
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ECM–oligourethene–silica hydrogels as a local drug release system of dexamethasone for stimulating macrophages†
Magdalena Rangel-Argoteab,
Jesús A. Claudio-Rizo b,
Laura E. Castellanob,
Arturo Vega-Gonzálezb,
José L. Mata-Mata*a and
Birzabith Mendoza-Novelo*b
b,
Laura E. Castellanob,
Arturo Vega-Gonzálezb,
José L. Mata-Mata*a and
Birzabith Mendoza-Novelo*b
aDepartamento de Química, DCNE, Universidad de Guanajuato, Noria alta s/n, 36050, Guanajuato, GTO, Mexico. E-mail: matamm@ugto.mx
bDepartamento de Ingenierías Química, Electrónica y Biomédica, DCI, Universidad de Guanajuato, Loma del Bosque 103, C. P. 37150, León, GTO, Mexico. E-mail: b.mendozanovelo@ugto.mx; Tel: +52-477-7885100 ext. 8464
First published on 7th February 2017
Abstract
Hydrogels based on an extracellular matrix (ECM) capable of delivering therapeutics in a controlled manner represent a platform to guide tissue regeneration. This work reports a novel approach wherein the incorporation of silica particles (SiP) inside ECM hydrogels supports the loading and releasing of dexamethasone (Dex). The biocomposite hydrogels, derived from porcine small intestine submucosa (SIS), water-soluble oligourethanes (PPU synthetized from polyethylene glycol and hexamethylene-diisocyanate) and SiP, delivered Dex at pH 7.4 and 37 °C in vitro. In this regard, the SiP (0, 5 and 15 wt%) accelerated the collagen polymerization and modified the collagen network structural parameters, while the oligourethane crosslinking regulated the mechanics and degradation of the material. The biocomposite hydrogels containing 15 wt% SiP showed controlled release of Dex for 11 days, obtaining a 79% release efficiency. As a consequence, the delivery of Dex from biocomposites was capable of enhancing cell metabolic activity and TGF-β1 secretion by macrophages. These composite collagen hydrogels combine structures and properties that make them promising templates for loading and delivering Dex that can modulate the macrophage response in a soft tissue engineering context.
1. Introduction
The design of bioactive hydrogels is a promising alternative for tailoring the structure and properties intended to improve the applications in soft tissue engineering.1–7 In this sense, the biomedical hydrogels need to be enriched in both a fast and efficient manner with therapeutic agents in order to enhance their biological performance. Previous reports have indicated that the hydrogels derived from ECM provide chemical, physical and biological characteristics to support the cell responses.8,9 However, the drug incorporation and its controlled release are limited in hydrogels based on type I collagen (main component of the ECM) due to their poor mechanical properties, fast degradation rate and fast drug release (so-called burst release).10,11 In this way, the chemical crosslinking2,8,12 and the addition of silica particles1,13–15 have demonstrated to be useful to enhance the loading and the controlled release of bioactive molecules in collagen-based hydrogels.The local delivery of dexamethasone (Dex), a corticosteroid, from 3D templates has shown to be capable of directing cell fate and regulating inflammation processes.16–18 Hydrogels based on PEG,19–21 poly(vinyl alcohol),16,22 hyaluronic acid,23 γ-cyclodextrin24 and chitosan25 have been tested as templates for loading/delivering Dex. In these systems, the gel porosity and mechanics, as well as the temporal changes in the gel properties, induced by both degradation and swelling, have facilitated the loading and controlled release of Dex. Besides, it has also been reported that the incorporation of Dex in hydrogels comprised of collagen–chitosan maintains the anti-inflammatory activity of Dex,26 however, it has not been studied the capacity of controlled release of Dex from these hydrogels. This work aims to establish the advantages offered by the SiP incorporation in ECM hydrogels to deliver Dex in a controlled manner.
Recently, we proposed a new method for preparation of biomedical hydrogels comprised of ECM, PPU and SiP.27 It was reported that the chemical structure of the oligourethanes governs the crosslinking density of collagen, and thereby tailors mechanics, swelling and degradation of hydrogels. Also, the incorporation of SiP in the ECM 3D network sustains the viability and function of macrophages.27–29 To take advantages of this new methodology regarding the tailored properties of biocomposite hydrogels, this paper describes a novel approach where Dex is simultaneously loaded during the formation of these biocomposite hydrogels and then released at physiological conditions. The content of the SiP (0, 5 and 15 wt%) in the PPU–ECM hydrogels is explored as a variable that decisively influences the polymerization rate, 3D network characteristics, swelling and biodegradation of the biocomposites, enhancing the controlled release of Dex and its effect on the in vitro macrophage response.
2. Experimental section
2.1. Materials
Poly(ethylene oxide) diol (PEG, Mn 1000 g mol−1), hexamethylene diisocyanate (HDI), sodium bisulfite (NaHSO3), tetraethylorthosilicate (TEOS), pepsin, type I collagenase, ethylenediaminetetraacetic acid (EDTA), dexamethasone sodium phosphate (Dex), 3-(4,5-dimethyl-thiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), 2,2-dihydroxy-1,3-indanedione (ninhydrin), as well as others salts and solvents were acquired from Sigma-Aldrich.2.2. Synthesis of blocked-oligourethanes coupled with colloidal silica
The oligourethanes were synthetized as previously described elsewhere.27 Briefly, melted PEG was reacted with HDI in a molar NCO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OH ratio of 4.0:1.0 for 2 h at 100 °C. The NCO end groups in the PUP product were blocked by the reaction with sodium bisulfite (40 wt% in water) for 2 h at 40 °C. Then, TEOS in ethanol was added to the reaction mixture and stirred (3 h, 25 °C) to achieve a full dispersion of colloidal silica into PUP solutions, followed by the addition of hydrogen peroxide solution (30 wt%, 25 °C). The concentration of TEOS in the oligourethane solution was varied in 0, 5 and 15 wt% (Table 1).
:OH ratio of 4.0:1.0 for 2 h at 100 °C. The NCO end groups in the PUP product were blocked by the reaction with sodium bisulfite (40 wt% in water) for 2 h at 40 °C. Then, TEOS in ethanol was added to the reaction mixture and stirred (3 h, 25 °C) to achieve a full dispersion of colloidal silica into PUP solutions, followed by the addition of hydrogen peroxide solution (30 wt%, 25 °C). The concentration of TEOS in the oligourethane solution was varied in 0, 5 and 15 wt% (Table 1).
| Composite hydrogel designation | Content of PPU/SiP/Dex (mg) | Turbidimetric kinetic parameters | ||
|---|---|---|---|---|
| tlag (min) | S (min−1) | t1/2 (min) | ||
| a Including 6 mg of total protein of hECM, n = 3, means + SD, * symbol indicates statistical significance (p < 0.05) in comparison with SIS. | ||||
| SIS | 0/0/0.4 | 9.4 ± 1.0 | 0.0109 ± 0.0012 | 13.0 ± 1.4 |
| SIS P0 | 1.2/0/0.4 | 4.7 ± 0.5* | 0.0159 ± 0.0017* | 11.0 ± 1.3 |
| SIS P5 | 1.2/0.3/0.4 | 3.5 ± 0.4* | 0.0185 ± 0.0020* | 9.8 ± 1.2* |
| SIS P15 | 1.2/0.9/0.4 | 9.4 ± 1.0 | 0.0168 ± 0.0018* | 9.2 ± 1.4* |
2.3. Extraction of type I collagen
Small intestine submucosa (SIS) was mechanically separated from porcine intestine, decellularized and characterized as previously described elsewhere.9 The SIS sections were lyophilized, cut into small fragments, and enzymatically digested (1 mg mL−1 pepsin in 0.01 M HCl, stirring for 72 h at room temperature, RT) to solubilize the collagen. The digested SIS (hECM) samples were stored at 4 °C.2.4. Preparation of Dex-loaded composite hydrogel
The collagen gelation was induced by adjusting the pH and salt concentration (pH = 7) of cold solubilized collagen (4 °C) followed by warming to 37 °C.9 In this work, the total protein concentration of 6 mg mL−1 was kept constant in all the hydrogel preparations.For the preparation of hybrid hydrogels, a constant concentration of 15 wt% of PUP coupled with 0, 5 or 15 wt% of silica was mixed with the hECM before gelation. The incorporation of Dex (0–800 μg) was performed on the mixture of the liquid precursors comprised of collagen, oligourethane and silica (Table 1). The hybrid hydrogels were routinely formed and aged for 18 h at 37 °C. The Fig. 1 schematizes the preparation of the active biocomposite hydrogels.
 | ||
| Fig. 1 Outline for the formation of biocomposite hydrogels as controlled release systems of dexamethasone. | ||
2.5. Determination of the Dex release
Each hydrogel disk (1.92 cm3) was prepared with a Dex initial loading amount (M0) of 400 μg. The release profiles of Dex were evaluated with respect to the incubation time in PBS (37 °C, pH 7.4). At specific times, 10 mL of PBS were retrieved and replaced with fresh PBS. The absorbance at 297 nm of PBS containing Dex was determined using a Thermo Scientific MultiSkan UV-Vis spectrophotometer. The amount of drug released at time t (Mt) was determined by a standard curve constructed in the range of 0 to 0.5 mg of Dex per mL of PBS (Fig. S3†). Total cumulative releasable Dex (M∞) was defined as the value of Mt at plateau. Release Dex amount data were changed to Dex release profiles according to the releasable Dex (Mt/M0 × 100) and fractional Dex release (Mt/M∞).30In this experiment set, the release of silica from the samples was also investigated. At each time point, the concentration of residual silica was determined using the Heteropoly Blue assay (Hach). The absorbance at 815 nm of PBS containing silica debris was determined using an UV-Vis spectrophotometer. The amount of released residual silica was determined by a standard curve constructed in the range of 0 to 1000 μg of silica per mL of PBS.
2.6. Physicochemical characterization of the composite hydrogels
The turbidity provides a measure of the relative degree of the collagen polymerization.31 The half time of gelation (t1/2) was defined as the time to reach 50% of the final Abs, the gelation rate (S) was defined as the slope of the linear region of the gelation curve, and the lag time (tlag) was defined as the intercept between the two initial linear regions. The Abs data were converted into turbidity values, τ, by using the relation τ = (Absln(10))/L; where L is the optical path length. Assuming that collagen fibers are randomly oriented, rigid rod-like and monodisperse in size, it is possible to infer the fibril diameter (d) and mass-length ratio (μ) using the wavelength (λ) dependence of turbidity as τλ5 = Aμ(λ2 − Bd2), where A = (88/15)π3 × cns(dn/dc)2 × 1/NA and B = (92/426)π2ns2; where c is the mass concentration of collagen, ns is the solvent refractive index (equal to 1.33), dn/dc is the specific refractive index increment (dn/dc = 0.186 cm3 g−1 for collagen), and NA is the Avogadro's constant.31,32 The μ was calculated from the slope of linear correlation for plotting λ5 versus λ2, while d was calculated from intercept with the y-axis.
2.7. Evaluation of the cell response
To assess the cell response to the Dex-loaded composite hydrogels, the metabolic activity of RAW-264.7 mouse macrophages and rat dermal fibroblasts, was checked by a MTT assay. Mouse macrophage cell (TIB-71) line was purchased from the American Type Culture Collection (ATCC). One day-1 neonatal Wistar rat was euthanized and primary dermal fibroblasts were isolated from skin tissue. The animal handling and care was performed in compliance with the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research of the National Academy of Sciences and with approval by the Institutional Committee of Bioethics in the Research of the University of Guanajuato. The skin tissue specimen was washed with PBS pH 7.2. The tissue was cut into ∼2 mm pieces and digested in Dulbecco's Modified Eagle Medium (DMEM) medium with 0.075% of type II collagenase, incubated at 37 °C, stirred slowly for 30 minutes. After that, it was added 0.125% of trypsin, stirred for 2.5 h at 37 °C. The cell suspension was filtered using a sterile filter of 100 μm pore size. The filtrate was centrifuged at 6000 rpm for 6 min. The pellet was resuspended in DMEM with 10% fetal bovine serum (FBS), antibiotic/antimycotic and transfer to Petri dish and incubated at 37 °C, 5% CO2. The cell cultured was checked after 48 h of incubation, discarded the old medium and changed for fresh medium. After the fibroblast reach 80% confluence, then frozen an aliquot of cell.The cells were seeded in culture plates and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C using Roswell Park Memorial Institute (RPMI) medium for macrophages or DMEM for fibroblasts, and 10% FBS. All the hydrogels were synthesized with sterile reactants and aged for 24 h into 24-well plates. In the first experiment set, the cells (1 mL of suspension, cell density of 3 × 104, 10% FBS) were seeded directly on each hydrogel and they were incubated for 3 and 7 days.
