
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting severe acute respiratory syndrome-coronavirus (SARS-CoV-1) with structurally diverse inhibitors: a comprehensive review
Maryam S. Hosseini-Zare
a,
Ramasamy Thilagavathi
b and
Chelliah Selvam
 *a
*a
aDepartment of Pharmaceutical and Environmental Health Sciences, College of Pharmacy and Health Sciences, Texas Southern University, Houston, TX-77004, USA. E-mail: selvam.chelliah@tsu.edu; Tel: +1-713-313-7552
bDepartment of Biotechnology, Faculty of Engineering, Karpagam Academy of Higher Education, Coimbatore, India
First published on 29th July 2020
Abstract
Coronaviruses, which were discovered in 1968, can lead to some human viral disorders, like severe acute respiratory syndrome (SARS), Middle East respiratory syndrome-related (MERS), and, recently, coronavirus disease 2019 (COVID-19). The coronavirus that leads to COVID-19 is rapidly spreading all over the world and is the reason for the deaths of thousands of people. Recent research has revealed that there is about 80% sequence homology between the coronaviruses that cause SARS and COVID-19. Considering this fact, we decided to collect the maximum available information on targets, structures, and inhibitors reported so far for SARS-CoV-1 that could be useful for researchers who work on closely related COVID-19. There are vital proteases, like papain-like protease 2 (PL2pro) and 3C-like protease (3CLpro), or main protease (Mpro), that are involved in and are essential for the replication of SARS coronavirus and so are valuable targets for the treatment of patients affected by this type of virus. SARS-CoV-1 NTPase/helicase plays an important role in the release of several non-structural proteins (nsps), so it is another essential target relating to the viral life cycle. In this paper, we provide extensive information about diverse molecules with anti-SARS activity. In addition to traditional medicinal chemistry outcomes, HTS, virtual screening efforts, and structural insights for better understanding inhibitors and SARS-CoV-1 target complexes are also discussed. This study covers a wide range of anti-SARS agents, particularly SARS-CoV-1 inhibitors, and provides new insights into drug design for the deadly SARS-CoV-2 virus.
 Maryam S. Hosseini-Zare | Maryam S. Hosseini-Zare received her M.S. in organic chemistry from Iran University of Sciences and Technology and joined the doctoral program of Texas Southern University's College of Pharmacy and Health Sciences in August 2017, beginning her current exploration of natural products and their anti-cancer activities under the supervision of Dr Chelliah Selvam in 2017. |
 Ramasamy Thilagavathi | Dr Thilagavathi Ramasamy obtained her bachelor's degree in pharmacy from The Tamil Nadu Dr M. G. R. Medical University, India. She joined the National Institute of Pharmaceutical Education and Research (NIPER, India) to obtain her master's degree in medicinal chemistry. Then she completed a PhD degree in pharmacy (specialization: medicinal chemistry) in 2005 at NIPER, India. She moved to the University of Alberta, Canada for postdoctoral research and then worked in anthrax drug discovery work at the University of Illinois at Chicago, USA. She worked as a research fellow at Curtin University of Technology, Perth, Australia. She worked on the synthesis of formacetal-modified RNA at the State University of New York, Binghamton, USA. Returning to India, she served as a Senior Research Scientist at Jubilant Biosys Ltd. Presently, she is working as a Professor and Head at the Department of Biotechnology, Faculty of Engineering, Karpagam Academy of Higher Education, India. Her current research includes chemoinformatics and computational drug discovery. |
 Chelliah Selvam | Dr Chelliah Selvam is an associate professor of medicinal chemistry at the Texas Southern University College of Pharmacy and Health Sciences. He studied Pharmacy at Dr MGR Medical University (India) and received his M.S. (Pharm) and PhD from the National Institute of Pharmaceutical Education and Research, India. Dr Selvam worked as a post-doc at the Universite Paris Descartes (2004–2007), Auckland Cancer Society Research Centre, the University of Auckland (2007–2008), the State University of New York at Binghamton (2008–2011), and South Dakota State University (2011–2012). His research interests focus on the design and synthesis of new chemical entities for the treatment of cancer, computer-aided drug design, and the synthesis of chemically modified RNA/siRNA for gene-silencing activity. |
Introduction
Virologists named a member of an unrecognized group of viruses a coronavirus in 1968 because of its appearance in electron microscopy images.1 Coronaviruses (CoVs) are the largest RNA viruses, belonging to the Coronaviridae family of enveloped, positive-sense viruses, and are about 120 nanometers in diameter.2 These viruses are divided into 4 genera: α-, β-, δ- and γ-coronaviruses. The β-coronaviruses are further divided into A, B, C, and D lineages.3 There are different varieties of these viruses due to their susceptibility to mutation and recombination, and they mainly infect humans, mammals, and birds. There are seven coronavirus variants that infect humans. HCoV-229E and HCoV-NL63 are α-coronaviruses, HCoV-OC43 and HCoV-HKU1 belong to the β-coronavirus lineage A, SARS-CoV-1 and SARS-CoV-2 belong to the β-coronavirus lineage B (β-B coronaviruses), and MERS-CoV belongs to the β-coronavirus lineage C.4–7 Among these, COVID-19 (SARS-CoV-2) and SARS-CoV-1 are the most closely related versions (about 80% sequence homology).8,9 Both are known to cause respiratory illness and are spread mainly via close person-to-person contact or via direct contact with infectious materials, such as respiratory secretions, from patients. In early 2003, SARS-CoV-1 infections were first reported in Asia, affecting thousands of people around the world, and 801 deaths10 occurred according to World Health Organization data. The re-emergence of a SARS disease with greater impact, as predicted by some, is now being bitterly experienced. The identification of molecular targets for SARS-CoV-1 and various efforts relating to the discovery of inhibitors have been made by the scientific community. In this study, we report a comprehensive review covering structurally diverse SARS-CoV-1 inhibitors.A coronavirus is made of spike glycoproteins or peplomers, which are essential for entering the host cells. The spike contains two subunits: S1, which binds to receptors on host cell surfaces; and S2, which fuses to cell membranes. ACE-2 is a type I transmembrane metallocarboxypeptidase11 and works as a cell membrane receptor for both SARS-CoV-1 and SARS-CoV-2.12–14 Studies have shown that the ACE2-binding affinity of the receptor-binding domain (RBD) in the S1 subunit of SARS-CoV-2 is 10 to 20 times higher than in the case of SARS-CoV-1, which may lead to higher infectivity and transmissibility of SARS-CoV-2 compared to SARS-CoV-1.15 Through the binding of the RBD in the S1 subunit of S proteins to ACE-2 of the host cells, interactions between heptad repeat 1 (HR1) and 2 (HR2) domains in the S2 subunit of S proteins occur, making a six-helix bundle (6-HB) fusion core. This fusion core may provide the opportunity for fusion and infection.16,17
SARS-CoV-1 generates several functional proteins through using SARS-CoV-1 NTPase/helicase18 in human host cells via the cleavage of its two overlapping “polyproteins”: pp1a (486 kDa) and pp1ab (790 kDa).19 Papain-like protease 2 (PL2pro) and 3C-like protease (3CLpro, also referred to as main protease, Mpro) cleave three sites in the polyproteins, respectively, to generate individual functional proteins. 3CLpro has a Cys–His catalytic dyad (Cys-145 and His-41), in which the cysteine thiol acts as a nucleophile that is crucial for the proteolytic process.20 This protein is generated from polyproteins via its own proteolytic activity21 and forms a homodimer that is catalytically active22 with one active site per subunit. Based on the crystal structure (Fig. 1a) of 3CLpro, this protein is made of an N-terminal finger (residues 1–8), a catalytic domain (residues 8–184; a surface view of which is shown in Fig. 1b), which has distinct features (the Cys–His catalytic dyad is represented in Fig. 1c), and a C-terminal domain (residues 201–306).23 Recently, studies have shown that SARS 3CL protease is sensitive to a loss of catalytic activity upon degradation at the 188Arg/189Gln site,24 which is depicted in a ball-and-stick model in Fig. 1d. Scientists have tried several approaches for the treatment of SARS, such as trying to find an effective vaccine,25,26 testing known antiviral drugs, and designing and synthesizing new drugs for the treatment of SARS-CoV-1 infections.27 Since, COVID-19, caused by the virus SARS-CoV-2, is recently rapidly spreading all over the world,6,28–31 we have tried to cover the topic of closely related SARS-CoV-1 inhibitors with diverse scaffolds. We hope that this paper helps scientists to find an effective inhibitor molecule for this fatal disease.
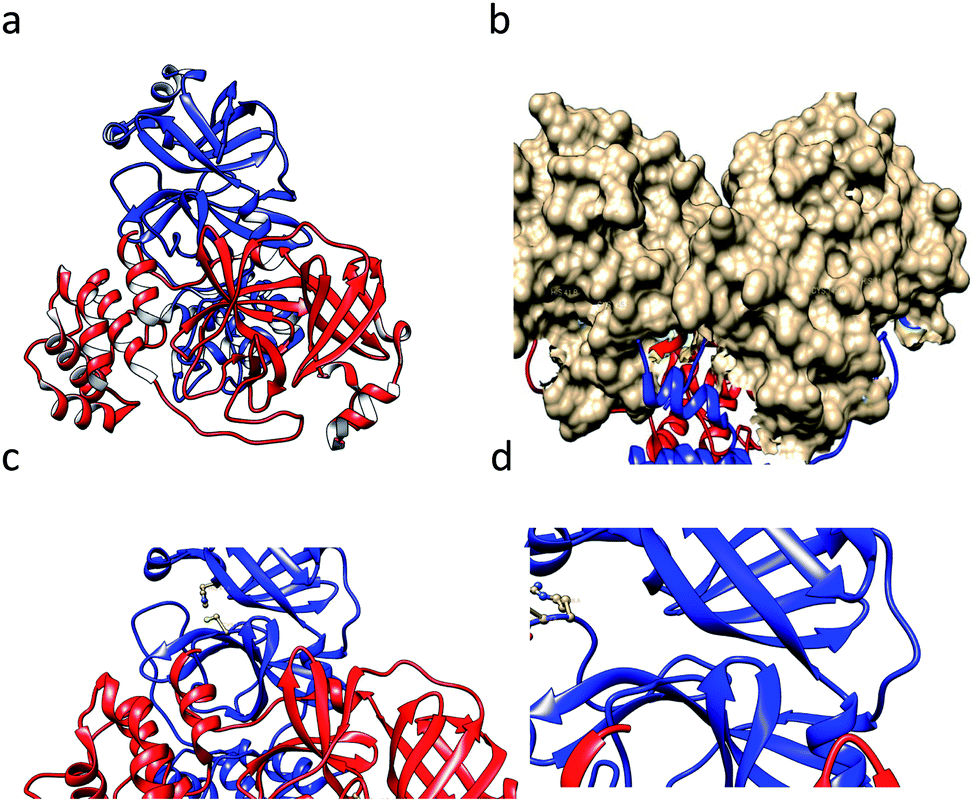 | ||
| Fig. 1 (a) The crystal structure of the SARS CoV-1-main protease (3CLpro) dimer (1ui1.pdb). (b) The surface of the catalytic domain. (c) The catalytic dyad residues Cys145 and His41 are shown in a ball-and-stick model. (d) The residues Arg188 and Gln89 are shown in a ball-and-stick model. The images were prepared using the tool Chimera. | ||
SARS-CoV-1 3C-like proteinase (3CLpro)
3CLpro is a cysteine protease, and it is a promising target for SARS-CoV-1 therapy because of its essential role in viral replication.32 This cysteine proteinase has Cys145 at the active site, which acts as a nucleophile, and His41 acts as a general acid-base in the proteolytic process and plays a crucial role in the regulation of the SARS life cycle.33 Researchers have assessed a wide variety of inhibitor classes, including covalently bonded peptides, non-peptide inhibitors, peptidomimetic analogs, and small molecule inhibitors.34–36 The discovery of SARS-CoV-1 main protease (Mpro) inhibitors, and associated challenges and structural aspects have been reported by Sirois et al.37 The enzyme active site or allosteric dimerization domain of this protein can be targeted by these inhibitors.38,39 Irreversible peptidomimetic structures, like the substrate-analog hexapeptidyl chloromethyl ketone (CMK)40 and TG-0205221,41 were used as the first generation of 3CLpro inhibitors and are mostly five residues in length. They form a covalent bond between the thiolate anion of the catalytic Cys145 residue and the reactive atom.40–45 The reactive parts include Michael acceptors,41,44,46 aldehydes,47 epoxy-ketones,48 halo-methyl ketones,49 and trifluoromethyl ketones.50 The structures of some of these analogues are listed in Fig. 2 (1–6).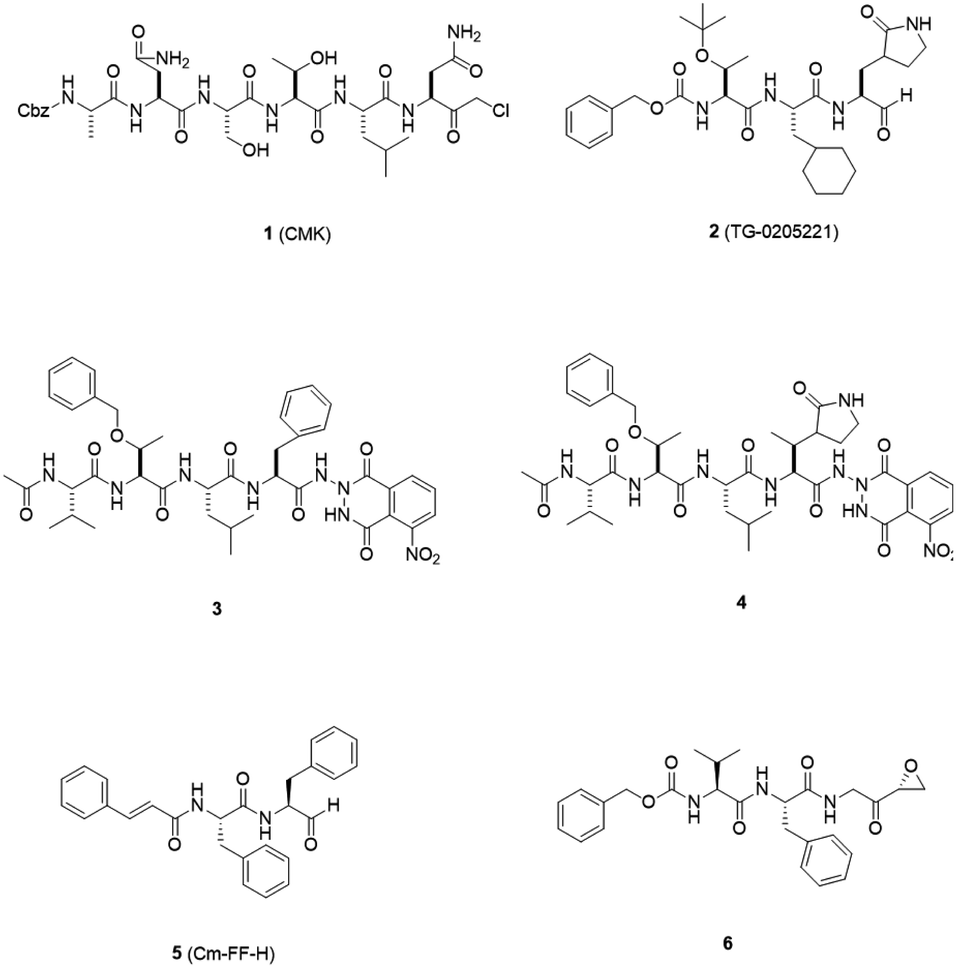 | ||
| Fig. 2 3CLpro inhibitors that form a covalent bond with 3CLpro via Michael acceptors, aldehydes, epoxy-ketones, and halo-methyl ketones. | ||
X-ray structural studies of the Mpro enzyme revealed critical molecular insight into the ligand-binding site interactions at the active site, and confirmed that P4-Boc-Ser is involved in making an important hydrogen bond in the S′4 subunit.46 Earlier efforts to find Mpro inhibitors led to the identification of a compound that made a covalent bond with CYS145.51 The inhibitory activity effects of TG-0205221 (2) were tested. A study of the crystal structure of this compound showed a unique binding mode comprising a covalent bond, hydrogen bonds, and numerous hydrophobic interactions. Analysis of the crystal structure of TG0205221 crystalized with Mpro from SARS-CoV-1 (2GX4.pdb) revealed the formation of a covalent bond with CYS145. This compound, by making hydrogen bonds between N–H on P2 with oxygen on Gln189, oxygen on P3 with N–H on Glu166, and N–H on P3 with oxygen on Glu166, contributed to the strong interaction with the protease.41 In addition, TG0205221 undergoes hydrogen bond interactions with His163 and Gly143 (Fig. 3).
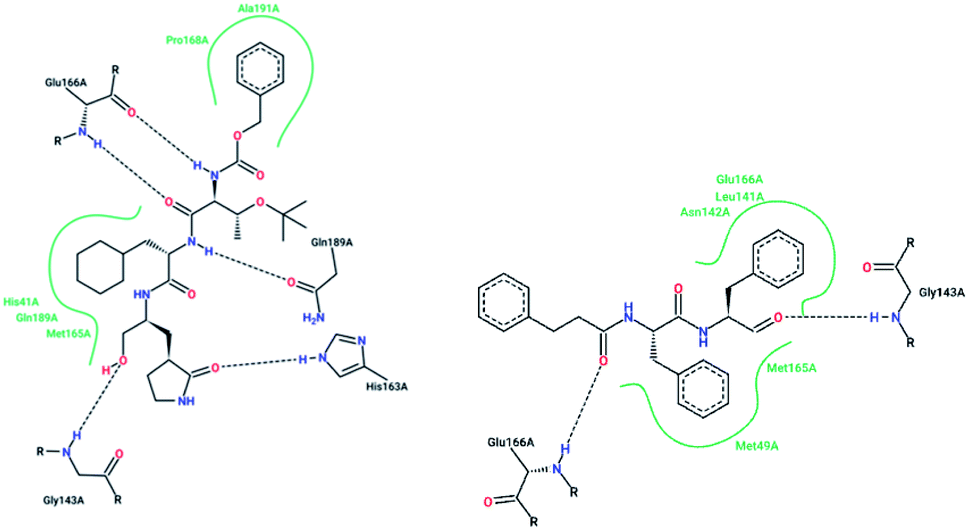 | ||
| Fig. 3 2-D interaction profiles of TG0205221 (2GX4.pdb) and Cm–FF–H (3SN8.pdb) with active site residues of SARS-CoV-1 3CLpro; hydrogen bonds are shown as broken lines, and hydrophobic interactions are shown in green. The picture was prepared using the “poseview” tool. | ||
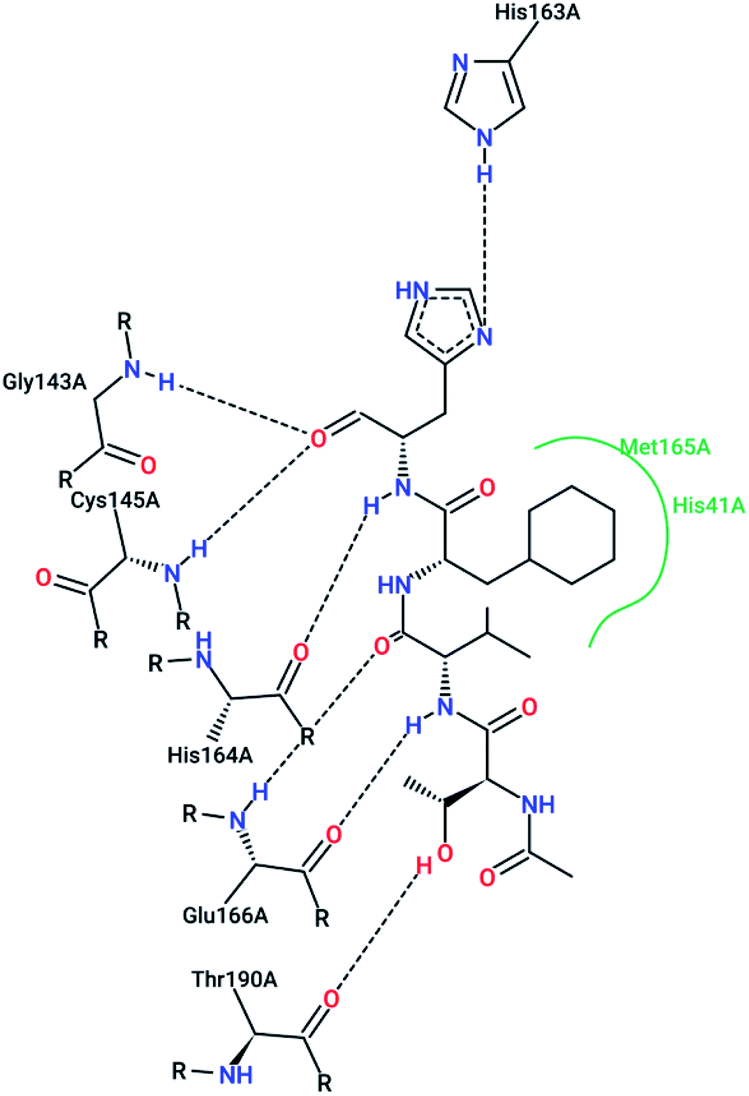
However, most covalent inhibitors are less attractive due to adverse drug responses, off-target side effects, toxicity, and lower potency.52–55 Therefore, recent studies have focused more on noncovalent protease inhibitors for the treatment of SARS-CoV-1 infections. Zhu et al.47 have reported peptide aldehyde inhibitors; one such type, Cm–FF–H, complexed with the SARS-CoV-1 protease structure (3SN8.pdb), leading to the interesting observation that the P1-phenyl alanine residue of the ligand bound to the relatively hydrophilic S1 pocket of the enzyme and still retained high inhibitory activity. A possible explanation could be the presence of the highly electrophilic aldehyde group of the inhibitor, which favors nucleophilic attack by catalytic Cys145 to form a thiohemiacetal (Fig. 3). Sydnes et al., after converting trifluoromethyl-b-amino alcohol to four tri- and tetra-glutamic acid and glutamine peptides, demonstrated the inhibitory activities of these compounds against SARS-CoV-1 3CLpro (7–10).50 Moreover, peptide aldehydes led to the discovery of reversible SARS-CoV-1 Mpro inhibitors (11, 12). Inhibitors bind to the active site of SARS-CoV-1 Mpro, which is located in a cleft between domains I and II. These derivatives of peptide aldehydes have a point mutation at either the P2 or P5 position compared to the lead structure and show 50 to 60 times more activity.56 Compound 13 is another peptide showing the existence of hydrogen bonding between the P1, P2, and P4 positions with the active site cleft of protease, which leads to the compact fitting of the tetra-peptide inhibitor.24 CYS145, Gly143, His164, Glu166, Thr190, and His163 are the active site residues involved in the hydrogen bond interactions with 13 (Fig. 5).
The potent tripeptidic Z-Val-Leu-Ala(pyrrolidone-3-yl)-2-benzothiazole, in which the P3 valine unit was substituted with a variety of distinct moieties, has shown moderate to good inhibition activities against 3CLpro. Analogs 14 and 15 are the most potent, with Ki values of 0.39 and 0.33 μM, respectively. Docking studies of 14 with SARS-CoV-1 3CL protease revealed that there is a hydrogen bond between the N-arylglycine unit and a backbone hydrogen bond donor at the S3 position.57 Additionally, 16 and 17 have shown potent inhibitory activities, with Ki values of 4.1 and 3.1 nM, respectively. These tripeptide-type SARS-CoV-1 3CL protease inhibitors have an electrophilic aryl ketone at the S2-site moiety and a benzothiazole warhead at the S10 position. A hydrogen bond was made at the cyclic lactam of the S1 site. The phenoxyacetyl moiety at the S4 site adopted a unique folding conformation.58 Compound 18 was designed as a potent dipeptide-type SARS-CoV-1 3CL protease inhibitor with a P3 scaffold, with a Ki value of 0.006 μM. SAR studies of this compound identified the rigid indole-2-carbonyl group as being one of the best P3 moieties. Moreover, it was revealed that methoxy substitution at the 4-position of the indole unit increased the inhibitory potency.59
Based on data gathered from high-throughput screening, α,β-epoxyketone tripeptide compounds were synthesized and tested against SARS-CoV-1 3CLpro. These compounds, as a result of irreversible bonds between the protease and one of the derivatives (19), resulted in inhibition activity with a k3/Ki of 0.002 μM−1 S−1. This derivative has a P3 L-phenylalanine residue and an R configuration at the C-2 of the epoxide group, which led to >50% inhibition of viral replication at 10 μM with no detectable cytotoxicity.48 Another peptide possessing P1-pyrrolidone and P1′-thiazole moieties showed potent SARS-CoV-1 3CLpro inhibition activity (20). Through using a modeling package (3D docking), it was revealed that nitrogen in thiazole made a hydrogen bond with His41.60
Aziridine- and oxirane-2-carboxylate derivatives were tested; those with trans-configured aziridine-2,3-dicarboxylates showed more activity against Mpro, while aziridine- and oxirane-2-carboxylic acid-containing compounds showed weak inhibitory activity. A docking study of analog 21 demonstrated that its active center is located close to the sulfur of Cys145. Moreover, the main part of this compound was in the S1 pocket of the enzyme and made hydrogen bonds with the amino acids of the B-chain and A-chain.61 In vitro studies of dipeptidyl N,N-dimethyl glutaminyl fluoromethyl ketones (fmk) showed their potent inhibitory activities against the protease. Among the synthesized derivatives, Z-Leu-Gln(NMe2)-fmk (22) showed less toxicity, more selectivity, and better inhibition activity, protecting Vero cells infected with strain 6109 with an EC50 value of 2.5 μM. In addition, this compound was inactive against rhinovirus type-2 in a cell-based assay.49 Trifluoromethyl ketones were tested against Mpro, and the results revealed that compounds with a benzyl group at the P1 site support the idea that the P2–P4 sites play an important role in determining binding affinity, although they are far from the active site. Analog 23 worked as the most effective derivative, with an IC50 value of 10 μM, and it exhibited a time-dependent decrease in enzyme activity.62 A series of keto-glutamine analogues with a phthalhydrazide group at the R-position worked as reversible inhibitors against SARS-CoV-1 3CLpro. Compound 24, which contains a tripeptide (Ac-Val-Thr-Leu), showed the best activity of these glutamine-based examples, with an IC50 value of 0.60 μM. The oxygen atoms on the nitro group of this compound formed hydrogen bonds with Asn142. Moreover, the phenyl ring formed an aromatic–aromatic stacking interaction with the phthalhydrazide group of the inhibitor (Fig. 4).42
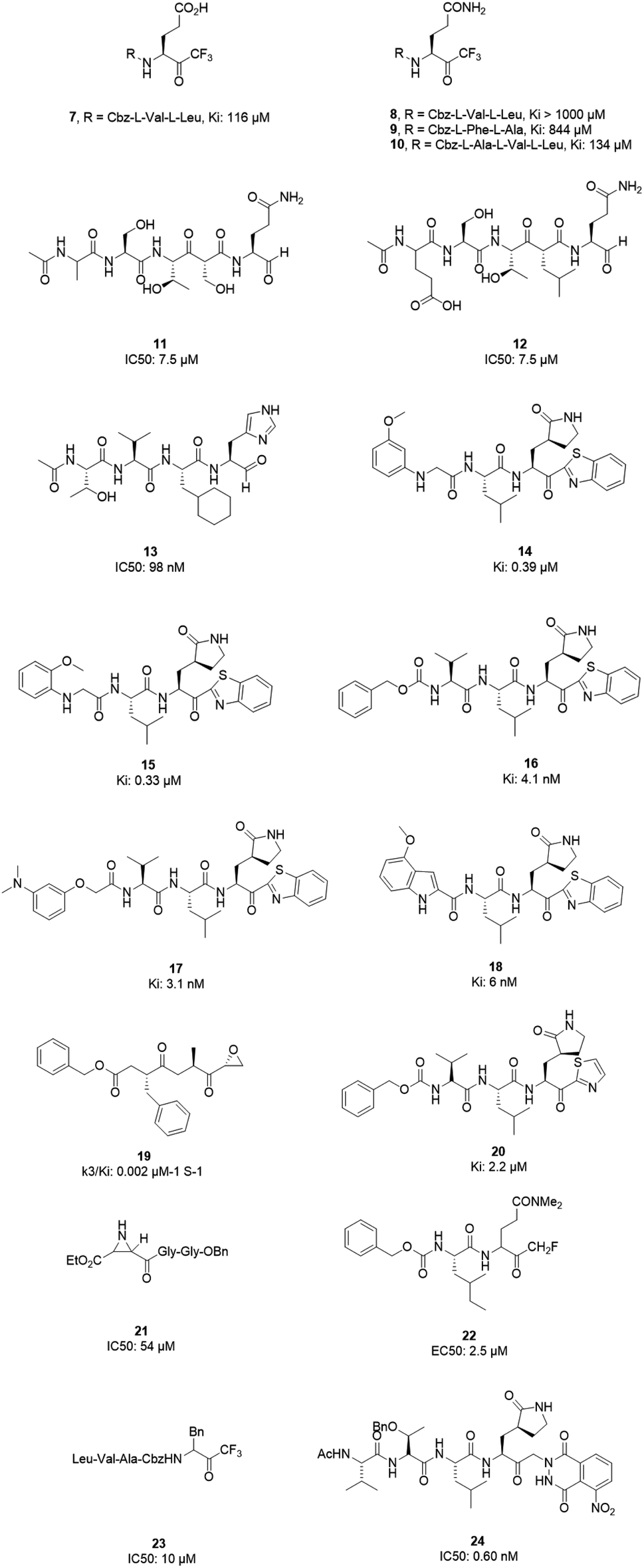 | ||
| Fig. 4 The chemical structures of peptidomimetic analogs of 3CLpro inhibitors. | ||
 | ||
| Fig. 5 The 2-D interaction profile of compound 3sna.pdb, 13, (3atw.pdb) with active site residues of SARS-CoV-1 3CLpro; hydrogen bonds are shown as broken lines, and hydrophobic interactions are shown in green. The picture was prepared using the “poseview” tool. | ||
Peptidic inhibitors exhibit good potency, but drawbacks such as in vivo instability and cell membrane impermeability should be kept in mind when converting these to drug molecules. There is a lot of interest in improving the poor pharmacokinetic profiles associated with peptidic inhibitors. The chemical modification of peptide linkages is one of the approaches for improving the PK profiles.
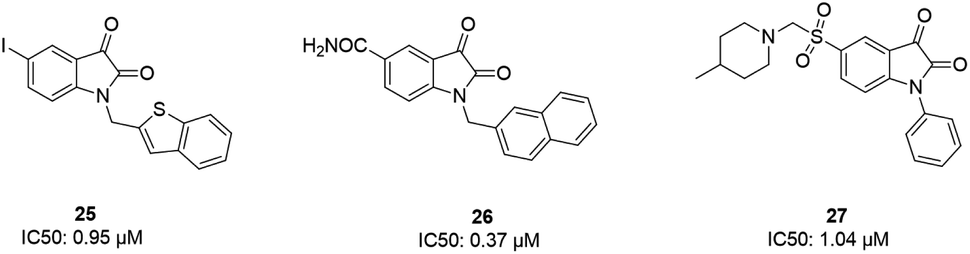
Some isatin derivatives showed potent and selective inhibitory activities against SARS coronavirus 3CL protease. A carbonyl group at the 2nd position and nitrogen at the 1st position are important for making hydrogen bonds. Substitution at the 5th position enables some derivatives to fit the S2 hydrophobic site. The 5-iodo substitution of isatin results in good potency (IC50: 0.95 μM) due to its fitting size and better binding affinity to the active site of the protease (25).63 Another isatin derivative (26) showed inhibitory activity, with an IC50 value of 0.37 μM, via undergoing a noncovalent reversible interaction with the active site of the protease. This compound showed more selective inhibition activity against 3CLpro compared to papain, chymotrypsin, and trypsin.64 Analog 27 was tested against SARS-CoV-1 3CL protease, resulting in potent inhibitory activity, with an IC50 value of 1.04 μM. From docking studies involving this compound, it was revealed that this compound made hydrogen bonds with Gly143 and Cys145 of the protease65 (Fig. 6).
 | ||
| Fig. 6 The chemical structures of isatin derivatives that can act as 3CLpro inhibitors. | ||
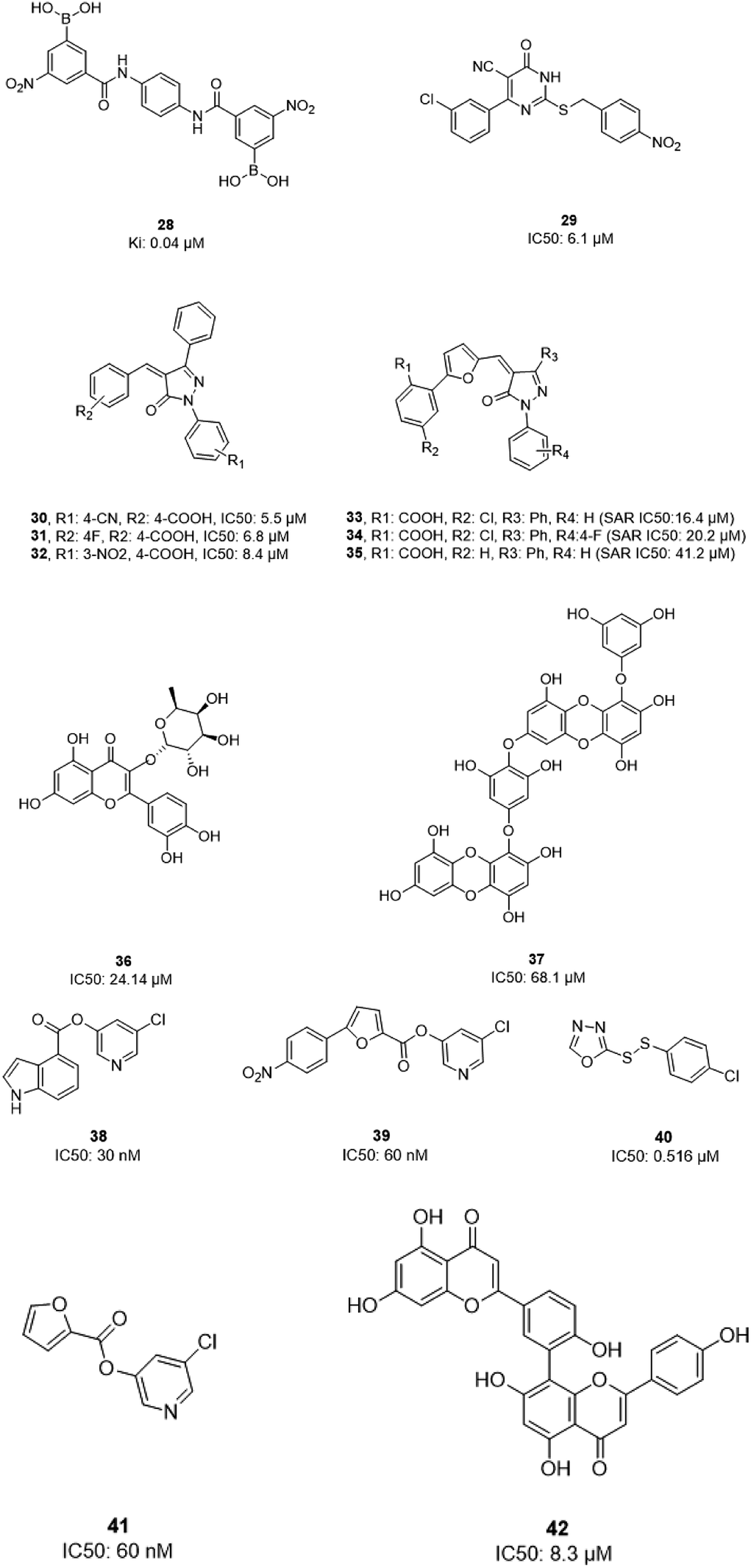
In 2004, Bacha et al. cloned and expressed full-length SARS-CoV-1 3CLpro using Escherichia coli and tested some newly synthesized fluorescently labeled substrates for their inhibitory activities. The results revealed that a compound containing boronic acid, which targeted a cluster of serine residues (Ser139, Ser144, and Ser147) of the protease, showed inhibitory activity via making a reversible bond with 3CLpro (28; Ki: 0.04 μM).66 2-(Benzylthio)-6-oxo-4-phenyl-1,6-dihydropyrimidine derivatives with an electron withdrawing substituent on the aryl group, like chloro, were more potent against SARS-CoV-1 3CLpro than those with an electron donating substituent, like methyl methoxy (29).67 Pyrazolone compounds were synthesized, and compounds with a 4-carboxylbenzylidene aryl ring attached to the C4 of pyrazolone showed SARS-CoV-1 3CLpro inhibition activities (30, 31, 32). Cytotoxicity studies of these compounds done via MTT assays showed no toxicity at 200 μM.68 In 2016, Kumar et al. reported that pyrazolone-core derivatives with a carboxyl group inhibited SARS-CoV-1 and MERS-CoV 3CLpro. Three analogs of pyrazolone (33, 34, 35) revealed that the presence of a carboxyl group at the R1 position led to H-bonds with His163 at the S1 subsite, which is important for protease specificity towards the conserved Gln residue.69 The natural compound quercetin-3-β-galactoside (36), effective for the treatment of allergies and preventing heart disease and cancer, was also identified as a 3CLpro inhibitor. Both molecular modeling and Q189A mutation studies revealed that Gln189 is important for the binding of quercetin-3-β-galactoside to SARS-CoV-1 3CLpro. Derivatives of this natural compound were synthesized, and the results showed the importance of the four hydroxy groups of quercetin for bioactivity.70 Quinone-methide triterpenes, including celastrol, pristimerin, tingenone, and iguesterin, which were derived from Triterygium regelii and dihydrocelastrol, were evaluated for SARS-CoV-1 3CLpro inhibitory activities and showed potency. On the other hand, dihydrocelastrol, which has a phenol moiety, showed low inhibitory activity.71 Phlorotannins isolated from the edible brown algae Ecklonia cava exhibited the significant blocking of the cleavage of SARS-CoV-1 3CLpro in cell-based assays. Among nine synthesized derivatives, analog 37 showed the most potent SARS-CoV-1 3CLpro trans/cis-cleavage inhibitory effects, with IC50 values of 2.7 and 68.1 μM, respectively, and no toxicity. Moreover, this compound made strong hydrogen bonds with the catalytic dyad (Cys145 and His41) of SARS-CoV-1 3CLpro.72 5-Chloropyridine-ester-derived compounds were tested against SARS-CoV-1 3CLpro, which demonstrated the critical effects of the position of carboxylate on the potency. Analog 38, with a 5-chloropyridinyl ester at position 4 of the indole ring, showed potent inhibitory activity, with an IC50 value of 30 nM, and antiviral activity, with an EC50 value of 6.9 μM. Docking studies of this compound revealed that the carbonyl group of the ester of this inhibitor formed a strong hydrogen bond with Cys145.55 Niu et al., via high-throughput screening, identified 3-chloropyridine acetate as an inhibitor of SARS-CoV-1. Data show that the 3-chloropyridine functionality, in the context of an ester compound, tends to cluster in the S1 specific pocket of SARS-CoV-1 Mpro. Additionally, the chemical properties of the leaving group on the carbonyl side of the inhibitor plays an important role in determining the rate of the subsequent water-mediated hydrolysis of the resultant acyl enzyme (39). In addition, analysis of the protease structure revealed that the S4 pocket has intrinsic flexibility, allowing for the accommodation of bulky groups, which could help to further optimize inhibitor–enzyme interactions.73 Unsymmetrical aromatic disulfides were tested against 3CLpro, leading to the identification of a reversible and non-competitive analog (40) with an IC50 value of 0.516 μM. Docking studies of compound 40 with the protease revealed hydrogen bonds between this compound and Cys145, Gly143, and Asn142.74 A halopyridinyl ester (41) showed potent inhibition activity against SARS 3CLpro, with an IC50 value of 60 nM. Electrospray mass spectrometry investigations proposed covalent bond formation between this inhibitor and the enzyme, which was important for its strong 3CLpro inhibition activity.41 Ethanol extract of Torreya nucifera leaves was evaluated against SARS-CoV-1 3CLpro and showed potent inhibition activity, and amentoflavone (42) showed good inhibitory activity75 (Fig. 7).
 | ||
| Fig. 7 The chemical structures of natural and synthesized 3CLpro inhibitors. | ||
The screening of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds with Gold, the docking program, led to the identification of compounds 43 and 44, which mimic the interaction of the peptide substrate with the active site of SARS-CoV-1 3CLpro. Their interactions revealed that the inhibitors occupied S1′, S1, and S2, while S4 was not occupied. Studies also showed the occupancy of S4 by a hydrophobic group, and hydrogen bonding sites, such as the backbone of Glu166 and Gln192 sidechains.76 Tsai et al., carried out structure-based virtual screening for the identification of SARS-CoV-1 3CLpro inhibitors. They found 21 active compounds with IC50 values ≤30 μM. They selected twenty-eight compounds from the family with IC50 values that ranged from 3 to 1000 μM for 3D-QSAR studies. Among all the screened compounds, analog 45 showed good inhibitory activity due to its thiazole and benzene rings, which can undergo strong reactions with Glu166, Leu167, Pro168, and Gln192 from the protease.77 Two natural compounds, tannic acid and 3-isotheaflavin-3-gallate (TF2B), which belong to the family of natural polyphenols found in tea, showed SARS-3CLpro inhibitory activities. Extracted compounds from Puer and black tea were more potent than those from green and oolong teas. 3-Isotheaflavin-3-gallate (TF2B) (46), tannic acid, and theaflavin-3,3-digallate (TF3) (47), a theaflavin compound in black tea, showed potent activities against SARS-3CLpro, with IC50 values of 7, 3 and 9.5 μM, respectively.78 (Fig. 9)
000 compounds with Gold, the docking program, led to the identification of compounds 43 and 44, which mimic the interaction of the peptide substrate with the active site of SARS-CoV-1 3CLpro. Their interactions revealed that the inhibitors occupied S1′, S1, and S2, while S4 was not occupied. Studies also showed the occupancy of S4 by a hydrophobic group, and hydrogen bonding sites, such as the backbone of Glu166 and Gln192 sidechains.76 Tsai et al., carried out structure-based virtual screening for the identification of SARS-CoV-1 3CLpro inhibitors. They found 21 active compounds with IC50 values ≤30 μM. They selected twenty-eight compounds from the family with IC50 values that ranged from 3 to 1000 μM for 3D-QSAR studies. Among all the screened compounds, analog 45 showed good inhibitory activity due to its thiazole and benzene rings, which can undergo strong reactions with Glu166, Leu167, Pro168, and Gln192 from the protease.77 Two natural compounds, tannic acid and 3-isotheaflavin-3-gallate (TF2B), which belong to the family of natural polyphenols found in tea, showed SARS-3CLpro inhibitory activities. Extracted compounds from Puer and black tea were more potent than those from green and oolong teas. 3-Isotheaflavin-3-gallate (TF2B) (46), tannic acid, and theaflavin-3,3-digallate (TF3) (47), a theaflavin compound in black tea, showed potent activities against SARS-3CLpro, with IC50 values of 7, 3 and 9.5 μM, respectively.78 (Fig. 9)
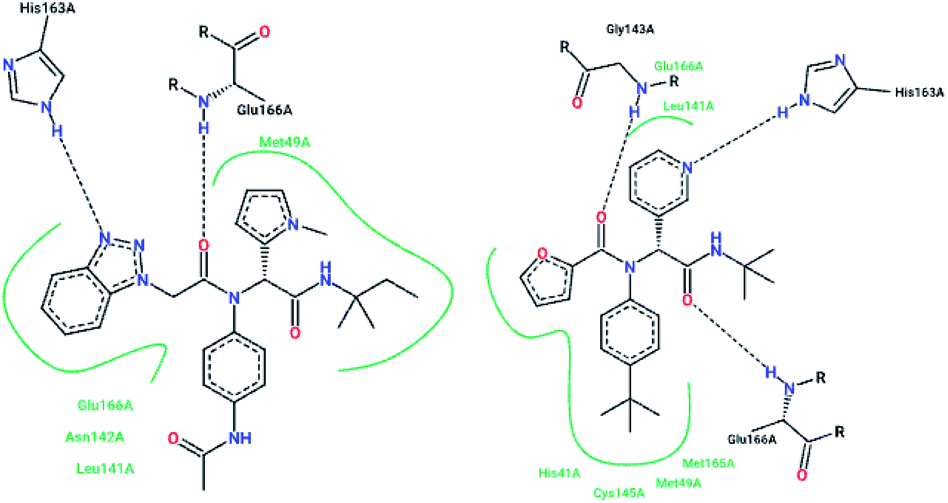
The high-throughput screening of NIH libraries for SARS 3CLpro inhibitors led to the identification of a noncovalent SARS-CoV-1 3CLpro inhibitor 48 (ML188) with an IC50 value of 1.5 μM, revealing a hydrogen bond interaction between the 3-pyridyl ring nitrogen of this compound and the active site His-163 sidechain located within the S1 subpocket. This analog specificity inhibits 3CLpro versus PLpro.79 In addition to compound 48, ML300 (49) shows inhibitory activity against 3CLpro. X-ray studies revealed the unique induced-fit reorganization of the S2–S4 binding pocket, with an IC50 value of 4.11 μM (Fig. 8). The optimization of these compounds led to the discovery of compound 50 with an IC50 value of 0.051 μM.80 A combination of virtual screening (VS) and high-throughput screening (HTS) techniques led to the identification of non-peptidic small molecule inhibitors against human SARS-CoV-1 3CLpro. 621000 compounds from the ZINC library were screened. This study resulted in the discovery of a new scaffold for further development (51).81 Mukherjee et al. carried out combined ligand and structure-based virtual screening against the Asinex platinum collection to identify inhibitors of the SARS-3CLpro enzyme, which resulted in activity against SARS-CoV-1 in whole-cell CPE assays. PJ207 (52) showed inhibitory activity against SARS-3CLpro, with an IC50 value of 39.4 μM and an EC50 value of 30 μM in virous infected cells. Hydrophobic interactions were identified between this analog and Thr25 and Leu27 of the S′1 site, and Met49 from S2, and Met165. Moreover, this analog showed additional hydrophobic interactions between the ethyl moiety of the ligand and Phe140 and Leu141.82 Virtual screening of noncovalent inhibitors led to the identification of calmidazolium (53), which is known as a calmodulin antagonist with SARS-3CLpro inhibition activity (Ki: 61 μM). The binding pose found by Dock4.0 for this compound was at the center of the cleft and in contact with the residue His41, Glu166, and Met165. This compound occupied approximately the S2, S1, and S′1 positions of the binding site. The biological activity of this compound is more related to the volume occupation of the pocket with hydrophobic interactions.83 A virtual screening and structure-based drug design study led to the discovery of the ZINC27332786 ligand (54). Molecular docking revealed that a pyridine group in this compound filled the S1 pocket with the formation of a hydrogen bond between the pyridine nitrogen and imidazole N–H of His163. The 1,2,4-triazole nitrogen forms a hydrogen bond with the backbone of Glu165/6. The hydrophobic S2 and S4 pockets were filled with two 2-chloro-6-fluorobenzyl rings. Another potent ligand was ZINC09411012 (55), which makes hydrogen bonds with His162, His/Gln163, Glu163, and Gln189.84 The screening of 50000 drug-like small molecules led to the identification of small molecules with SARS-3CLpro inhibitor activities (56, 57, 58, 59, and 60).85 A class of stable benzotriazole esters86 was reported, acting as noncovalent, competitive, and irreversible inhibitors against 3CLpro. Studies have shown that active site Cys145 was acylated, assisted by the catalytic dyad. The most potent compound was 61, with a Ki value of 7.5 nM. Docking studies revealed that the NH group of the indole moiety of this compound formed a hydrogen bond with the side chain OH of Thr25.87 Zhang et al. attempted structure-based design for the treatment of SARS-CoV-1 3CLpro, in which existing drugs used for the treatment of HIV, and psychotic and parasite diseases, like lopinavir, ritonavir, niclosamide, promazine, PNU, and UC2, were screened. The results showed that these drugs demonstrated SARS-CoV-1 3CLpro inhibition and could be used as templates for designing SARS-CoV-1 3CLpro inhibitors.88 104 small molecules were identified via the screening of 50240 compounds. Three analogs (62, 63, and 64) exhibited SARS-CoV-1 Mpro, helicase (Hel), and spike (S) protein-angiotensin-converting enzyme 2 (ACE2)-mediated viral entry, respectively. Compound 64 formed hydrogen bonds with Cys145 and His41.89 Active metal-conjugated inhibitors were tested against 3CLpro and analyzed crystallographically. Data revealed that the zinc ion in the zinc-centered complex 65 played an important role in targeting the catalytic residues via binding to the His41 and Cys145 catalytic dyad to yield zinc-centered tetrahedral geometry. Zinc atoms in this complex were chelated by two nitrogen and two oxygen atoms (Fig. 9).90
 | ||
| Fig. 8 The 2-D interaction profiles of the HTS hit compounds 49 (ML300; 4mds.pdb) and 48 (ML188; 3v3m.pdb) in the active site residue of SARS-CoV-1 3CLpro; hydrogen bonds are shown with broken lines, and hydrophobic interactions are shown in green. The picture was prepared using the “poseview” tool. | ||
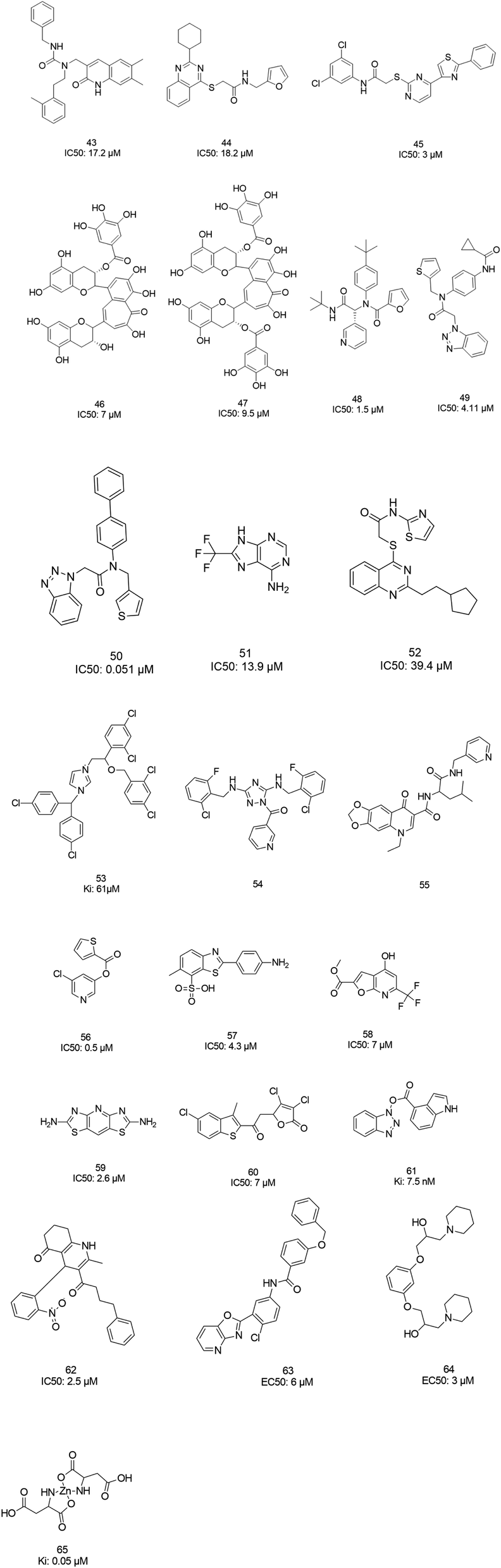 | ||
| Fig. 9 The chemical structures of 3CLpro inhibitors identified via virtual screening. | ||
Mechanism of epoxy ketone, boronic acid, and alpha-keto derivatives
It is interesting to examine the mechanism of action of protease inhibitors. Peptidyl epoxy ketones inhibit 3CLpro via the S-alkylation of the active site cysteine. Similarly, amino acid residues that can donate an electron pair to boron in protease inhibitors with boronic acid moieties undergo the chemical reaction91 that is shown in Fig. 10. Popular warheads, such as α-ketoacids, α-ketoesters, and α-ketoamides, are aldehyde substitutes, having the potential to form a tetrahedral transition state analog when an enzyme nucleophile is added.92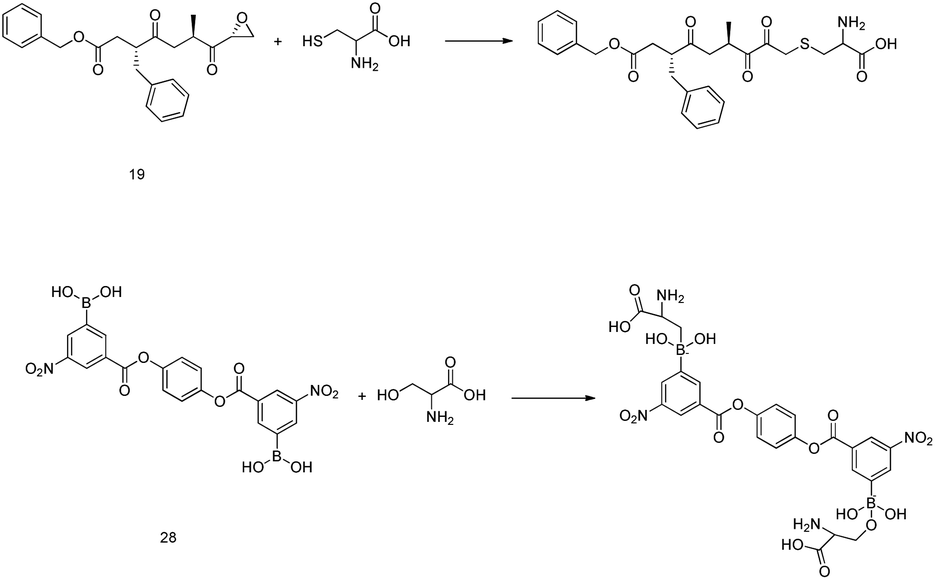 | ||
| Fig. 10 The reaction mechanisms of epoxy ketone and boronic acid derivatives. | ||
SARS-CoV-1 papain-like protease (PLpro)
The papain-like protease (PLpro), an essential enzyme for viral replication, has emerged as a drug target for SARS-CoV-1. SARS-CoV-1 PLpro has a catalytic triad, formed by Cys112, His273, and Asp287, and the presence of a zinc ion in the finger domain of PLpro, which is coordinated by four cysteine residues, is important for catalysis.27 The structures of inhibitors cocrystallized with PLpro enzymes, as solved via X-ray crystallography, provide information about the interaction profile.MeOH extracts of the fruits of the Paulownia tomentosa led to the identification of many small molecules that all contained a 3,4-dihydro-2H-pyran moiety with PLpro inhibitory activity. Compounds with a dihydro 2H-pyran group isolated from this natural source showed better inhibition properties than the parent compounds (66).93 Tanshinones derived from Salvia miltiorrhiza were tested against PLpro. All synthesized derivatives acted as time-dependent inhibitors of PLpro. Compound 67 was shown to be a simple reversible slow-binding inhibitor with mixed-type and selective inhibition properties.94 The inhibitory activities of Broussonetia papyrifera-derived polyphenols against 3CLpro and PLpro were tested, which resulted in potent compounds being isolated. Among these isolated compounds, analog 68 exhibited more noncompetitive inhibitory activity, with an IC50 value of 3.7 μM.95 Ethanol extracts of Psoralea corylifolia seeds led to the identification of six aromatic compounds with PLpro inhibitory activities in a dose-dependent manner, with IC50 values ranging from 4.2–38.4 μM. Compounds 69 and 70 were more potent inhibitors, with a reversible type I mechanism, based on analysis of KI and KIS values.96 Diarylheptanoids from Alnus japonica showed dose-dependent and reversible inhibition activities against SARS PLpro. Structure–activity analysis of one of the isolated diarylheptanoids (71) revealed the importance of the α,β-saturated carbonyl moiety in the molecule for its inhibitory activity.97 The high-throughput screening of a diverse chemical library and lead optimization resulted in the identification of a series of SARS PLpro inhibitors. Analogs 72 and 73 were more potent and showed nearly equivalent enzymatic inhibition and antiviral activity. Docking studies of these two compounds revealed that the flexible piperidine ring formed hydrogen bonds with the backbone carbonyl oxygen atoms of α-ketoacids, α-ketoesters, and α-ketoamides at Tyr269.98 Moreover, from the same high-throughput screening and optimization study, 74 was identified as a potent SARS inhibitor.99 The sensitive fluorescence-based high-throughput screening of 50080 compounds led to the identification of noncovalent inhibitors for PLpro. Compound 75 inhibited SARS-CoV-1 viral replication in Vero E6 cells with an EC50 value of 15 μM and showed no associated cytotoxicity (Fig. 11). The X-ray crystal structure of the compound 75-PLpro complex revealed that it occupied the S4–S3 subunits of the enzyme and induced loop closure that shuts down catalysis at the active site.100 Hydrogen bond interactions between 75 (GRL0617) and the Asp165 and Gln270 residues of the SARS-CoV-1 PLpro active site are shown in Fig. 12.
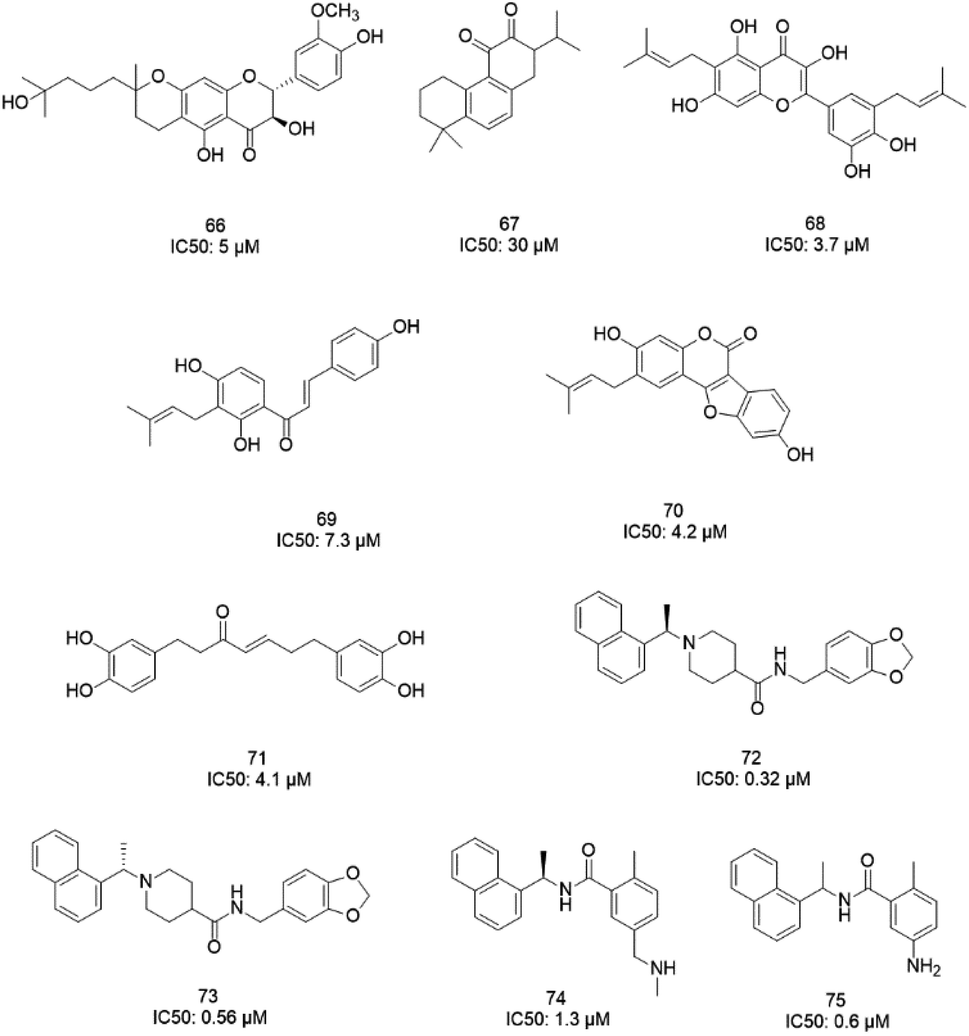 | ||
| Fig. 11 The chemical structures of PLpro inhibitors. | ||
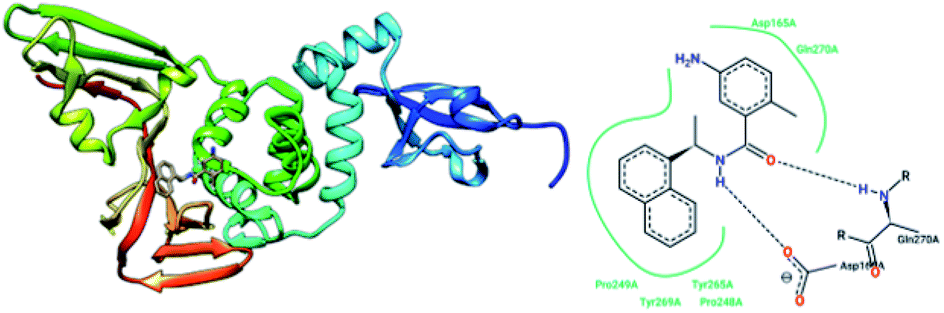 | ||
| Fig. 12 The X-ray structure of SARS-CoV-1 PLpro in complex with compound 75 (GRL0617; 3E9S.pdb) and a 2-D interaction profile of GRL0617 and the active site residues of SARS-CoV-1 PLpro; hydrogen bonds are shown as broken lines, and hydrophobic interactions are shown in green. The picture was prepared using the “poseview” tool. | ||
Other inhibitors
Molecular dynamic simulations and docking techniques were used for screening 29 approved and experimental drugs against SARS CoV proteinase. The results revealed that some existing HIV-1 protease inhibitors had high binding to SARS CoV proteinase. The carbonyl oxygen of L-700,417 (76) formed hydrogen bonds with the N–H atom of the imidazole ring of His41 and the amide nitrogen of Glu165.101 A series of bismuth complexes showed SARS coronavirus helicase ATPase and duplex unwinding activities at micromolar concentrations.102 Aryl diketoacids (ADK) are metal chelators and served as anti-viral agents that could target enzymes such as HIV-1 integrase103 and anti-HCV;104 they were evaluated as SARS CoV (SCV) NTPase/helicase (Hel) inhibitors. These analogs were tested via mimicking the binding mode of the bismuth complexes. These complexes are in competition with the Zn2+ ion binding sites in SCV Hel, which disrupts and inhibits both the NTPase and helicase activities. Results revealed that ADK analogs selectively inhibit the duplex DNA-unwinding activity without having a significant impact on the helicase ATPase activity.105 Based on SAR studies, Wang et al. proposed a pharmacophore model of SARS-CoV-1 NTPase/helicase, which constituted of a diketoacid core, a hydrophobic arylmethyl substituent, and a free catechol unit (77) (Fig. 13).106 | ||
| Fig. 13 The chemical structures of inhibitors with diverse mechanisms. | ||
Natural analogs
Glycyrrhizin (GL) (78), a natural and major pentacyclic triterpenoid glycoside from licorice root extracts,107 has SARS-CoV-1 inhibitory properties. However, Hoever et al. tested some derivatives of this compound and found that the amide groups in GL, the conjugation of GL with two amino acids of the enzyme, and free 30-COOH function increased the GL activity 70-fold against SARS-CoV-1.108 Several natural compounds were tested against SARS helicase, nsP13, and the hepatitis C virus (HCV) helicase, NS3h. Results revealed that none of the tested compounds inhibited the DNA unwinding activity or ATPase activity of the human HCV helicase protein, but the SARS-CoV-1 helicase protein was inhibited potently by myricetin (79) and scutellarein (80), which affected the ATPase activity but not the unwinding activity of nsP13. Moreover, these two compounds did not exhibit cytotoxicity against normal breast epithelial MCF10A cells.109 Nucleoside analogs with 6-chloropurine as the nucleobase were tested against SARS-CoV-1, and two compounds, 81 and 82, with better potency than the known anti-SARS-CoV-1 agents mizoribine and ribavirin were identified.110 Stilbene derivatives are known to exhibit a wide range of activities,111 such as anti-SARS activity. Li et al. evaluated the effects of some derivatives of stilbene on SARS-CoV-1-induced cytopathicity in Vero E6 cell cultures, resulting in the discovery of the analogs 83 and 84 as potent SARS-CoV-1 inhibitors with no significant cytotoxicity effects (Fig. 13).112Common polyphenol-type inhibitors from natural products that we discussed here are well-known frequent-hitters, which are reported to have many pharmacological activities. In general, the lack of selectivity and low potency of these analogues against the given target make them less attractive prospects.
Conclusions and future perspectives
The emergence of SARS, and recently COVID-19, which arise from β-B coronaviruses, have led to the deaths of hundreds of people all over the world.5 Since there is still no effective drug for the treatment of COVID-19, and since SARS and COVID-19 arise from the same type of coronavirus and show 80% sequence homology,8,9 we have reviewed compounds that were active against SARS-CoV-1 and their specific interactions with the virus molecular targets. The first generation of peptidomimetic structures of 3CLpro inhibitors are useful for the treatment of SARS, making irreversible and covalent bonds with enzymes.40,41 Studies have demonstrated that covalent inhibitors lead to adverse drug responses, off-target side effects, toxicity, and lower potency.53 Moreover, isatin, a chemical compound that is derived from indole, showed potent and selective inhibitory activity against 3CLpro, with potency in the micromolar to nanomolar range.62–64 Furthermore, natural compounds from the Paulownia tree and Salvia miltiorrhiza were tested against PLpro, and potent and selective PLpro inhibitory activities were revealed.93,94In this paper, we present the most promising derivatives described in the literature for further optimization efforts. Among peptidomimetics inhibitors, compounds 16, 17, 18, and 24 are the most potent peptidomimetics and exhibited excellent cellular potencies against 3CLpro, among the examples in the reviewed literature.42,58,59 In addition, analog 37 was the most potent SARS-CoV-1 3CLpro trans/cis-cleavage inhibitor, with IC50 values of 2.7 and 68.1 μM, respectively, and no toxicity.70 An isatin analog, 26, underwent noncovalent, reversible interactions with the active site of the protease and showed more selective inhibitory activity against 3CLpro compared to papain, chymotrypsin, and trypsin.64 ML188 (48) is a promising example of a noncovalent SARS-CoV-1 CLpro inhibitor with an IC50 value of 1.5 μM, which specifically inhibited 3CLpro versus PLpro.79 Moreover, ML300 is another noncovalent 3CLpro inhibitor with an IC50 value of 4.11 μM.80 Compound 65 was identified as a PLpro inhibitor, which inhibits viral replication in Vero E6 cells without any significant cytotoxicity.98 Myricetin and scutellarein (79 and 80) inhibited SARS-CoV-1 helicase protein activity, potently, via affecting ATPase activity without exhibiting cytotoxicity against normal breast epithelial MCF10A cells.89 Compounds 81 and 82, which are nucleoside analogs that have 6-chloropurine as the nucleobase, showed more potency than mizoribine and ribavirin. Derivatives of Aryl diketoacids (ADK) selectively inhibited duplex DNA-unwinding activity without having a significant impact on helicase ATPase activity.105 Compounds 83 and 84, stilbene analogs, acted as potent SARS-CoV-1 inhibitors with no significant cytotoxicity.112 In conclusion, despite the huge efforts of researchers, an effective drug for the treatment of SARS has not been found. We hope that this paper will help medicinal chemists to identify novel anti-SARS-CoV-2 inhibitors and subsequently lead to the discovery of an effective therapy for COVID-19.
Conflicts of interest
The authors declare that there are no conflicts of interest.References
- L. S. Oshiro, J. H. Schieble and E. H. Lennette, J. Gen. Virol., 1971, 12, 161–168 CrossRef CAS PubMed.
- A. R. Fehr and S. Perlman, Coronaviruses, 2015, 1282, 1–23 CrossRef CAS PubMed.
- M. Letko, A. Marzi and V. Munster, Nat. Microbiol., 2020, 5, 562–569 CrossRef CAS PubMed.
- T. Pillaiyar and S. Meenakshisundaram, Drug Discovery Today, 2020, 25, 668–688 CrossRef CAS PubMed.
- K. Nakagawa, K. G. Lokugamage and S. Makino, Adv. Virus Res., 2016, 96, 165–185 CAS.
- R. J. d. Groot, S. C. Baker, R. S. Baric, C. S. Brown, C. Drosten, L. Enjuanes, R. A. M. Fouchier, M. Galiano, A. E. Gorbalenya, Z. A. Memish, S. Perlman, L. L. M. Poon, E. J. Snijder, G. M. Stephens, P. C. Y. Woo, A. M. Zaki, M. Zambon and J. Ziebuhr, J. Virol., 2013, 87, 7790–7792 CrossRef PubMed.
- S. v. Boheemen, M. d. Graaf, C. Lauber, T. M. Bestebroer, V. S. Raj, A. M. Zaki, A. D. M. E. Osterhaus, B. L. Haagmans, A. E. Gorbalenya, E. J. Snijder and R. A. M. Fouchiera, mBio, 2012, 3, e00473 Search PubMed.
- H. Lee, H. Lei, B. D. Santarsiero, J. L. Gatuz, S. Cao, A. J. Rice, K. Patel, M. Z. Szypulinski, I. Ojeda, A. K. Ghosh and M. E. Johnson, ACS Chem. Biol., 2015, 10, 1456–1465 CrossRef CAS PubMed.
- A. Grifoni, J. Sidney, Y. Zhang, R. H. Scheuermann and B. Peters, Cell Host Microbe, 2020, 27, 671–680 CrossRef CAS PubMed.
- T. R. Tong, Recent Pat. Anti-Infect. Drug Discovery, 2006, 1, 297–308 CrossRef CAS PubMed.
- A. J. Turner, S. R. Tipnis, J. L. Guy, G. Rice and N. M. Hooper, Can. J. Physiol. Pharmacol., 2002, 80, 346–353 CrossRef CAS PubMed.
- I. Hamming, W. Timens, M. L. C. Bulthuis, A. T. Lely, G. J. Navis and H. van Goor, J. Pathol., 2004, 203, 631–637 CrossRef CAS PubMed.
- P. Verdecchia, C. Cavallini, A. Spanevello and F. Angeli, Eur. J. Intern. Med., 2020, 76, 14–20 CrossRef CAS PubMed.
- C. Lei, K. Qian, T. Li, S. Zhang, W. Fu, M. Ding and S. Hu, Nat. Commun., 2020, 11, 1–5 Search PubMed.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 581, 221–224 CrossRef CAS PubMed.
- Z. Li, A. C. Tomlinson, A. H. Wong, D. Zhou, M. Desforges, P. J. Talbot, S. Benlekbir, J. L. Rubinstein and J. M. Rini, eLife, 2019, 8, e51230 CrossRef CAS PubMed.
- S. Belouzard, J. K. Millet, B. N. Licitra and G. R. Whittaker, Viruses, 2012, 4, 1011–1033 CrossRef CAS PubMed.
- J. A. Tanner, R. M. Watt, Y.-B. Chai, L.-Y. Lu, M. C. Lin, J. S. M. Peiris, L. L. M. Poon, H.-F. Kung and J.-D. Huang, J. Biol. Chem., 2003, 278, 39578–39582 CrossRef CAS PubMed.
- T. Muramatsu, C. Takemoto, Y.-T. Kim, H. Wang, W. Nishii, T. Terada, M. Shirouzu and S. Yokoyama, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12997–13002 CrossRef CAS PubMed.
- Y.-S. Wu, W.-H. Lin, J. T.-A. Hsu and H.-P. Hsieh, Curr. Med. Chem., 2006, 13, 2003–2020 CrossRef CAS PubMed.
- V. Thiel, K. A. Ivanov, Á. Putics, T. Hertzig, B. Schelle, S. Bayer, B. Weißbrich, E. J. Snijder, H. Rabenau, H. W. Doerr, A. E. Gorbalenya and J. Ziebuhr, J. Gen. Virol., 2003, 84, 2305–2315 CrossRef CAS PubMed.
- J. Shi, J. Sivaraman and J. Song, J. Virol., 2008, 82, 4620–4629 CrossRef CAS PubMed.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld and Z. Rao, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS PubMed.
- K. Akaji, H. Konno, H. Mitsui, K. Teruya, Y. Shimamoto, Y. Hattori, T. Ozaki, M. Kusunoki and A. Sanjoh, J. Med. Chem., 2011, 54, 7962–7973 CrossRef CAS PubMed.
- S. Jiang, Y. He and S. Liu, Emerging Infect. Dis., 2005, 11, 1016–1020 CrossRef CAS PubMed.
- Y.-Q. Cheng, ChemBioChem, 2006, 7, 471–477 CrossRef CAS PubMed.
- Y. M. Báez-Santos, S. E. St John and A. D. Mesecar, Antiviral Res., 2015, 115, 21–38 CrossRef PubMed.
- M. Pachetti, B. Marini, F. Benedetti, F. Giudici, E. Mauro, P. Storici, C. Masciovecchio, S. Angeletti, M. Ciccozzi, R. C. Gallo, D. Zella and R. Ippodrino, J. Transl. Med., 2020, 18, 179 CrossRef CAS PubMed.
- S. A. Khan, K. Zia, S. Ashraf, R. Uddin and Z. Ul-Haq, J. Biomol. Struct. Dyn., 2020, 1–10 Search PubMed.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, Y. Wang, Q. Wang, Y. Xu, M. Li, X. Li, M. Zheng, L. Chen and H. Li, Acta Pharm. Sin. B, 2020, 10, 766–788 CrossRef PubMed.
- N. Bung, S. R. Krishnan, G. Bulusu and A. Roy, ChemRxiv, 2020 DOI:10.26434/chemrxiv.11998347.v2.
- H. Yang, M. Bartlam and Z. Rao, Curr. Pharm. Des., 2006, 12, 4573–4590 CrossRef CAS PubMed.
- B. Liu and J. Zhou, J. Comput. Chem., 2005, 26, 484–490 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- M. S. Lall, Y. K. Ramtohul, M. N. G. James and J. C. Vederas, J. Org. Chem., 2002, 67, 1536–1547 CrossRef CAS PubMed.
- L. S. Zalman, M. A. Brothers, P. S. Dragovich, R. Zhou, T. J. Prins, S. T. Worland and A. K. Patick, Antimicrob. Agents Chemother., 2000, 44, 1236–1241 CrossRef CAS PubMed.
- S. Sirois, R. Zhang, W. Gao, H. Gao, Y. Li, H. Zheng and D. Wei, Curr. Comput.-Aided Drug Des., 2007, 3, 191–200 CrossRef CAS.
- P. Wei, K. Fan, H. Chen, L. Ma, C. Huang, L. Tan, D. Xi, C. Li, Y. Liu, A. Cao and L. Lai, Biochem. Biophys. Res. Commun., 2006, 339, 865–872 CrossRef CAS PubMed.
- L. Ding, X.-X. Zhang, P. Wei, K. Fan and L. Lai, Anal. Biochem., 2005, 343, 159–165 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- S. Yang, S.-J. Chen, M.-F. Hsu, J.-D. Wu, C.-T. K. Tseng, Y.-F. Liu, H.-C. Chen, C.-W. Kuo, C.-S. Wu, L.-W. Chang, W.-C. Chen, S.-Y. Liao, T.-Y. Chang, H.-H. Hung, H.-L. Shr, C.-Y. Liu, Y.-A. Huang, L.-Y. Chang, J.-C. Hsu, C. J. Peters, A. H.-J. Wang and M.-C. Hsu, J. Med. Chem., 2006, 49, 4971–4980 CrossRef CAS PubMed.
- R. Jain, H. Pettersson, J. Zhang, K. Aull, P. Fortin, C. Huitema, L. Eltis, J. Parrish, M. James, D. Wishart and J. Vederas, J. Med. Chem., 2004, 47, 6113–6116 CrossRef CAS PubMed.
- J. Zhang, H. I. Pettersson, C. Huitema, C. Niu, J. Yin, M. N. G. James, L. D. Eltis and J. C. Vederas, J. Med. Chem., 2007, 50, 1850–1864 CrossRef CAS PubMed.
- A. Ghosh, K. Ratia, B. Santarsiero, B. Harcourt, P. Rota, S. Baker, M. E. Johnson, A. D. Mesecar and A. Ghosh, J. Med. Chem., 2005, 48, 6767–6771 CrossRef CAS PubMed.
- X. Xue, H. Yu, H. Yang, F. Xue, Z. Wu, W. Shen, J. Li, Z. Zhou, Y. Ding, Q. Zhao, X. C. Zhang, M. Liao, M. Bartlam and Z. Rao, J. Virol., 2008, 82, 2515–2527 CrossRef CAS PubMed.
- A. K. Ghosh, K. Xi, V. Grum-Tokars, X. Xu, K. Ratia, W. Fu, K. V. Houser, S. C. Baker, M. E. Johnson and A. D. Mesecar, Bioorg. Med. Chem. Lett., 2007, 17, 5876–5880 CrossRef CAS PubMed.
- L. Zhu, S. George, M. F. Schmidt, S. I. Al-Gharabli, J. Rademann and R. Hilgenfeld, Antiviral Res., 2011, 92, 204–212 CrossRef CAS PubMed.
- D. H. Goetz, Y. Choe, E. Hansell, Y. T. Chen, M. McDowell, C. B. Jonsson, W. R. Roush, J. McKerrow and C. S. Craik, Biochemistry, 2007, 46, 8744–8752 CrossRef CAS PubMed.
- H.-Z. Zhang, H. Zhang, W. Kemnitzer, B. Tseng, C. Jindrich, M. Michaelis, H. W. Doerr and S. X. Cai, J. Med. Chem., 2006, 49, 1198–1201 CrossRef CAS PubMed.
- M. O. Sydnes, Y. Hayashi, V. K. Sharma, T. Hamada, U. Bacha, J. Barrila, E. Freire and Y. Kiso, Tetrahedron, 2006, 62, 8601–8609 CrossRef CAS PubMed.
- A. Paasche, A. Zipper, S. Schäfer, J. Ziebuhr, T. Schirmeister and B. Engels, Biochemistry, 2014, 53, 5930–5946 CrossRef CAS PubMed.
- A. K. Ghosh, K. Xi, M. E. Johnson, S. C. Baker and A. D. Mesecar, Annu. Rep. Med. Chem., 2006, 41, 183–196 CAS.
- A. Tuley and W. Fast, Biochemistry, 2018, 57, 3326–3337 CrossRef CAS PubMed.
- B. Turk, Nat. Rev. Drug Discovery, 2006, 5, 785–799 CrossRef CAS PubMed.
- A. K. Ghosh, G. Gong, V. Grum-Tokars, D. C. Mulhearn, S. C. Baker, M. Coughlin, B. S. Prabhakar, K. Sleeman, M. E. Johnson and A. D. Mesecar, Bioorg. Med. Chem. Lett., 2008, 18, 5684–5688 CrossRef CAS PubMed.
- S. I. Al-Gharabli, S. T. A. Shah, S. Weik, M. F. Schmidt, J. R. Mesters, D. Kuhn, G. Klebe, R. Hilgenfeld and J. Rademann, ChemBioChem, 2006, 7, 1048–1055 CrossRef CAS PubMed.
- P. Thanigaimalai, S. Konno, T. Yamamoto, Y. Koiwai, A. Taguchi, K. Takayama, F. Yakushiji, K. Akaji, Y. Kiso, Y. Kawasaki, S.-E. Chen, A. Naser-Tavakolian, A. Schön, E. Freire and Y. Hayashi, Eur. J. Med. Chem., 2013, 65, 436–447 CrossRef CAS PubMed.
- S. Konno, P. Thanigaimalai, T. Yamamoto, K. Nakada, R. Kakiuchi, K. Takayama, Y. Yamazaki, F. Yakushiji, K. Akaji, Y. Kiso, Y. Kawasaki, S.-E. Chen, E. Freire and Y. Hayashi, Bioorg. Med. Chem., 2013, 21, 412–424 CrossRef CAS PubMed.
- P. Thanigaimalai, S. Konno, T. Yamamoto, Y. Koiwai, A. Taguchi, K. Takayama, F. Yakushiji, K. Akaji, S.-E. Chen, A. Naser-Tavakolian, A. Schön, E. Freire and Y. Hayashi, Eur. J. Med. Chem., 2013, 68, 372–384 CrossRef CAS PubMed.
- T. Regnier, D. Sarma, K. Hidaka, U. Bacha, E. Freire, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2009, 19, 2722–2727 CrossRef CAS PubMed.
- E. Martina, N. Stiefl, B. Degel, F. Schulz, A. Breuning, M. Schiller, R. Vicik, K. Baumann, J. Ziebuhr and T. Schirmeister, Bioorg. Med. Chem. Lett., 2005, 15, 5365–5369 CrossRef CAS PubMed.
- Y.-M. Shao, W.-B. Yang, T.-H. Kuo, K.-C. Tsai, C.-H. Lin, A.-S. Yang, P.-H. Liang and C.-H. Wong, Bioorg. Med. Chem., 2008, 16, 4652–4660 CrossRef CAS PubMed.
- L.-R. Chen, Y.-C. Wang, Y. W. Lin, S.-Y. Chou, S.-F. Chen, L. T. Liu, Y.-T. Wu, C.-J. Kuo, T. S.-S. Chen and S.-H. Juang, Bioorg. Med. Chem. Lett., 2005, 15, 3058–3062 CrossRef CAS PubMed.
- L. Zhou, Y. Liu, W. Zhang, P. Wei, C. Huang, J. Pei, Y. Yuan and L. Lai, J. Med. Chem., 2006, 49, 3440–3443 CrossRef CAS PubMed.
- W. Liu, H.-M. Zhu, G.-J. Niu, E.-Z. Shi, J. Chen, B. Sun, W.-Q. Chen, H.-G. Zhou and C. Yang, Bioorg. Med. Chem., 2014, 22, 292–302 CrossRef CAS PubMed.
- U. Bacha, J. Barrila, A. Velazquez-Campoy, S. A. Leavitt and E. Freire, Biochemistry, 2004, 43, 4906–4912 CrossRef CAS PubMed.
- R. Ramajayam, K.-P. Tan, H.-G. Liu and P.-H. Liang, Bioorg. Med. Chem. Lett., 2010, 20, 3569–3572 CrossRef CAS PubMed.
- R. Ramajayam, K.-P. Tan, H.-G. Liu and P.-H. Liang, Bioorg. Med. Chem., 2010, 18, 7849–7854 CrossRef CAS PubMed.
- V. Kumar, K.-P. Tan, Y.-M. Wang, S.-W. Lin and P.-H. Liang, Bioorg. Med. Chem., 2016, 24, 3035–3042 CrossRef CAS PubMed.
- L. Chen, J. Li, C. Luo, H. Liu, W. Xu, G. Chen, O. W. Liew, W. Zhu, C. M. Puah, X. Shen and H. Jiang, Bioorg. Med. Chem., 2006, 14, 8295–8306 CrossRef CAS PubMed.
- Y. B. Ryu, S.-J. Park, Y. M. Kim, J.-Y. Lee, W. D. Seo, J. S. Chang, K. H. Park, M.-C. Rho and W. S. Lee, Bioorg. Med. Chem. Lett., 2010, 20, 1873–1876 CrossRef CAS PubMed.
- J.-Y. Park, J. H. Kim, J. M. Kwon, H.-J. Kwon, H. J. Jeong, Y. M. Kim, D. Kim, W. S. Lee and Y. B. Ryu, Bioorg. Med. Chem., 2013, 21, 3730–3737 CrossRef CAS PubMed.
- C. Niu, J. Yin, J. Zhang, J. C. Vederas and M. N. G. James, Bioorg. Med. Chem., 2008, 16, 293–302 CrossRef CAS PubMed.
- L. Wang, B.-B. Bao, G.-Q. Song, C. Chen, X.-M. Zhang, W. Lu, Z. Wang, Y. Cai, S. Li, S. Fu, F.-H. Song, H. Yang and J.-G. Wang, Eur. J. Med. Chem., 2017, 137, 450–461 CrossRef CAS PubMed.
- Y. B. Ryu, H. J. Jeong, J. H. Kim, Y. M. Kim, J.-Y. Park, D. Kim, T. T. H. Naguyen, S.-J. Park, J. S. Chang, K. H. Park, M.-C. Rho and W. S. Lee, Bioorg. Med. Chem., 2010, 18, 7940–7947 CrossRef CAS PubMed.
- P. Mukherjee, P. Desai, L. Ross, E. L. White and M. A. Avery, Bioorg. Med. Chem., 2008, 16, 4138–4149 CrossRef CAS PubMed.
- K.-C. Tsai, S.-Y. Chen, P.-H. Liang, I.-L. Lu, N. Mahindroo, H.-P. Hsieh, Y.-S. Chao, L. Liu, D. Liu, W. Lien, T.-H. Lin and S.-Y. Wu, J. Med. Chem., 2006, 49, 3485–3495 CrossRef CAS PubMed.
- C.-N. Chen, C. P. C. Lin, K.-K. Huang, W.-C. Chen, H.-P. Hsieh, P.-H. Liang and J. T.-A. Hsu, Altern. Med., 2005, 2, 209–215 Search PubMed.
- J. Jacobs, V. Grum-Tokars, Y. Zhou, M. Turlington, S. A. Saldanha, P. Chase, A. Eggler, E. S. Dawson, Y. M. Baez-Santos, S. Tomar, A. M. Mielech, S. C. Baker, C. W. Lindsley, P. Hodder, A. Mesecar and S. R. Stauffer, J. Med. Chem., 2013, 56, 534–546 CrossRef CAS PubMed.
- M. Turlington, A. Chun, S. Tomar, A. Eggler, V. Grum-Tokars, J. Jacobs, J. S. Daniels, E. Dawson, A. Saldanha, P. Chase, Y. M. Baez-Santos, C. W. Lindsley, P. Hodder, A. D. Mesecar and S. R. Stauffer, Bioorg. Med. Chem. Lett., 2013, 23, 6172–6177 CrossRef CAS PubMed.
- H. Lee, A. Mittal, K. Patel, J. L. Gatuz, L. Truong, J. Torres, D. C. Mulhearn and M. E. Johnson, Bioorg. Med. Chem., 2014, 22, 167–177 CrossRef CAS PubMed.
- P. Mukherjee, F. Shah, P. Desai and M. Avery, J. Chem. Inf. Model., 2011, 51, 1376–1392 CrossRef CAS PubMed.
- Z. Liu, C. Huang, K. Fan, P. Wei, H. Chen, S. Liu, J. Pei, L. Shi, B. Li, K. Yang, Y. Liu and L. Lai, J. Chem. Inf. Model., 2005, 45, 10–17 CrossRef CAS PubMed.
- M. Berry, B. C. Fielding and J. Gamieldien, Viruses, 2015, 7, 6642–6660 CrossRef CAS PubMed.
- J. E. Blanchard, N. H. Elowe, C. Huitema, P. D. Fortin, J. D. Cechetto, L. D. Eltis and E. D. Brown, Chem. Biol., 2004, 11, 1445–1453 CrossRef CAS PubMed.
- K. H. G. Verschueren, K. Pumpor, S. Anemüller, S. Chen, J. R. Mesters and R. Hilgenfeld, Chem. Biol., 2008, 15, 597–606 CrossRef CAS PubMed.
- C.-Y. Wu, K.-Y. King, C.-J. Kuo, J.-M. Fang, Y.-T. Wu, M.-Y. Ho, C.-L. Liao, J.-J. Shie, P.-H. Liang and C.-H. Wong, Chem. Biol., 2006, 13, 261–268 CrossRef CAS PubMed.
- X. W. Zhang and Y. L. Yap, Bioorg. Med. Chem., 2004, 12, 2517–2521 CrossRef CAS PubMed.
- R. Y. Kao, W. H. W. Tsui, T. S. W. Lee, J. A. Tanner, R. M. Watt, J.-D. Huang, L. Hu, G. Chen, Z. Chen, L. Zhang, T. He, K.-H. Chan, H. Tse, A. P. C. To, L. W. Y. Ng, B. C. W. Wong, H.-W. Tsoi, D. Yang, D. D. Ho and K.-Y. Yuen, Chem. Biol., 2004, 11, 1293–1299 CrossRef CAS PubMed.
- C.-C. Lee, C.-J. Kuo, M.-F. Hsu, P.-H. Liang, J.-M. Fang, J.-J. Shie and A. H.-J. Wang, FEBS Lett., 2007, 581, 5454–5458 CrossRef CAS PubMed.
- G. F. Whyte, R. Vilar and R. Woscholski, J. Chem. Biol., 2013, 6, 161–174 CrossRef PubMed.
- M. Siklos, M. BenAissa and G. R. J. Thatcher, Acta Pharm. Sin. B, 2015, 5, 506–519 CrossRef PubMed.
- J. K. Cho, M. J. Curtis-Long, K. H. Lee, D. W. Kim, H. W. Ryu, H. J. Yuk and K. H. Park, Bioorg. Med. Chem., 2013, 21, 3051–3057 CrossRef CAS PubMed.
- J.-Y. Park, J. H. Kim, Y. M. Kim, H. J. Jeong, D. W. Kim, K. H. Park, H.-J. Kwon, S.-J. Park, W. S. Lee and Y. B. Ryu, Bioorg. Med. Chem., 2012, 20, 5928–5935 CrossRef CAS PubMed.
- J.-Y. Park, H. J. Yuk, H. W. Ryu, S. H. Lim, K. S. Kim, K. H. Park, Y. B. Ryu and W. S. Lee, J. Enzyme Inhib. Med. Chem., 2017, 32, 504–512 CrossRef CAS PubMed.
- D. W. Kim, K. H. Seo, M. J. Curtis-Long, K. Y. Oh, J.-W. Oh, J. K. Cho, K. H. Lee and K. H. Park, J. Enzyme Inhib. Med. Chem., 2014, 29, 59–63 CrossRef CAS PubMed.
- J.-Y. Park, H. J. Jeong, J. H. Kim, Y. M. Kim, S.-J. Park, D. Kim, K. H. Park, W. S. Lee and Y. B. Ryu, Biol. Pharm. Bull., 2012, 35, 2036–2042 CrossRef CAS PubMed.
- A. K. Ghosh, J. Takayama, K. V. Rao, K. Ratia, R. Chaudhuri, D. C. Mulhearn, H. Lee, D. B. Nichols, S. Baliji, S. C. Baker, M. E. Johnson and A. D. Mesecar, J. Med. Chem., 2010, 53, 4968–4979 CrossRef CAS PubMed.
- A. K. Ghosh, J. Takayama, Y. Aubin, K. Ratia, R. Chaudhuri, Y. Baez, K. Sleeman, M. Coughlin, D. B. Nichols, D. C. Mulhearn, B. S. Prabhakar, S. C. Baker, M. E. Johnson and A. D. Mesecar, J. Med. Chem., 2009, 52, 5228–5240 CrossRef CAS PubMed.
- K. Ratia, S. Pegan, J. Takayama, K. Sleeman, M. Coughlin, S. Baliji, R. Chaudhuri, W. Fu, B. S. Prabhakar, M. E. Johnson, S. C. Baker, A. K. Ghosh and A. D. Mesecar, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16119–16124 CrossRef CAS PubMed.
- E. Jenwitheesuk and R. Samudrala, Bioorg. Med. Chem. Lett., 2003, 13, 3989–3992 CrossRef CAS PubMed.
- N. Yang, J. A. Tanner, Z. Wang, J.-D. Huang, B.-J. Zheng, N. Zhu and H. Sun, Chem. Commun., 2007, 4413–4415 RSC.
- D. J. Hazuda, P. Felock, M. Witmer, A. Wolfe, K. Stillmock, J. A. Grobler, A. Espeseth, L. Gabryelski, W. Schleif, C. Blau and M. D. Miller, Science, 2000, 287, 646–650 CrossRef CAS PubMed.
- J. Kim, K.-S. Kim, H. S. Lee, K.-S. Park, S. Y. Park, S.-Y. Kang, S. J. Lee, H. S. Park, D.-E. Kim and Y. Chong, Bioorg. Med. Chem. Lett., 2008, 18, 4661–4665 CrossRef CAS PubMed.
- C. Lee, J. M. Lee, N.-R. Lee, B.-S. Jin, K. J. Jang, D.-E. Kim, Y.-J. Jeong and Y. Chong, Bioorg. Med. Chem. Lett., 2009, 19, 1636–1638 CrossRef CAS PubMed.
- C. Lee, J. M. Lee, N.-R. Lee, D.-E. Kim, Y.-J. Jeong and Y. Chong, Bioorg. Med. Chem. Lett., 2009, 19, 4538–4541 CrossRef CAS PubMed.
- R.-Y. Huang, Y.-L. Chu, Z.-B. Jiang, X.-M. Chen, X. Zhang and X. Zeng, Cell. Physiol. Biochem., 2014, 33, 375–388 CrossRef CAS PubMed.
- G. Hoever, L. Baltina, M. Michaelis, R. Kondratenko, L. Baltina, G. A. Tolstikov, H. W. Doerr and J. Cinatl, J. Med. Chem., 2005, 48, 1256–1259 CrossRef CAS PubMed.
- M.-S. Yu, J. Lee, J. M. Lee, Y. Kim, Y.-W. Chin, J.-G. Jee, Y.-S. Keum and Y.-J. Jeong, Bioorg. Med. Chem. Lett., 2012, 22, 4049–4054 CrossRef CAS PubMed.
- M. Ikejiri, M. Saijo, S. Morikawa, S. Fukushi, T. Mizutani, I. Kurane and T. Maruyama, Bioorg. Med. Chem. Lett., 2007, 17, 2470–2473 CrossRef CAS PubMed.
- S. K. Ko, W. K. Whang and I. H. Kim, Arch. Pharmacal Res., 1995, 18, 282–288 CrossRef CAS.
- Y.-Q. Li, Z.-L. Li, W.-J. Zhao, R.-X. Wen, Q.-W. Meng and Y. Zeng, Eur. J. Med. Chem., 2006, 41, 1084–1089 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |