
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow iMALDI can improve clinical diagnostics
R.
Popp
a,
M.
Basik
b,
A.
Spatz
b,
G.
Batist
b,
R. P.
Zahedi
 bc and
C. H.
Borchers
*abc
bc and
C. H.
Borchers
*abc
aUniversity of Victoria Genome British Columbia Proteomics Centre, University of Victoria, Victoria, British Columbia V8Z 7X8, Canada. E-mail: christoph@proteincentre.com
bGerald Bronfman Department of Oncology, Jewish General Hospital, McGill University, 5100 de Maisonneuve Boulevard West, Suite 720, Montreal, Quebec H4A 3T2, Canada
cSegal Cancer Proteomics Centre, Lady Davis Institute, Jewish General Hospital, McGill University, 3755 Côte-Sainte-Catherine Road, Montreal, Quebec H3T 1E2, Canada
First published on 1st May 2018
Abstract
Protein mass spectrometry (MS) is an indispensable tool to detect molecular signatures that can be associated with cellular dysregulation and disease. Despite its huge success in the life sciences, where it has led to novel insights into disease mechanisms and the identification of potential protein biomarkers, protein MS is rarely used for clinical protein assays. While conventional matrix-assisted laser desorption/ionization (MALDI) MS is not compatible with complex samples, liquid chromatography-MS (LC-MS)-based assays may be too complex and may lack the robustness and ease of automation required for routine use in the clinic. Therefore, clinical protein assays are dominated by immunohistochemistry and immunoassays which, however, often lack standardization and fully depend on antibody specificity. Immuno-MALDI (iMALDI) MS may overcome these hurdles by utilizing anti-peptide antibodies for the specific enrichment of targeted analytes and on-target detection of the captured analytes, thus combining the unique properties of MS for the unambiguous detection and quantitation of analytes with a workflow that can be fully automated. Here we discuss the requirements for clinical protein assays, the pitfalls of existing methods, how iMALDI has been successfully used to quantify endogenous peptides and proteins from clinical samples, as well as its potential as a powerful tool for companion diagnostics in the light of precision medicine.
The demand for clinical diagnostic assays
In the era of precision medicine there is a huge demand for novel, robust, and cost-effective diagnostic and prognostic assays that allow the measurement of molecular signatures, thus allowing detection of the early stages of disease and, ideally, guiding therapy. In general, molecular markers must either represent known biology, such as disease mechanisms, or must correlate with a measurable outcome, such as tumour shrinkage/growth or overall survival.1 For instance, HER2, a member of the human epidermal growth factor receptor (ErbB) family with tyrosine kinase activity, is overexpressed in 25–30% of human breast cancers.2,3 HER2 overexpression can be associated with aggressive disease and poor prognosis,4 whereas HER2 gene amplification has been associated with resistance to certain therapeutic approaches.5,6 A variety of HER2 assays are available, determining either HER2 protein expression using immunohistochemistry (IHC) with monoclonal or polyclonal antibodies, or analysing the HER2 gene status using (fluorescence) in situ hybridization (ISH/FISH).7 Besides HER2, there are only a few proteins that have become FDA-approved protein tumour markers and are currently in use in clinical practice.8 There are, however, more than 50 biomarkers that are currently listed by the FDA as “pharmacogenomics biomarkers” that may serve to select the treatment for patients in a variety of fields such as oncology, infectious disease, psychiatry, gastroenterology, rheumatology, neurology, cardiology, or haematology.In the clinic, proteins are mainly measured using IHC or immunoassays (IA) such as the enzyme-linked immunosorbent assay (ELISA). Whereas IHC measurements provide and visualize relevant spatial information, the reason for the predominant use of IA is because they (i) are well-established and accepted in the field, (ii) easy-to-use, (iii) do not require sophisticated and expensive equipment that is complicated to operate, (iv) do not require extensive training of operators, (v) provide a high throughput due to parallelization, and (vi) provide high sensitivity.9 The sensitivity, however, depends on the antibody and sample (matrix effects) at hand.
In general, however, antibodies can lack specificity and in the past have led to the unnecessary treatment of healthy individuals.10 They may fail to recognize an antigen for instance due to unusual post-translational modification (PTM) patterns or, indeed, may fail to detect small but potentially disease-relevant differences between antigens (e.g., due to PTM). Antibodies may also suffer from matrix-dependent cross-reactivity and autoantibodies may strongly interfere with IA, as has been reported for thyroglobulin.11 High concentrations of antigen may lead to saturation effects and consequent underestimation of the antigen levels in a biospecimen (the Hook effect). A general issue of detection based solely on antibodies is that the final (quantitative) readout is never unambiguously connected to the validated identification of the molecular target within the analysis. Ideally, antibodies recognizing different epitopes of the same molecule would be used in parallel to at least partially address this issue. This strategy, however, further increases the cost per sample, which is inherently high for immunoassays, and has the disadvantage that the multiplexing capabilities of antibody-based detection is limited and sample consumption can be high.9
As for IA, IHC can suffer from antibody specificity issues. In addition, the fixation process can lead to molecular deformation that can hamper recognition by antibodies, particularly for glutaraldehyde-based fixation.12 Additionally, sample fixation has the potential to induce artificial patterns particularly for signalling PTMs such as protein phosphorylation, or, conversely, may fail to preserve these. In IHC, results are often interpreted in a semi-quantitative manner, and are manually ranked based on staining intensity which introduces subjectivity and variability into the assays,13 with a continuing need for improved standardization.14,15 Recently, Morales-Betanzos et al. demonstrated that IHC methods for detecting programmed death-ligand 1 (PD-L1), an important companion diagnostics for immune checkpoint therapies, can suffer severely from interference by endogenous PD-L1 glycosylation patterns that prevent proper epitope recognition, leading to underestimation of PD-L1 expression levels.16
As a consequence of the aforementioned issues, even for well-established cancer biomarkers, the results obtained using different assay platforms may differ17 – a problem that also occurs in typical blood tests which have slightly varying reference ranges based on the methodology applied.
Targeted mass spectrometry and clinical assays
Some of the problems faced in current assays used to measure protein expression in clinical samples can be overcome by protein mass spectrometry (MS). In particular, multiple reaction monitoring-based (MRM)-based quantitative MS has received widespread attention in recent years as a powerful tool for quantifying proteins in biological and clinical samples.18 Although it requires expensive equipment that needs to be routinely monitored and maintained through the use of quality control standards, the costs for individual samples are comparably low, particularly owing to the high multiplexing capacity of this technique which allows the quantitation of 100s of proteins with good linearity over several orders of magnitude and without major sample consumption. In standard “bottom-up” protein MS workflows, proteins are extracted from biological samples and subsequently proteolytically cleaved into peptides.19 These peptides are the surrogates for estimating protein abundances and allow the determination of relative or absolute changes in protein concentration by liquid chromatography-MS (LC-MS). The precision of MS can be substantially improved through the use of stable isotope-labelled standard (SIS) reference peptides. These share the same physicochemical properties as their endogenous variants, but have a defined mass shift (usually >3 Da) due to the incorporation of 13C, 15N, or 18O atoms. Spiking a SIS peptide at a defined amount into a sample will allow absolute quantitation of the corresponding endogenous peptide by comparing their elution profiles (see Fig. 1). The SIS approach substantially improves quantitative precision and can reduce CVs to below 10%. An improved procedure is based on the use of two SIS peptide isotopologues for each endogenous peptide to be quantified. The second SIS peptide (SIS2) will be spiked at equimolar amounts into the ‘unknown’ sample and aliquots used to generate an external calibration curve with increasing amounts of SIS1 – importantly, without interference from the endogenous peptide.20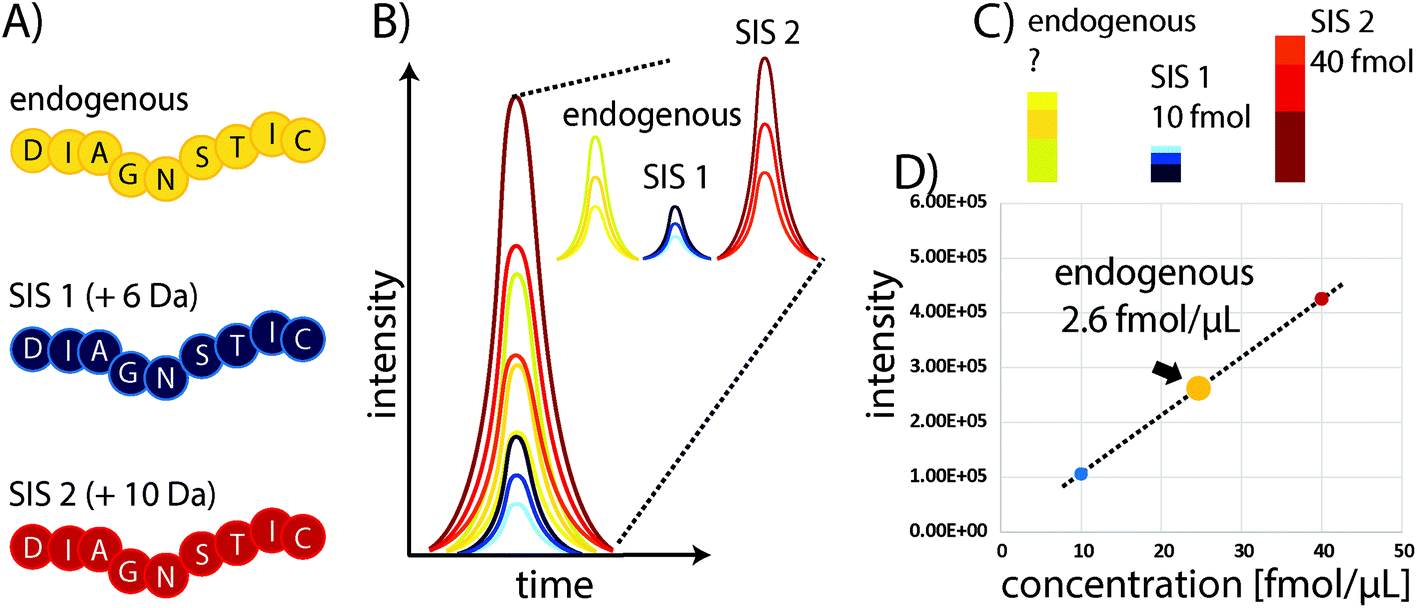 | ||
| Fig. 1 Protein quantitation using surrogate peptides and SIS reference peptides to generate an internal two-point calibration curve. (A) The endogenous target peptide and corresponding SIS peptides share the same sequences and physicochemical properties, but vary in mass (here indicated mass shifts of +6 and +10 Da). (B) When spiked into a sample the peptide variants will co-elute and the relative intensities of specific fragment ions of the same precursor are consistent (shown here for three “transitions”). The resulting elution profiles reflect the relative peptide abundances. (C) By spiking in known amounts of the SIS peptides (D) a calibration curve allows the precise determination of the concentration of the endogenous peptide. | ||
Although targeted MS methods provide high sensitivity and allow the detection and quantitation of attomole amounts of peptide, they still can be insufficient in the case of highly complex samples, such as plasma or extremely low-abundance targets.21 Therefore, Anderson and colleagues introduced the Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) method,22 which combines anti-peptide antibody-based immunoprecipitation to reduce sample complexity and enrich the target of interest with subsequent elution and LC-MS detection. Thus, the typical issues with antibody-based assays are avoided, as both precise quantitation and unambiguous identification are conducted using MS (see Fig. 1).
Importantly, the use of anti-peptide antibodies has several advantages over anti-protein antibodies as they are neither dependent on protein folding nor limited to protein surface areas, and consequently are less prone, for instance, to PTM-induced steric changes and hindrance. Although once set up, targeted LC-MS workflows are straightforward and can be well-standardized and partially automated at both the experimental and data analysis levels, implementing LC-MS as a routine analysis tool in clinical environments is very challenging. Even with dedicated and well-trained operators LC-MS is not a particularly robust method and although multiplexing allows quantifying a large number of targets per sample, the actual sample throughput is comparably low as the actual LC-MS measurement can take up to one hour – although this strongly depends on the type of instrumentation, the number and nature of targets, and the complexity of the samples.
Another technique that combines antibodies with MS is mass cytometry (MC). MC may complement IHC and allows sensitive and multiplexed spatial analysis of many samples. Here, antibodies are conjugated with elemental isotopes, which then are detected with high sensitivity by inductively-coupled plasma (ICP) MS. The same limitations regarding antibodies and fixation, however, also apply to MC.
Why is iMALDI a powerful tool for clinical assays?
In the light of the advantages and limitations of current assays, we strongly believe, that another technology that combines immunoprecipitation with matrix-assisted laser desorption/ionization (MALDI)23 mass spectrometry (iMALDI) has the potential to overcome current hurdles that hinder the implementation of MS-based assays in the clinic.24–28 Instead of eluting bound peptides from anti-peptide antibodies into a vial, followed by final sample preparation steps such as desalting and then LC-MS analysis, in iMALDI peptides are directly eluted from the antibody using the MALDI matrix on to the MALDI plate.29 This is an elegantly simple and rapid procedure that minimizes sample losses due to adsorption onto vials or tubing. When the matrix is dried on the target, the analytes (i.e. endogenous target peptide and SIS variants) are incorporated into matrix crystals and can be readily analysed (see Fig. 2).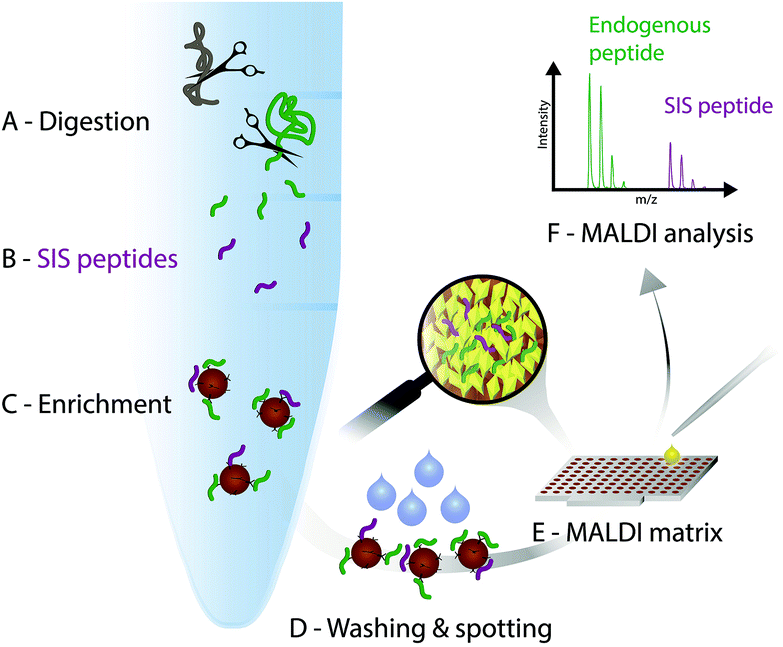 | ||
| Fig. 2 iMALDI workflow. (A) Digestion of proteins in a particular sample type, such as cell lysate. (B) Addition of a known amount of a stable isotope-labelled standard (SIS) peptide analogous to the endogenous (END) target peptide. (C) The END and SIS peptides are co-captured from the digest, leading to enrichment of both the END and SIS peptides with antibodies coupled to magnetic beads. (D) Washing of the bead-antibody–peptide complexes and spotting onto a MALDI plate. (E) Application of the acidic MALDI matrix solution elutes the target peptides, followed by co-crystallization of the peptides with the matrix molecules. (F) MALDI-TOF analysis. Quantitation is based on the ratio of the END to the SIS forms of the peptide. | ||
A detailed description on how to effectively develop an iMALDI method for protein quantitation via proteolytic peptides has been published previously.29 Briefly, after selection of a target peptide sequence that is unique in the proteome of interest, antibodies targeting the sequence can either be generated following previously published methods,30 or purchased if commercially available. After proteolytic digestion of the sample, such as blood plasma or tissue lysate, a SIS variant of the endogenous target peptide is spiked into the sample. Both peptides are enriched by the peptide-specific antibody which is coupled to magnetic beads. After washing off non-specifically bound molecules, the bead-antibody-peptide complexes are spotted onto a MALDI plate and allowed to dry. An acidic MALDI matrix solution then elutes the peptides off the beads. The peptides co-crystallize with the matrix molecules, followed by MALDI-TOF analysis. Notably, TOF/TOF instruments with MS/MS capability allow sequence identification of the target peptides.
MALDI instruments are in general more robust and less expensive than LC-MS instrumentation but offer comparable sensitivity, with analysis times of only seconds per analysis. They are generally not well-suited for complex samples, but – importantly – enrichment using a specific anti-peptide antibody simplifies the sample and eliminates the typical need for an LC separation to reduce sample complexity. The iMALDI procedure can be almost fully automated, with the only manual step being the transfer of the MALDI plate into the MS. Importantly, MALDI instrumentation is already routinely used in many clinics as an important tool for microbiology,31,32 such that the required instrumentation and know-how to run and operate it are already present. Thus, if specific antibodies are available and the enrichment procedure is optimized and standardized to obtain maximum recovery and robustness, iMALDI is the ideal combination of IA and MS for the precise quantitation of protein targets with high throughput and high precision at low cost. Further, it should be noted that if antibodies are not available, antibody development for affinity-MS applications is required, which can be costly. However, compared to immunoassays such as sandwich ELISA or Western blots, only one instead of two antibodies is required, thereby significantly reducing assay development time and cost.
How MS can serve as companion diagnostics in the clinics
We have developed iMALDI assays for various applications. For example, Jiang et al. developed an iMALDI assay targeting the epidermal growth factor receptor (EGFR).33 The assay achieved a sensitivity of low attomole levels and was successfully applied to the quantitation of EGFR from breast cancer cell lines and a tumour biopsy sample. In addition, an iMALDI assay for the detection of Francisella tularensis via its IglC protein was developed and applied to nasal swab samples from mice, inoculated with F. tularensis, and spiked into human plasma. The target protein was detected from as low as 800 colony forming units (CFU) spiked into plasma.34The application of iMALDI assays has further been expanded to include the quantitation of Angiotensin I (Ang I) from human plasma. The quantitation of Ang I is relevant for the determination of plasma renin activity (PRA) which is crucial for the diagnosis of primary aldosteronism, a form of often treatable secondary hypertension.35 While our initial experiments focussed on optimizing the quantitation of either Ang I itself,36 or on multiplexed quantitation of Ang I and Ang II using a cross-reactive antibody,37 follow-up studies have been performed to assess the suitability of the developed Ang I iMALDI assay in determining PRA values in human plasma samples. Camenzind et al. demonstrated a strong correlation between the iMALDI method and clinically used radioimmunoassay (RIA) and LC-MS methods by analysing 64 patient samples, with R2 values of 0.94 and 0.95 for iMALDI vs. RIA and iMALDI vs. LC-tandem MS (MS/MS), respectively.38 Furthermore, CVs of the technical replicates were <5%, while inter- and intra-day replicates showed CVs of 17%. The advantages of iMALDI over RIA – i.e., specificity, no need for radionucleotides, and shorter analysis times than LC-MS were demonstrated.
In a next step, the iMALDI Ang I assay was further optimized and automated on a liquid handling robot to improve precision and throughput.39 Furthermore, MALDI analysis, previously performed on large MALDI instruments, was transferred to a small benchtop MALDI-TOF instrument, which has been FDA-cleared for clinical microbial identification.40 A method comparison on 188 human patient samples (Fig. 3) demonstrated excellent correlation to a clinically-employed LC-MS41 method with an R2 value of >0.98, while achieving 7.5-fold faster analysis than LC-MS/MS. Additionally, the sensitivity and linear range of the iMALDI assay was found suitable to cover low, medium, and high PRA patient samples, as defined by Alderman et al.42 CVs across the linear range were found to be <10% when the MALDI analysis was carried out in reflector mode, and <15% when carried out in the linear mode, further demonstrating the high precision of MALDI analysis coupled with immuno-enrichment and the use of SIS peptides. It was further concluded that – when considering one replicate per patient sample, up to 744 patient samples could be run per day, thereby highlighting the suitability of iMALDI as a high throughput technique. A modified version of the assay with simplified sample preparation that does not require a complex and expensive liquid handling system, termed BEARS, for “bead-extractor assisted ready-to-use reagent system”, has been published recently.43
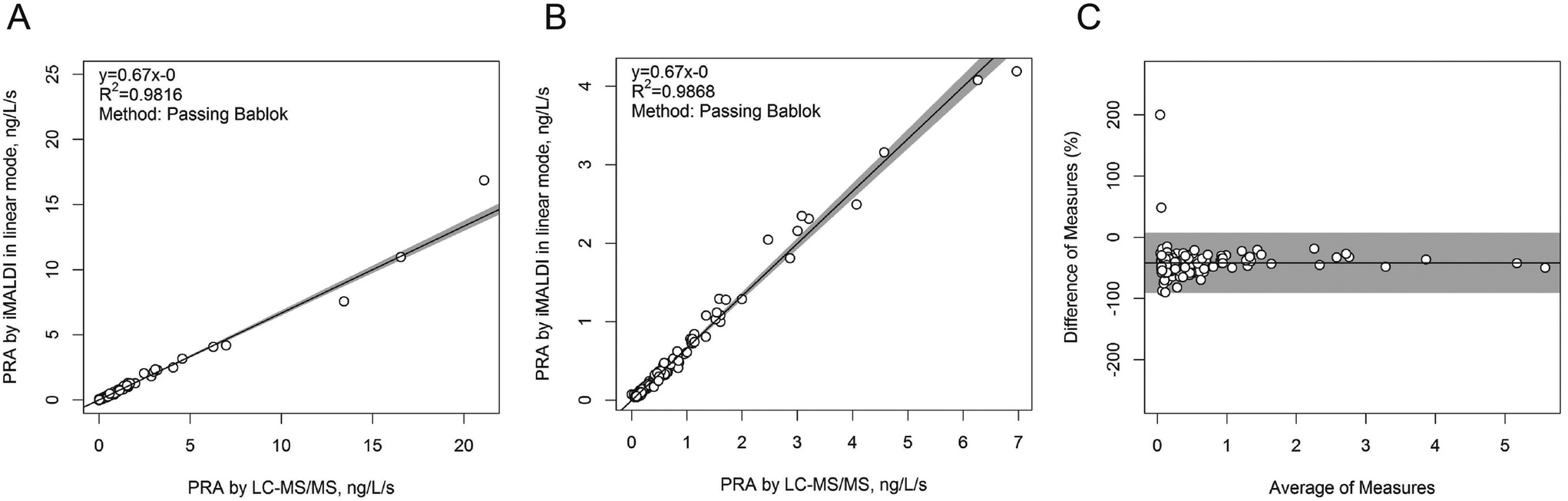 | ||
| Fig. 3 Method comparison of plasma renin activity determination by iMALDI in linear MALDI mode vs. LC-MS/MS. Passing-Bablok linear regression calculations for all 188 patient samples analysed (A), for the 151/188 samples that fell within the linear range of the iMALDI assay (B), and for the difference plot for the 151/181 patient samples (C) indicate excellent correlation of iMALDI and LC-MS/MS. Reprinted (adapted) with permission from Popp, et al., Biochim. Biophys. Acta, 2015, 1854, 547–558.39 Copyright 2015 Elsevier. | ||
Most recently, we have been developing iMALDI assays for the quantitation of cell signalling pathway proteins linked to the development and progression of cancer. The goal of these assays was two-fold: (i) to improve the patient stratification for targeted cancer therapies by adding the protein layer to genomic and transcriptomic-based stratification strategies, and (ii) to improve upon the drawbacks of currently used approaches to assess signalling pathway proteins. These drawbacks include non-specificity, difficulties in multiplexing, and semi-quantitative results (in the case of IHC, which is commonly used in a clinical setting), and the long analysis times and the complexity of LC-multiple-reaction monitoring (LC-MRM) based methods, commonly found in research settings.
To achieve these goals, we have developed iMALDI assays that target the C-terminal tryptic peptides of the Protein Kinase B (AKT) isoforms AKT1 (466RPHFPQFSYSASGTA480) and AKT2 (468THFPQFSYSASIRE481).44 These particular peptides contain key phosphorylation sites known to be involved in full kinase activation.45,46 Furthermore, AKT is a key node of the PI3K/AKT/mTOR pathway, one of the most commonly dysregulated cell signalling pathways involved in cancer development and progression and therefore a prime target for therapeutic development.47
The AKT1 and AKT2 iMALDI assays achieved limits of detection (LODs) of 0.02 and 0.05 fmol μg−1 total lysate protein, respectively, while requiring only 10 μg total lysate protein per enrichment. Furthermore, CVs were consistently below 15% while covering a linear range of 0.05 to 2.0 fmol μg−1. We were able to quantify AKT1 and AKT2 from various cell lines, including MDA-MB-231 breast cancer cells, as well as flash frozen tumour tissue lysates (see Fig. 4). In the next step, the assays will be expanded for the quantitation of phosphorylation stoichiometry, and for the analysis of extracts from formalin-fixed paraffin-embedded (FFPE) tissue samples. Additionally, the sensitivity and specificity of the AKT assays will be investigated on tumour samples obtained from patient-derived mouse xenografts (PDX) prior to and after drug treatment, and the results will be correlated with known clinical outcomes.
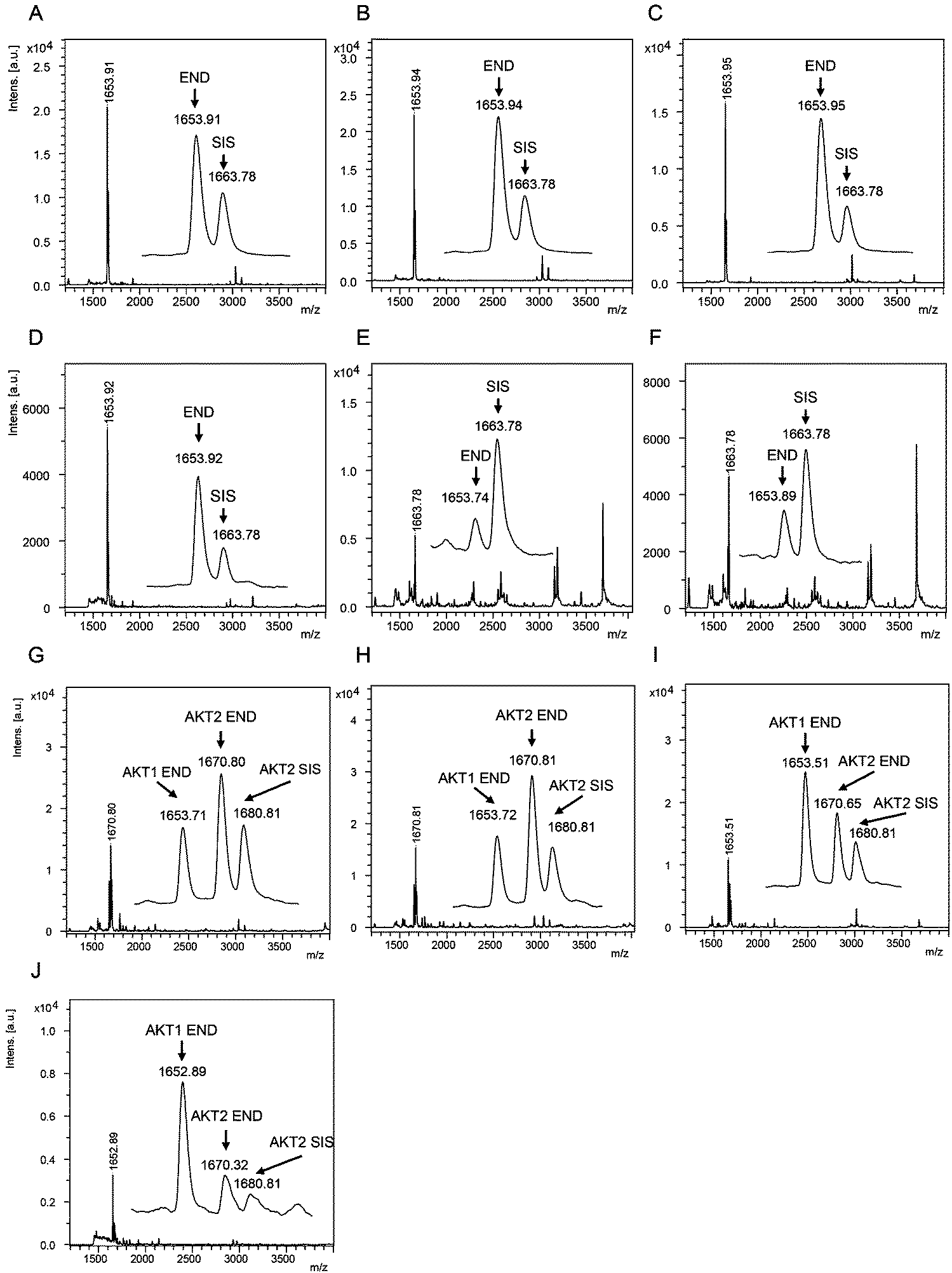 | ||
| Fig. 4 (A–F) Mass spectra showing the capture of endogenous AKT1 peptide RPHFPQFSYSASGTA at m/z 1653.9 and the corresponding AKT1 SIS peptide at 2 fmol per well (m/z 1663.8) from (A) MDA-MB-231 parental and (B) EGF-induced breast cancer cells, and flash frozen tumour lysates from (C) a HCT116 colon cancer mouse xenograft and (D–F) breast tumour samples. Mass spectra acquired from iMALDI analysis of AKT2 (THFPQFSYSASIRE) of (G) parental and (H) EGF-induced MDA-MB-231 breast cancer cells, (I) HCT116 colon cancer mouse xenograft tumour, and (J) a breast tumour. Per replicate digestion and capture, 10 μg total lysate were used. END = endogenous peptide; SIS = stable isotope-labeled standard peptide. Reprinted (adapted) with permission from Popp, et al., Anal. Chem., 89(19), 10592–10600.44 Copyright 2017 American Chemical Society. | ||
Conclusions
The enormous potential for implementing MS-based protein assays into the clinic is in stark contrast with the number of clinical assays that have currently been established. Although MS has undoubtedly revolutionized our understanding of biochemical pathways and complexity, its implementation into routine clinical environments is still lagging. While LC-MS based technologies might be too complex and lack robustness for routine clinical use, iMALDI is a promising alternative that can be fully automated and parallelized to meet the requirements for clinical assays. We have shown in various applications that the iMALDI technology is an accurate, precise, and highly sensitive technique with unique advantages, offering the potential to be used for companion diagnostics both during drug development and also for guiding patient stratification decisions and patient monitoring.In the future, the iMALDI technology will be expanded to target additional signalling-pathway proteins to assess signalling-pathway activity based on selected representative nodes in a network-wide fashion.
Conflicts of interest
CHB holds the patent for iMALDI, and is the CSO of MRM Proteomics, Inc. The other authors declare that they have no conflicts.Acknowledgements
We are grateful to Genome Canada and Genome British Columbia for financial support (project codes 264PRO, 204PRO for operations, and 214PRO for technology development). We would also like to thank Genome Canada, Genome British Columbia, and Genome Quebec for support for this project (project code 183AKT-GAPP). CHB is also grateful for support from the Leading Edge Endowment Fund (University of Victoria) and for support from the Segal McGill Chair in Molecular Oncology at McGill University (Montreal, Quebec, Canada). CHB, AS, MB, and GB are grateful for support from the Warren Y. Soper Charitable Trust and the Alvin Segal Family Foundation to the Jewish General Hospital (Montreal, Quebec, Canada). AS, MB, and GB are also grateful for support from Genome Canada's Business-Led Networks of Centres of Excellence program, Exactis Innovations, and the Fonds de recherche du Québec – Santé, Axe cancer du sein et de l'ovaire (FRSQ-ACSO).Notes and references
- R. L. Schilsky, J. H. Doroshow, M. Leblanc and B. A. Conley, Clin. Cancer Res., 2012, 18, 1540–1546 CrossRef PubMed.
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl, L. Fehrenbacher, J. M. Wolter, V. Paton, S. Shak, G. Lieberman and D. J. Slamon, J. Clin. Oncol., 1999, 17, 2639–2648 CrossRef CAS PubMed.
- J. Andersson, B. Linderholm, G. Greim, B. Lindh, H. Lindman, J. Tennvall, L. Tennvall-Nittby, D. Pettersson-Skold, A. Sverrisdottir, M. Soderberg, S. Klaar and J. Bergh, Acta Oncol., 2002, 41, 276–281 CrossRef PubMed.
- D. J. Slamon, G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich and W. L. McGuire, Science, 1987, 235, 177–182 CAS.
- M. D. Pegram, R. S. Finn, K. Arzoo, M. Beryt, R. J. Pietras and D. J. Slamon, Oncogene, 1997, 15, 537–547 CrossRef CAS PubMed.
- C. Carlomagno, F. Perrone, C. Gallo, M. De Laurentiis, R. Lauria, A. Morabito, G. Pettinato, L. Panico, A. D'Antonio, A. R. Bianco and S. De Placido, J. Clin. Oncol., 1996, 14, 2702–2708 CrossRef CAS PubMed.
- G. Pauletti, W. Godolphin, M. F. Press and D. J. Slamon, Oncogene, 1996, 13, 63–72 CAS.
- A. K. Fuzery, J. Levin, M. M. Chan and D. W. Chan, Clin. Proteomics, 2013, 10, 13 CrossRef PubMed.
- T. G. Cross and M. P. Hornshaw, J. Appl. Bioanal., 2016, 2, 108–116 CrossRef.
- A. N. Hoofnagle and M. H. Wener, J. Immunol. Methods, 2009, 347, 3–11 CrossRef CAS PubMed.
- C. A. Spencer, M. Takeuchi, M. Kazarosyan, C. C. Wang, R. B. Guttler, P. A. Singer, S. Fatemi, J. S. LoPresti and J. T. Nicoloff, J. Clin. Endocrinol. Metab., 1998, 83, 1121–1127 CAS.
- C. B. Saper, J. Histochem. Cytochem., 2009, 57, 1–5 CrossRef CAS PubMed.
- A. D. Powers and S. P. Palecek, J. Healthcare Eng., 2012, 3, 503–534 CrossRef PubMed.
- N. S. Goldstein, S. M. Hewitt, C. R. Taylor, H. Yaziji, D. G. Hicks and S. Members of Ad-Hoc Committee On Immunohistochemistry, Appl. Immunohistochem. Mol. Morphol., 2007, 15, 124–133 CrossRef CAS PubMed.
- R. A. Walker, Histopathology, 2006, 49, 406–410 CrossRef CAS PubMed.
- C. A. Morales-Betanzos, H. Lee, P. I. Gonzalez Ericsson, J. M. Balko, D. B. Johnson, L. J. Zimmerman and D. C. Liebler, Mol. Cell. Proteomics, 2017, 16, 1705–1717 CAS.
- M. L. Rawlins and W. L. Roberts, Clin. Chem., 2004, 50, 2338–2344 CAS.
- M. A. Gillette and S. A. Carr, Nat. Methods, 2013, 10, 28–34 CrossRef CAS PubMed.
- H. Steen and M. Mann, Nat. Rev. Mol. Cell Biol., 2004, 5, 699–711 CrossRef CAS PubMed.
- A. LeBlanc, S. A. Michaud, A. J. Percy, D. B. Hardie, J. Yang, N. J. Sinclair, J. I. Proudfoot, A. Pistawka, D. S. Smith and C. H. Borchers, J. Proteome Res., 2017, 16, 2527–2536 CrossRef CAS PubMed.
- F. Weiss, B. H. van den Berg, H. Planatscher, C. J. Pynn, T. O. Joos and O. Poetz, Biochim. Biophys. Acta, 2014, 1844, 927–932 CrossRef CAS PubMed.
- N. L. Anderson, N. G. Anderson, L. R. Haines, D. B. Hardie, R. W. Olafson and T. W. Pearson, J. Proteome Res., 2004, 3, 235–244 CrossRef CAS PubMed.
- F. Hillenkamp, M. Karas, R. C. Beavis and B. T. Chait, Anal. Chem., 1991, 63, 1193A–1203A CrossRef CAS PubMed.
- K. Sparbier, T. Wenzel, H. Dihazi, S. Blaschke, G. A. Muller, A. Deelder, T. Flad and M. Kostrzewa, Proteomics, 2009, 9, 1442–1450 CrossRef CAS PubMed.
- J. Jiang, C. E. Parker, K. A. Hoadley, C. M. Perou, G. Boysen and C. H. Borchers, Proteomics: Clin. Appl., 2007, 1, 1651–1659 CrossRef CAS PubMed.
- D. R. Mason, J. D. Reid, A. G. Camenzind, D. T. Holmes and C. H. Borchers, Methods, 2012, 56, 213–222 CrossRef CAS PubMed.
- J. Jiang, C. E. Parker, J. R. Fuller, T. H. Kawula and C. H. Borchers, Anal. Chim. Acta, 2007, 605, 70–79 CrossRef CAS PubMed.
- J. D. Reid, D. T. Holmes, D. R. Mason, B. Shah and C. H. Borchers, J. Am. Soc. Mass Spectrom., 2010, 21, 1680–1686 CrossRef CAS PubMed.
- B. Shah, J. D. Reid, M. A. Kuzyk, C. E. Parker and C. H. Borchers, Methods Mol. Biol., 2013, 1023, 97–120 CAS.
- S. Hoeppe, T. D. Schreiber, H. Planatscher, A. Zell, M. F. Templin, D. Stoll, T. O. Joos and O. Poetz, Mol. Cell. Proteomics, 2011, 10, M110.002857 Search PubMed.
- A. Croxatto, G. Prod'hom and G. Greub, FEMS Microbiol. Rev., 2012, 36, 380–407 CrossRef CAS PubMed.
- P. Seng, C. Abat, J. M. Rolain, P. Colson, J. C. Lagier, F. Gouriet, P. E. Fournier, M. Drancourt, B. La Scola and D. Raoult, J. Clin. Microbiol., 2013, 51, 2182–2194 CrossRef CAS PubMed.
- J. Jiang, C. E. Parker, K. A. Hoadley, C. M. Perou, G. Boysen and C. H. Borchers, Proteomics: Clin. Appl., 2007, 1, 1651–1659 CrossRef CAS PubMed.
- J. Jiang, C. E. Parker, J. R. Fuller, T. H. Kawula and C. H. Borchers, Anal. Chim. Acta, 2007, 605, 70–79 CrossRef CAS PubMed.
- C. Catena, G. Colussi, E. Nadalini, A. Chiuch, S. Baroselli, R. Lapenna and L. A. Sechi, Arch. Intern. Med., 2008, 168, 80–85 CrossRef CAS PubMed.
- J. D. Reid, D. T. Holmes, D. R. Mason, B. Shah and C. H. Borchers, J. Am. Soc. Mass Spectrom., 2010, 21, 1680–1686 CrossRef CAS PubMed.
- D. R. Mason, J. D. Reid, A. G. Camenzind, D. T. Holmes and C. H. Borchers, Methods, 2012, 56, 213–222 CrossRef CAS PubMed.
- A. G. Camenzind, J. G. van der Gugten, R. Popp, D. T. Holmes and C. H. Borchers, Clin. Proteomics, 2013, 10, 20 CrossRef PubMed.
- R. Popp, D. Malmstrom, A. G. Chambers, D. Lin, A. G. Camenzind, J. G. van der Gugten, D. T. Holmes, M. Pugia, M. Jaremek, S. Cornett, D. Suckau and C. H. Borchers, Biochim. Biophys. Acta, 2015, 1854, 547–558 CrossRef CAS PubMed.
- Bruker, Bruker Announces FDA Clearance for Second, Expanded Claim for the MALDI Biotyper CA System, https://www.bruker.com/nc/news-records/single-view/article/bruker-announces-fda-clearance-for-second-expanded-claim-for-the-maldi-biotyper-ca-system.html, accessed 07-May-2016.
- J. G. Van der Gugten and D. T. Holmes, in Clinical Applications of Mass Spectrometry in Biomolecular Analysis: Methods and Protocols, ed. U. Garg, Humana Press Inc, Totowa, 2016, vol. 1378, pp. 243–253 Search PubMed.
- M. H. Alderman, H. W. Cohen, J. E. Sealey and J. H. Laragh, Am. J. Hypertens., 2004, 17, 1–7 CrossRef CAS PubMed.
- H. Li, R. Popp, M. Chen, E. M. MacNamara, D. Juncker and C. H. Borchers, Anal. Chem., 2017, 89, 3834–3839 CrossRef CAS PubMed.
- R. Popp, H. Li, A. LeBlanc, Y. Mohammed, A. Aguilar-Mahecha, A. G. Chambers, C. Lan, O. Poetz, M. Basik, G. Batist and C. H. Borchers, Anal. Chem., 2017, 89, 10592–10600 CrossRef CAS PubMed.
- P. Liu, M. Begley, W. Michowski, H. Inuzuka and M. Ginzberg, Nature, 2014, 508, 541–545 CrossRef CAS PubMed.
- S. J. Humphrey, S. B. Azimifar and M. Mann, Nat. Biotechnol., 2015, 33, 990–995 CrossRef CAS PubMed.
- D. A. Fruman and C. Rommel, Nat. Rev. Drug Discovery, 2014, 13, 140–156 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |