
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-small platinum-based coordination nanoparticles for radiotherapy†‡
Riya
George
 *a,
Lucile
Fétiveau
a,
Erika
Porcel
*b,
Farah
Savina
b,
Charles
Bosson Bapaume
b,
Diana
Dragoe
a,
François
Brisset
a,
Hynd
Remita
c,
Sandrine
Lacombe
b and
Laure
Catala
*a
*a,
Lucile
Fétiveau
a,
Erika
Porcel
*b,
Farah
Savina
b,
Charles
Bosson Bapaume
b,
Diana
Dragoe
a,
François
Brisset
a,
Hynd
Remita
c,
Sandrine
Lacombe
b and
Laure
Catala
*a
aUniversité Paris-Saclay, CNRS, Institut de chimie moléculaire et des matériaux d’Orsay, 91405, Orsay, France. E-mail: laure.catala@universite-paris-saclay.fr; riya.george@universite-paris-saclay.fr
bUniversité Paris-Saclay, CNRS, Institut des Sciences Moléculaires d’Orsay, 91405, Orsay, France. E-mail: erika.porcel@universite-paris-saclay.fr
cUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, 91405, Orsay, France
First published on 9th October 2023
Abstract
Radiation therapy is an effective and localised method for cancer treatment and its efficacy is considerably improved by using radioenhancing agents. Nanoparticles containing high atomic number (Z) elements have emerged as an appealing class of enhancers. This work reports the first example of radioenhancers based on ultrasmall platinum (Pt)-polycyanometallate nanoparticles. Importantly, both novel nano-enhancers (Pt[Pt(CN)4] and Pt[Fe(CN)6]) are prepared in fairly large quantities using a water-based, surfactant-free, green route. The post-coating of the inorganic cores by different biocompatible polymers is straightforward and allows probing of the influence of the coating on the radiotherapeutic efficiency of the nanosystems. The structural/chemical integrity is maintained after high doses of γ-radiation and high colloidal stability is obtained in biological media. At 250 μM total metal concentration, these nanoparticles are non-toxic to cells, but when coupled with γ-radiation, they amplify apoptosis of HeLa cells by 20% at 4.5 Gray, which rises to 60% at larger doses. Furthermore, irradiation studies conducted on a model DNA probe evidence that the amount of lethal Double Strand Breaks (DSB) is significantly enhanced in the presence of these novel Pt-based radioenhancers. The radiotherapeutic efficiency of these versatile coordination nanoparticles obtained using a cheap, fast and green process opens interesting possibilities in the field of nanomedicine.
Introduction
Even in the 21st century, cancer remains one of the biggest healthcare challenges with 19.3 million new cases detected in 2020, resulting in 10 million deaths all across the globe.1 There is widespread ongoing research to develop targeted, safe, and patient-specific treatment methods. Radiation therapy is applied to more than 50% of cancer patients to eliminate cancerous tissues in a localised way while minimizing the chances of recurrence in the affected area. A major advantage of radiotherapy is that tumours located deep within the body could also be targeted using high energy X-rays, γ-rays or ion beams as their penetration depth is greater than 10 cm.2 However, a high dose of these ionizing radiations can damage the healthy tissues and organs adjacent to the tumour, and cause adverse side-effects like severe fatigue, radiodermatitis and in some cases, even second cancers due to mutations generated during the process.3 Therefore, several radio-enhancers have been developed over the past two decades to maximize cancer cell killing while working within a safe range of therapeutic doses of radiation. These agents need to be stable, non-toxic to normal tissues and fairly resistant to metabolic breakdown.4Irradiating cells internalized with high-Z elements generate a cascade of secondary electrons.5 These electrons are brought about by the ionization of heavy metal atoms, which induce a significant increase in the amount of reactive oxygen species (ROS) produced upon water radiolysis. When these radicals are produced in close proximity to the cancer cells, their proliferation is stalled, causing apoptosis and subsequent elimination. In 2004, J. F. Hainfield et al. provided the first proof that gold nanoparticles (NPs) significantly improved the in vivo efficacy of radiation therapy. They demonstrated that the ultra-small size of these particles proves beneficial for bio-distribution and relatively high concentrations of the heavy metal could be incorporated without toxicity.6 The potential of metallic NPs in enhancing the biological efficiency of radiation was realised, and this resulted in an exponential increase in the development of varied particles that can improve the performance of conventional radiation therapy. Additionally, nanoparticles provide a relatively large surface area and are flexible towards size, shape, and charge modifications, which proves to be advantageous for drug encapsulation and delivery to target sites.7 Few of the NPs developed for radiotherapy like gold (CYT-6091 CytImmune),8 gadolinium-based (AGuiX, NHTheraguix)9 and hafnium oxide-based nanoparticles (NBTXR3, Nanobiotix)10 have reached the clinical trial phase. Besides, precious-metal-based radio-enhancers, rare-earth element-based, semiconductor, and even non-metallic NPs have been explored over the past years.11–15
However, most synthesis methods require multiple steps, organic solvents, reducing agents or surfactants for controlling the nanoparticles’ growth. Subsequent functionalization and additional steps of purification are often required to make them water-soluble and biocompatible.16 Such complications could be avoided by using a radiolytic method for synthesis and as Leza et al. showed, monodisperse Pt NPs were prepared in a one-pot manner using γ-radiation. These particles efficiently enhance the effect of both proton and medical carbon beams on cancer cells.17,18 However, the scarce availability of ionizing sources (cobalt-60 radiation, accelerated electrons) to perform the synthesis of these nanoparticles reduces their development. It is therefore important to propose green, one-pot synthetic procedures for the generation of novel radio-enhancing agents.
With this aim in mind, new microporous coordination nanoparticles related to Prussian Blue (PB) and polycyanometallate networks have been explored for improving radiation therapy. PB is a famous pigment and the oldest microporous coordination polymer. It is an FDA-approved compound known as Radiogardase® and is used for radioisotope decontamination.19 Nanoparticles of PB and related polycyanometallate coordination networks with different compositions have been studied for their magnetic and optical properties, applications in catalysis, batteries as well as electrochromic devices.20,21 Besides, many new and exciting biomedical applications of PB-based nanostructures have been discovered and their applicability in photothermal therapy (PTT), chemotherapeutic drug delivery and multimodal imaging (MRI, PET, PAI) has been demonstrated over the past decade.22 The synthesis of such coordination NPs using a fast and green surfactant-free, water-based method while maintaining a sub-10 nm size has been reported.23–26 These nanoparticles gather more advantages: (i) a high proportion of metal atoms are at the surface (50% for 4 nm NPs) of the particle due to its porous nature; (ii) the combination of different metals in precise proportions is possible; (iii) their post-coating by polymers/molecules without affecting the core size and shape is straightforward and allows evaluation of the coating's impact; (iv) the presence of zeolitic water in the PB FCC structure may participate in radioenhancement.
Indeed, it was recently demonstrated that the microporosity of a nanostructure could amplify the production of ROS and facilitate their diffusion after irradiation. Li et al. showed for the first time that the porous structure of 200 nm MIL-100 iron (Fe) nanoMOFs not only serves as cargo to release drug molecules but also participates as efficient radiation enhancers due to the presence of well-dispersed Fe centres.27 Unlike nanoMOFs, PB and related polycyanometallate nanoparticles are stable even in diluted conditions and do not degrade. It was hypothesised that, since such polycyanometallate-based NPs are also intrinsically porous, irradiation in the presence of water could increase the production of reactive radicals. In 2020, Ren et al. reported 200 nm hollow mesoporous PB NPs doped with radio-sensitizing bismuth nanodots for achieving combined photo-thermal and radiation therapy.28 Still, the application of ultrasmall polycyanometallate coordination compounds for radiation therapy remains a widely unexplored field. Therefore, a new family of radioenhancers made up of porous Pt-containing coordination networks is proposed in this work. Two novel Pt-based nanostructures of Pt[Fe(CN)6] and Pt[Pt(CN)4] are reported in this study. The use of DNA plasmid as a model system to understand the molecular scale mechanisms occurring in the presence of our NPs and radiation is also discussed. The effect of radiation beams on HeLa cells in the presence of these coordination nanosystems is examined.
