
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vancomycin mimicry: towards new supramolecular antibiotics
Alister J.
Flint
and
Anthony P.
Davis
 *
*
University of Bristol, School of Chemistry, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: anthony.davis@bristol.ac.uk
First published on 27th September 2022
Abstract
Vancomycin is the best-known of the glycopeptide group antibiotics (GPAs), a family of agents which operate by binding the C-terminal deptide D-Ala–D-Ala. This anionic epitope is an interesting target because it plays a central role in bacterial cell wall synthesis, and is not readily modified by evolution. Accordingly, vancomycin has been in use for >60 years but has only provoked limited resistance. Agents which mimic vancomycin but are easier to synthesise and modify could serve as valuable weapons against pathogenic bacteria, broadening the scope of the GPAs and addressing the resistance that does exist. This article gives an overview of vancomycin's structure and action, surveys past work on vancomycin mimicry, and makes the case for renewed effort in the future.
 Alister J. Flint | Alister Flint gained a MSci in Chemistry from University College London in 2019. After completing his undergraduate degree, he joined the Davis group at the University of Bristol as a PhD student. His doctoral work focuses on the recognition of biologically relevant anions in water with a particular emphasis on the recognition properties of vancomycin-group antibiotics. |
 Anthony P. Davis | Tony Davis gained a B.A. in Chemistry from Oxford University in 1977, and a D. Phil. in 1979. After postdoctoral work in Oxford and ETH Zürich, he was appointed in 1982 as a Lecturer at Trinity College, Dublin. In September 2000 he moved to the University of Bristol, where he is Professor of Supramolecular Chemistry in the School of Chemistry. His research focuses on the development of supramolecular systems with potential for biological applications, especially carbohydrate receptors and transmembrane anion transporters. He has co-founded two companies to exploit discoveries in carbohydrate recognition and sensing; Ziylo, which was sold in 2018 to Novo Nordisk, and Carbometrics, which continues to work in the area. Since 2019 he has been Chair of the Editorial Board of Organic and Biomolecular Chemistry. |
1. Introduction
Peptides are ubiquitous biological signalling molecules that provide structurally diverse, modifiable fingerprints for molecular recognition.1 Sequence-selective peptide recognition controls many fundamental biological processes involved in gene expression, cell-signalling and infection.2,3 The ability to interact with these systems using synthetic receptors represents a promising area of biomedical research, particularly in settings where biomodulation using small-molecules has limited efficacy, such as affecting protein–protein interactions or where resistance to small-molecules evolves rapidly.4–7One of the most studied bioactive peptides is the C-terminal D-Alanyl–D-Alanine (D-Ala–D-Ala) sequence which functions as a recognition motif coordinating various enzyme catalysed processes in the biosynthesis of bacterial cell-wall peptidoglycan (Fig. 1).8,9
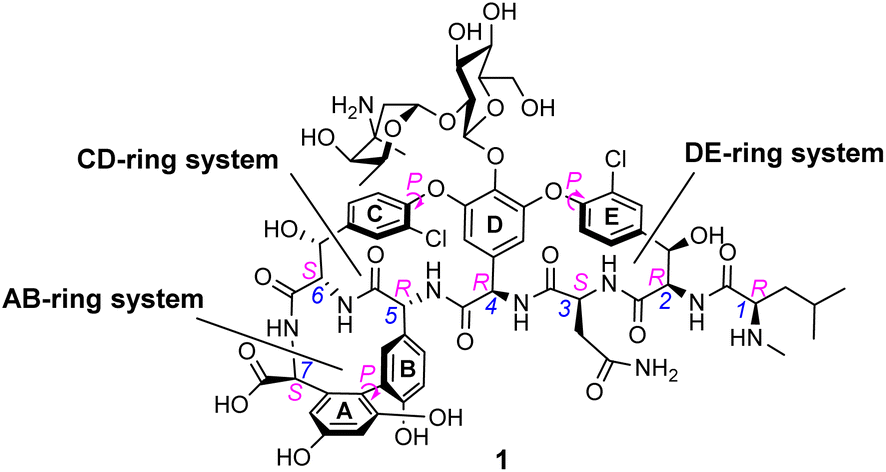 | ||
| Fig. 1 Formula of vancomycin 1 highlighting various chiral elements (magenta) and heptapeptide residue numbering (blue). The three macrocyclic ring systems are labelled according to their aromatic substituents A–E. | ||
The D-Ala–D-Ala carboxylate is also a substrate of the naturally occurring glycopeptide-group antibiotics (GPAs), and selective recognition of the D-Ala–D-Ala sequence is understood to be central to their mode of action.10 For example, the medicinally important GPA vancomycin 1 (Fig. 1) binds to N-acyl-D-Ala–D-Ala carboxylates with reported association constants (Ka) of ca. 105 M−1 in water.10–12 The GPAs are unusually small natural receptors, comparable in size to synthetic host molecules. For this reason, vancomycin mimicry has become a popular target for supramolecular chemists interested in binding polar species in water.
Vancomycin has attracted interest from a variety of researchers due to its unique biological and chemical properties. Clinicians first used vancomycin during the 1950s to treat Gram-positive bacterial infections in patients with β-lactam allergies or where resistance meant β-lactams were no longer effective. Such was the urgency for alternative therapies to treat penicillin resistant infections that vancomycin was introduced to the clinic decades before its chemical structure or mechanism of action were fully elucidated.13
Gradually, it was understood that vancomycin disrupts extracellular peptidoglycan assembly by inhibiting the key glycan cross-linking step and to a lesser extent glycan polymerisation.14–16 Peptidoglycan is a highly cross-linked three-dimensional matrix comprising a glycan co-polymer of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues cross-linked by short peptides.14–16 The peptidoglycan layer is continually regenerated by incorporating new Lipid II monomers which are generated within the bacterial cell and translocated across the membrane into the periplasmic space by a recyclable bactoprenol residue (Fig. 2). Once translocated into the periplasmic space, Lipid II monomers are incorporated into the growing glycan chain by formation of a 1,4-β-glycosyl linkage, catalysed by the glycosyltransferase domain of membrane-bound penicillin-binding proteins (PBP). This process also releases the bactoprenol residue which is recycled. The extended glycan co-polymer is subsequently cross-linked by the transpeptidase domain of PBPs which catalyse formation of an amide linkage between short peptides attached to GlcNAc residues on neighbouring glycan chains.
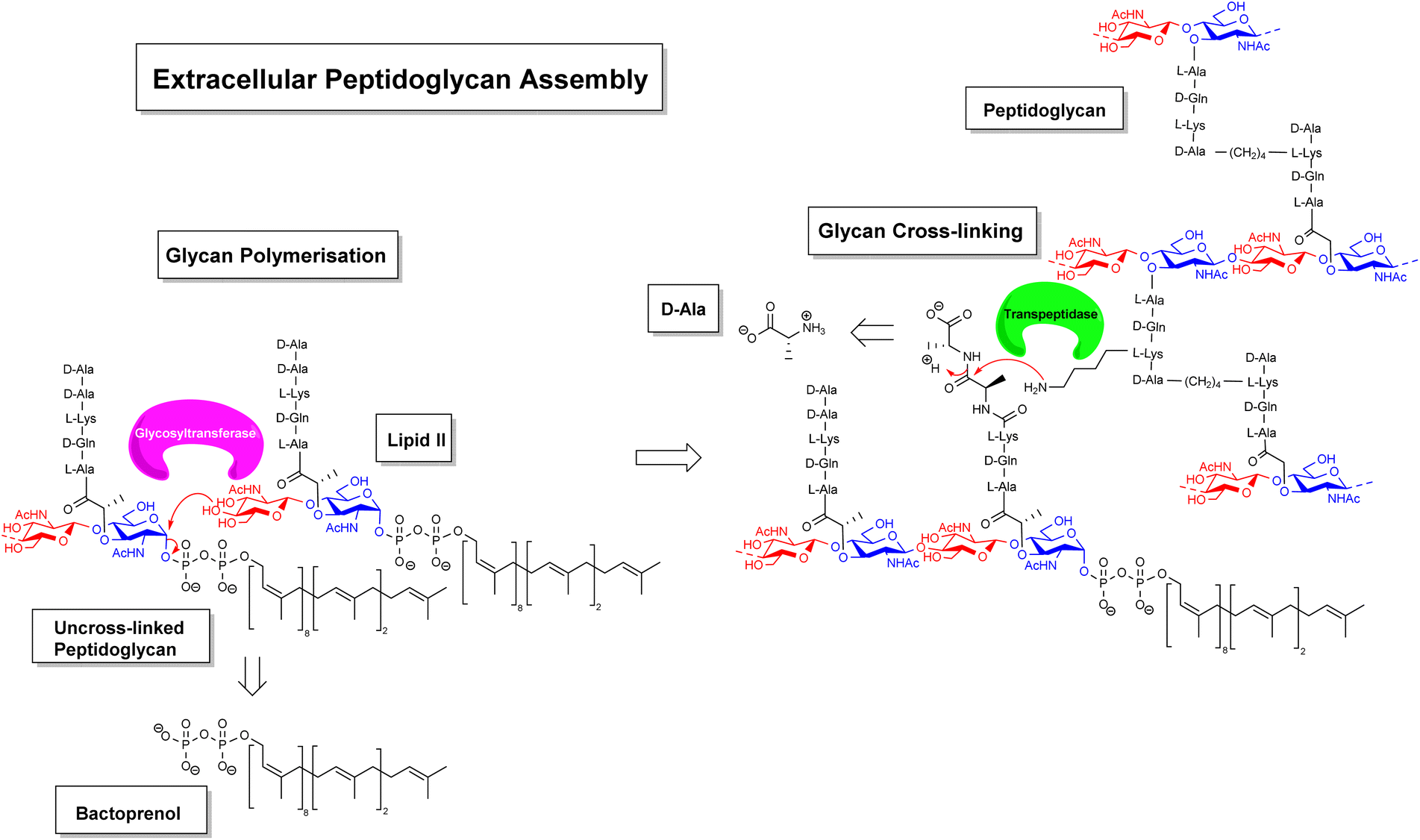 | ||
| Fig. 2 Schematic highlighting extracellular stages of peptidoglycan biosynthesis inhibited by vancomycin and other GPAs. | ||
Glycan cross-linking is essential for strengthening the peptidoglycan superstructure against fluctuating osmotic pressures experienced by the cell-wall matrix. Both glycan polymerisation and cross-linking are mediated by recognition of the C-terminal D-Ala–D-Ala sequence. Vancomycin binds to the D-Ala–D-Ala sequence blocking recognition by PBPs. This process inhibits glycan cross-linking and to a lesser extent glycan polymerisation steps in peptidoglycan biosynthesis, weakening the overall cell-wall assembly with respect to osmotic pressure and leading to fatal lysis of the bacterial cell.
Vancomycin's mechanism of action is distinct from other antibiotics targeting peptidoglycan biosynthesis.17 For example, β-lactam antibiotics such as methicillin inhibit glycan cross-linking by irreversibly acylating an active site serine residue in the transpeptidase domain of penicillin-binding proteins.18 Penicillin-binding proteins are directly transcribed meaning they can undergo rapid drug-induced remodelling through genetic mutation which has resulted in resistance to β-lactam antibiotics (e.g. methicillin-resistant S. aureus, MRSA).19 Vancomycin is not susceptible to this resistance mechanism, as expression of the D-Ala–D-Ala sequence is highly conserved. Because the use of the sequence is controlled by several different gene products, modification is dependent on multiple mutations and the development of resistance is therefore much more difficult. As a result, vancomycin has demonstrated exceptional clinical longevity and is still valuable after >60 years of use.20
Once vancomycin's mode of action and chemical structure were elucidated, it became a focus of interest for both synthetic and supramolecular chemists. The former were drawn to the challenging structure and the potential for making analogues,21–23 while the latter saw the possibility of synthetic hosts which would mimic vancomycin's ability to bind D-Ala–D-Ala and thus its antibiotic activity.24,25 The potential here was clear, as vancomycin analogues or mimics could possess lower susceptibility towards resistance compared to conventional small molecule agents while overcoming some of the problems associated with the natural antibiotic. Major limitations of vancomycin and other GPAs relate to poor pharmacokinetic properties requiring parenteral administration for blood stream and complicated skin infections (which are common indications for use), and lack of activity against Gram-negative bacteria.26 Vancomycin mimicry presents an opportunity to develop synthetic structures better suited to medicinal chemistry optimisation and large-scale total-synthesis. Despite Nicolaou's publication in 1999 of the first successful total synthesis of vancomycin,21 commercial production of various GPAs including vancomycin is still limited to batch fermentation and semi-synthesis restricting the scope of structural tuning.
Although Vancomycin resistance has been slow to develop, it has arrived in recent years with the Van-genes, acquired by S. aureus and Enterococcal spp. from less harmful vancomycin-producing bacteria. These genes are responsible for the expression of alternative PBP recognition motifs D-Ala–D-Ser and D-Ala–D-Lac27 which form low affinity complexes with vancomycin due to repulsive steric and electrostatic interactions respectively, rendering the antibiotic ineffective.28 Vancomycin resistance has thus become a pressing concern, and one of the most promising applications of vancomycin mimicry is the development of synthetic host molecules targeting dual recognition of D-Ala–D-Ala and the low-affinity recognition motifs responsible for resistance. This work has the potential to produce next-generation antibiotics with unrivalled tolerance to antibiotic-resistance. This review discusses aspects of vancomycin's structure and function related to peptide recognition and highlights current approaches to vancomycin mimicry with synthetic host molecules.
2. Vancomycin – a model for peptide recognition in water
2.1 Structure
In biological terms, vancomycin is a remarkably small peptide receptor (1449.27 Da), orders of magnitude smaller than penicillin binding protein 2A (ca. 149 kDa) which carries out glycan polymerisation and cross-linking in MRSA (Fig. 3). Vancomycin's chemical structure is based on a heptapeptide which has acquired a tricyclic architecture through connections formed between aromatic side-chains (Fig. 1). To this is added a disaccharide, linked by O-glycosylation to the central residue, composed of a glucosyl unit and the amino-sugar vancosamine.29 The five aromatic amino residues present in the heptapeptide backbone are cross-linked forming a 12-membered biaryl-linked AB-ring system and two 16-membered biaryl ether-linked CD- and DE-ring systems (for labelling see Fig. 1). The Ar–Ar and Ar–O–Ar systems are conformationally fixed and exhibit planar chirality having right-handed P-configurations.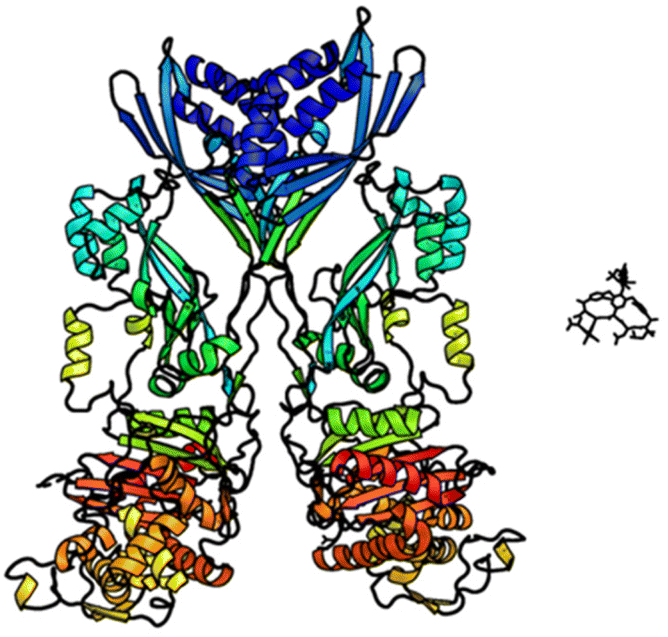 | ||
| Fig. 3 Crystal structures of penicillin binding protein 2a and vancomycin showing relative sizes. | ||
Vancomycin is relatively stable towards atropisomerism which requires temperatures above 140 °C and extended reaction times producing less biologically active conformers.30 The three-dimensional structure of vancomycin's D-Ala–D-Ala binding site was determined from NMR studies and X-ray crystal structures to resemble a hydrophobic cleft preorganised by residue cross-linking with inward facing polar functionality, including an anion-binding centre at one end (Fig. 4).31,32 Residue cross-linking maintains binding site preorganisation which minimises entropic losses accompanying complex formation and promotes substrate selectivity. Calorimetric binding studies have shown that vancomycin's complexation of the D-Ala–D-Ala sequence is enthalpically driven at physiologically relevant temperatures with heat change sufficient to compensate for entropy lost during complexation.10,16 Vancomycin binds to N-acyl-D-Ala–D-Ala carboxylates reversibly with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex stoichiometry through non-covalent interactions. Complexation follows a two-step induced-fit binding process in which the substrate is initially bound in a weak complex which undergoes conformational rearrangement to form a second higher affinity complex.33
:1 complex stoichiometry through non-covalent interactions. Complexation follows a two-step induced-fit binding process in which the substrate is initially bound in a weak complex which undergoes conformational rearrangement to form a second higher affinity complex.33
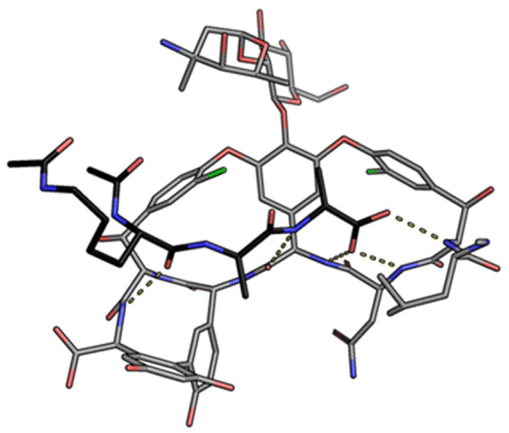 | ||
| Fig. 4 Crystal structure of vancomycin in complex with Ac–Lys(Ac)–D-Ala–D-Ala, a water-soluble analogue of the Lipid II peptide side chain. | ||
2.2 Interactions with the substrate
Vancomycin appears to bind its substrate through a combination of hydrophobic and hydrogen bonding interactions (Fig. 4). Hydrophobic interactions have been identified between the aromatic residues of vancomycin and the methyl side groups carried by the D-Ala–D-Ala sequence. In addition to entropic contributions, hydrophobic effects may also be driven enthalpically by the liberation of trapped water molecules (non-classical hydrophobic effect).34,35 This phenomenon is enhanced in concave binding sites such as vancomycin's where concave surface topography prevents water molecules forming a full complement of four H-bonds creating so-called high-energy water which is more easily displaced from the binding site.36Vancomycin and N-acyl-D-Ala–D-Ala also show H-bonding complementarity, forming five intermolecular H-bonds which may be viewed as two distinct secondary structure elements (Fig. 5). One is a β-sheet comprising two H-bonds between vancomycin residues four and seven and the substrate, and the second is a group of three H-bonds donated from residues two to four to the substrate carboxylate in a nest sub-structure (Fig. 6).37 These interactions display cooperativity and loss of a single H-bond reduces binding activity 10-fold in water.38 The acyl group at the N-terminus of the D-Ala–D-Ala unit supplies a H-bond acceptor. In the natural substrate this acyl group is an L-lysyl residue, and many studies of vancomycin binding employ the bis-acetylated Ac-L-Lys(Ac)-D-Ala–D-Ala 2. The lysine side-chain may make some contribution to binding.39
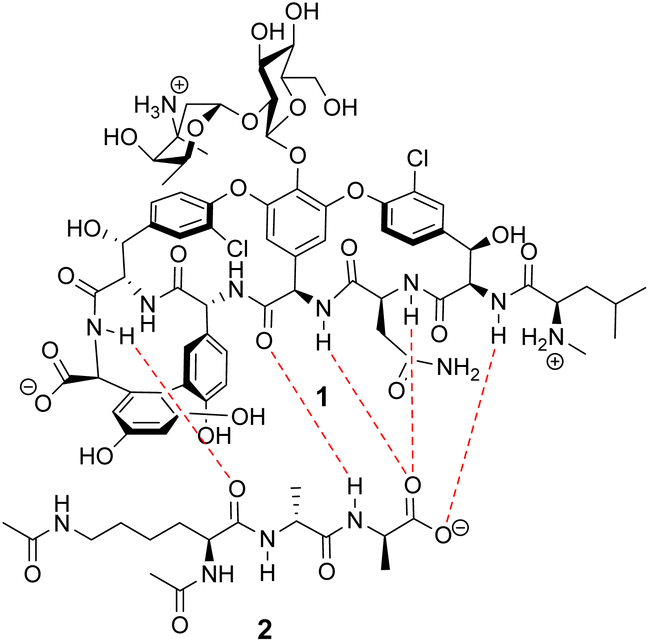 | ||
| Fig. 5 H-bonding (red) interaction between vancomycin 1 and water soluble substrate Ac–L-Lys(Ac)–D-Ala–D-Ala 2 at physiological pH (ca. 7.4). | ||
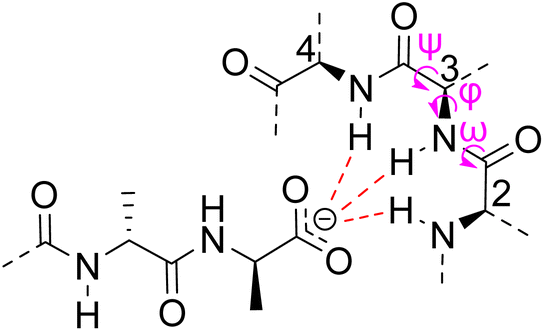 | ||
| Fig. 6 Schematic representation of vancomycin's carboxylate binding site formed from heptapeptide residues 2, 3 and 4 and mainchain dihedral bond angles ω ψ φ. | ||
The D-Ala–D-Ala sequence carries a C-terminal carboxylic acid which is deprotonated at physiological pH (ca. 7.4) presenting a handle for strong electrostatic interactions. Carboxylate recognition in water is challenging due to the heavily solvated nature of the substrate and the presence of competitor anions in biological media. Synthetic host molecules tend to use cationic moieties to bind carboxylates in water through ion-pairing.40 However, ionic interactions are not directional and often produce poor substrate selectivity over competing anions. They also suffer from higher desolvation energies in water relative to charge-neutral binding sites. In comparison, carboxylate recognition in Nature is often accomplished using charge neutral nest sub-structures which are less strongly hydrated. Vancomycin contains a carboxylate-binding nest formed by heptapeptide residues two, three and four which together donate three H-bonds to the substrate carboxylate moiety.41 These are understood to be the strongest directional interactions in the complex and likely benefit from their proximity to the hydrophobic DE-ring system which destabilises water within the binding site.36
Nest structures are short peptide sequences found in proteins and peptides which bind polar or charged substrates through multidentate interactions. Nest structures form when at least two adjacent amino residues within a peptide strand possess main-chain dihedral bond angles (φ,ψ) of approximately 90, 0° (or −90, 0°) and 70, 40° (or −70, −40°) respectively creating a small, preorganised concavity for multiple H-bonding interactions between inward facing amide N–H bonds and an anionic or partially charged (δ−) substrate moiety.42 Precise arrangement of recognition motifs within nest structures promotes substrate selectivity and affinity. For example, the presence of opposite R,S,R chirality in the vancomycin carboxylate nest is essential for effective complexation.37,38,43,44 Nest structures play an important role in biological recognition phenomena in water where individual H-bonds are weakened by competing interactions with solvent molecules and chelation of the substrate may be necessary to overcome hydration.
2.3 N-Methyl leucine – the role of charge
Vancomycin has net positive charge owing to protonation of the vancosamine and N-methyl leucine residues and deprotonation of the C-terminus carboxylic acid at physiological pH. Positive charge on the N-methyl leucine residue is also thought to contribute to substrate recognition through ion-pairing interactions with the substrate C-terminus carboxylate as evidenced by complexation induced changes in chemical shift in solution NMR studies.45,46 However, the cationic N-methyl ammonium residue projects away from the carboxylate-anion in X-ray crystal structures of the complex formed between vancomycin and D-Ala–D-Ala and is not considered part of the carboxylate nest structure.31 Instead, the N-methyl leucine residue is thought to participate in recognition and orientation of the substrate in a weakly bound pre-equilibrium complex before rearranging to the strongly bound equilibrium complex conformer seen in crystal structures.2.4 Dimer formation
During their pioneering ligand binding studies, Perkins and Nieto observed concentration-dependent aggregation of vancomycin and attributed this to the formation of dimers.11 With the advent of more advanced NMR techniques, Williams and co-workers were able to characterise the molecular processes underpinning GPA dimerization and its role in the GPA mechanism of action.16 In the case of vancomycin, dimerization and ligand binding were shown to be mutually cooperative. Vancomycin's dimer binds to simple cell-wall analogues carrying the D-Ala–D-Ala sequence more strongly than monomeric vancomycin while monomers of the cell-wall analogue-vancomycin complex dimerize more strongly than unbound vancomycin. Vancomycin dimerization is minimal below concentrations of 10−4 M and the binding constants reported here (Kaca. 105 M−1) were recorded at these low concentrations.10 Vancomycin and other GPAs are concentrated at the cell-wall where aggregation is thought to potentiate antibiotic activity.Vancomycin dimerization was independently verified by X-ray crystal structures showing vancomycin-peptide complexes in face-to-face and back-to-back conformations stabilised by two and four H-bonds respectively (Fig. 9).47,48 Dimerization is thought to strengthen intermolecular interactions within and between individual host–guest complexes by polarising amide functionality involved in H-bonding.16 The bound ligand is also thought to stabilise dimer formation by reducing conformational motion in the monomeric complex. In this way, dimerization and ligand binding operate cooperatively. It is also important to recognise that interactions between the vancomycin dimer and peptidoglycan chains are strengthened by the chelate effect as once the dimer is bound, subsequent peptide–dimer interactions are intramolecular (Fig. 7).49 This effect may explain why vancomycin is less biologically active than some GPAs such as eremomycin which dimerize more strongly, despite vancomycin having higher affinities for cell-wall analogues in solution.53 Essentially, the chelate effect is not observed unless the cell-wall analogue carries multiple peptide ligands.10 Covalently-linked GPA dimers have been reported which display improved antibiotic activity.50
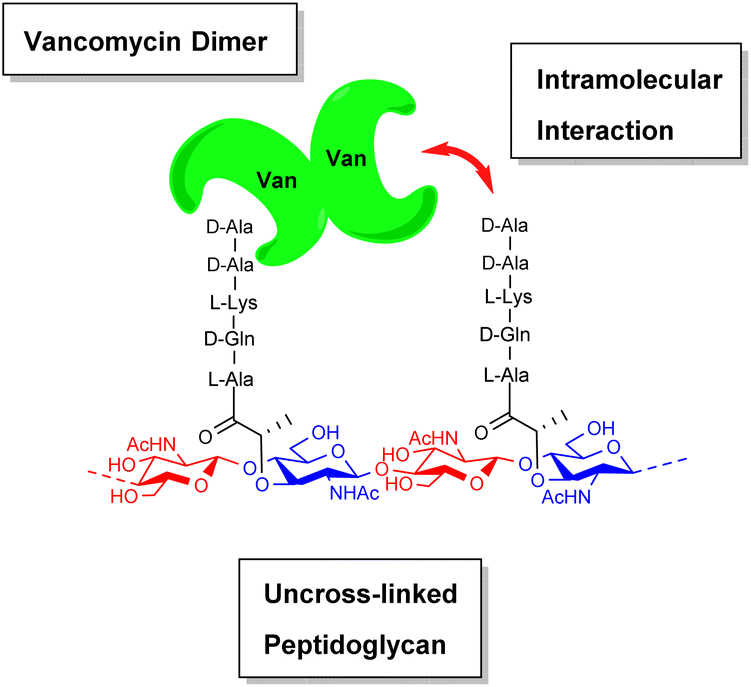 | ||
| Fig. 7 Schematic highlighting intramolecular interactions between vancomycin dimer and peptidoglycan. | ||
Interestingly, the sugar moieties present on amino residues four and six of various GPA scaffolds play important roles in dimerization.51 For example, vancomycin's disaccharide moiety present on amino residue four provides a steric constraint which enforces conformational homogeneity within larger supramolecular assemblies containing multiple interacting dimeric subunits, and removal of the disaccharide leads to a significant decrease in antibiotic activity.52,53
Significantly, dimerization is not essential for the antibiotic activity of all GPAs. Teicoplanin 3 does not dimerize and instead carries a lipophilic moiety attached to the glucosamine residue which is thought to embed in the cell membrane (Fig. 8).54 The enhanced activity here may be due to the fact that interactions between membrane bound Lip II and teicoplanin are effectively intramolecular and benefit from the chelate effect.49
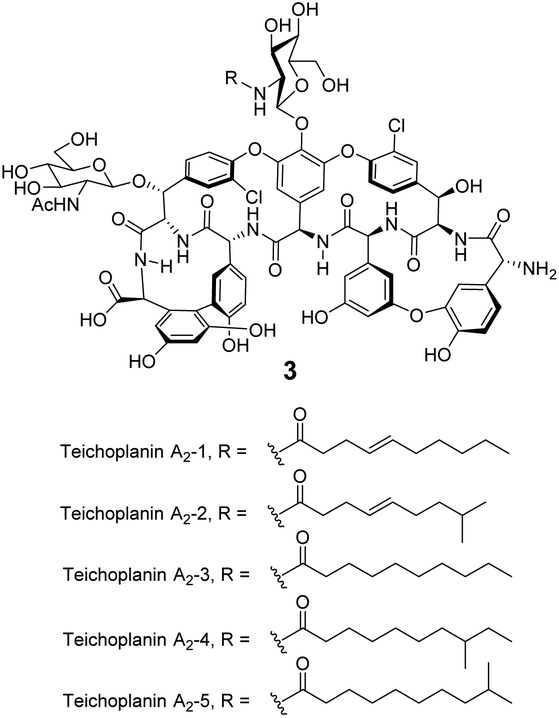 | ||
| Fig. 8 Formula of teicoplanin 3 which is composed of a mixture of structural analogues. Each carries a different lipophilic moiety attached to the glucosamine residue. | ||
2.5 Substrate selectivity
The substrate selectivity of vancomycin is unrivalled by current state-of-the-art synthetic peptide receptors which typically target peptide substrates carrying large hydrophobic (e.g. Leu, Val, Phe) or ionisable (e.g. Lys, Glu) side-groups.4,55 Vancomycin displays exceptional selectivity towards D-Ala–D-Ala over other amino residue sequences (sequence-selectivity) and stereoisomers of the same sequence (stereoselectivity).56 An important implication of this selectivity is that vancomycin binds to its target peptide without severely disrupting the host eukaryote's metabolism at therapeutic doses, as eukaryotic peptides are composed predominantly of L-amino acids which do not interact strongly with vancomycin.Substrate selectivity during molecular encapsulation processes is largely governed by the relative volumes and geometries of the host cavity and substrate. The occupied cavity volume packing coefficient describes the proportion of a cavity's volume occupied by a substrate. Rebek and co-workers defined the optimal packing coefficient for hydrophobic cavities as 0.55, rising to 0.70 where complexation is stabilised by strong intermolecular forces.57 Cavity volume is an important consideration in molecular design that can be used to promote substrate selectivity. Vancomycin's cavity is optimised for D-Ala–D-Ala and larger guests clash with the binding site while smaller guests may lack the necessary structural features to form strong interactions. Computational modelling can help to estimate cavity volume and packing co-efficiencies. Jelfs and co-workers have developed open-source python codes to calculate structural properties of molecular pores including cavity volume which may be applicable in computer aided design.43
Chirality adds an extra level of complexity to the recognition process. Stereoselectivity requires a receptor to implement an asymmetric three-dimensional arrangement of functionality complementary to the desired substrate stereoisomer by induction or preorganisation of the binding site.58 Jurczak and co-workers recently demonstrated that stereoselectivity in chiral carboxylate recognition is also controlled by the solvent composition with increasing water content reducing stereoselectivity.60 In vancomycin's case, the inherent chirality of the binding site and its overall preorganisation produce excellent stereoselectivity for D-Ala–D-Ala over L-Ala containing stereoisomers in water.43,56
3. Supramolecular vancomycin mimicry
This section describes the design of synthetic molecules which attempt to mimic the structure and/or function of glycopeptide-group antibiotics (GPAs). Three general approaches to design (biomimicry, combinatorial and structure-based design) are discussed with each sub-section further divided into receptors designed to function in organic solvents or water.Water presents an especially challenging environment for molecular recognition while also being the most important for biological applications.59 The challenge stems mainly from the polarity and H-bonding potential of water molecules, which interact strongly with each other and also with polar functional groups on substrates and receptors. The interactions with the binding partners inhibit complex formation through competition, so that polar interactions such as hydrogen bonding are generally weak in water. On the other hand, the strong water–water interaction provides a driving force for binding in that weaker interactions, e.g. with hydrocarbons, are avoided – the basis of the hydrophobic effect. The effect of water is thus a complex factor in receptor design which can be difficult to manage, especially for polar substrates.
The choice of solvent can affect both binding strength and selectivity. Jurczak and co-workers investigated solvent-dependence on recognition of chiral carboxylates using acyclic charge-neutral receptors.60 Increasing water content from 0.5 to 5% in acetonitrile dramatically reduced both binding affinity and stereoselectivity. Interestingly, the dielectric constant of the solvent was not considered the predominant factor and Gutmann's donor number which describes the Lewis basicity of the solvent appeared to correlate more closely with observed trends in stereoselectivity.
Based on this understanding, solvent is clearly an important consideration in receptor design. For example, charged binding sites may perform better in less competitive organic solvents while charge-neutral binding sites rich in hydrophobic residues may be better suited to more polar solvents, particularly water. Vancomycin has evolved to function in the presence of water, and vancomycin mimics should ultimately aim to do the same if they are to function as antibiotics. Therefore, examples discussed here which were designed to function only in organic solvents are included for perspective and should be considered stepping-stones towards more evolved, water compatible systems.
3.1 Biomimetic design
The earliest examples of vancomycin mimicry were structural mimics of the GPA-class.61 The term biomimetic was later coined to describe their close structural resemblance to existing GPAs.62 The majority of early biomimetic structures were developed as simplified model systems to demonstrate improved synthetic methodologies for the construction of biaryl and biaryl ether linkages present in GPAs and their binding properties were overlooked. For example, Boger and co-workers developed conditions to control atropisomerism and epimerization of vancomycin's three macrocyclic ring systems and heptapeptide backbone producing biomimetic CD-ring 4, DE-ring 5 and ABCD-rings 6 (Fig. 10).22,63 These methodologies were later applied in the total synthesis of vancomycin's aglycon 7.64 Although binding studies were not carried out, biological activities were determined against selected strains of S. aureus, and E. faecium. The biomimetic substructures 4–6 were inactive while vancomycin's aglycon 7 displayed >10-fold reduction in antibiotic activity compared to the complete glycopeptide, revealing the disaccharide's role in biological activity.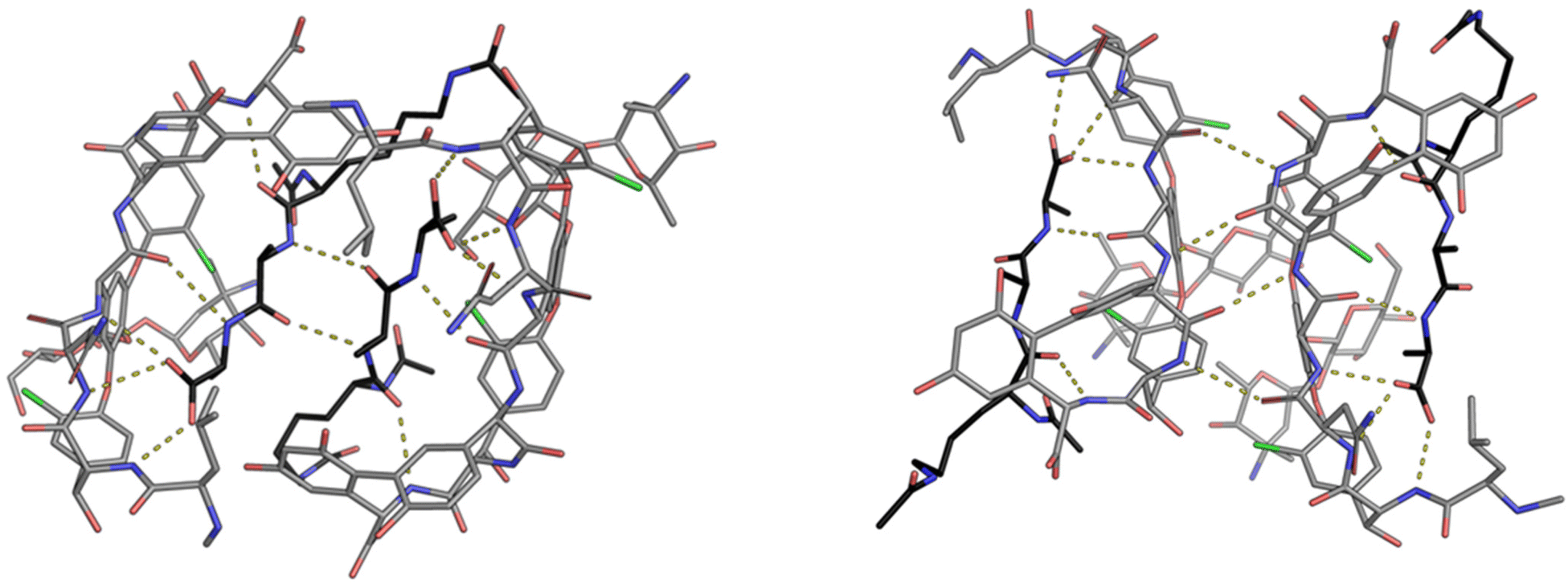 | ||
| Fig. 9 Crystal structures of vancomycin in complex with Ac–Lys(Ac)–D-Ala–D-Ala–OH highlighting the face-to-face dimer (left) stabilised by two H-bonds and back-to-back dimer (right) stabilised by four H-bonds. Vancomycin is highlighted with grey backbone and Ac–Lys(Ac)–D-Ala–D-Ala–OH with black.25 | ||
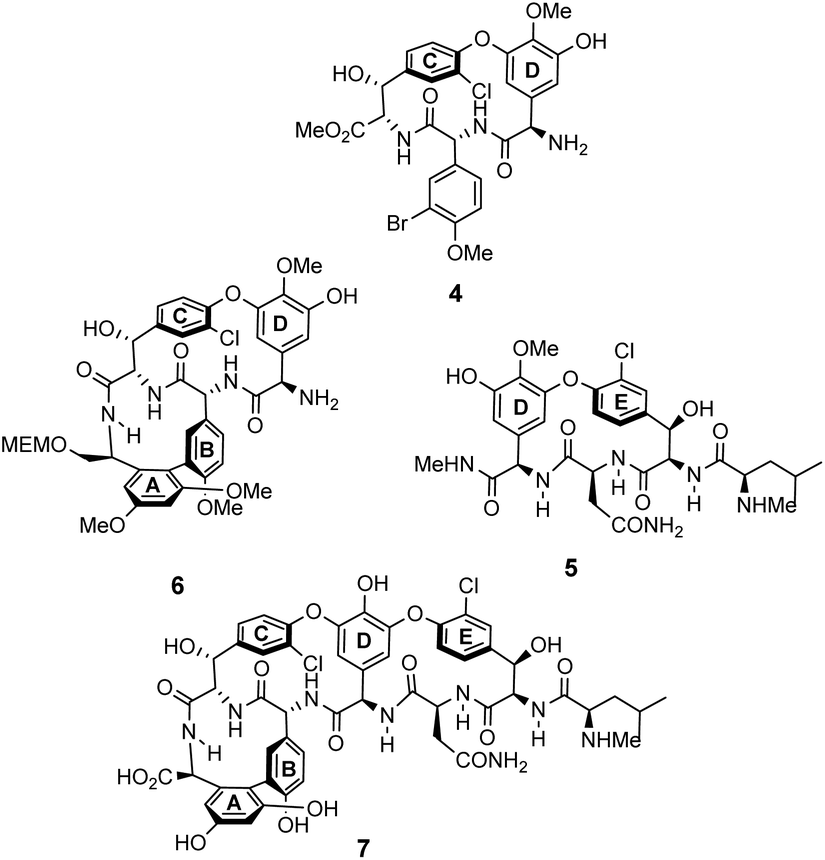 | ||
| Fig. 10 Examples of biomimetic CD-ring 4, DE-ring 5 and ABCD-ring systems 6 alongside vancomycin's aglycon 7. | ||
 | ||
| Scheme 1 Synthesis of biomimetic carboxylate binding site 14 based on vancomycin's DE-ring system. | ||
NMR studies monitoring changes in key proton resonances indicated that the macrocyclic sub-unit of 14 is restricted from rotation about the aryl ether linkage which may preorganise the binding site. Binding properties were tested towards various achiral carboxylic acid substrates in deuterated chloroform and an approximate binding constant was reported for cyanoacetic acid (Kaca. 580 M−1). Proton resonance shifts indicated the substrate interacts with the receptor's amide residues and aromatic ring system, with carboxylate–ammonium ion pairing interactions appearing the predominant factor in the strength of complexation. Substrate recognition therefore requires efficient proton exchange which may explain why no binding activity was detected for substrates carrying carboxylic acids with a pKa value exceeding four.
Pieters also developed receptors mimicking vancomycin's DE-ring system.66 Controlling atropisomerism during CsF-induced SNAr macrocyclization of linear precursor 15 was problematic and receptors 16–19 were isolated and studied as diastereomeric mixtures differing in orientation of the nitro-substituent (Scheme 2). Receptor 16 was tested as a mixture of stereoisomers which did not display stereoselectivity towards enantiomers of Ac–Ala. Interestingly, 16 displayed similar affinity for Ac–D-Ala and Ac–D-Lac in acetonitrile (Kaca. 103) which may have resulted from free rotation about the C-terminus methyl amide permitting dual donor–acceptor H-bonding with the variable NH/O position.
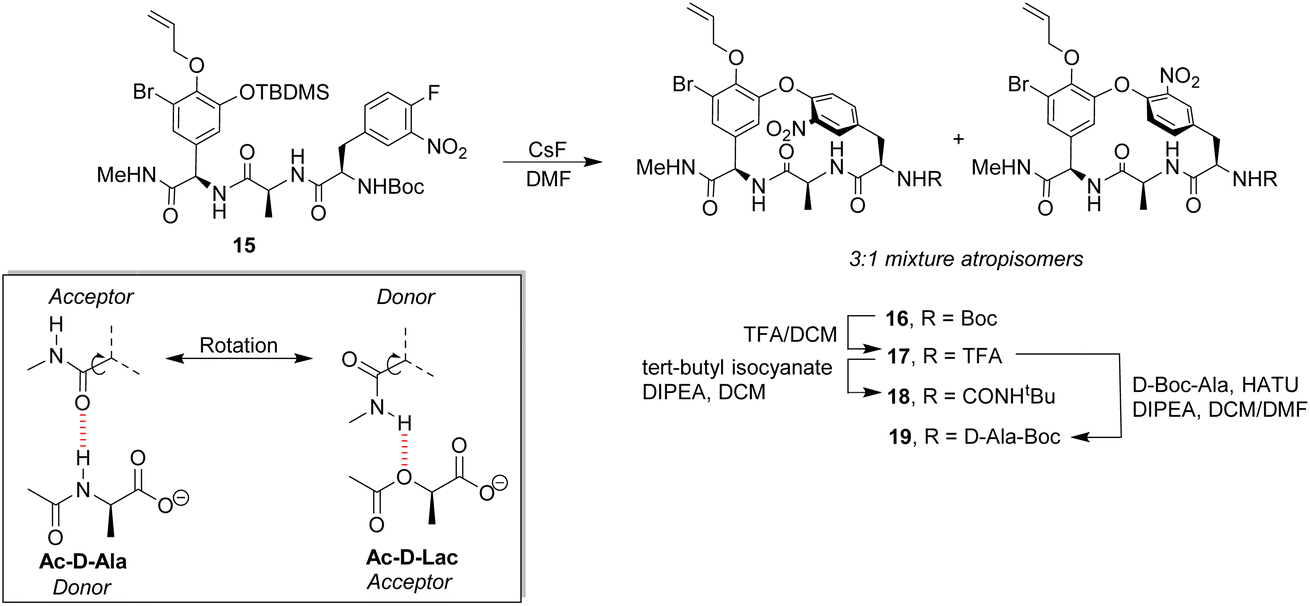 | ||
| Scheme 2 CsF-induced SNAr macrocyclization of linear precursor 15 to biomimetic receptors 16–19 and schematic of methyl amide donor–acceptor H-bonding to Ac–D-Ala/Lac. | ||
Pieters and co-workers later designed a small library of DE-ring system mimics 20–22 to investigate how branched hydrophobic, aromatic and basic residues affect recognition within the carboxylate nest sub-unit (Fig. 11).67 However, racemization during solid-phase synthesis was problematic and only receptor 20 was separated into the four component diastereomers. One of these displayed promising binding activity towards Ac–D-Ala in acetonitrile (Ka 7.3 × 103 M−1), though the absolute stereochemistry of this compound was not determined.
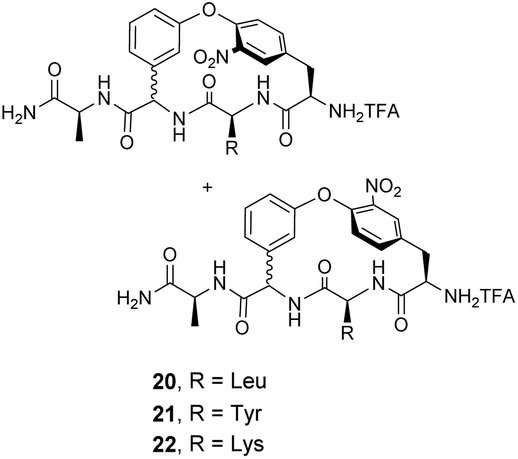 | ||
| Fig. 11 Biomimetic receptors 20–22. | ||
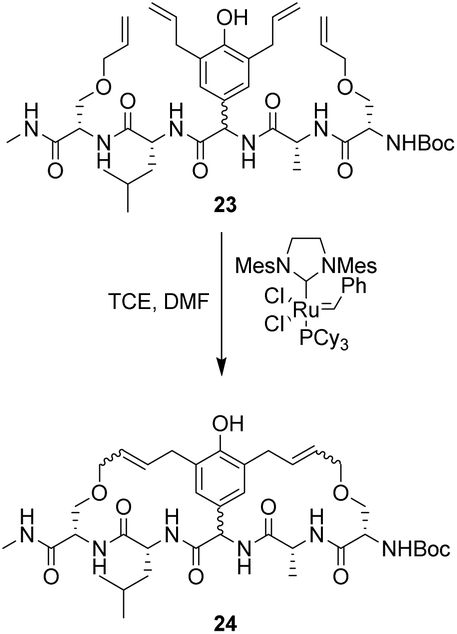 | ||
| Scheme 3 Synthesis of 24 using tandem ring-closing metathesis produced a mixture of stereoisomers. | ||
However, this approach produced an inseparable mixture of diastereomers with different E/Z and R/S stereochemistry. Attempts to access optically pure linear precursor 23 were hampered by uncontrolled racemization at the central hydroxyphenyl glycine residue during the Stille-coupling of precursor 25 which gave a racemic mixture of 26 (Scheme 4). In later work an amide was installed at the C-terminus of a related substrate, which supressed racemization.70
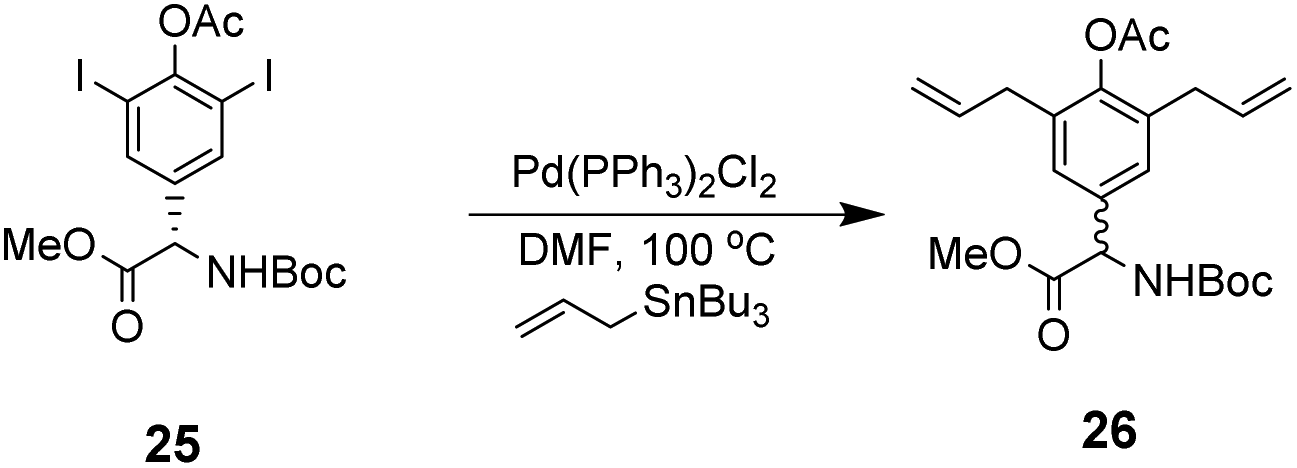 | ||
| Scheme 4 Racemization of 25 during Stille coupling. | ||
To control E/Z-isomerism, alkynes were chosen as alternative linkers to assemble biomimetic DE-ring systems 28 and 29 (Scheme 5).69 The intention was to stereoselectively reduce the alkynes in subsequent steps. Unfortunately, macrocyclization of linear precursors 27 and 30 using Sonogashira and amide coupling reactions respectively gave low yields and the products displayed limited solubility in most organic solvents leading to poor recovery. Low reaction productivity was likely due to the strained nature of the alkyne-linked ring system which was supported by formation of less-strained products such as cyclic dimer 31 (Fig. 12). Experience with alkyne functionality led Liskamp and co-workers to explore other alkyne-derived linkers such as 1,4- and 1,5-triazoles.70–72 Two series of DE-ring mimics of varying ring size were constructed from linear precursor 32 using azide–alkyne cycloadditions (AAC) to access 1,4-triazoles 33 using a Cu(I)-catalyst and 1,5-triazoles 34 using a Ru(II)-catalyst (Scheme 6).70 For the 1,4-triazole series, only n = 4 could be constructed using Cu-catalysed AAC macrocyclization. Ring strain was thought to be the limiting factor as dimers were again observed.
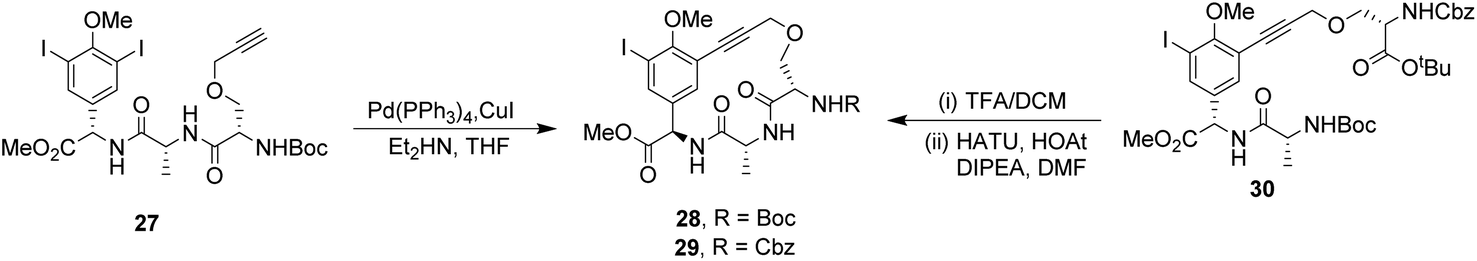 | ||
| Scheme 5 Synthesis of alkyne-linked vancomycin DE-ring mimics 28 and 29 and by-product cyclic dimer 30. | ||
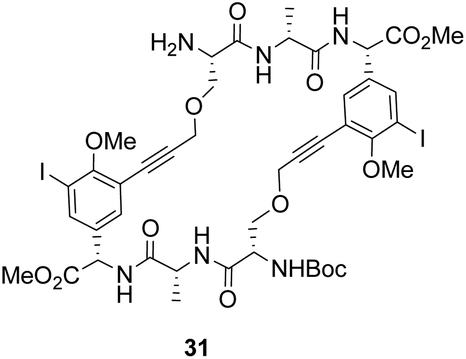 | ||
| Fig. 12 By-product cyclic dimer 31. | ||
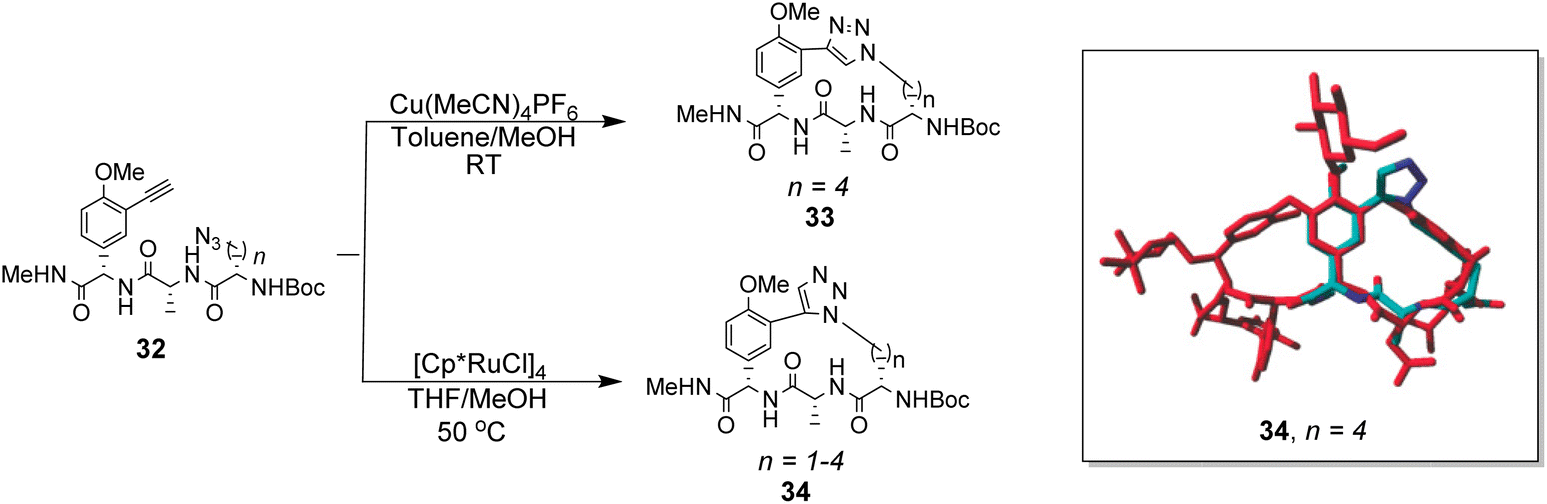 | ||
| Scheme 6 Synthesis of 1,4- and 1,5-triazole linked DE-ring mimics 33 (n = 4) and 34 (n = 1–4) from linear precursor 32 using Cu-catalysed and Ru-catalysed azide–alkyne cycloadditions. Energy minimisation acquired with AMBER99 forcefield shows 34 (n = 4) in blue superimposed onto balhimycin shown in red. | ||
Computational modelling suggested that 34 (n = 4) was the variant which most closely resembled the DE-ring of vancomycin-related GPA balhimycin. This insight led to the design of 35, employing the 1,5-triazole linker in a bicyclic mimics of vancomycin's ABC-ring system (Fig. 13).72 An alkene was used to link the AB-ring section which produced E/Z isomers which were separated using HPLC. Interestingly, 35 was isolated in 33% diastereomeric excess favouring the Z-alkene geometry, perhaps due to reduced ring strain. Both isomers were tested for binding activity towards Ac–Lys(Ac)–D-Ala–D-Ala and Ac–Lys(Ac)–D-Ala–D-Lac in aqueous citrate buffer (pH 5.1) where they performed similarly (Table 1). Both receptors displayed 100-fold lower binding activity towards Ac–Lys(Ac)–D-Ala–D-Ala compared to vancomycin (Kaca. 103). Similar binding activities were observed towards Ac–Lys(Ac)–D-Ala–D-Lac which could indicate little or no interaction with the variable NH/O position. Binding constants to stereoisomeric substrates with L-components were not reported meaning stereoselectivities could not be determined.
 | ||
| Fig. 13 Biomimetic receptors 35–37. | ||
| Binding constants (M−1) | |||||
|---|---|---|---|---|---|
| Guest | Vancomycin | 35 (Z) | 35 (E) | 36 | 37 |
| Binding constants determined by isothermal titration calorimetry in aqueous citrate buffer (pH = 5.1).71,72 | |||||
| Ac–Lys(Ac)–D-Ala–D-Ala | 3.47 ± 0.12 × 105 | 2.35 ± 0.36 × 103 | 4.06 ± 0.84 × 103 | 2.46 ± 0.29 × 103 | 1.26 ± 0.24 × 104 |
| Ac–Lys(Ac)–D-Ala–D-Lac | 2.19 ± 0.12 × 103 | 2.17 ± 0.30 × 103 | 1.21 ± 0.86 × 103 | 3.14 ± 0.38 × 103 | 3.28 ± 0.31 × 103 |
| Ac–D-Ala–D-Ala | — | — | — | 2.15 ± 0.24 × 103 | — |
| Ac–D-Ala | — | — | — | 3.33 ± 0.12 × 103 | — |
Another bicyclic system resembling vancomycin's CDE-ring system, 36 (Fig. 13) was assembled using 1,5-triazoles to link the two macrocycles.71 Receptor 36 was also tested in aqueous citrate buffer (pH = 5.1) and displayed similar affinities for mono- and dipeptide substrates (Kaca. 103) suggesting the receptor interacts primarily with the C-terminal residue. Again, minimal differences in binding activity were detected for peptide substrates terminating in D-Ala or D-Lac. Neither bicyclic system 35 nor 36 displayed substantial in vitro antibiotic activity against a strain of vancomycin-sensitive S. aureus. It was envisaged that components of 35 and 36 could be combined to form a complete tricyclic mimic of vancomycin.
Combining the Ru-catalyst metathesis and azide–alkyne cycloaddition ligation strategies developed to prepared 35 and 36, Liskamp and co-workers successfully synthesised a tricyclic mimic of vancomycin's complete tricyclic aglycon 37 which was isolated as a single stereoisomer (Fig. 13).73 In solution binding studies, 37 displayed superior binding activity towards two cell-wall analogues compared to 35 and 36 (Table 1). However, 37 still displayed ca. 30-fold lower binding activity compared to vancomycin. Tricyclic mimic 37 did show activity in vitro against a strain of vancomycin-susceptible S. aureus, but still fell some way short of vancomycin itself (minimum inhibitory concentration 37 = 37.5 μg mL−1, vancomycin = 2 μg mL−1).
Boger and co-workers developed a series of structures closely related to vancomycin which are collectively called ‘maxamycins’.20 Although these may be considered derivatives of vancomycin they are also, in a sense, new biomimetic designs. The major objective was to bypass VanA-type resistance by modifying the core to bind D-Ala–D-Lac. This was achieved in prototype aglycon 38, where an amidine replaces an amide (Fig. 14). The amidine residue exists in equilibrium between protonated and deprotonated states under physiological pH. In the protonated state, the amidinium can donate positive charge to the oxygen lone pair of the terminal D-Lac residue present in the D-Ala–D-Lac depsipeptide of VanA bacteria. Additionally, when deprotonated, the amidine behaves as a H-bond acceptor, donating a lone pair to the terminal D-Ala N–H moiety present in most bacteria.74
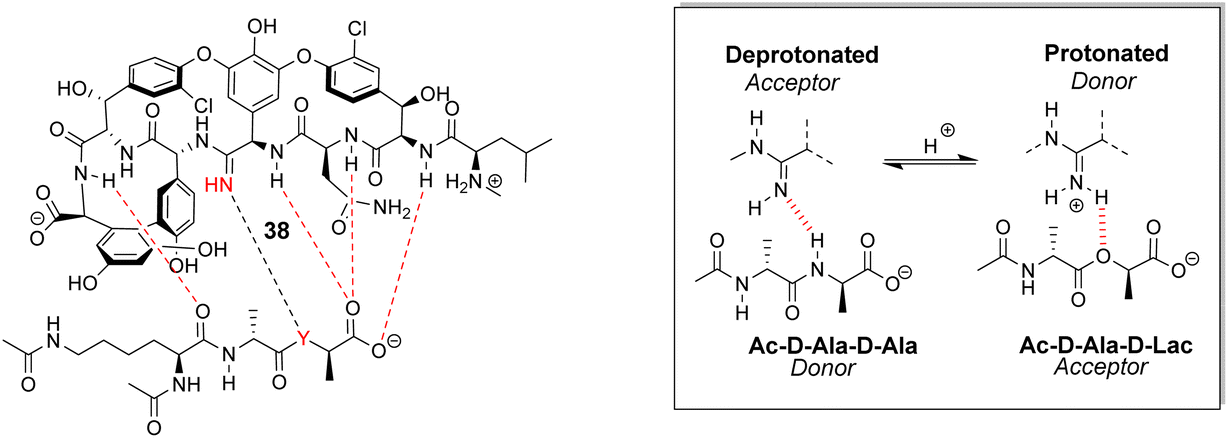 | ||
| Fig. 14 Dual acceptor-donor H-bonding activity of receptor 38 to cell-wall analogues carrying D-Ala–D-Ala (Y = NH) or D-Ala–D-Lac (Y = O). | ||
This dual propensity allows members of the maxamycin group to form strong complexes with either the D-Ala–D-Ala or D-Ala–D-Lac sequence. Aglycon 38 displays binding activity towards both D-Ala–D-Ala and D-Ala–D-Lac in aqueous citrate buffer (Table 2), and promising antibiotic activity against VanA-type vancomycin resistant bacteria while retaining effectiveness against vancomycin-sensitive bacteria (Table 3).20 Work is currently focused on streamlining a scalable synthesis to produce quantities necessary for preclinical development.75 This work represents a breakthrough towards next-generation biomimetic GPAs with improved activity against vancomycin-resistant bacteria.
| Binding constants (M−1) | ||
|---|---|---|
| Substrate | Vancomycin | 38 |
| Ac–Lys(Ac)–D-Ala–D-Ala | 4.4 × 105 | 7.3 × 104 |
| Ac–Lys(Ac)–D-Ala–D-Lac | 4.3 × 102 | 6.9 × 104 |
| MIC (μg mL−1) | ||
|---|---|---|
| Strain | Vancomycin | 38 |
| E. faecalis (VanA) | 250 | 0.31 |
| E. faecium (VanA) | 250 | 0.31 |
3.2 Combinatorial design
Combinatorial design is radically different from the biomimetic approach to identifying vancomycin mimics. Instead of targeting very specific structures which may be difficult to access, libraries of potential receptors are generated through straightforward procedures and screened for binding properties. The libraries are generally based on scaffolds which confer “receptor-like” architectures, but success is based more on chance than on rational design. The prospects improve as libraries become larger, and a key development was the one-bead one-compound (OBOC) approach which allows the synthesis and screening of very large libraries indeed.77,78The OBOC method uses split-mix solid phase synthesis to rapidly generate many chemically diverse structures in parallel from a relatively small pool of precursors. Each unique structure is attached to a separate resin bead which minimises interference between different library members during screening. Binding studies are carried out by screening the entire compound library with a chemical probe containing a ligand of interest attached to a dye-label and residual bead staining after washing taken to indicate binding activity. Stained beads carrying structures of interest, referred to as hit compounds, are decoded to determine the structure attached. Hit compounds are then re-synthesised and validated in quantitative binding studies. In principle, the solid phase synthesis used to generate the libraries can be adapted to give a variety of structural types, but peptides dominate in practice due to the availability of amino acid precursors and the reliability of amide coupling chemistry on resin beads.79
The application of OBOC to receptor design was pioneered by W. C. Still and co-workers.78 Their methodology included an ingenious and powerful tagging scheme for identifying beads; however, this was not widely adopted and most work in the area has relied on more convenient methods such as Edman degradation or mass spectrometry. Still's group showed that OBOC methods could lead to sequence-selective receptors for peptides, at least in organic solvents, paving the way for applications in vancomycin mimicry.80,81
600 M−1) and Ds–D-Ala–D-Lac (Ka 10100 M−1) in chloroform.
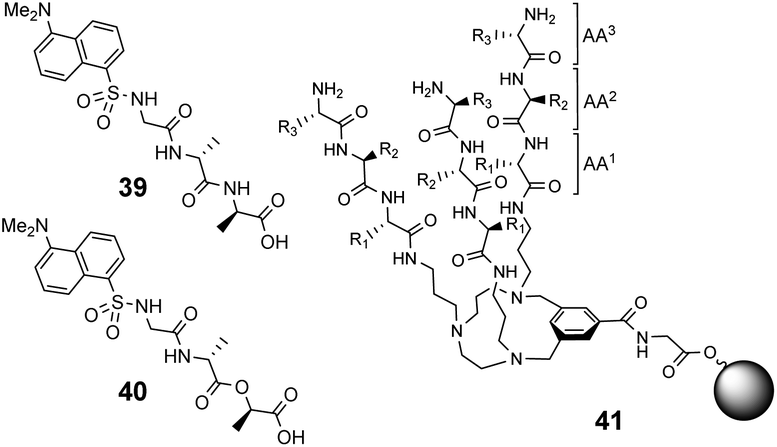 | ||
| Fig. 15 Dansyl-labelled probes 39 and 40 and vancomycin mimetic combinatorial library 41 containing triazacyclophane (TAC)-based tripeptides attached to O-glycine functionalised ArgoGel™ resin. | ||
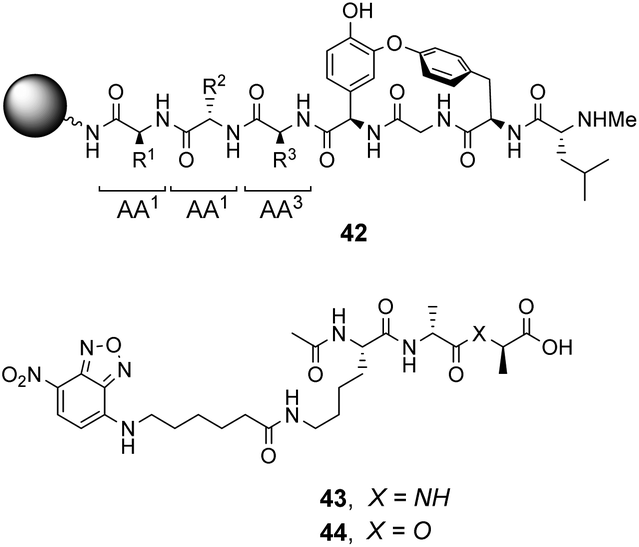 | ||
| Fig. 16 Schematic of biomimetic combinatorial library 42 generated on ArgoGel™ resin beads and fluorescent NBD-labelled substrates 43 and 44. | ||
Analysis of structure-binding relationships showed a high-frequency of aromatic and cyclic amino residues in the most fluorescent library beads, which is understandable given that hydrophobic interactions dominate molecular recognition in water. Receptors 45 and 46 are based on the most frequently identified amino residues and displayed improved affinity towards the D-Ala–D-Lac sequence compared to vancomycin in aqueous binding studies (Fig. 17).
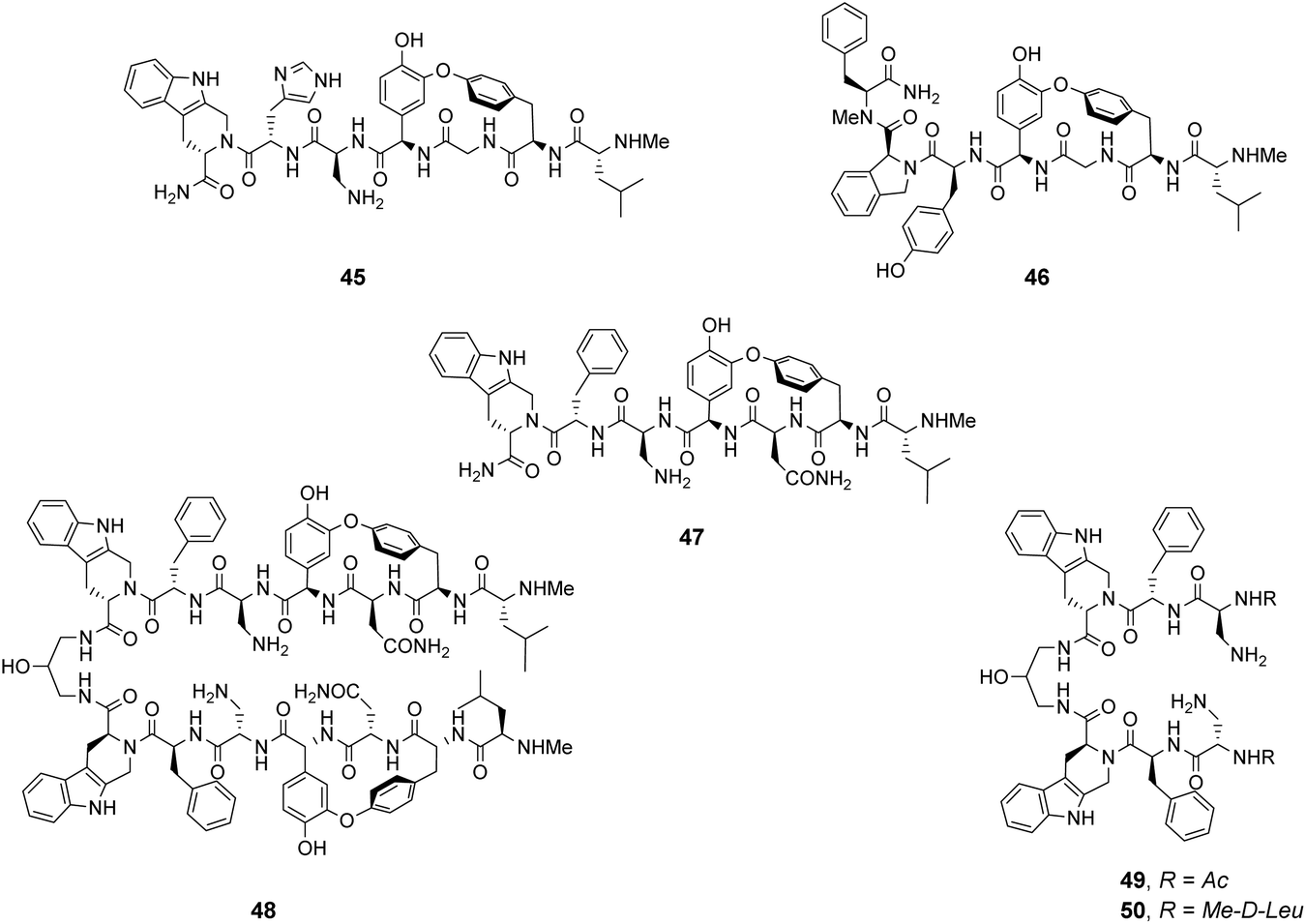 | ||
| Fig. 17 Combinatorial receptors 45–47 mimicking vancomycin's DE-ring. Dimer 48 was based on the most biologically active library member. Two control compounds 49 and 50 were used to interrogate the mechanism of action. | ||
In search of biologically active structures, Ellman and co-workers synthesised another large compound library directly screening for biological activity against strains of methicillin-resistant S. aureus (MRSA) and VanA-type vancomycin-resistant E. faecium (VRE).84 Compound 47 was the most potent, displaying 4-fold higher activity against VRE compared to vancomycin. Covalently linked dimers were prepared based on 47 as dimerization is known to improve the activity of GPAs against VRE attributed to avidity for D-Ala–D-Lac.85 Dimer 48 displayed 60-fold higher activity against VRE compared to vancomycin. To probe the mechanism of action, dimers 49 and 50 lacking the intended carboxylate nest were prepared as controls as these compounds were thought unlikely to bind cell-wall peptides. Both displayed similar antibiotic activity to 47 and it is therefore unclear whether D-Ala–D-Lac recognition is involved in the biological activity of the compounds identified.
Over a period of two decades, Schmuck and co-workers developed the guanidiniocarbonyl-pyrrole (GCP) moiety as a remarkably effective motif for binding carboxylates in water.86 As part of this work they applied combinatorial methodology in a search for vancomycin mimics, investigating a small, focused library of form 51 containing 512 linear GCP-terminated tripeptides (Fig. 18). The GCP moiety provided a carboxylate binding site and promoted anti-parallel β-sheet alignment by orienting the substrate. This was intended to induce side-chain interactions between the receptor and different tetrapeptide substrates amplifying substrate selectivity.87 The library was encoded using the IRORI™-radio frequency tagging technology; this generally employs canisters containing many resin beads allowing appreciable quantities of individual molecules to be generated, tagged with their synthesis history.88
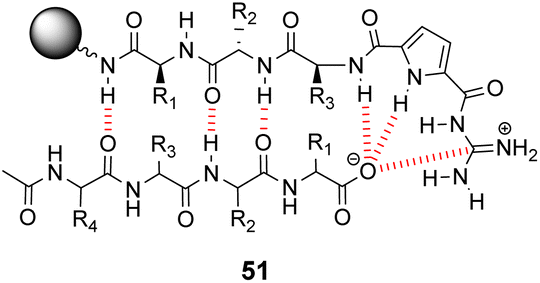 | ||
| Fig. 18 GCP-based receptor library 51 generated on TentaGel®-NH2 resin and anti-parallel β-sheet alignment with tetrapeptide substrates. | ||
The compound library was prepared from eight proteinogenic amino acids which were selected to survey the effects of charge and polarity on substrate recognition in water. Screening was carried out using a quantitative fluorescence assay which provided approximate binding constants with impressive accuracy. The best hit compound 52 was selected from library screening with 53, a fluorescent dansyl-labelled analogue of the cell-wall C-terminal tetrapeptide (Fig. 19). Binding properties were validated in an aqueous solution-based UV-titration assay in tris-buffer (pH 6.15) which showed that complexation of an unlabelled substrate 54 (Ka 15400 M−1) was in good agreement with the on-bead assay (Ka 17100 M−1). A statistical quantitative structure–activity relationship analysis (qSAR) was carried out to rationalise trends in binding properties among library members which demonstrated that H-bonding and electrostatic interactions predominantly controlled binding activity.
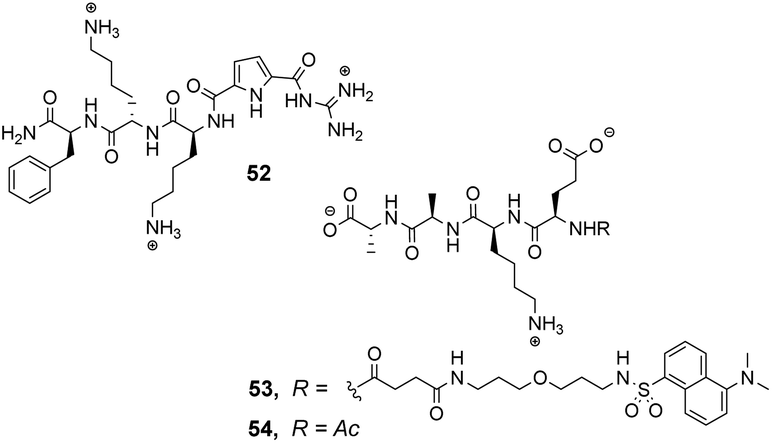 | ||
| Fig. 19 Hit compound 52 and probe molecules 53 and 54. | ||
Later studies explored the substrate selectivity of hit compound 52 and revealed it displays good selectivity over the inverse amino residue sequence (Ac–D-Ala–D-Ala–L-Lys–D-Glu) with an associated binding constant approximately three-fold lower compared to 54 in the same solution-based UV-assay (Ka 4500 M−1).89 However, another study surveying a larger substrate scope showed that receptor 52 displays a preference for tetrapeptides containing a higher frequency of anionic glutamate residues binding Ac–D-Glu–D-Glu–D-Glu–D-Glu most strongly in water (Ka 23700 M−1) with sequence-dependent stereoselectivity also identified.90 These results were consistent with the earlier qSAR analysis where H-bonding and electrostatic interactions were identified as the predominant interactions in substrate recognition.
Kilburn and co-workers prepared compound libraries of form 55 which contained tweezer-like receptors with guanidium-based carboxylate binding sites and two tripeptide side-arms which were envisaged to interact with substrate inducing selectivity (Fig. 20).91 Screening was carried out in aqueous media with four different substrates including D-Ala–D-Ala, each of which was labelled with Disperse Red dye. However, the library showed limited selectivity for the substrates which stained >10% of the beads present.
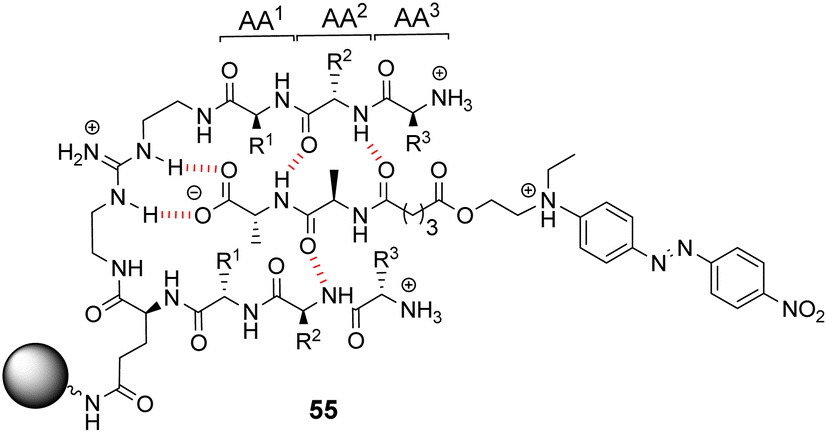 | ||
| Fig. 20 Guanidinium-based tweezer library 55 on TentaGel®-NH2 resin highlighting interactions with Dispersed Red dye-labelled substrate. | ||
Building on earlier work in organic solvents, Liskamp and co-workers investigated diverse compound libraries based on diazacyclophane (DAC) and triazacyclophane (TAC) scaffolds in biologically relevant aqueous phosphate buffer (pH 7).92,93 The DAC-scaffold library 56 was generated by attaching two dipeptides to each library member, varying the dipeptide sequence for each compound in the library (Fig. 21). The TAC-scaffolds took two forms. Firstly, the group investigated library 57 in which each library member was functionalised with three dipeptides, again varying the dipeptide residue sequence for each compound in the library.92 Note also that two libraries of form 57 were generated by varying the TAC-ring size. Binding studies for the homofunctionalized DAC and TAC libraries 56 and 57 were carried out using dansyl-labelled 39 (Ds–D-Ala–D-Ala) and 40 (Ds–D-Ala–D-Lac) and fluorescent FITC-labelled peptidoglycan. In general, hit compounds contained a high frequency of basic and hydrophobic residues. Selected hit compounds from libraries 56 and 57 displayed comparable binding affinity and selectivity for the substrates tested which demonstrated that the additional third arm present in 57 does not contribute substantially to binding activity in water.
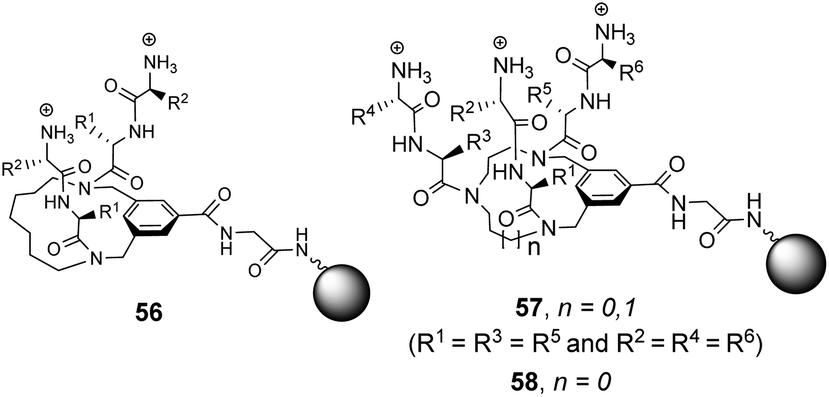 | ||
| Fig. 21 Compound libraries 56–58 carrying two or three dipeptides attached to an O-glycine functionalised ArgoGel™-NH2 resin. | ||
A second three-armed TAC-library 58 was also investigated by generating TAC-scaffolds carrying three different dipeptides. Similar structure-binding activity relationships to those identified for 56 and 57 were noted.93 In both studies, library screenings were carried out using probes carrying a dansyl fluorophore and interactions between the dye-label and the compound library may have biased the screening results.
Liskamp and co-workers did investigate the influence of dye-labelling on library screening in water using a large combinatorial library of cyclotribenzylene (CTB)-based tripodal receptors in aqueous phosphate buffer (pH = 7).94
The CTB moiety was chosen as it can be functionalised easily at three positions and provides a preorganised hydrophobic concavity. Library 61 was constructed by varying two of the tripeptide arms using a split-and-mix procedure while the third tripeptide arm, composed of three L-Ala residues, was kept the same and used to attached library members to the resin (Fig. 22). Three different fluorescent labels were investigated, attached to D-Ala–D-Ala and D-Ala–D-Lac; dansyl in 39 and 40, NBD in 59 and Disperse Red in 60. Also studied was a fluorescein (FITC) labelled peptidoglycan. There were clear differences in binding activity based on the identity of the dipeptide and the fluorescent probe indicating that interactions with the label were significant.
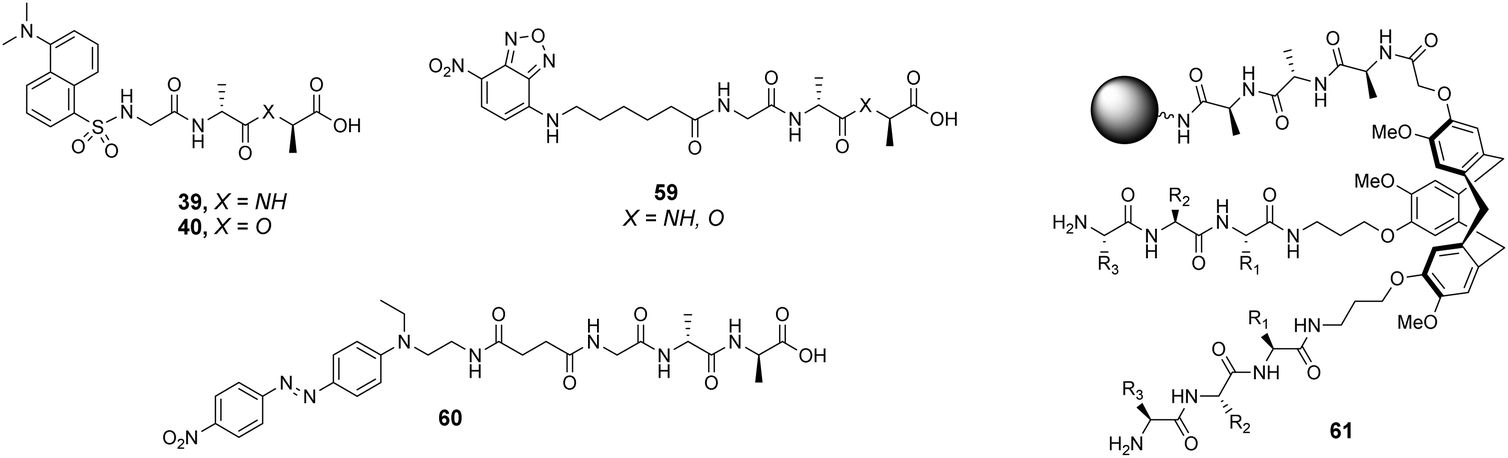 | ||
| Fig. 22 Selected fluorescent labelled probes 39, 40 and 59, 60 and cyclotribenzylene-based scaffold 61 attached to ArgoGel™-NH2 resin. | ||
In general, selectivity was best using FITC-labelled peptidoglycan with 44% of hit compounds selected containing the consensus sequence Lys–AA2–Lys. A receptor based on this sequence (AA1 = Lys, AA2 = Leu, AA3 = Lys) was tested with the four fluorescent labels carrying D-Ala–D-Ala and compared to a control compound (AA1 = Ser, AA2 = Ala, AA3 = Glu) selected from one of the non-fluorescent beads. No fluorescence was detected for the control while the hit compound displayed fluorescent staining with each substrate confirming that binding was due principally to interactions with the tripeptide sequence rather than non-specific interactions with the fluorophore. Unfortunately, the selected hit compound did not inhibit growth of a strain of S. aureus.
3.3 Rational design
In contrast to the empiricism of combinatorial design, rational design aspires to construct receptors individually based on a priori knowledge of chemical structure and function. Molecular fragments or functional groups which are predicted to interact favourably with functionality present in the substrate are chosen and linked together in an arrangement which complements the substrate geometry. Computational molecular modelling is frequently used to assist the process by providing conformational geometries and binding energies derived from ab initio calculations.95 Structure-based designs need not relate to vancomycin structurally, increasing the range of architectures that can be used. This also allows for designs to be biased, for example, towards synthetically accessible or easily derivatisable molecules.:1 enantioselectivity for Ac–L-Ala.99
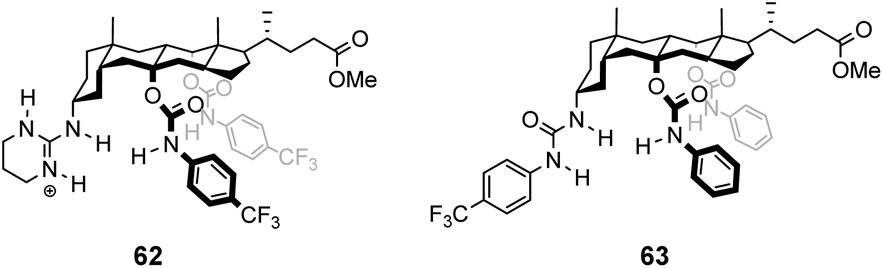 | ||
| Fig. 23 Molecular structures of cholapods 62 and 63. | ||
Kilburn and co-workers prepared chiral receptors targeting stereoselective recognition of cell-wall related peptides (Fig. 24).100 An early prototype 64 was based on a chiral hydrophobic ring system derived from L-phenylalanine and included a diamido-pyridine moiety which provided a carboxylate binding site with two H-bond donors. The receptor displayed moderate stereoselectivity towards Cbz–L-Ala–L-Ala dipeptide in chloroform. However, the monocyclic system was considered too flexible and bicyclic systems such as 65 were developed to increase stereoselectivity by preorganising the binding site (Fig. 24). Receptor 65 also featured a diamido-pyridine moiety and two chiral selectors derived from L-glutamic acid 5-methyl ester close to the carboxylate binding site. Receptor 65 displayed impressive stereoselectivity and affinity (Ka 33000 M−1) towards Cbz–L-Ala–L-Ala dipeptide, albeit in chloroform. Presumably, the enantiomer of 65 derived from D-glutamic acid 5-methyl ester would bind Cbz–D-Ala–D-Ala with equivalent stereoselectivity and affinity.
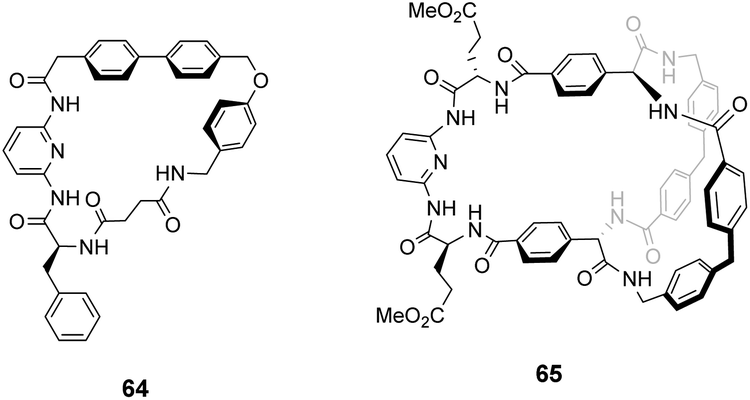 | ||
| Fig. 24 Macrocycles 64 and 65. | ||
Ballester and co-workers designed two macrocyclic receptors 67 and 69 featuring two peptide strands preorganised into parallel or antiparallel alignment using benzophenone sub-units as macrocyclic linkages (Scheme 7).101 It was envisaged that the peptide strands would mimic β-sheet secondary structures found in Nature and that a guest peptide could occupy the inter-strand space, aligning itself with the adjacent β-strands forming a three-strand β-sheet conformation stabilised by multiple H-bonds.
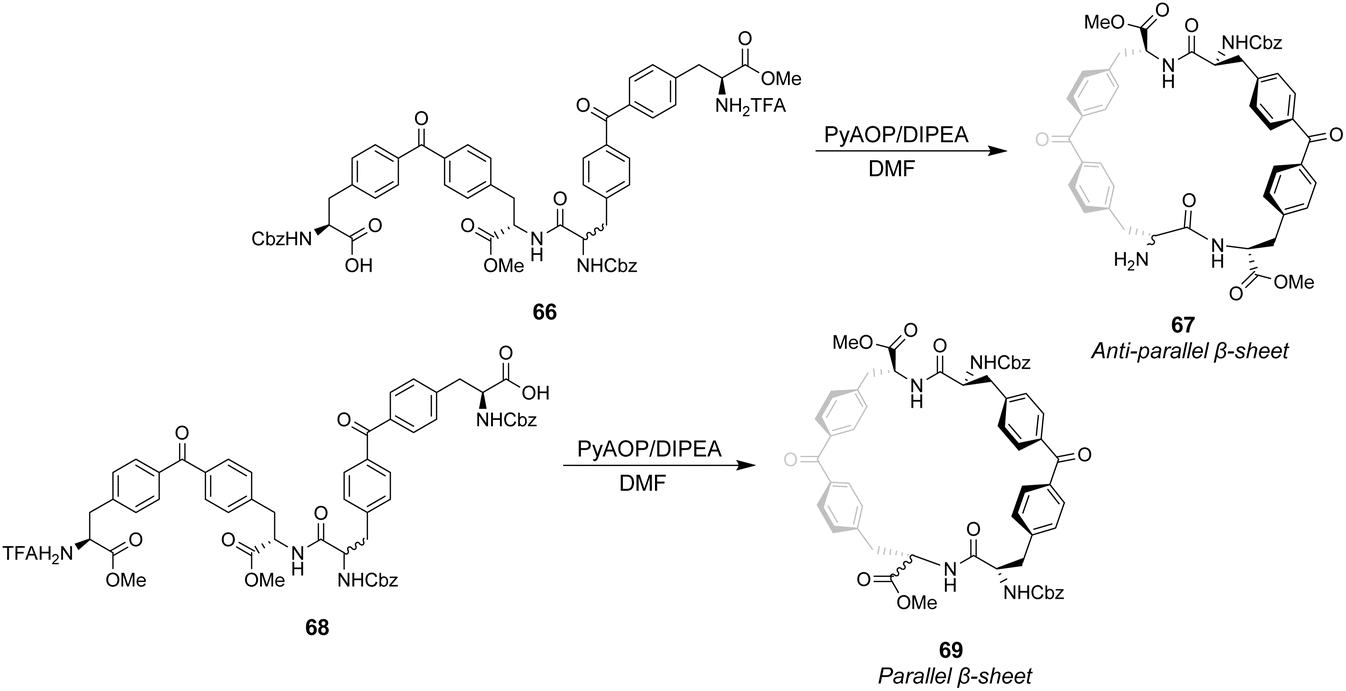 | ||
| Scheme 7 Synthesis of receptors 67 and 69 from linear precursors 66 and 68. | ||
In addition, computational modelling predicted that the benzophenone sub-units which span the inter-strand space would provide suitable hydrophobic cavities for inclusion of methyl side groups carried by the D-Ala–D-Ala sequence.
The desired macrocycles were assembled from linear precursors 66 and 68 which were used as diastereomeric mixtures due to epimerization at an earlier step in their synthesis. This was not problematic as the diastereomeric mixtures of receptors 67 and 69 produced were separable by HPLC. Binding studies were carried out in chloroform on the all-S diastereomers of receptors 67 and 69 and their linear precursors 66 and 68 (Table 4). Receptor 69 performed poorly with low binding constants and minimal stereoselectivity reported while receptor 67 displayed stronger affinity but only modest stereoselectivity for the D-Ala–D-Ala sequence. Surprisingly, linear precursor 68 displayed the highest binding affinity for D-Ala–D-Ala while both 66 and 68 displayed greater stereoselectivity for D-Ala–D-Ala compared to their respective macrocyclic derivatives which was attributed to increased conformational flexibility relative to the macrocyclic structures. Moreover, exo-cyclic conformations was posited for 67 and 69 based on observations from NMR experiments which position the substrate on an external surface.
| Binding constants (M−1) | ||||
|---|---|---|---|---|
| Guest | 66 | 67 | 68 | 69 |
| D-Ala–D-Ala | 6309 | 6606 | 8318 | 1047 |
| L-Ala–L-Ala | 1202 | 3311 | 4365 | 912 |
| D-Ala–L-Ala | 436 | 1047 | 190 | 1148 |
| L-Ala–D-Ala | 794 | 1445 | 436 | 759 |
Ungaro and co-workers developed a bicyclic peptidocalix[4]arene 70 featuring a chiral macrocyclic bridge segment derived from L-alanine which were intended to promote stereoselectivity through steric interactions with chiral substrates (Fig. 25).102,103 The calix[4]arene scaffold was selected as it could be functionalised easily and is known to have good binding properties in water due to its preorganised hydrophobic cavity.
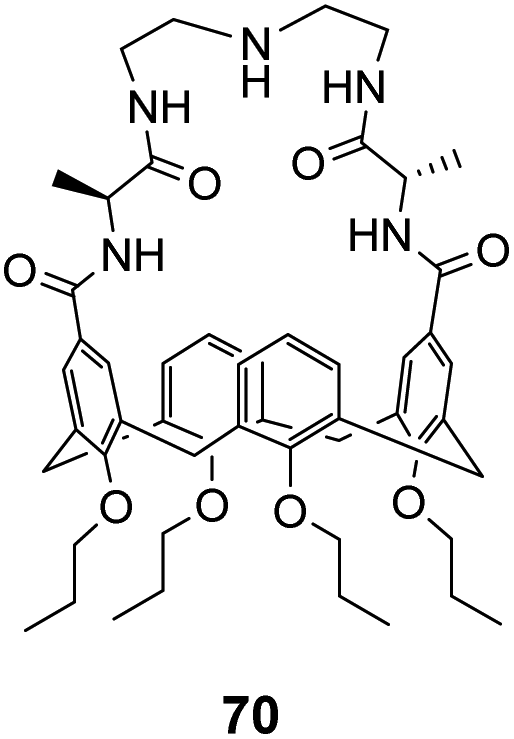 | ||
| Fig. 25 Peptidocalix[4]arene 70. | ||
Receptor 70 was initially tested in antimicrobial assays where it showed promising activity in vitro against Gram positive bacteria including a strain of MRSA.102 Binding activity was later tested towards substrates carrying alanine and alanyl-alanine C-terminal sequences in chloroform, where 70 displayed moderate binding activity (Kaca. 103 M−1) though with minimal stereoselectivity.103 Most likely, the chiral alanine residues were too small and distant from the binding site to induce greater discrimination.
As discussed previously, Hamilton and co-workers designed a biomimetic receptor 14 based on the vancomycin carboxylate nest which operated primarily through ion pairing and displayed moderate binding activity towards achiral carboxylates such as cyanoacetic acid (Kaca. 580 M−1) in chloroform. In later work, the group applied a structure-based design approach to develop charge-neutral receptors inspired by the carboxylate nest of the vancomycin-related GPA ristocetin, which binds carboxylates using primarily H-bonds and hydrophobic interactions (Fig. 26).104,105
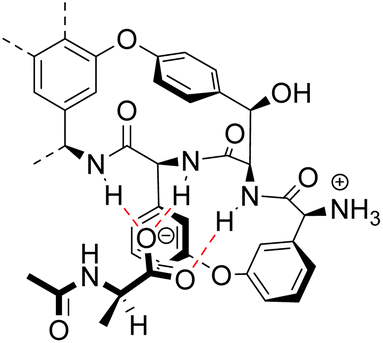 | ||
| Fig. 26 The ristocetin carboxylate nest in complex with Ac–D-Ala. | ||
The multidentate binding mechanism of ristocetin was mimicked in a series of synthetic charge-neutral receptors assembled from (1R,2R)-trans-1,2-diaminocyclohexane 71 and various N-protected amino acids using CDI-mediated amide couplings (Scheme 8).105
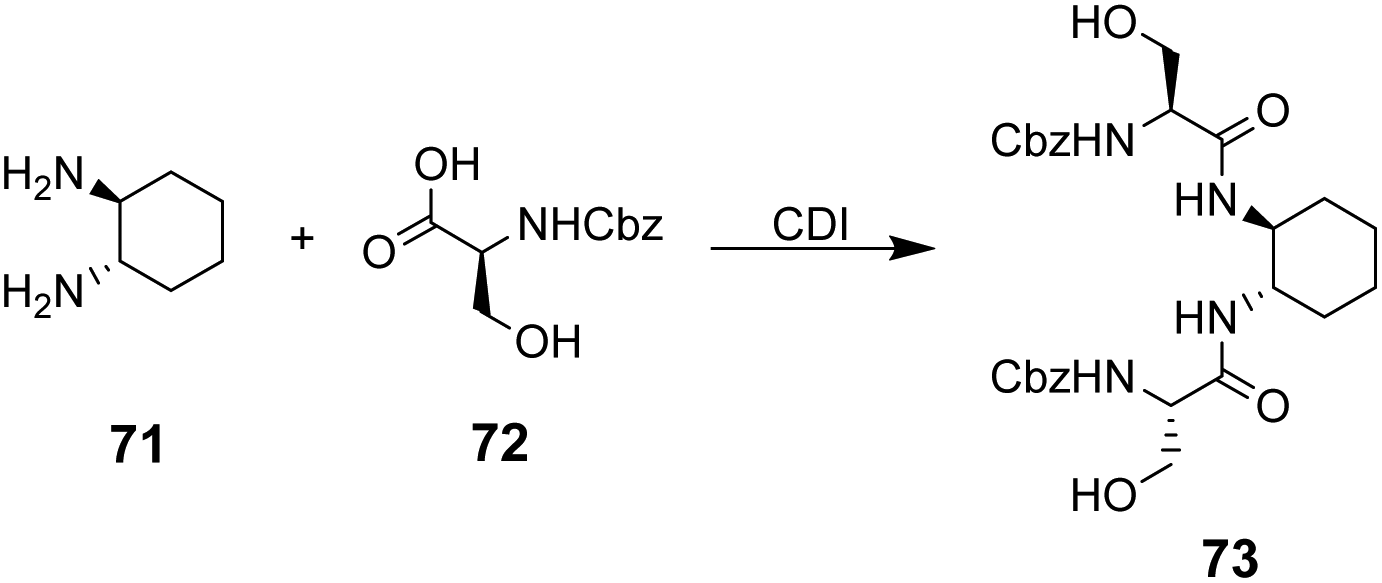 | ||
| Scheme 8 Synthesis of receptor 73 from (1R,2R)-trans-1,2-diaminocyclohexane 71 and N-protected L-serine 72. | ||
Amide N–H bonds were separated from one another by two carbon atoms mimicking the arrangement found in ristocetin, as this was considered an important structural characteristic for binding activity. The synthetic systems were straightforward to assemble and several analogues were made to probe structure–activity relationships within the synthetic carboxylate binding site. The best performing receptor 73 was assembled using N-protected L-serine residues 72 and displayed promising binding activity (Ka 2.7 × 105 M−1) towards achiral tetrabutylammonium acetate in acetonitrile (Scheme 8). Tetrabutylammonium acetate was shown to interact most strongly with the urethane N–H and serine side chain O–H and removal of these residues independently led to substantial losses in binding activity which was taken as evidence of a cooperative multidentate H-bonding recognition mechanism.
100 M−1) and Ac–D-Ala–D-Lac (Ka 18600 M−1) in 10% DMSO/aqueous solution.106 Substitution of alanine or lactate residues for valine or glycine in the substrate produced dipeptides with significantly lower binding affinities. In the case of valine the bulky isopropyl side group was presumed to destabilise the complex, while glycine possesses greater conformational flexibility and was thought to form minimal hydrophobic interactions.
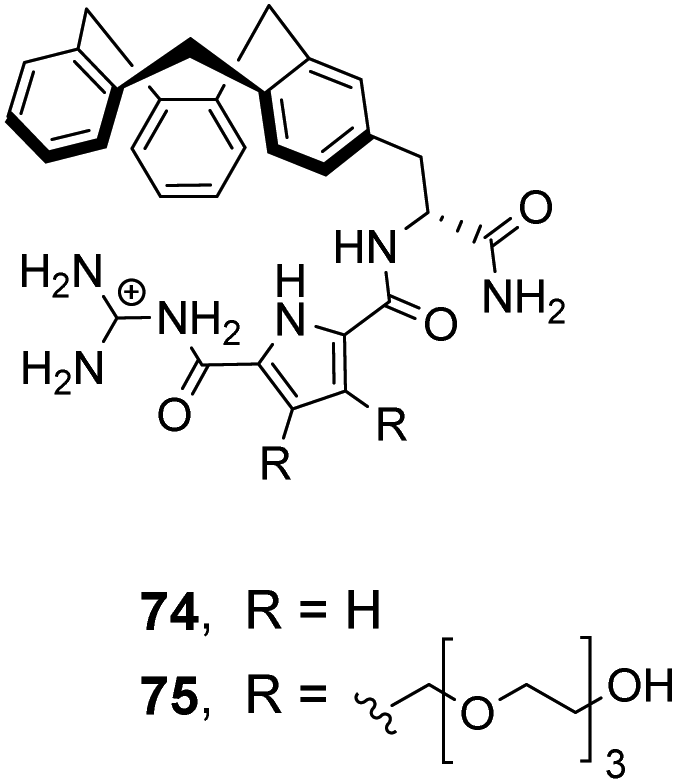 | ||
| Fig. 27 GCP-based receptors 74 and 75. | ||
In order to study binding in pure water, without organic co-solvent, a second receptor 75 was developed with water-solubilising PEG chains. Receptor 75 displayed similar binding properties to 74 in water at sub-millimolar concentrations.106 However, at higher receptor concentrations gelation was problematic prohibiting structural studies with NMR and only UV and fluorescence could be used to monitor binding as these methods are more sensitive. Disappointingly, neither 74 nor 75 displayed stereoselectivity towards the substrates tested.
Finally, an early attempt by Diederich and co-workers employed cyclic receptor 76, with a cyclophane ring system to preorganise the binding site (Fig. 28).25 Quaternary ammonium and aromatic moieties were positioned as handles for electrostatic and hydrophobic interactions respectively. X-ray crystal structures confirmed an open cavity conformation in the solid phase and Monte Carlo simulations predicted inclusion of aliphatic carboxylates. However, when tested in aqueous media the macrocycle did not appear include Ac–D-Ala–D-Ala or other substrates. Instead, binding was detected outside the cavity with affinity governed principally by ion-pairing.
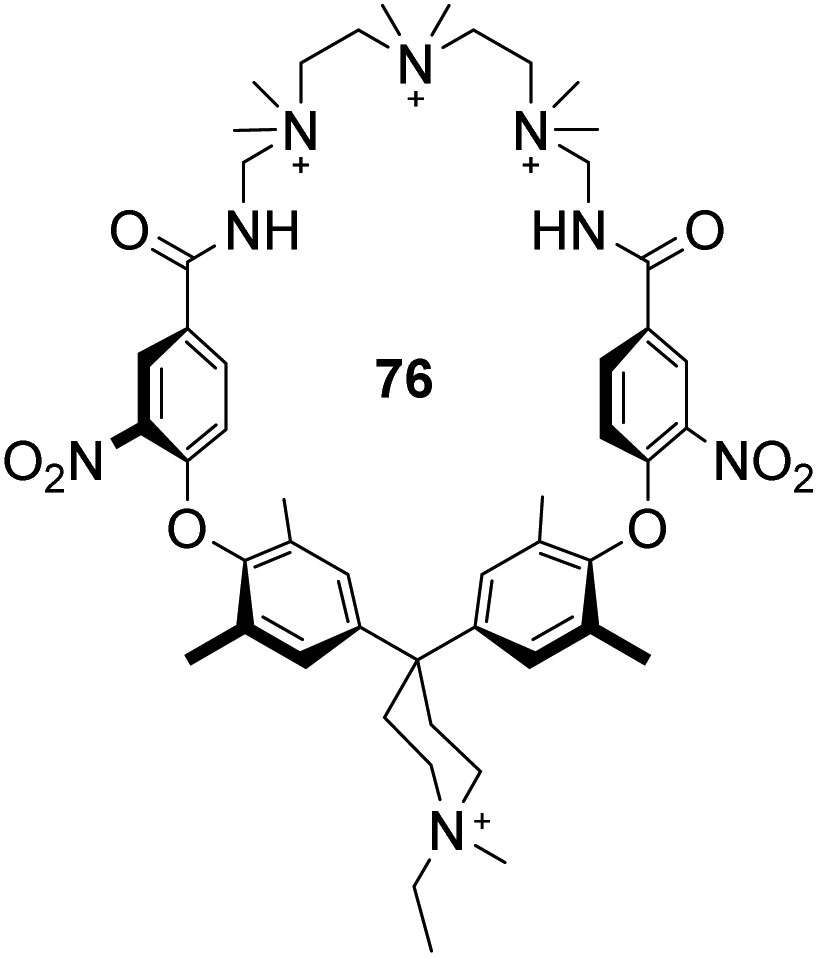 | ||
| Fig. 28 Polyammonium cyclophane 76. | ||
4. Conclusions
Anti-microbial resistance (AMR) is recognised as one of the major threats to humanity. The search for new antibiotics proceeds apace, but it is likely that most new discoveries will provoke resistance before too long. Agents which are intrinsically less likely to generate resistance are therefore especially valuable, and in this regard GPAs such as vancomycin seem to be exceptional. By binding a key intermediate, C-terminal D-Ala–D-Ala, they address a target which is specific to bacterial metabolism and very difficult for bacteria to change. The resistance which has eventually developed, via the change to D-Ala–D-Lac, presents another target which seems likely to be long-lasting. New agents which bind D-Ala–D-Lac have obvious importance, but even D-Ala–D-Ala remains a target of interest. The GPAs have limitations (e.g. lack of oral activity, inactive against Gram-negative organisms) which are not easy to address. Alternatives which are synthetically accessible, and therefore tuneable, could be extremely valuable.Unfortunately, vancomycin mimicry is a very difficult challenge. Binding polar molecules in water is problematic in itself, because of the complex effects of the solvent. Further to this, it is likely that extreme selectivity will be required. Even if a molecule binds D-Ala–D-Ala/Lac quite strongly, it will have no effect on bacteria if it is occupied by other components of the biological medium. Moreover, binding to other species could lead to bad side-effects. The only approach which has worked so far is to base designs very closely on vancomycin itself, as achieved by Boger and co-workers. This, however, raises problems of scaling which may be hard to solve.
The idea of designing radically new vancomycin analogues has quite a long history – many of the attempts described in this review are more than 20 years old. Momentum seems to have stalled, presumably because the problem is now perceived as too difficult. Nonetheless, our ability to design and construct molecules continues to improve as computational methods develop and new synthetic methodology appears. Moreover combinatorial chemistry remains a tool with great potential – for example, there are opportunities to expand the design scope to cyclic and peptidomimetic compound libraries.107–111 Recent experience in another area suggests that the highly selective binding of polar molecules in water is not an unrealistic goal.112 Antimicrobial resistance is a persistent problem, and even if success takes many years, efforts will not be wasted. It would be good to think that the supramolecular chemistry community will not forget this issue, and that eventually a solution will be found.
Conflicts of interest
There are no conflicts to declare.References
- P. Tompa, N. E. Davey, T. J. Gibson and M. M. Babu, Mol. Cell, 2014, 55, 161–169 CrossRef CAS PubMed.
- N. Sewald and H.-D. Jakubke, Peptides: chemistry and biology, John Wiley & Sons, 2015 Search PubMed.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- S. van Dun, C. Ottmann, L. G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960–13968 CrossRef CAS.
- D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2494 CrossRef CAS PubMed.
- A. J. Egan, J. Errington and W. Vollmer, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS.
- P. Schumann, Methods Microbiol., 2011, 38, 101–129 CAS.
- M. Rekharsky, D. Hesek, M. Lee, S. O. Meroueh, Y. Inoue and S. Mobashery, J. Am. Chem. Soc., 2006, 128, 7736–7737 CrossRef CAS PubMed.
- M. Nieto and H. R. Perkins, Biochem. J., 1971, 123, 773–787 CrossRef CAS PubMed.
- M. Nieto and H. R. Perkins, Biochem. J., 1971, 123, 789–803 CrossRef CAS PubMed.
- D. P. Levine, Clin. Infect. Dis., 2006, 42, S5–S12 CrossRef CAS PubMed.
- D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005, 105, 425–448 CrossRef CAS PubMed.
- K. Nicolaou, C. N. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096–2152 CrossRef PubMed.
- D. H. Williams and B. Bardsley, Angew. Chem., Int. Ed., 1999, 38, 1172–1193 CrossRef PubMed.
- P. Sarkar, V. Yarlagadda, C. Ghosh and J. Haldar, MedChemComm, 2017, 8, 516–533 RSC.
- L. H. Otero, A. Rojas-Altuve, L. I. Llarrull, C. Carrasco-López, M. Kumarasiri, E. Lastochkin, J. Fishovitz, M. Dawley, D. Hesek and M. Lee, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16808–16813 CrossRef.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- Z.-C. Wu and D. L. Boger, Acc. Chem. Res., 2020, 53, 2587–2599 CrossRef CAS PubMed.
- K. Nicolaou, H. J. Mitchell, N. F. Jain, N. Winssinger, R. Hughes and T. Bando, Angew. Chem., Int. Ed., 1999, 38, 240–244 CrossRef CAS.
- D. L. Boger, R. M. Borzilleri, S. Nukui and R. T. Beresis, J. Org. Chem., 1997, 62, 4721–4736 CrossRef CAS.
- D. A. Evans, M. R. Wood, B. W. Trotter, T. I. Richardson, J. C. Barrow and J. L. Katz, Angew. Chem., Int. Ed., 1998, 37, 2700–2704 CrossRef CAS PubMed.
- N. Pant and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 2002–2003 CrossRef CAS.
- B. Hinzen, P. Seiler and F. Diederich, Helv. Chim. Acta, 1996, 79, 942–960 CrossRef CAS.
- A. Marsot, A. Boulamery, B. Bruguerolle and N. Simon, Clin. Pharmacokinet., 2012, 51, 1–13 CrossRef CAS PubMed.
- P. Courvalin, Clin. Infect. Dis., 2006, 42, S25–S34 CrossRef CAS PubMed.
- P. J. Stogios and A. Savchenko, Protein Sci., 2020, 29, 654–669 CrossRef CAS PubMed.
- C. M. Harris, H. Kopecka and T. M. Harris, J. Am. Chem. Soc., 1983, 105, 6915–6922 CrossRef CAS.
- D. L. Boger, S. Miyazaki, S. H. Kim, J. H. Wu, S. L. Castle, O. Loiseleur and Q. Jin, J. Am. Chem. Soc., 1999, 121, 10004–10011 CrossRef CAS.
- M. Schäfer, T. R. Schneider and G. M. Sheldrick, Structure, 1996, 4, 1509–1515 CrossRef.
- Y. Nitanai, T. Kikuchi, K. Kakoi, S. Hanamaki, I. Fujisawa and K. Aoki, J. Mol. Biol., 2009, 385, 1422–1432 CrossRef CAS PubMed.
- P. H. Popieniek and R. Pratt, J. Am. Chem. Soc., 1991, 113, 2264–2270 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- M. B. Hillyer and B. C. Gibb, Annu. Rev. Phys. Chem., 2016, 67, 307–329 CrossRef CAS.
- H. Molinari, A. Pastore, L. Y. Lian, G. E. Hawkes and K. Sales, Biochemistry, 1990, 29, 2271–2277 CrossRef CAS PubMed.
- C. C. McComas, B. M. Crowley and D. L. Boger, J. Am. Chem. Soc., 2003, 125, 9314–9315 CrossRef CAS PubMed.
- Z. G. Jia, M. L. O'Mara, J. Zuegg, M. A. Cooper and A. E. Mark, FEBS J., 2013, 280, 1294–1307 CrossRef CAS PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- E. J. Milner-White, J. W. Nissink, F. H. Allen and W. J. Duddy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1935–1942 CrossRef PubMed.
- J. D. Watson and E. J. Milner-White, J. Mol. Biol., 2002, 315, 171–182 CrossRef CAS.
- D. H. Williams and J. P. Waltho, Biochem. Pharmacol., 1988, 37, 133–141 CrossRef CAS.
- M. Miklitz and K. E. Jelfs, J. Chem. Inf. Model., 2018, 58, 2387–2391 CrossRef CAS PubMed.
- F. Wang, H. Zhou, O. P. Olademehin, S. J. Kim and P. Tao, ACS Omega, 2018, 3, 37–45 CrossRef CAS PubMed.
- H. Molinari, A. Pastore, L. Y. Lian, G. E. Hawkes and K. Sales, Biochemistry, 1990, 29, 2271–2277 CrossRef CAS PubMed.
- Y. Nitanai, T. Kikuchi, K. Kakoi, S. Hanamaki, I. Fujisawa and K. Aoki, J. Mol. Biol., 2009, 385, 1422–1432 CrossRef CAS PubMed.
- P. J. Loll, A. Derhovanessian, M. V. Shapovalov, J. Kaplan, L. Yang and P. H. Axelsen, J. Mol. Biol., 2009, 385, 200–211 CrossRef CAS PubMed.
- D. A. Beauregard, A. J. Maguire, D. H. Williams and P. E. Reynolds, Antimicrob. Agents Chemother., 1997, 41, 2418–2423 CrossRef CAS PubMed.
- M. A. T. Blaskovich, K. A. Hansford, M. S. Butler, Z. Jia, A. E. Mark and M. A. Cooper, ACS Infect. Dis., 2018, 4, 715–735 CrossRef CAS PubMed.
- J. P. Mackay, U. Gerhard, D. A. Beauregard, R. A. Maplestone and D. H. Williams, J. Am. Chem. Soc., 1994, 116, 4573–4580 CrossRef CAS.
- J. Kaplan, B. D. Korty, P. H. Axelsen and P. J. Loll, J. Med. Chem., 2001, 44, 1837–1840 CrossRef CAS PubMed.
- U. Gerhard, J. P. Mackay, R. A. Maplestone and D. H. Williams, J. Am. Chem. Soc., 1993, 115, 232–237 CrossRef CAS.
- M. S. Butler, K. A. Hansford, M. A. T. Blaskovich, R. Halai and M. A. Cooper, J. Antibiot., 2014, 67, 631–644 CrossRef CAS PubMed.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2494 CrossRef CAS PubMed.
- H. Perkins, Pharmacol. Ther., 1982, 16, 181–197 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- S. Wasiłek and J. Jurczak, J. Org. Chem., 2020, 85, 11902–11907 CrossRef.
- A. V. R. Rao, M. K. Gurjar, K. L. Reddy and A. S. Rao, Chem. Rev., 1995, 95, 2135–2167 CrossRef CAS.
- M. C. Monnee, A. J. Brouwer, L. M. Verbeek, A. M. van Wageningen and R. M. Liskamp, Bioorg. Med. Chem. Lett., 2001, 11, 1521–1525 CrossRef CAS.
- D. L. Boger, S. L. Castle, S. Miyazaki, J. H. Wu, R. T. Beresis and O. Loiseleur, J. Org. Chem., 1999, 64, 70–80 CrossRef CAS PubMed.
- D. L. Boger, S. Miyazaki, S. H. Kim, J. H. Wu, S. L. Castle, O. Loiseleur and Q. Jin, J. Am. Chem. Soc., 1999, 121, 10004–10011 CrossRef CAS.
- M. J. Mann, N. Pant and A. D. Hamilton, J. Chem. Soc., Chem. Commun., 1986, 158–160 RSC.
- R. J. Pieters, Tetrahedron Lett., 2000, 41, 7541–7545 CrossRef CAS.
- C. J. Arnusch and R. J. Pieters, Eur. J. Org. Chem., 2003, 3131–3138 CrossRef CAS.
- T. Hefziba, D. T. Rijkers, J. Kemmink, H. W. Hilbers and R. M. Liskamp, Org. Biomol. Chem., 2004, 2, 2658–2663 RSC.
- H. T. ten Brink, D. T. Rijkers and R. M. Liskamp, J. Org. Chem., 2006, 71, 1817–1824 CrossRef CAS.
- J. Zhang, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Org. Lett., 2011, 13, 3438–3441 CrossRef CAS PubMed.
- J. Zhang, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Chem. Commun., 2013, 49, 4498–4500 RSC.
- X. Yang, L. P. Beroske, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Tetrahedron Lett., 2017, 58, 4542–4546 CrossRef CAS.
- X. Yang, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Bioorg. Med. Chem. Lett., 2022, 73, 128887–128891 CrossRef CAS.
- A. Okano, R. C. James, J. G. Pierce, J. Xie and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 8790–8793 CrossRef CAS PubMed.
- M. J. Moore, S. Qu, C. Tan, Y. Cai, Y. Mogi, D. Jamin Keith and D. L. Boger, J. Am. Chem. Soc., 2020, 142, 16039–16050 CrossRef CAS PubMed.
- J. Xie, A. Okano, J. G. Pierce, R. C. James, S. Stamm, C. M. Crane and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 1284–1297 CrossRef CAS.
- K. S. Lam, M. Lebl and V. Krchňák, Chem. Rev., 1997, 97, 411–448 CrossRef CAS PubMed.
- W. C. Still, Acc. Chem. Res., 1996, 29, 155–163 CrossRef CAS.
- N. Srinivasan and J. D. Kilburn, Curr. Opin. Chem. Biol., 2004, 8, 305–310 CrossRef CAS PubMed.
- A. Borchardt and W. C. Still, J. Am. Chem. Soc., 1994, 116, 373–374 CrossRef CAS.
- Y. A. Cheng, T. Suenaga and W. C. Still, J. Am. Chem. Soc., 1996, 118, 1813–1814 CrossRef CAS.
- M. C. F. Monnee, A. J. Brouwer, L. M. Verbeek, A. M. A. van Wageningen and R. M. J. Liskamp, Bioorg. Med. Chem., 2001, 11, 1521–1525 CrossRef CAS.
- R. Xu, G. Greiveldinger, L. E. Marenus, A. Cooper and J. A. Ellman, J. Am. Chem. Soc., 1999, 121, 4898–4899 CrossRef CAS.
- K. A. Ahrendt, J. A. Olsen, M. Wakao, J. Trias and J. A. Ellman, Bioorg. Med. Chem., 2003, 13, 1683–1686 CrossRef CAS.
- U. N. Sundram, J. H. Griffin and T. I. Nicas, J. Am. Chem. Soc., 1996, 118, 13107–13108 CrossRef CAS.
- M. Giese, J. Niemeyer and J. Voskuhl, ChemPlusChem, 2020, 85, 985–997 CrossRef CAS PubMed.
- C. Schmuck, M. Heil, J. Scheiber and K. Baumann, Angew. Chem., Int. Ed., 2005, 44, 7208–7212 CrossRef CAS PubMed.
- A. W. Czarnik, Curr. Opin. Chem. Biol., 1997, 1, 60–66 CrossRef CAS.
- C. Schmuck and M. Heil, Chem. – Eur. J., 2006, 12, 1339–1348 CrossRef CAS PubMed.
- C. Schmuck and P. Wich, Angew. Chem., Int. Ed., 2006, 45, 4277–4281 CrossRef CAS PubMed.
- K. B. Jensen, T. M. Braxmeier, M. Demarcus, J. G. Frey and J. D. Kilburn, Chem. – Eur. J., 2002, 8, 1300–1309 CrossRef CAS.
- M. C. Monnee, A. J. Brouwer and R. M. Liskamp, QSAR Comb. Sci., 2004, 23, 546–559 CrossRef CAS.
- C. Chamorro, J.-W. Hofman and R. M. J. Liskamp, Tetrahedron, 2004, 60, 8691–8697 CrossRef CAS.
- C. Chamorro and R. M. J. Liskamp, Tetrahedron, 2004, 60, 11145–11157 CrossRef CAS.
- B. P. Hay, Chem. Soc. Rev., 2010, 39, 3700–3708 RSC.
- F. Ulatowski and J. Jurczak, Asian J. Org. Chem., 2016, 5, 715–723 CrossRef CAS.
- P. Niedbała, K. Dąbrowa, S. Wasiłek and J. Jurczak, Molecules, 2021, 26, 6417 CrossRef PubMed.
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC.
- L. J. Lawless, A. G. Blackburn, A. J. Ayling, M. N. Pérez-Payán and A. P. Davis, J. Chem. Soc., Perkin Trans. 1, 2001, 1329–1341 RSC.
- P. D. Henley, C. P. Waymark, I. Gillies and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2000, 1021–1031 RSC.
- A. M. Castilla, M. M. Conn and P. Ballester, Beilstein J. Org. Chem., 2010, 6, 5 Search PubMed.
- A. Casnati, M. Fabbi, N. Pelizzi, A. Pochini, F. Sansone, R. Unguro, E. Di Modugno and G. Tarzia, Bioorg. Med. Chem., 1996, 6, 2699–2704 CrossRef CAS.
- L. Frish, F. Sansone, A. Casnati, R. Ungaro and Y. Cohen, J. Org. Chem., 2000, 65, 5026–5030 CrossRef CAS PubMed.
- T. R. Herrin, A. M. Thomas, T. J. Perun, J. C. Mao and S. W. Fesik, J. Med. Chem., 1985, 28, 1371–1375 CrossRef CAS PubMed.
- J. S. Albert and A. D. Hamilton, Tetrahedron Lett., 1993, 34, 7363–7366 CrossRef CAS.
- C. Schmuck, D. Rupprecht and W. Wienand, Chem. – Eur. J., 2006, 12, 9186–9195 CrossRef CAS.
- A. La Venia, B. Lemrová and V. Krchňák, ACS Comb. Sci., 2013, 15, 59–72 CrossRef CAS.
- S. L. Giudicessi, J. M. Gurevich-Messina, M. C. Martínez-Ceron, R. Erra-Balsells, F. Albericio, O. Cascone and S. A. Camperi, ACS Comb. Sci., 2013, 15, 525–529 CrossRef CAS.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- N. K. Bashiruddin and H. Suga, Curr. Opin. Chem. Biol., 2015, 24, 131–138 CrossRef CAS PubMed.
- Z.-M. Wu, S.-Z. Liu, X.-Z. Cheng, W.-Z. Ding, T. Zhu and B. Chen, Chin. Chem. Lett., 2016, 27, 1731–1739 CrossRef CAS.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |