L) distribution, where l is the smallest distance between the metallic domain and one of the QR tips (Fig. 2). Interestingly, whatever the irradiation time (20 min, 1 h, or 2 h) and the associated QR shortening, the l/L ratio remained nearly constant, around 0.22–0.26. Since the gold particle is assumed to be located over the fixed CdSe seed, this observation is in line with an equal relative rate of CdS destruction at both ends of the rod (i.e. with l′ being the length of the longer arm, we have 1/l dl/dt ≈ 1/l′ dl′/dt). Eventually, we monitored the size of the gold domain. Its diameter remains constant around 3.8 nm between 20 min and 1 h but was reduced at ∼2.3 nm for an irradiation time of 2 h. The latter observation is attributed to the partial redissolution by oxidation of the metallic particles. We also found some gold domains at the tips of the QRs (l/L = 0), maybe after the degraded QR broke apart (Fig. S4, ESI†). To summarize, an irradiation time between 20 and 30 min induces an efficient photoreduction of the chloro-Au(I) salt as a single gold domain on the QR surface while preventing semiconductor oxidation.
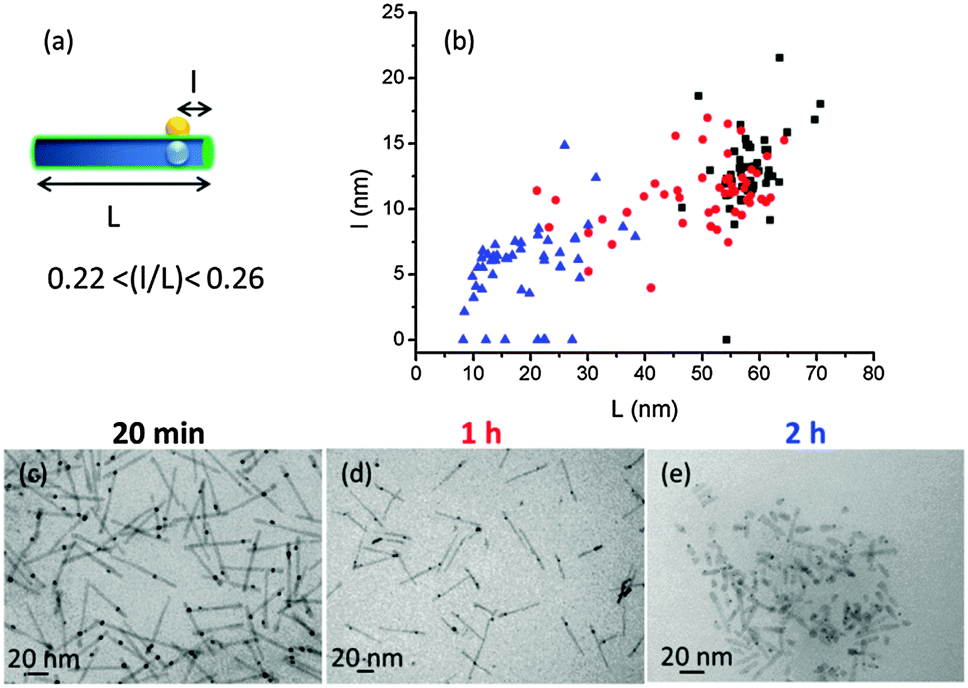 | ||
| Fig. 2 (a) Scheme of the HNPs and (b) localization of the gold domain for three different irradiation times (20 min, 1 h, and 2 h, in black, red, and blue respectively). Data were extracted from TEM images (c) after 20 min, (d) after 1 h, and (e) after 2 h. Statistics over 100 particles in total. | ||
We next examined how the nature of the peptide ligands could modulate the kinetics of photoreduction. By replacing the peptide derivative C3E6D by C3A11E4CO2H, which bears an additional alkyl chain (Fig. S5a, ESI†), the reduction kinetics were much slower and the plasmon band could only be detected after 1 h of irradiation (Fig. S5b1, ESI†). Furthermore, a fluorescence enhancement occurred during the first 45 min of irradiation, i.e. before gold reduction could be observed (Fig. S5b2, ESI†). As a similar phenomenon was observed in the absence of chloro-Au(I), it can be attributed to the partial desorption of the cysteine derivatives. This process may be related to the oxidation of cysteine into cystine by the photogenerated holes, as previously reported.27 For longer irradiation times, a fluorescence quenching was observed corresponding to the subsequent reduction of the gold salt (Fig. S5c, ESI†), similarly to that observed with C3E6D. In addition, the stable absorbance at 400–490 nm is consistent with the expected better passivation of the QR surface, which avoids the semi-conductor photooxidation. The latter observation is in line with a lower resazurin photoreduction rate for C3A11E4CO2H compared to C3E6D (Fig. S1b, ESI†). On this basis QRs capped with the C3E6D peptide were selected for the following experiments.
To better understand the growth of the gold domain, a systematic investigation of the role of each reagent was performed at a fixed irradiation time (20 min). The different experimental conditions are summarized in Table S1 (ESI†) and the resulting heterostructured nanoparticles were characterized in terms of optical (UV-visible and fluorescence) and morphological properties (Fig. S6 and S7, ESI†). The above-described HNPs (Fig. 1 and 2) were prepared according to the conditions of sample 1. When chloro-Au(I) was introduced with a concentration 10 times higher than the reference one (sample 2 vs. 1), the nucleation of multiple discrete gold domains on the QR surface was observed (Fig. S7a, ESI†). In the absence of TEA (sample 3), no HNP was seen after light irradiation (Fig. S7b, ESI†). Additionally, in the absence of ascorbic acid (sample 4), HNPs with a higher polydispersity in gold size, location and homonucleation were obtained (Fig. S7c, ESI†). In the absence of both TEA and AA (sample 5), the high degradation of the QRs due to their photooxidation was striking (Fig. S7d, ESI†). Overall, all reactants were found to be necessary to control the photochemical synthesis of well-defined single-domain HNPs, favoring localized nucleation while avoiding polynucleation and limiting QR degradation.
The opportunity to control the structure of metal/semi-conductor particles in water opens the route towards their functionalization with a broad range of biomolecules. DNA is well known as a smart molecular tool to direct the self-assembly of nanomaterials since (i) the thermodynamics of association between complementary strands is well understood; (ii) oligonucleotides derived with various reactive groups are readily available. However, the final hybrid nanoarchitecture depends on the localization of DNA on the nanoparticles. In this context, our strategy is based on the pre-functionalization of chloro-Au(I) with an oligonucleotide to drive its grafting specifically on the gold domain at a given distance from the tip of the HNP. Taking advantage of the high affinity of Au(I) towards sulphur, gold(I) thiolates can be easily synthesized by treating phosphine gold chloride with a thiol-bearing molecule under basic conditions.28 Here, the substitution of the chloride ion by a thiol-derived DNA1 ligand (at pH 10.5) led to the formation of the DNA1–Au(I) complex (Fig. 3a). DNA1–HNPs were next synthesized from a DNA1–Au(I)/chloro-Au(I) mixture in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio, under sample 1 conditions (see ESI† and Table S1) and using a 20 min irradiation time (Fig. 3b). After thorough washing of the HNPs and systematic removal of soluble DNA present in the supernatant, the efficiency of DNA grafting was evaluated by UV spectroscopy. Fig. 3c shows the expected UV signature of the aromatic nucleotides centered at 260 nm, thereby proving the presence of DNA on HNPs. The presence of DNA does not affect the strong quenching of the fluorescence as reported in Fig. 1b. Importantly, monodisperse DNA–HNPs were observed under TEM with a single discrete gold domain present on each QR (Fig. 3d).
:4 molar ratio, under sample 1 conditions (see ESI† and Table S1) and using a 20 min irradiation time (Fig. 3b). After thorough washing of the HNPs and systematic removal of soluble DNA present in the supernatant, the efficiency of DNA grafting was evaluated by UV spectroscopy. Fig. 3c shows the expected UV signature of the aromatic nucleotides centered at 260 nm, thereby proving the presence of DNA on HNPs. The presence of DNA does not affect the strong quenching of the fluorescence as reported in Fig. 1b. Importantly, monodisperse DNA–HNPs were observed under TEM with a single discrete gold domain present on each QR (Fig. 3d).
 | ||
| Fig. 3 (a) Chemical modification of chloro-Au(I) with DNA. (b) Scheme of DNA grafting via reduction of DNA–Au(I) on the QR, yielding DNA–HNPs. (c) UV spectra of DNA–HNPs obtained for different DNA–Au(I):QR ratios: 1:30 (solid line) and 1:3 (dotted lines). (d) TEM image of the DNA–HNPs synthesized in a 1:30 DNA–Au(I):QR ratio. | ||
Eventually, we investigated the possibility of taking advantage of the molecular recognition between complementary oligonucleotides through base pairing to demonstrate the precise location of DNA on the HNP surface. Specific assembly was triggered by mixing the DNA1–HNPs and gold NPs (15 nm in diameter) bearing the complementary DNA2 single strand, in equimolar proportions. Those two complementary DNA sequences were previously successfully used to control interactions between spherical silica particles.29 Under such conditions, a particle population with a significant increase of the average hydrodynamic diameter could be detected by dynamic light scattering (DLS) (Fig. S8, ESI†). TEM images evidence the formation of enlarged structures (Fig. 4a). An affinity competition assay was next performed, where addition of a 100-fold excess of free DNA1 induced the disassembly of the nanoparticle conjugates (see Fig S9a, ESI†). On the other hand, HNPs and AuNPs bearing non-complementary DNA sequences were hardly co-localized in the same area on the TEM grid while DLS did not evidence the association of the two sets of particles (Fig. S8 and S9b, ESI†). Altogether, these data support the occurrence of specific base-pairing interactions taking place at the surface of DNA-grafted HNPs synthesized by a one-step process. In a final control experiment, the starting QRs were processed in the presence of the thiolated oligonucleotide DNA1 instead of the DNA1–Au(I) complex and then mixed with gold NPs bearing the complementary DNA2 sequence. Under these conditions no particle association could be observed (Fig. 4b) showing the absence of spontaneous DNA grafting at the QR surface.
 | ||
| Fig. 4 TEM images of (a) the DNA1–HNPs interacting with complementary DNA2–AuNPs, and (b) the QRs processed with thiolated DNA1, thus lacking the gold nucleus, interacting with DNA2–AuNPs. | ||
As a conclusion we have reported a new strategy for the simultaneous functionalization of semi-conductive quantum rods with gold nanodomains and DNA in water. Such aqueous conditions should make our method broadly applicable to a wide variety of biomolecules of interest provided that the corresponding Au(I)–phosphine complex can be prepared. In parallel, the expected location of the binding site along the quantum rod length, on the gold nucleus, offers an easy access to Janus heterostructured nanoparticles, with promising applications in the development of photoactive surfaces and interfaces.
V.M. is thankful to the Direction Générale de l'Armement (DGA) for its financial support. C.A. thanks the ANR-Retour post-doctorant for financial support (ANR-09-RPDOC-023-01).
Notes and references
- U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed.
- G. Menagen, J. E. Macdonald, Y. Shemesh, I. Popov and U. Banin, J. Am. Chem. Soc., 2009, 131, 17406–17411 CrossRef CAS PubMed.
- A. Vaneski, A. S. Susha, J. Rodríguez-Fernández, M. Berr, F. Jäckel, J. Feldmann and A. L. Rogach, Adv. Funct. Mater., 2011, 21, 1547–1556 CrossRef CAS PubMed.
- R. Costi, G. Cohen, A. Salant, E. Rabani and U. Banin, Nano Lett., 2009, 9, 2031–2039 CrossRef CAS PubMed.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrovic, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- L. Carbone, A. Jakab, Y. Khalavka and C. Sönnichsen, Nano Lett., 2009, 9, 3710–3714 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- N. Zhao, J. Vickery, G. Guerin, J. I. Park, M. A. Winnik and E. Kumacheva, Angew. Chem., Int. Ed., 2011, 50, 4606–4610 CrossRef CAS PubMed.
- A. Salant, E. Amitay-Sadovsky and U. Banin, J. Am. Chem. Soc., 2006, 128, 10006–10007 CrossRef CAS PubMed.
- R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schuller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2014, 9, 74–78 CrossRef CAS PubMed.
- H. Zhu, N. Song, H. Lv, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2012, 134, 11701–11708 CrossRef CAS PubMed.
- M. Kazes, D. Y. Lewis, Y. Ebenstein, T. Mokari and U. Banin, Adv. Mater., 2002, 14, 317–321 CrossRef CAS.
- (a) A. Sitt, F. D. Sala, G. Menagen and U. Banin, Nano Lett., 2009, 9, 3470–3476 CrossRef CAS PubMed; (b) A. Sitt, I. Hadar and U. Banin, Nano Today, 2013, 8, 494–513 CrossRef CAS PubMed.
- A. Dif, E. Henry, F. Artzner, M. Baudy-Floc'h, M. Schmutz, M. Dahan and V. Marchi-Artzner, J. Am. Chem. Soc., 2008, 130, 8289–8296 CrossRef CAS PubMed.
- T. Bizien, P. Even-Hernandez, M. Postic, E. Mazari, S. Chevance, A. Bondon, C. Hamon, D. Troadec, L. Largeau, C. Dupuis, C. Gosse, F. Artzner and V. Marchi, Small, 2014, 10, 3707–3716 CrossRef CAS PubMed.
- M. H. Stewart, K. Susumu, I. L. Medintz, J. B. Delehanty, B. C. Mei, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2010, 132, 9804–9813 CrossRef CAS PubMed.
- (a) J. Aldana, Y. A. Wang and X. Peng, J. Am. Chem. Soc., 2001, 123, 8844–8850 CrossRef CAS PubMed; (b) K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed.
- R. Costi, A. E. Saunders, E. Elmalem, A. Salant and U. Banin, Nano Lett., 2008, 8, 637–641 CrossRef CAS PubMed.
- M. Berr, A. Vaneski, A. S. Susha, J. Rodriguez-Fernandez, M. Doblinger, F. Jackel, A. L. Rogach and J. Feldmann, Appl. Phys. Lett., 2010, 97, 093108 CrossRef PubMed.
- H. Shen, W. Xu and P. Chen, Phys. Chem. Chem. Phys., 2010, 12, 6555–6563 RSC.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- Y. Ma, J. Zeng, W. Li, M. McKiernan, Z. Xie and Y. Xia, Adv. Mater., 2010, 22, 1930–1934 CrossRef CAS PubMed.
- M. J. Berr, P. Wagner, S. Fischbach, A. Vaneski, J. Schneider, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2012, 100, 223903 CrossRef PubMed.
- T. Mokari, C. G. Sztrum, A. Salant, E. Rabani and U. Banin, Nat. Mater., 2005, 4, 855–863 CrossRef CAS PubMed.
- S. Tamang, G. Beaune, I. Texier and P. Reiss, ACS Nano, 2011, 5, 9392–9402 CrossRef CAS PubMed.
- M. K. Corbierre and R. B. Lennox, Chem. Mater., 2005, 17(23), 5691–5696 CrossRef CAS.
- (a) J. Wu, T. Coradin and C. Aimé, J. Mater. Chem. B, 2013, 1, 5353–5359 RSC; (b) J. Wu, J. Silvent, T. Coradin and C. Aimé, Langmuir, 2012, 28, 2156–2165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc05148g |
| This journal is © The Royal Society of Chemistry 2015 |