Experimental section
Materials
Potassium ferrocyanide (K4[Fe(CN)6]·3H2O), potassium tetra chloroplatinate (K2[PtCl4]), potassium tetracyanoplatinate (K2[Pt(CN)4]·3H2O), and dextran were purchased from Aldrich. Polyvinylpyrrolidone (PVP) (Thermofischer, average MW 3500), Agarose D5 (Type D-5-DNA Grade from EUROMEDEX), Tris-Acetate-EDTA (TAE, 50x), Tris EDTA (TE)(1x), DNA (pBR322, 0.5 μg μL−1, from molecular biology), 6xDNA loading dye (#R0611, molecular biology), Ethidium Bromide (EB, ref: EU0070, 10 mg mL−1, molecular biology Grade, from EUROMEDEX).Preparation of nanoparticles
The conditions for the most optimized sample are reported. For obtaining PtFe NPs, K4[Fe(CN)6]·3H2O (10 mM) and K2[PtCl4] (10 mM) solutions were prepared in distilled water and heated to reach the temperature of 55 °C. K2[PtCl4] solution was added in one go into the K4[Fe(CN)6]·3H2O solution under vigorous stirring. The mixture turned yellow in colour and the reaction was run for 1 h while maintaining the temperature. The solution was cooled down to room temperature and turned dark green in colour.In the case of PtPt NPs, the precursor solutions K2[Pt(CN)4]·3H2O (10 mM) and K2[PtCl4] (10 mM) were prepared in water, and heated until the activation temperature of 85 °C was reached. K2[PtCl4] was quickly added to the metal cyanide solution under constant stirring. The initial colour of the solution mixture was light brown, but as the reaction progressed (1 h, 85 °C) it became a colourless solution. The reaction was stopped after 1 h and the solution was brought to room temperature.
Polymer coating
PVP solution (150 mM) was prepared in water and added to preformed NP solution under constant stirring, maintaining a ratio of 30 (PVP monomer to metal). After 30 minutes, the solution was ultra-filtered and washed twice with distilled water to remove excess reactants. This solution was utilised for characterization and biological tests. A similar process was followed for coating the NPs with dextran (125 mM).Powders of NPs were obtained by the addition of acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with respect to the colloidal solution of PtFe and acetone/methanol for PtPt), followed by centrifugation at 2 °C until a pellet was formed. They were dried under a vacuum for several hours.
:1 with respect to the colloidal solution of PtFe and acetone/methanol for PtPt), followed by centrifugation at 2 °C until a pellet was formed. They were dried under a vacuum for several hours.
Physicochemical characterization
The hydrodynamic diameter and zeta potential of each prepared sample were determined using Zetasizer NanoZS (Malvern Instruments, UK) equipped with backscattering mode. Multiple batches of the sample were analyzed with standard disposable polystyrene and DTS 1070 cuvettes, and the mean values are reported with corresponding standard deviation. A transmission electron microscope (TEM JEOL 1400, Japan) operating at an accelerating voltage of 120 kV, was used to observe the size and dispersity of the sample. Formvar-coated copper grids were glow-discharged before placing a few drops of PtFe and PtPt NP solution. The characteristic chemical bonds making up the structure of the nanoparticles were verified using an FT-IR spectrophotometer (PerkinElmer Spectrum 100) by preparing KBr pellets. UV-Visible absorption spectra (Cary Win UV), energy dispersive spectra (EDS SAMx) and X-ray photoelectron spectra (Thermofisher Scientific K-Alpha) were also used for the characterization of the NPs.Irradiation studies
Cobalt-60 gamma source (1.25 meV) at the Institute of Physical Chemistry (ICP), University Paris-Saclay was used as the radiation source for the experiments. PtFe and PtPt NP solutions synthesised with PVP and dextran coating along with their precursors: K4[Fe(CN)6], K4[Fe(CN)6] + PVP, K4[Fe(CN)6] + dextran, K2[Pt(CN)4]·3H2O and K2[Pt(CN)4]·3H2O + PVP were analysed before and after irradiation to evaluate changes occurring in presence of high energy beams. Each sample was degassed with N2O and irradiated for an hour at a 4 kGy h−1 dose rate.Radio-sensitizing effect on DNA: sample preparation and analysis
The plasmid pBR322 (Fisher Scientific in 10 mM Tris-HCl, 1 mM EDTA) was used as a probe to represent biomolecules, in order to understand the radio-enhancing effect of prepared nanoparticles. It consists of circular double-stranded DNA of 4361 base pairs (2.83 × 106 Da) with 95% in native supercoiled (S) conformation and approximately 5% in the relaxed (R) conformation. R arises as a result of single-strand breaks (SSB) in DNA while double-strand breaks (DSB) are caused by two breaks in two strands separated by less than 10 base pairs. DSB leads to linear (L) conformation in the plasmid and this form is absent in the native product.Each sample was prepared by using 10 μg of DNA (10 μL) in 123 μL Tris-EDTA buffer solution. The plasmid was incubated with each nanoparticle solution (24 μL) for one hour to ensure binding. The stock solution was diluted to attain a total metal (Fe + Pt/Pt) concentration of 5.3 × 10−6 mol L−1 and the final volume was adjusted to 180 μL using Milli-Q water (18.2 MΩ cm at 25 °C). A control sample was prepared simultaneously containing the plasmid with 24 μL of water to present the effect of radiation in the absence of radio-enhancers. Each sample was split into 6 aliquots (18 μL each) and exposed to γ-radiation with the applied dose ranging between 0–158 Gy.
The damaging effect of irradiation was quantified by analysis of linear, relaxed and supercoiled conformation in the sample using agarose gel electrophoresis. 14 well (1% agarose) gels were prepared in Tris-Acetate-EDTA (TAE) buffer and 9 μL of each irradiated plasmid solution was loaded in the wells together with a blue dye. Due to the difference in volume, the three forms migrated different distances by electrophoresis (80 V, 3 h, 4 °C). The gel was stained using ethidium bromide (EB) solution, washed twice, studied under a UV transilluminator (λ = 302 nm) and recorded by a CCD camera. The images were examined to calculate amounts of L, S and R conformations present in each irradiated sample. Since S binds 1.47 times less to EB than R and L, normalization was done and respective fractions of R (R′), S (S′) and L (L′) were calculated.
| Total = 1.47 × S + R + L |
| R′ = R/total |
| S′ = 1.47 × S/total |
| L′ = L/total |
| DSB per plasmid = L′/(1 − L′) | (1) |
Cell viability studies
HeLa and primary dermal fibroblast cells purchased commercially from the American Type Culture Collection (CCL-2TM, LGC Standards S.a.r.l. – France Office), were cultivated in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies) enriched with 4.5 g L−1 glucose and supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS), 1% antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin) and 1% non-essential amino acids (all supplements from PAA Laboratories) and cultured at 37 °C in 5% CO2 atmosphere.The toxicity of PtFe and PtPt nanoparticles was evaluated in both systems: healthy primary Fibroblast cells and cervical cancer-derived HeLa cells, using a colony formation assay. The cells were incubated with nanoparticle solutions containing total metal concentrations in the range of 0.1–1.0 × 10−3 mol L−1 for 6 hours. The colonies formed were fixed and stained using 0.5% methylene blue 50% (v/v) methanol solution and counted manually. The control sample containing cells incubated with the same amount of water was used to determine the toxicity of NPs at each concentration. Each data point was triplicated to incorporate a margin of experimental error.
Uptake quantification
HeLa cells (2 × 105) were incubated with PtFe and PtPt nanoparticles for 6 h at a total metal concentration of 2.5 × 10−4 mol L−1. At the end of the incubation period, the culture medium was removed; the cells were rinsed with PBS solution and harvested after trypsinization. The cells were then counted using LUNA counter and centrifuged for 7 min at 200 × g. The supernatant was removed and the cell pellets were stored in glass tubes. Before analysis, the pellets were dissolved in aqua regia and diluted using ultrapure water. The quantification of platinum internalized within the cells was performed at UT2A (Pau, France) by inductively coupled plasma (ICP) analysis.Radio-sensitizing effect on cells
The impact of radiation on cell survival in the presence of prepared NPs was evaluated using a colony formation assay. HeLa cells were cultured in T25 flasks and maintained at 37 °C overnight, in a humidified 95% air–5% CO2 atmosphere. They were then incubated for 6 hours with an enriched culture medium containing PtFe and PtPt NPs at 2.5 × 10−4 mol L−1 total metal concentration. The cells with the nanoparticles, as well as the control containing an equivalent amount of water, were irradiated using the Cs-137 (0.63 meV) source at Institute Curie. The dose supplied ranges from 0–7.5 Gy. After the exposure, the cells were seeded onto Petri dishes to start the standard 14 day culture time. Colonies formed were fixed, stained, and counted to determine survival fractions at each dose and the data obtained was normalized against the corresponding control. Each measurement was performed in triplicate and the average was used to calculate dose enhancement and amplification factors in the presence of high-Z nanoparticles.Results and discussion
Synthesis and characterization of Pt-based NPs
The synthesis of (Pt[Fe(CN)6] and Pt[Pt(CN)4]) self-standing nanoparticles is described in the experimental section. Hereafter, for the sake of simplicity, they will be referred to as PtFe and PtPt NPs. A few studies have reported on colloids of Pt/polycyanometallate-based particles mainly for catalysis purposes, but stabilizers were used to stop their evolution to cyanogels.20,30In our case, the one-step reaction was carried out in pure water under easily reproducible conditions. A Pt(II) salt was used in the experimental procedure to incorporate a high-Z metal in the NP. Once the activation temperature was reached, the addition of K2PtCl4 to the corresponding polycyanometallate solution (either K4[Fe(CN)6] or K2[Pt(CN)4]) triggered the gradual substitution of chloro (–Cl) ligands by the cyano (–CN) ligands. This was followed by a change in the colour of the reaction mixture depending on the precursors used. The reaction time, temperature, and ratio of precursors added play a crucial role in the final structure and composition of the NPs. The colloidal solutions were fully stable over several months.
TEM micrographs reveal homogenous PtFe and PtPt nanoparticles. The size distribution histograms (Fig. 1) calculated by averaging the sizes of 200 NPs reveal that PtFe particles have a core average diameter of 5.2 ± 1.0 nm and PtPt is much smaller at 1.2 ± 0.4 nm. Due to the intense absorption of the green-coloured PtFe colloidal solution in the laser absorption region, the hydrodynamic diameter obtained was not reliable. DLS measurements indicated that the hydrodynamic diameter of synthesized PtPt NPs was 9.9 ± 3.9 nm which appeared to increase by ∼3 nm in three days. However, the addition of PVP after the synthesis, stops the growth and the sub-10 nm size is maintained (ESI,‡ Fig. S1). The zeta potential of PVP-coated PtFe and PtPt in water was around −23 ± 5 mV and −27 ± 6 mV respectively (ESI,‡ Fig. S2). The presence of polycyanometallate complex at the surface of the NPs explains the negative charge and their long-term stability in the colloidal solution. The strong electrostatic repulsions prevent aggregation of the particles and help maintain their size and shape in polar solvents as well as biologically relevant media like PBS (ESI,‡ Fig. S3).
 | ||
| Fig. 1 TEM micrographs of PtFe NPs (left) and PtPt NPs (right) at different magnifications, with their corresponding size distribution charts. | ||
Spectroscopic analysis
PtFe and PtPt NPs were recovered as powders (experimental section) and characterised by infrared, energy dispersive (EDS) and X-ray photoelectron (XPS) spectroscopies. We present in this section the PVP-coated samples that were used in the biological tests.Infrared spectroscopy is a powerful characterization technique to probe the nanoparticles’ composition. The cyanide stretch (v(CN)) in the 2000–2200 cm−1 range depends highly on its chemical environment, especially the linked metal centre (M–CN). It was used to ascertain changes occurring during the synthesis. In the PtFe NP spectra (Fig. 2), the peak at 2109 cm−1 is attributed to the vibrations of the bridging cyanide between the two metals (FeII–CN–PtII), while the shoulder at 2130 cm−1 corresponds to terminal FeIII–CN. A peak is also observed at 2072 cm−1, which is a characteristic of PB (FeII–CN–FeIII) pairs. UV-Visible absorption spectra recorded on the colloidal solution of PtFe confirm the formation of FeIII during the synthesis (ESI,‡ Fig. S4). The intense absorption peak in the 300–400 nm region corresponds to charge transfer within FeII–CN–PtII pairs forming the coordination network, while the broad band around 750–820 nm indicates the presence of PB pairs. Comparing their molar absorption coefficients with PB, it is deduced that the number of such FeII–CN–FeIII pairs formed is extremely small, amounting to roughly 3.5%. Therefore, the oxidation of FeII to FeIII during the synthesis is minimal but explains the green colour of the PtFe colloidal solution due to the presence of these two bands.
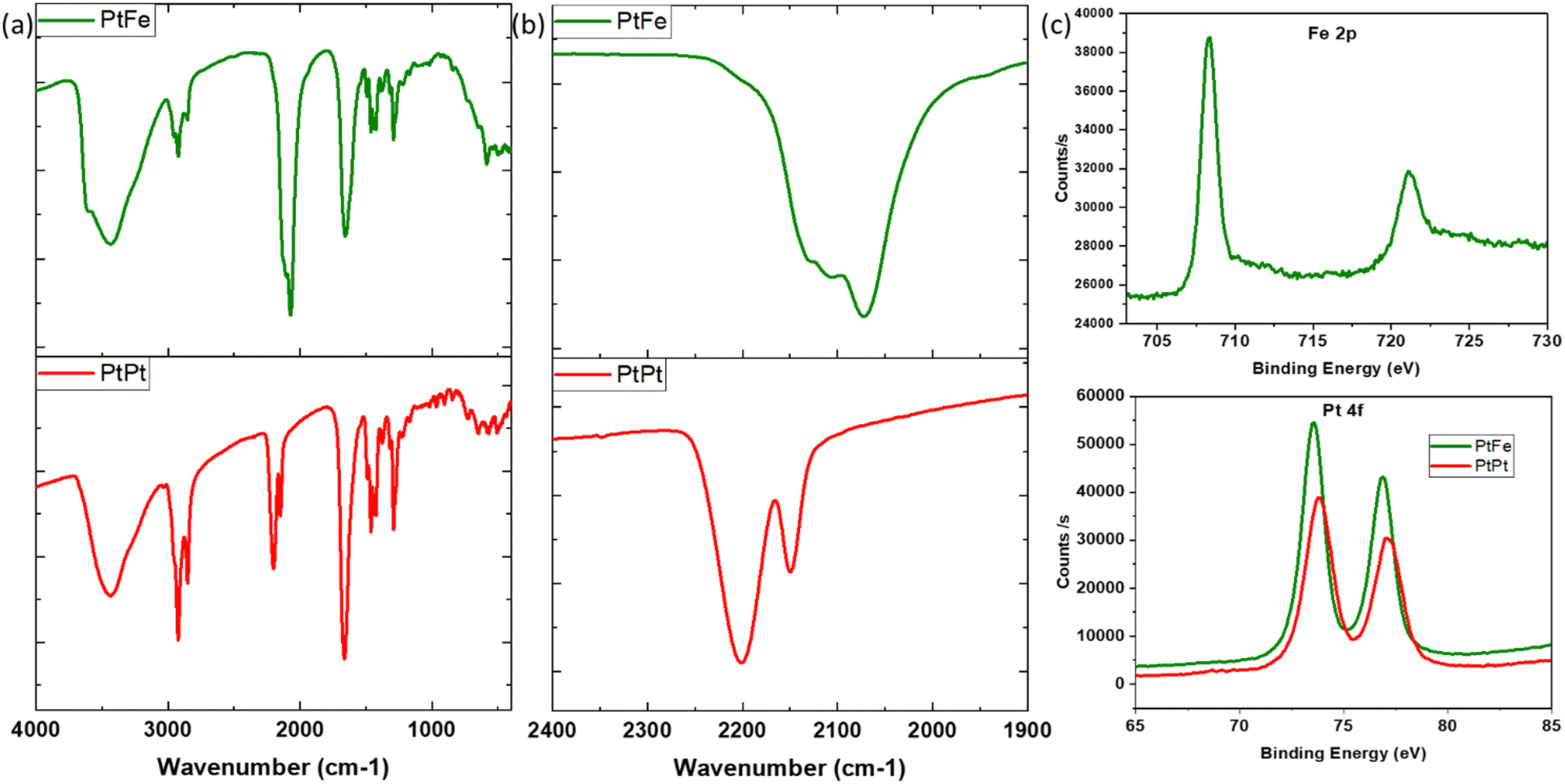 | ||
| Fig. 2 (a) IR spectra of PVP coated PtFe (green) and PtPt (red) NPs, (b) focus on IR peaks in the CN region and (c) Fe-2p core level peaks from XPS spectra of PtFe NP (top) and Pt-4f region of PtFe NP (green) and PtPt NP (red). | ||
The IR analysis of PtPt NPs shows a clear shift of about 25 cm−1 with respect to its tetracyanoplatinate(II) precursor (ESI,‡ Fig. S4). The peak at 2150 cm−1 corresponds to terminal PtII–CN stretch and the intense vibration of bridging cyanide (PtII–CN–PtII) is detected at 2201 cm−1. For both PtFe and PtPt, the characteristic vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1660 cm−1, ternary CN at 2854 cm−1 and symmetric CH2 stretch at 2920 cm−1 confirm the presence of the PVP coating.
O stretch at 1660 cm−1, ternary CN at 2854 cm−1 and symmetric CH2 stretch at 2920 cm−1 confirm the presence of the PVP coating.
The formula unit of these novel nanosystems was determined using EDS to be close to K1.2Cl0.4Pt[Fe(CN)6]0.7 for PtFe and K0.1Cl0.1Pt[Pt(CN)4] for PtPt NPs. XPS analysis of the deposited PtFe powder provides a clear indication of iron in its +II oxidation state with its 2p edge located at 708.3 eV (Fig. 2c), and the 3% of FeIII was not detected. The Pt binding energy (BE) changed to slightly lower values compared to tetracyanoplatinate (73.9 to 73.5 eV), which is attributed to PtII linked through the N end of the cyanide bridge. The analysis of PtPt powder shows that platinum retains its +II oxidation state in the NP with binding energy peaks centred at 73.8 and 77.1 eV. Almost no potassium was found, which is in agreement with the EDS data (ESI,‡ Fig. S8b).
In a nutshell, the results suggest that the structure of PtFe (Fig. 3 left) is similar to that of a PB analogue with a large number of vacancies in ferrocyanide moieties (30%) due to incomplete replacement of some –Cl ligands around Pt(II) in the NPs. Regarding the PtPt NPs, they are made of stacked sheets of [PtPt(CN)4] where each [Pt(CN)4] is surrounded by 4 Pt(II) atoms, with very little amount of –Cl ligands remaining, probably at the surface (Fig. 3 right). X-ray powder diffraction on both samples revealed no peaks as expected, because PtFe NPs contain defects and PtPt NPs are below 3 nm in size.
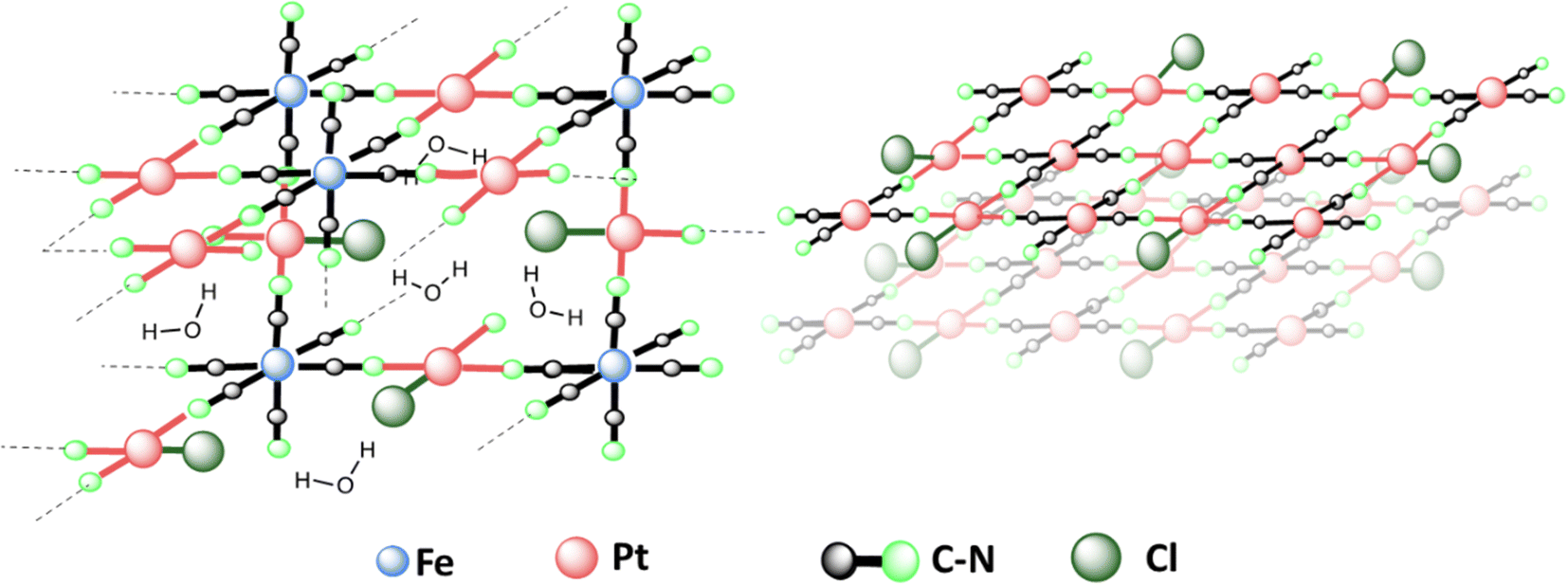 | ||
| Fig. 3 Proposed structure of PtFe (left) and PtPt (right) NPs based on spectroscopic analysis results. PtFe has a 3D structure similar to Prussian Blue, with large number of vacancies due to incomplete replacement of Cl ligands with CN. PtPt has a 2D stacked structure due to tetracyanoplatinate coordination with 4 other Pt(II). | ||
Evaluation of stability in physiological media and irradiation
Both colloidal solutions are stable at room temperature and can be stored over long periods without change in composition at 4 °C when coated with PVP. The consequence of exposure to biological media was analysed by dispersing the nanoparticles in Foetal Bovine Serum (FBS). The evolution was studied using UV-visible spectroscopy at the normal body temperature of 37 °C. PtFe NPs underwent very minimal change over the course of 15 hours in FBS (ESI,‡ Fig. S9). The minor peaks at 414 nm and 772 nm which corresponds to a small amount of FeIII present in the NP, experience a slight decline over time. This is due to the high affinity of phosphate present in the serum towards ferric ions. However, the major band representing FeII–CN–PtII that is integral for the nanoparticle structure undergoes no degradation. Similarly, the PtII–CN–PtII band around 306 nm in PtPt NPs remains unaffected over 15 hrs at 37 °C. These studies provide an indication that these polycyanometallate-based coordination NPs remain stable during their transport within the human body.Similar tests were run in artificial lysosomal fluid (ALF) and the evolution of the absorption spectra was studied for a duration of 6 hours (ESI,‡ Fig. S10). The major peaks of both NPs did not change even in this acidic medium. A small increase in intensity was seen in the peaks corresponding to FeIII, which is attributed to the slight oxidation of FeII under such conditions. Altogether, it was concluded that these novel nanoparticles are not damaged by the cellular medium and could be used for further tests.
In order to evaluate the viability of the NPs as radio-enhancing agents, their stability under irradiation was investigated. Both the solutions with NPs and the controls (precursors), in the presence of PVP and dextran polymers were exposed to a gamma-radiation dose of 4 kGy. Upon irradiation, reactive oxygen species (ROS) are generated in solution. The ROS play a crucial role in enhancing the effect of radiation in living cells. They are capable of oxidising FeII to FeIII and it is substantiated by the appearance of a new band around 425 nm after irradiation of ferrocyanide control solution due to the LMCT band of ferricyanide moiety (ESI,‡ Fig. S11). However, in the presence of dextran polymer, this evolution was not observed indicating no oxidation had taken place. This evidenced that dextran is capable of scavenging the ROS formed and therefore is not appropriate for coating nanoparticles developed for radiation therapy. Since PtFe and PtPt nanoparticles could easily be post-coated, the synthesis was optimised to include PVP polymer. The presence of PVP does not hinder ROS generation as evidenced by the LMCT band in the irradiated ferrocyanide + PVP sample (ESI,‡ Fig. S11b). DLS, TEM and UV-visible spectral analysis performed before and after radiation showed that PtFe and PtPt nanostructures are not significantly altered. Thus, they remain stable during the course of radiation treatment. Due to the ROS-scavenging nature of the polymer, the NPs coated with dextran were discarded and further experiments were only performed using PVP-coated NPs.
Radio-induction of nanoscale damage
It has been hypothesised that high-energy radiation beams induce irreparable damage to cell organelles, proteins and nucleic acids by producing ROS, during water radiolysis. These nanometric damages on biomolecules lead to mutations, cell death, and subsequent elimination, and is the most commonly reported working principle behind radiotherapy. In order to study the impact of these new NPs to induce such complex damage on biomolecules, plasmids were used as bio-nanoprobes. They were incubated with PtFe and PtPt NPs ([M] = 5.3 × 10−6 mol L−1) and subjected to varying doses of radiation. The intact form of the plasmid exists in a supercoiled conformation. Breaks in this circular double-stranded model-DNA diminishes the constraints in the structure and leads to the formation of relaxed (R′) and linear conformations (L′). The three forms of DNA were separated by gel electrophoresis and the detected amount of each enables quantification of single and double-strand breaks. As described in the experimental section, the amount of DSBs were calculated using the amount of L′ that rose out of irradiating plasmid pBR322.The number of lethal, nanometric DSBs generated as a function of radiation dose (0–160 Gy) is presented in Fig. 4. As expected, the number of nanosize breaks rises with an increase in the dose of radiation applied to the samples. But, in the presence of PtFe and PtPt NPs, the amount of damage is significantly enlarged and the sensitizer enhancing ratio (SER) was used to quantify the efficiency of the NPs. It was calculated using the following equation:
| SER = DSBNP/DSBcontrol | (2) |
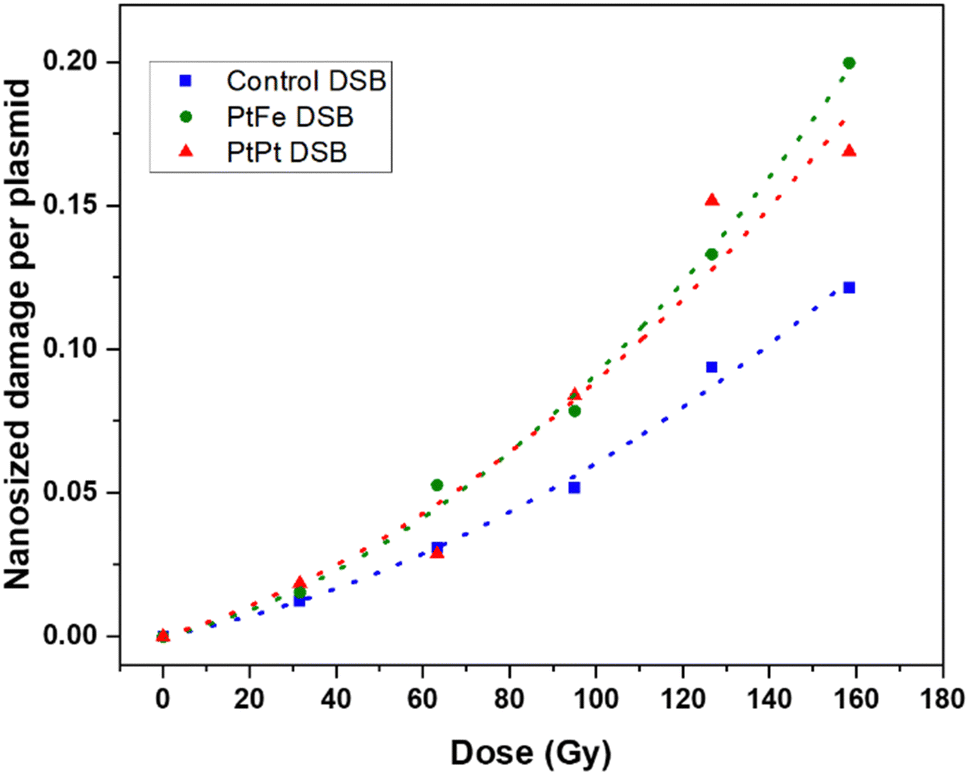 | ||
| Fig. 4 Nanometric damages induced by gamma radiation on plasmid nanoprobes without NPs (blue) and in the presence of PtFe (green) and PtPt (red) NPs and their respective fitted curves. | ||
It is interesting to see that with PtPt, irrespective of the dose supplied, an almost constant enhancement in double-strand breaks in the range of 46–49% was obtained (Table 1). On the other hand, PtFe NPs produce a comparatively smaller effect at low doses (SER 1.34 at 30 Gy) and sequentially intensifies the breaks to the model DNA with increasing dose of radiation (1.59 at 160 Gy). The aim of adding radio-enhancers is to maximize the amount of damage to the cancerous cells, instigate quick apoptosis and minimize the possibility of regeneration while applying a therapeutic dose of radiation. Therefore, even an amplification of 30% in DSBs, in the presence of these porous coordination nanoparticles is very significant. It is important to note that the plasmid used for the experiment is a representative model for different organelles present in the cell and not specifically for DNA in the nucleus. As shown by several groups most NPs do not penetrate or enter the cell nucleus, but complex damage to cytoplasmic organelles also proves detrimental to the cell.31,32 Here, we have demonstrated that at a minimal concentration of 5.3 × 10−6 mol L−1, both nanoparticles amplified the effect of radiation, indicating clearly that they could act as radiation-enhancing agents. However, in contrast to cells, once the strand breaks occur after irradiation, no repair mechanisms happen within the plasmid. Therefore as a next step, the effect of NP and radiation on living cells needed to be understood.
| Dose (Gy) | SER FePt | SER PtPt |
|---|---|---|
| 30 | 1.34 | 1.49 |
| 65 | 1.44 | 1.48 |
| 95 | 1.51 | 1.48 |
| 125 | 1.55 | 1.47 |
| 160 | 1.59 | 1.46 |
Cytotoxicity, uptake and radiation effects of nanoparticles in cells
The primary fibroblasts are used as a model of healthy cells with normal metabolism, while HeLa cells provide an indication of the effect of NPs towards cancerous cells. The effect of a range of different metal concentrations ([M]) in PtFe and PtPt NPs was tested in both cell lines with an incubation time of 6 h (Fig. 5). The columns represent average survival fractions with respective average deviation values. According to the international standard for biological evaluation of medical devices, a sample is considered non-cytotoxic when even the highest concentration provides 70% cell survival.33 The fibroblast cell viability is close to 80% at the highest metal concentration ([M]) of 1 × 10−3 M for both nanoparticles indicating their safety for medical applications, towards healthy cells. HeLa cells appear more sensitive to the presence of nanoparticles as they reach the same limit at 5.0 × 10−4 M concentration. Therefore, a lower and safer total metal concentration of 2.5 × 10−4 mol L−1 was chosen for further experiments with HeLa cells, as the cancer cell-killing effect rising specifically from radiation enhancement needed to be quantified.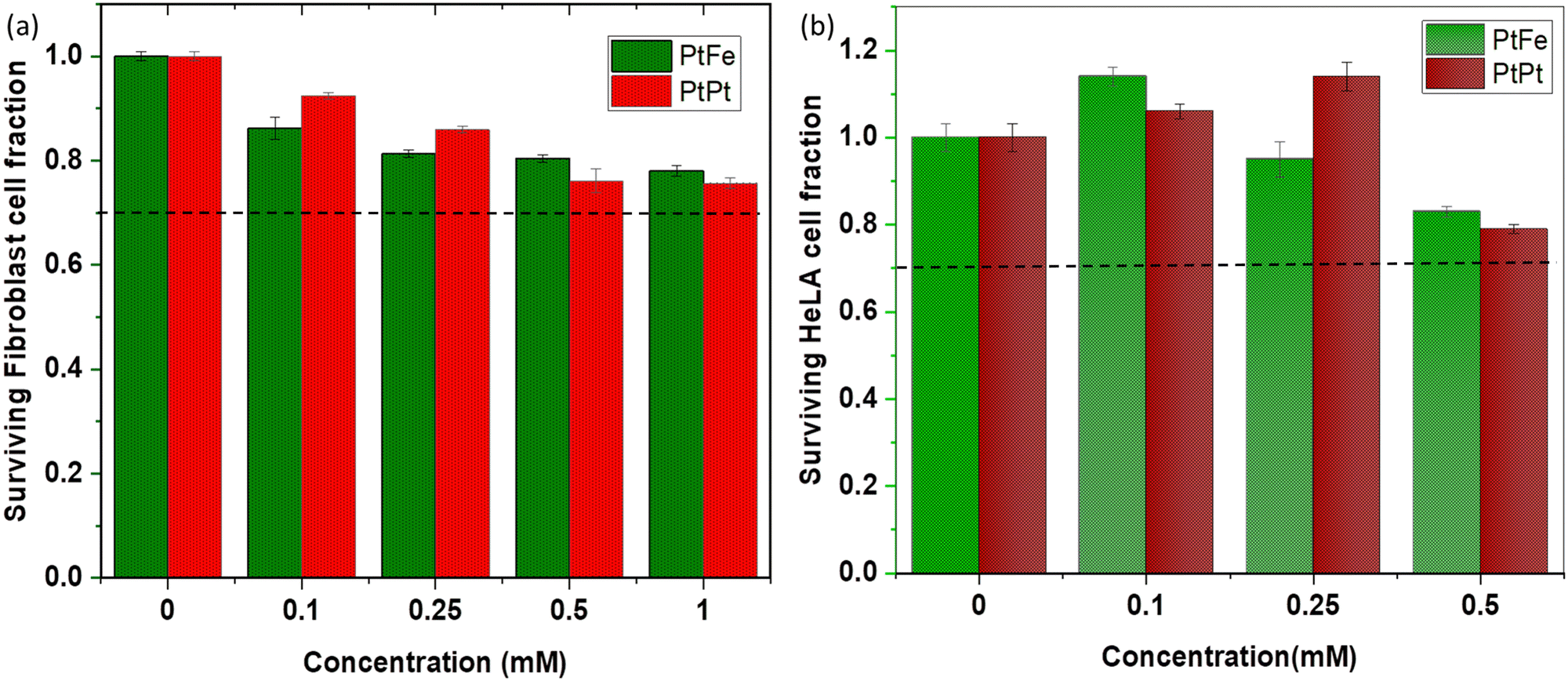 | ||
| Fig. 5
In vitro toxicity results of PtFe and PtPt NPs evaluated by a clonogenic assay using (a) fibroblast cells and (b) HeLa cells after 6 h incubation. The total metal concentration: [Pt + Fe]/[Pt], was considered for the experiment and it lies in the range of 0–1 × 10−3 M. The black dotted line corresponds to the safety limit according to ISO-10933. | ||
ICP-OES analysis of HeLa (ca. 1.3 × 106) cells incubated with PtFe NPs for 6 h showed that a total of 73 ng of Pt was internalized. Therefore, 0.28 fmol of this high-Z element was taken up by one cell. In the case of PtPt NPs, 182 ng of platinum was detected in ca. 1.8 × 106 HeLa cells, meaning the amount of Pt internalized was approximately 0.52 fmol per cell. The impact of these particles in cell killing was studied using γ-rays from a Cesium-137 source (0.63 meV). The cancer cells incubated with NPs were irradiated for different time intervals at a dose rate of 0.75 Gy min−1. The colonies formed at the end of the standard 14 day incubation period helped determine the survival fraction (SF) at each dose.
The colony count data were fitted using a linear-quadratic model (LQM) (SF vs. dose) using eqn (3):
| SF(D) = e−(αD + βD2) | (3) |
The percentage of surviving cells sequentially decreased with increasing the radiation dose but as presented in Fig. 6, this decline is significantly amplified in the presence of PtFe and PtPt NPs. This result proves the radioenhancing property of these novel polycyanometallate-based NPs. The effectiveness of NPs as radioenhancers was quantified using Dose Enhancement Factor (DEF) which gives a measure of the efficiency of in vitro cell killing at a given dose point (D). It was calculated using the following equation:
| DEF = SFcontrol,fit/SFNP,fit | (4) |
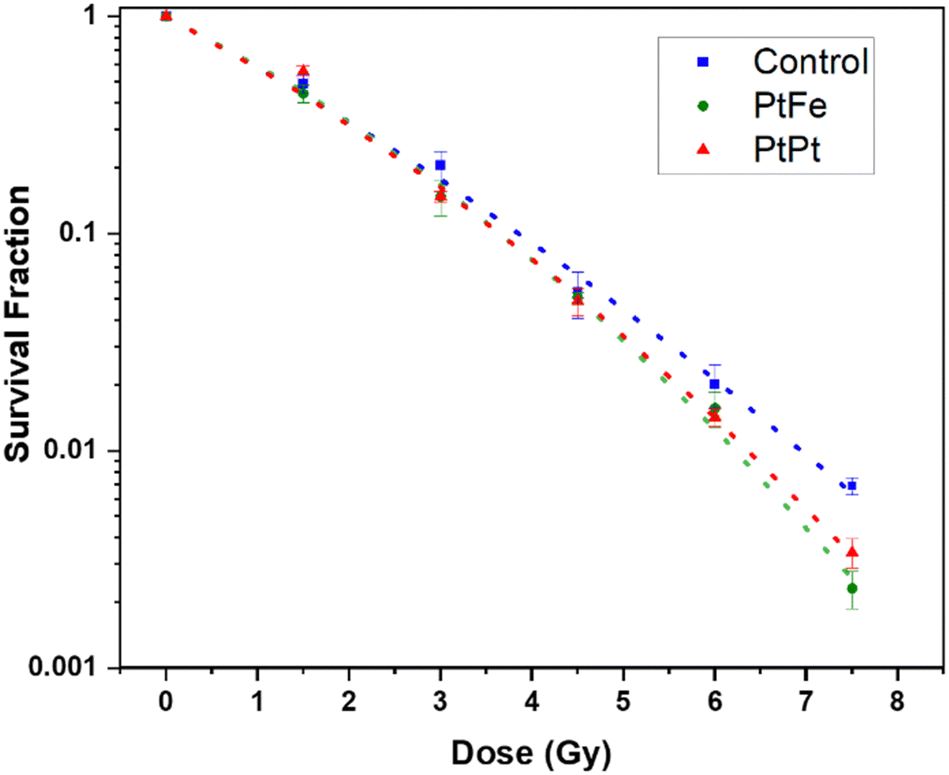 | ||
| Fig. 6 Survival fraction of HeLa cells after Cs-137 irradiation without NPs (blue), in the presence of 2.5 × 10−4 M PtFe (green) and PtPt (red) NPs fit using LQM (dotted lines) as a function of dose. | ||
| Sample | α (Gy−1) | β (Gy−2) | α/β (Gy) |
|---|---|---|---|
| Control | 0.515 ± 0.050 | 0.021 ± 0.007 | 24.30 ± 9.44 |
| PtFe | 0.464 ± 0.050 | 0.044 ± 0.008 | 10.57 ± 2.21 |
| PtPt | 0.500 ± 0.049 | 0.034 ± 0.007 | 14.38 ± 3.56 |
Compared to the control, the presence of Pt increases the number of radicals formed in the solution. It is possible that, due to the microporosity of the NP, the generated ROS quickly disperses through the biological system causing a markedly high amount of sub-lethal damages that is transformed into a final lethal effect in the cell. It has to be noted that as seen with plasmid experiments earlier, despite the presence of a lower amount of high-Z element in PtFe NP, they produce similar and sometimes even better amplification of radiation damage when compared to PtPt NPs.
This could be due to two reasons. The γ-radiation interacts with Fe atoms as well and following a combination of various phenomena (activation, de-excitation and emission of electrons) also leads to the production of radicals that help with cell killing.27 Secondly, as compared to the 2D sheets that form PtPt NP, the PtFe nanostructure is a Prussian Blue-like structure full of defects and vacancies that can be occupied by water molecules. Therefore, more reactive radicals are formed within and around PtFe NP and they have higher freedom of mobility to undergo faster diffusion throughout the cell media. This is a further indication that the microporosity of nanostructures used in radiation therapy heavily affects the final results. It can be concluded from the data that even at low concentrations, both the nanoparticles cause the death of targeted cells and positively improve the lethality of γ-radiation. As the results from the clonogenic assay and plasmid experiments are in agreement, it can be said that the complex damages occurring to the biomolecules after gamma radiation are the root cause of the amplification of cancer cell death.
Conclusions
The present study reported a green, cheap and fast synthetic procedure of two novel Pt-containing polycyanometallate-based nanostructures. A complete characterization revealed that these ultra-small, negatively charged, coordination nanoparticles are stable in water, buffer as well as physiological media like FBS and ALF. These PtFe and PtPt NPs are easily post-coated with polymers depending on the targeted applications and they remained intact upon irradiation, which suggested their applicability as radioenhancers. A marked increase in lethal double-strand breaks on plasmids is observed in the presence of these newly synthesised NPs. Plasmid DNA was chosen as a nanoprobe to quantify complex molecular damage and the efficiency of NPs to amplify this damage when combined with radiation. This complex damage can occur in any biomolecule of the cytoplasm, in the vicinity of the NP. The in vitro tests revealed that they were non-toxic to primary fibroblast and HeLa cells in a good range of concentrations. However, irradiation of the cells containing the NPs with an ionizing beam induces strong cytotoxicity, with the amplification factor rising close to 60%. Therefore, our cyanide-bridged PtFe and PtPt NPs may serve as effective agents that amplify the damage to cancer cells at therapeutic doses. Interestingly, the PtFe shows an improved radioenhancing effect compared to its PtPt counterpart and it could be due to its more porous, 3D Prussian Blue-like structure. The role of zeolitic water as well as porosity will be further investigated. We envisage imparting MRI contrast properties by coordination of paramagnetic ions (Mn(II), Fe(III)) at the surface (as shown by some of us on PB) for image-guided treatments.Author contributions
RG: investigation, conceptualization, methodology, data curation, validation, writing – original draft and editing. LF: methodology, investigation. EP: methodology, in vitro experiments, resources, supervision, writing – review and editing. FS and CB: in vitro experiments. FB: high-resolution TEM and EDS. DD: XPS measurements. HR and SL: funding acquisition, project administration, resources, writing – review and editing. LC: conceptualization, methodology, funding acquisition, project administration, resources, supervision, writing – draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the DIM RESPORE, NanoTherad and INanoTheRad, Paris Saclay University and CNRS for their financial support. The present work has benefited from Imagerie-Gif core facility supported by I’Agence Nationale de la Recherche (ANR-11-EQPX-0029/Morphoscope, ANR-10- INBS-04/FranceBioImaging; ANR-11-IDEX-0003-02/Saclay Plant Sciences).Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71(3), 209–249 CrossRef PubMed.
- M. Chalmers, Phys. World, 2003, 16(8), 32 CrossRef.
- C. B. Dracham, A. Shankar and R. Madan, Radiat Oncol J., 2018, 36(2), 85–94 CrossRef PubMed.
- G. E. Adams, Br. Med. Bull., 1973, 29(1), 48–53 CrossRef CAS PubMed.
- K. Kobayashi, N. Usami, E. Porcel, S. Lacombe and C. Le Sech, Mutat. Res., 2010, 704(1–3), 123–131 CAS.
- J. F. Hainfeld, D. N. Slatkin and H. M. Smilowitz, Phys. Med. Biol., 2004, 49(18), N309–N315 CrossRef CAS PubMed.
- A. Z. Wilczewska, K. Niemirowicz, K. H. Markiewicz and H. Car, Pharmacol Rep., 2012, 64(5), 1020–1037 CrossRef CAS PubMed.
- S. K. Libutti, G. F. Paciotti, A. A. Byrnes, H. R. Alexander Jr, W. E. Gannon, M. Walker, G. D. Seidel, N. Yuldasheva and L. Tamarkin, Clin. Cancer Res., 2010, 16(24), 6139–6149 CrossRef CAS PubMed.
- C. Verry, S. Dufort, J. Villa, M. Gavard, C. Iriart, S. Grand, J. Charles, B. Chovelon, J. L. Cracowski, J. L. Quesada, C. Mendoza, L. Sancey, A. Lehmann, F. Jover, J. Y. Giraud, F. Lux, Y. Crémillieux, S. McMahon, P. J. Pauwels, D. Cagney, R. Berbeco, A. Aizer, E. Deutsch, M. Loeffler, G. Le Duc, O. Tillement and J. Balosso, Radiother. Oncol., 2021, 160, 159–165 CrossRef CAS PubMed.
- S. Bonvalot, P. L. Rutkowski, J. Thariat, S. Carrère, A. Ducassou, M. P. Sunyach, P. Agoston, A. Hong, A. Mervoyer, M. Rastrelli, V. Moreno, R. K. Li, B. Tiangco, A. C. Herraez, A. Gronchi, L. Mangel, T. Sy-Ortin, P. Hohenberger, T. de Baère, A. Le Cesne, S. Helfre, E. Saada-Bouzid, A. Borkowska, R. Anghel, A. Co, M. Gebhart, G. Kantor, A. Montero, H. H. Loong, R. Vergés, L. Lapeire, S. Dema, G. Kacso, L. Austen, L. Moureau-Zabotto, V. Servois, E. Wardelmann, P. Terrier, A. J. Lazar, J. V. M. G. Bovée, C. Le Péchoux and Z. Papai, Lancet Oncol., 2019, 20(8), 1148–1159 CrossRef CAS PubMed.
- R. Crapanzano, V. Secchi and I. Villa, Appl. Sci., 2021, 11(15), 7073 CrossRef CAS.
- Y. Li, K. H. Yun, H. Lee, S. H. Goh, Y. G. Suh and Y. Choi, Biomaterials, 2019, 197, 12–19 CrossRef CAS PubMed.
- A. Mignot, C. Truillet, F. Lux, L. Sancey, C. Louis, F. Denat, F. Boschetti, L. Bocher, A. Gloter, O. Stéphan, R. Antoine, P. Dugourd, D. Luneau, G. Novitchi, L. C. Figueiredo, P. C. de Morais, L. Bonneviot, B. Albela, F. Ribot, L. Van Lokeren, I. Déchamps-Olivier, F. Chuburu, G. Lemercier, C. Villiers, P. N. Marche, G. Le Duc, S. Roux, O. Tillement and P. Perriat, Chemistry, 2013, 19(19), 6122–6136 CrossRef CAS PubMed.
- E. Gharibshahi, E. Saion, A. Ashraf and L. Gharibshahi, Appl. Radiat. Isot., 2017, 130, 211–217 CrossRef CAS PubMed.
- Y. Zhang, X. Han, Y. Liu, S. Wang, X. Han and C. Cheng, Mater. Adv., 2022, 3, 3709–3725 RSC.
- Z. Bao, M. He, H. Quan, D. Jiang, Y. Zheng, W. Qin, Y. Zhou, F. Ren, M. Guo and C. Jiang, RSC Adv., 2016, 6, 35124–35134 RSC.
- X. Yang, D. Salado-Leza, E. Porcel, C. R. González-Vargas, F. Savina, D. Dragoe, H. Remita and S. Lacombe, Int. J. Mol. Sci., 2020, 21(5), 1619 CrossRef CAS PubMed.
- D. Salado-Leza, E. Porcel, X. Yang, L. Štefančíková, M. Bolsa-Ferruz, F. Savina, D. Dragoe, J. L. Guerquin-Kern, T. D. Wu, R. Hirayama, H. Remita and S. Lacombe, Nanotechnol., Sci. Appl., 2020, 13, 61–76 CrossRef CAS PubMed.
- Center for Drug Evaluation and Research. Approval Letter for Radiogardase (insoluble Prussian Blue), 2003, 21–626.
- Z. Y. Liu, G. T. Fu, L. Zhang, X. Y. Yang, Z. Q. Liu, D. M. Sun, L. Xu and Y. W. Tang, Sci. Rep., 2016, 6, 32402 CrossRef CAS PubMed.
- S. Y. Lin, Y. Q. Yao, L. Zhang, J. J. Feng and A. J. Wang, Mater. Today Energy, 2021, 20, 100701 CrossRef CAS.
- Z. Qin, Y. Li and N. Gu, Adv. Healthcare Mater., 2018, 7(20), 1800347 CrossRef PubMed.
- G. Paul, Y. Prado, N. Dia, E. Rivière, S. Laurent, M. Roch, L. V. Elst, R. N. Muller, L. Sancey, P. Perriat P, O. Tillement, T. Mallah and L. Catala, Chem. Commun., 2014, 50(51), 6740–6743 RSC.
- L. Catala and T. Mallah, Coord. Chem. Rev., 2017, 346, 32–61 CrossRef CAS.
- G. Fornasieri, M. Aouadi, E. Delahaye, P. Beaunier, D. Durand, E. Rivière, P.-A. Albouy, F. Brisset and A. Bleuzen, Materials, 2012, 5, 385–403 CrossRef CAS PubMed.
- L. Fétiveau, G. Paul, A. Nicolas-Boluda, J. Volatron, R. George, S. Laurent, R. Muller, L. Sancey, P. Mejanelle, A. Gloter, F. Gazeau and L. Catala, Chem. Comm., 2019, 55, 14844–14847 RSC.
- X. Li, E. Porcel, M. Menendez-Miranda, J. Qiu, X. Yang, C. Serre, A. Pastor, D. Desmaële, S. Lacombe and R. Gref, ChemMedChem, 2020, 15(3), 274–283 CrossRef CAS PubMed.
- C. Ren, Y. Cheng, W. Li, P. Liu, L. Yang, Q. Lu, M. Xu, F. Tan, J. Li and N. Li, Biomater. Sci., 2020, 8, 1981–1995 RSC.
- M. Spotheim-Maurizot, M. Charlier and R. Sabattier, Int. J. Radiat. Biol., 1990, 57(2), 301–313 CrossRef CAS PubMed.
- C. M. Burgess, N. Yao and A. B. Bocarsly, J. Mater. Chem., 2009, 19, 8846–8855 RSC.
- L. Štefanciková, S. Lacombe, D. Salado, E. Porcel, E. Pagáčová, O. Tillement, F. Lux, D. Depeš and M. Falk, J. Nanobiotechnol., 2016, 14, 63 CrossRef PubMed.
- E. Pagáčová, L. Štefančíková, F. Schmidt-Kaler, G. Hildenbrand, T. Vičar, D. Depeš, J.-H. Lee, F. Bestvater, S. Lacombe, E. Porcel, S. Roux, F. Wenz, O. Kopečná, I. Falková, M. Hausmann and M. Falk, Int. J. Mol. Sci., 2019, 20(3), 588 CrossRef PubMed.
- Biological evaluation of medical devices-ISO-10993-5-2009.
Footnotes |
| † In tribute to Dr Géraldine Carrot. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00516j |
| This journal is © The Royal Society of Chemistry 2023 |