In another experiment, hydrogels were soaked in non-supplemented culture medium (2 mL), at 37 °C for 48 h, in order to extract soluble compounds. The supernatants (media conditioned with hydrogels) were supplemented with 10% of FBS and used directly (100%) or after dilution to 50% or 25% with fresh supplemented RPMI. Then, cells were cultured in the presence of the supernatants for 1 day. Finally, the cellular activity was quantified by the MTT assay. The cellular activity of cells growing in microplates (without materials) was used as a control. To assess the stimulation of macrophages by Dex-loaded composite hydrogels, the production of cytokines transforming growth factor-beta 1 (TGF-β1) and monocyte chemoattractant protein (MCP-1, also called CCL-2) was measured in the culture medium supernatants (after 3 and 7 days of culture) by ELISA kits (eBioscience) according to the manufacturer's specifications.
2.8. Statistical analysis
The mean and standard deviation (SD) are presented for each data set. Data sets were compared using analysis of variance (ANOVA). The difference of the means was checked with a Sidak–Holm post hoc test and was considered statistically significant at level p < 0.05.3. Results
3.1. Capacity of the Dex loading in the composite hydrogels
The liquid precursors comprised of SIS hydrolysate and oligourethanes with dispersed SiP had an adequate viscosity for mixing, allowing the volume measurements to be repeated easily. A first criterion of interest is the loading capacity of Dex in the hydrogels without impeding gel formation. It was observed that 400 μg of Dex per gel (total volume of 1 cm3) allow obtaining manageable composite hydrogels, but a higher amount did not allow gelation of the materials (Fig. S1†). Thus, all the results reported below were obtained with biocomposites formulated with 400 μg the Dex per each gel.3.2. Modification of type I collagen fibrillogenesis
The gelation kinetic parameters measured by turbidimetry can be used to elucidate the 3D network characteristics obtained by collagen polymerization in combination with SiP and Dex.33 Fig. 3a illustrates the effect of the SiP content on the changes of the absorbance at 406 nm. All-four turbidimetric curves had a sigmoidal shape. Table 1 shows the parameters of the turbidimetric gelation kinetics of Dex-loaded composite hydrogels. The nucleation stage of collagen polymerization can be observed at tlag of 9.4, 4.7, 3.5 and 9.4 min for SIS, SIS P0, SIS P5 and SIS P15, respectively. In the case of SIS P15 composite, the tlag did not show significant difference compared to SIS hydrogel. The polymerization stage, where the collagen fibrils grow into larger fibers producing an increment in the Abs values, was afterward appreciated.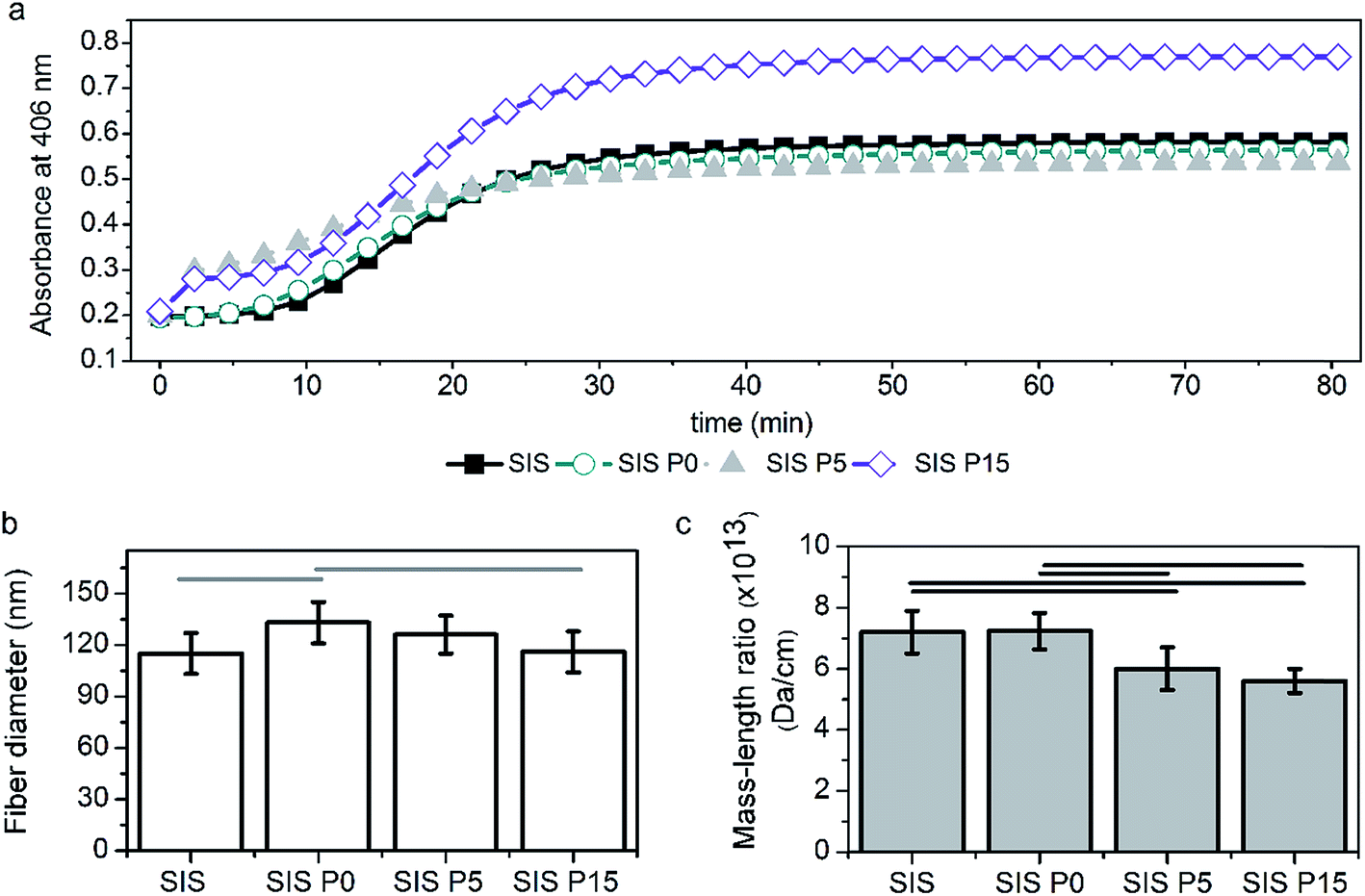 | ||
| Fig. 2 Type I collagen polymerization analyzed by turbidimetry. (a) Changes of absorbance with respect to time, indicating the gelation process at pH 7 and 37 °C for different formulations. The effect of the formulation on the fiber size (b) and mass/length ratio (c) in the formed collagen network. The legend expresses the SIS hydrogels modified with oligourethane without silica (SIS P0), with 5% of silica (SIS P5), or 15% of silica (SIS P15). The difference of the means is significant (p < 0.05) between all marked groups. | ||
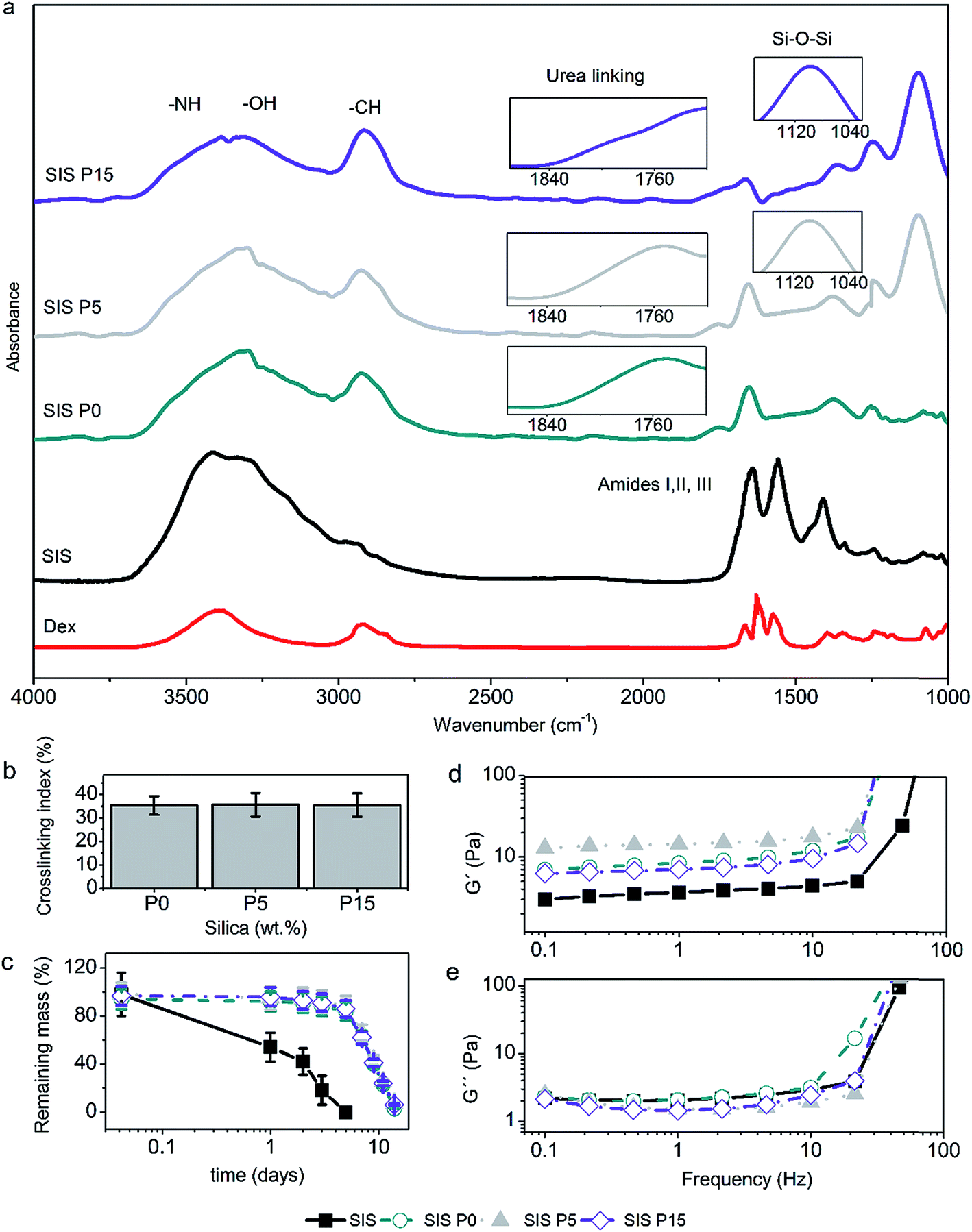 | ||
| Fig. 3 Physicochemical properties of Dex-loaded composite hydrogels. (a) ATR-FTIR spectra, showing the vibrational signal of urea bonds and siloxane bonds (inset). (b) Crosslinking index of the composite hydrogels, measured as the percentage of blocked collagen-amine. (c) Mass loss profiles under the action of type I collagenase (14 U per gel), where the mass loss produced in SIS hydrogel is statistically different (p < 0.05) compared to SIS P0, SIS P5 and SIS P15 materials. Data are expressed as mean values ± SD, n = 3. (d) Changes of storage modulus (G′) or (e) loss modulus (G′′) with respect to the oscillation frequency. | ||
The polymerization rate (S) characterized this stage, showing values statistically different for SIS P0, SIS P5 and SIS P15 compared to SIS. The t1/2 was decreased significantly for formulations with SiP and Dex. A higher SiP content increased the gelation rate and thus t1/2 was decreased. The fiber diameter (Fig. 3b) and mass/length ratio (Fig. 3c) of 3D-collagenous network were decreased with the increment of the SiP content.
3.3. Structure and properties of Dex-loaded composite hydrogels
All four hydrogels showed the characteristic IR bands of the stretching of N–H and O–H, stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide I), and deformation of N–H (amide II) in the regions of 3400–3200, 1700–1600, and 1650–1550 cm−1, respectively, as illustrated in Fig. 3a. The IR bands of the Dex were not observed in the spectra of the biocomposites, due to that the signals could be superimposed with the signals of the major components of the composite hydrogels, i.e., type I collagen and oligourethanes. On the other hand, the IR bands revealed the urea linkages formed by crosslinking of collagen with the end-isocyanate of the oligourethanes. This was observed as a shoulder in the amide I region at wavenumber around 1710–1740 cm−1 (Fig. 3a inset). A characteristic intense band due to stretching of C–H bonds around 2900 cm−1 was observed as well, which was more intense for formulations with oligourethanes. Moreover, the presence of SiP was confirmed by an intense band around 1060 cm−1 due to stretching of siloxane bonds (Si–O–Si) (Fig. 3a inset).
O (amide I), and deformation of N–H (amide II) in the regions of 3400–3200, 1700–1600, and 1650–1550 cm−1, respectively, as illustrated in Fig. 3a. The IR bands of the Dex were not observed in the spectra of the biocomposites, due to that the signals could be superimposed with the signals of the major components of the composite hydrogels, i.e., type I collagen and oligourethanes. On the other hand, the IR bands revealed the urea linkages formed by crosslinking of collagen with the end-isocyanate of the oligourethanes. This was observed as a shoulder in the amide I region at wavenumber around 1710–1740 cm−1 (Fig. 3a inset). A characteristic intense band due to stretching of C–H bonds around 2900 cm−1 was observed as well, which was more intense for formulations with oligourethanes. Moreover, the presence of SiP was confirmed by an intense band around 1060 cm−1 due to stretching of siloxane bonds (Si–O–Si) (Fig. 3a inset).
The crosslinking index, defined as the extent of amine groups that have reacted with end-isocyanate groups, did not have a dependence on the SiP content, as shown in Fig. 3b. It can be assumed that SiP were dispersed inside the composite hydrogels, and they were not able to form chemical crosslinking linkages. Therefore, the reticulation of the collagen matrix was due uniquely to the reaction with the oligourethanes.
The analysis for mass loss of composite hydrogels after the incubation with type I collagenase (Fig. 3c) indicated that the rate of enzymatic degradation had not a direct relationship with the SiP content. The Dex-loaded SIS hydrogel was degraded completely after 5 days, while composite hydrogels were degraded completely after 14 days. The Dex-loaded SIS P0, SIS P5 and SIS P15 composite hydrogels exhibited similar degradation profiles without statistically significant variations of the mass loss over time.
The evaluation of the viscoelastic response of composite hydrogels indicated that the storage modulus (G′) of Dex-loaded composite hydrogels at 1 Hz increased 3.2, 3.6 and 2.8 times, for SIS P0, SIS P5 and SIS P15, respectively, compared to the SIS hydrogel (Fig. 3d). All materials displayed G′ values higher than the loss modulus (G′′) (Fig. 3e), which means that all samples featured a gel-like behavior. The G′ for composite hydrogels free of Dex was previously reported around 45 Pa at 0.1 Hz;27 the present results indicated that the Dex incorporation decreased the elastic modulus in a 73%.
3.4. Microstructure of Dex-loaded composite hydrogels
The morphology of the cross-section of all four lyophilized composites was evaluated by scanning electron microscopy, as shown in Fig. 4a–d. The Dex-loaded composites exhibited a microstructure with interconnected pores. The analysis of the pore sizes revealed mean sizes of 298 ± 54, 213 ± 36, 221 ± 73 and 219 ± 58 μm for sponges derived from SIS, SIS P0, SIS P5 and SIS P15, respectively. The pore size for SIS was statistically significant versus SIS P0, which can be related to the oligourethane crosslinking. The pore size was not significantly varied with the silica content. At higher magnification can be seen that the composite sponges were formed by a network with interconnected pores, where the fibers had a mean diameter of 128 ± 21 nm (Fig. 4e). The EDX analysis for all-four composite sponges (data not shown) indicated that the elemental composition of the materials was mainly C, N, O; albeit Si was also detected. Fig. 4f shows that the fibrous surface of the pore walls was coated by silica particles, but it was not possible to distinguish the size of the silica particle by SEM. However, TEM analysis revealed the ultrastructure of the SiP dispersed in the composites, as shown in Fig. 4g–i, which allows observing not only the particle size and shape of the single particle, but also particle porosity. The uniform size distribution and mesoporous structure of the particles can be clearly observed from TEM images. A population of mesoporous siliceous nanoparticles with particle diameters and pore size of 86–98 and 5–19 nm, respectively, was appreciated. More TEM images of silica nanoparticles of the composite hydrogels are given in the ESI (Fig. S2†). | ||
| Fig. 4 Microstructure of Dex-loaded biocomposites. Representative SEM micrographs indicating the pore size for composite sponges: SIS (a), SIS P0 (b), SIS P5 (c) and SIS P15 (d). Representative SEM micrograph shows the typical fibrilar network in the biocomposites (e) and silica particles deposited on fiber surface (f). Representative TEM micrographs of material after calcination of biocomposites show the population of meso-nanosilica particles (g). Magnification zones illustrate the diameter (h) and pore size (i) of mesoporous nanosilica particles. | ||
3.5. Swelling and release of Dex and silica from composite hydrogels
Fig. 5a illustrates the effect of the SiP content on the water absorption of Dex-loaded composite hydrogels. The SIS hydrogel absorbed around 117% of its initial weight after 8 day incubation. The SIS P0 hydrogel showed a lower ability to absorb water, absorbing 109% of water after 10 days. However, the SIS P5 and SIS P15 composite hydrogels exhibited a higher water uptake than SIS P0 and SIS alone; SIS P5 firstly absorbed around 111% after 2 days and 117% after 7 days, while SIS P15 first absorbed 118% after 2 days and 122% after 7 days. Besides, a plateau was reached after 9 days with a maximum water uptake around 122%, for SIS, SIS P5 and SIS P15.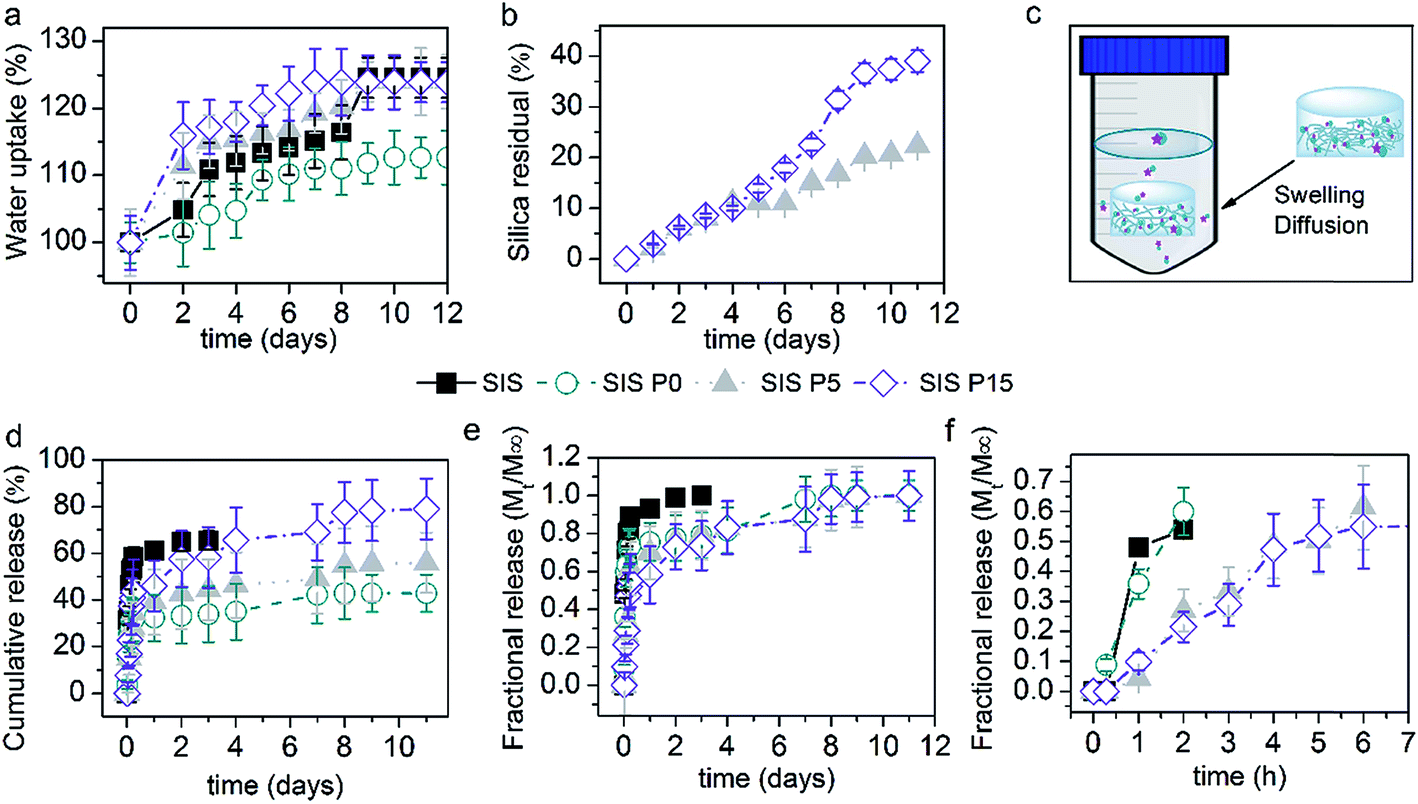 | ||
| Fig. 5 Dex-release profiles from biocomposite hydrogels. (a) Variation of water uptake produced in materials with respect to time. SIS P5 and SIS P15 materials are statistically different (p < 0.05) compared to SIS P0. (b) Silica debris release from composite hydrogels. (c) Schematic description of the experimental setup for releasing Dex and silica debris. The cumulative release (d) and fractional release (e) profiles of Dex from biocomposite hydrogels. Fractional release at shorter time periods (f). The cumulative release of Dex produced in SIS P15 materials is statistically different (p < 0.05) compared to SIS P0 and SIS P5 after 1 day of incubation. The cumulative release of Dex from 1 to 3 days for SIS is significant (p < 0.05) compared to SIS P0, SIS P5 and SIS P15. Data are expressed as mean values ± SD, n = 3. | ||
Silica debris was released from SIS P5 and SIS P15 biocomposites, as shown in Fig. 5b. The released amount reached a maximum of 22 and 39% of loaded-initially silica after 11 days for SIS P5 and SIS P15, respectively. Fig. 5c schematizes the experimental setup for releasing of Dex and silica debris. Fig. 5d and e show the release efficiency profiles (Mt/M0 × 100) and fractional release profiles (Mt/M∞) of Dex from biocomposite collagen hydrogels, respectively. The Dex released from SIS P0, SIS P5 and SIS P15 hydrogels exhibited a biphasic pattern characterized by fast initial release in the first 12 h, followed by a slower release phase. In the first 6 h, 47, 27, 18 and 23% of loaded-initially Dex was efficiently released from SIS, SIS P0, SIS P5 and SIS P15, respectively. For SIS alone hydrogel, 262 ± 4 μg mL−1 of Dex was released after 3 days; afterwards, the disintegration of the material was observed, which made it difficult to quantify the Dex due to the interferences with the dissolved gel components. For SIS P0, SIS P5 and SIS P15 hydrogels, 135 ± 12, 177 ± 13 and 232 ± 23 μg mL−1 of Dex were released after 3 days, respectively. Ultimately, results indicated that 43, 56 and 79% of loaded-initially Dex was efficiently released from SIS P0, SIS P5 and SIS P15 hydrogels after 9 days, respectively. The SIS P5 and SIS P15 composites performed a delayed fractional release of Dex, as shown in Fig. 5f. However, this controlled release was not observed for SIS alone or SIS P0 hydrogels.
3.6. Effect of the Dex-loaded hydrogels on the cell response
In first instance, the biological response to composite hydrogels releasing Dex was assessed in terms of the metabolism of fibroblasts and macrophages cultured on the materials. The metabolic activity of macrophages was increased when cells were cultured 3 days in all four materials compared to the control, as shown in Fig. 6a. However, the metabolic activity of fibroblasts after this same elapsed time was decreased, as shown in Fig. 6b. After 7 days of culture, the metabolic activity of macrophages or fibroblasts was increased in both the controls and composite materials, compared to the 3 day culture. This metabolic activity was not significantly modified in composite materials compared to controls after 7 day culture, but was drastically decreased in SIS alone materials.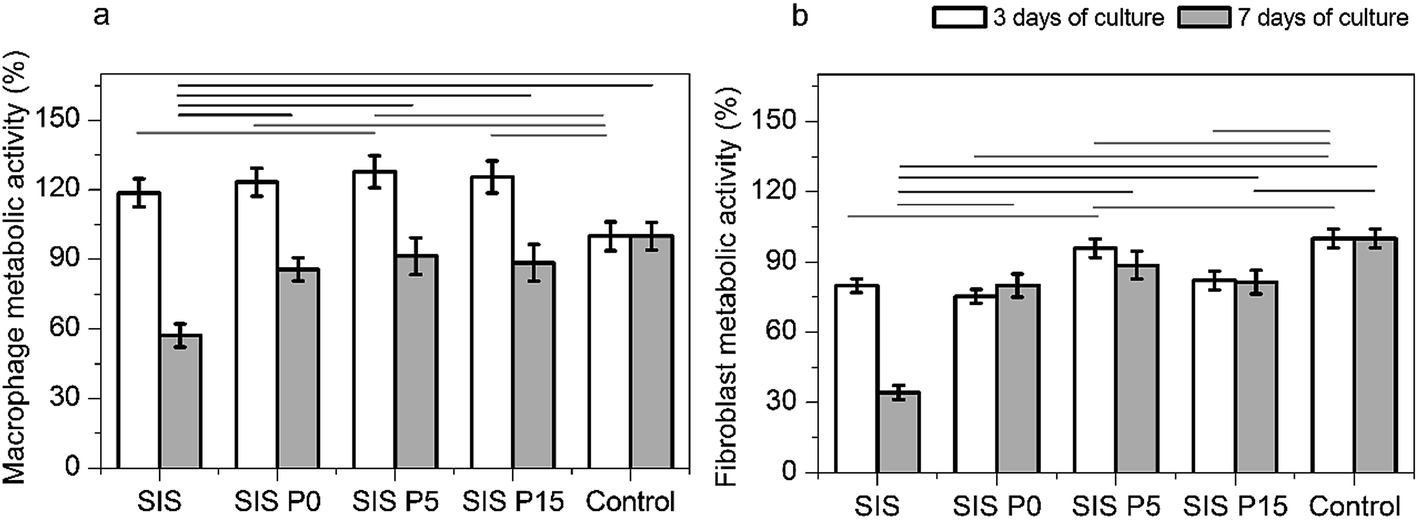 | ||
| Fig. 6 Evaluation of the cell responses. Metabolic activity of macrophage (a) and fibroblasts (b) after 3 and 7 days of culture on composite hydrogels. Data are expressed as mean values ± SD, n = 3. The difference of the means is significant (p < 0.05) between all marked groups. | ||
In another experiment set, the effect of soluble products from Dex-loaded composite hydrogels on macrophage metabolic activity was evaluated. The metabolic activity of macrophages cultured with products extracted from all-four materials was higher in comparison with cells cultured without extracts, as shown in Fig. S4.† However, this effect was ameliorated when cells were stimulated with extracts diluted to 50 and 25% with fresh medium. These results suggest that products extracted from Dex-loaded composite hydrogels, were responsible of the enhanced metabolic activity of macrophages.
The secretion of cytokines TGF-β1 and CCL-2 (involved in angiogenesis and inflammation processes) confirmed the activation of macrophages by Dex-loaded composite hydrogels, as shown in Fig. 7. All four Dex-loaded composite hydrogels showed a higher TGF-β1 secretion than the control (cells without materials). During the first 3 days of culture, the macrophages cultured in SIS and SIS P5 hydrogels secreted the highest amount of TGF-β1 (Fig. 7a) compared to other composite hydrogels. However, after 7 day culture, the TGF-β1 secretion by macrophages cultured in SIS hydrogels and controls was significantly decreased. The secretion of TGF-β1 by macrophages cultured on composite hydrogels was maintained with the culture time. The macrophages seeded on all four Dex-loaded hydrogels did not show a significantly different secretion of CCL-2, compared to control after 3 day culture (Fig. 7b). The CCL-2 production was significantly diminished from 3 to 7 days of culture in cells cultured in the Dex-loaded materials, as well as in the control. These results suggest that the Dex-loaded biocomposites were suitable to stimulate the secretion of TGF-β1 by macrophages, but, meanwhile, were not proficient to stimulate to the secretion of CCL-2.
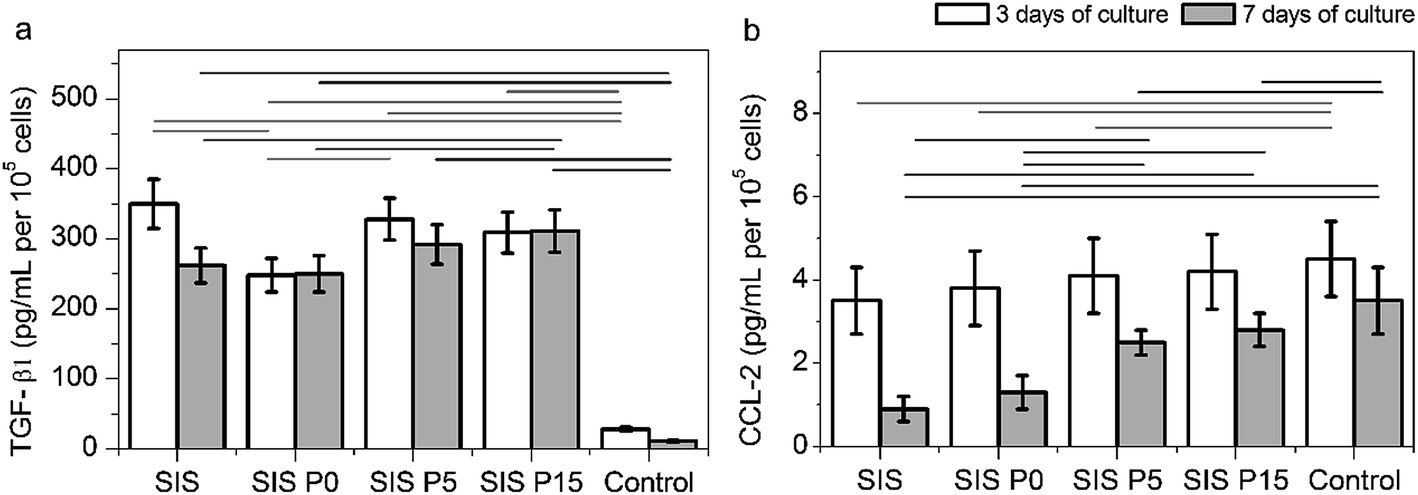 | ||
| Fig. 7 Analysis for the macrophage activation. The secretion of TGF-β1 (a) and CCL-2 (b) by macrophages cultured on composite hydrogels. Data are expressed as mean values ± SD, n = 3. The difference of the means is significant (p < 0.05) between all marked groups. | ||
4. Discussion
The loading of therapeutics in hydrogels based on collagen is being explored in strategies intended to modulate the inflammation processes and to control infections in the wound healing materials field.1,26,33 The limitation of collagen-based systems is the therapeutic bioavailability for long-term applications. For instance, the “burst release” phenomenon leads to excessive initial therapeutic delivery and also reduces the effective lifetime of the device.34 This phenomenon is favored in collagen systems, due to their poor mechanical stability, rapid degradation and high susceptibility to hydrolysis.35 With this scenario, efficient strategies are needed to regulate the hydrophobicity/hydrophilicity ratio, to enhance mechanical stability and to control degradation rate, ensuring the biocompatibility of the collagen hydrogels.36,37The therapeutic dose of Dex used in conventional formulations has been reported in 0.5 mg per mL.38 Strategies to reduce the burst effect have been designed with polymeric materials such as PGLA,22,39 PCL40 and PEG;41 as well as with silica materials.42 This work reports an alternative to incorporate Dex in hydrogels comprised of type I collagen, oligourethanes and silica particles, where collagen is the major component. Thus, these hybrid hydrogels are studied as vehicles of controlled release of a therapeutic molecule capable of influence the immune cells (Fig. 1).
The crosslinking of the collagenous network produced by the oligourethanes is important to regulate the structure and properties of the composite hydrogels. The incorporation of silica particles allows the loading of Dex up to 400 μg of Dex per each cm3 of gel. According to turbidimetry results, this concentration of Dex does not modify significantly the formation process of 3D polymeric network. Similarly, the crosslinking density, degradation, and swelling do not vary significantly in hydrogels with and without Dex.27 However, the storage modulus and pore size in composite hydrogels loaded with Dex are lower compared to Dex-free materials.27 The presence of Dex inside composite hydrogels diminishes the longitudinal alignment of the collagen molecules due to the reduced formation of entanglement points, altering the fibrous network structure and decreasing its elasticity (G′). Similar evidences are reported in systems based on collagen loaded with dopamine43 and γ-cyclodextrin hydrogels with drug loaded micelles.44 On the other hand, the increment of SiP content influences the collagen polymerization and the structural network parameters (Fig. 2 and Table 1). It has been reported that negatively charged silica particles interact with positively charged collagen amines accelerating or inhibiting the fibrillogenesis according to the silica concentration.45 The urea linkages produced by the addition of the free collagen-amines with the PUP end-isocyanates validate the collagen crosslinking, while the identification of the siloxane bonds (Si–O–Si) ensures the presence of silica dispersed in the 3D matrix. The increment in the storage moduli has a direct relationship with the crosslinking extent produced by the oligourethane, and this relation does not depend on the SiP content (Fig. 3d). The improvement in the storage modulus is associated with the crosslinking inside collagenous network.46 A laminar structure with interconnected pores is found for all-four materials (Fig. 4). The maintained typical microstructure for collagenous materials can enhance the cell attachment, proliferation and migration in tissue engineering strategies, allowing the diffusion of oxygen and nutriments, and release of therapeutics.47 The removal of the organic matter from the composites confirms the presence of mesoporous silica nanoparticles (Fig. 4g). The silica particles accelerate the collagen polymerization but do not affect the pore size and the mechanical properties of the hybrid hydrogels. It has been reported that the mechanical properties of silica–collagen materials are increased when silica bears reactive moieties that can covalently bond to amine or carboxylic groups of collagen.45 In this work, the increment in the storage modulus has a direct relationship with the crosslinking extent produced by the oligourethane, but not with the SiP content (Fig. 3d).
It has been reported that the mesoporous silica nanoparticles have a high surface area and strong adsorbent capacity, providing stable sites for the adsorption of diverse drugs,41,48 consequently they represent an opportunity to regulate the burst release in collagen-based materials. Also, the silica particles are not covalently linked with the collagen, and therefore the silica debris with trapped drug can diffuse through the gel network. The water uptake and degradation of ECM-hydrogels contribute with the absorption of wound exudate, prevention of scar tissue formation, and regularization of drugs release.49–51 In this work, a direct relation between water uptake capacity and silica content is found (Fig. 5a), while the degradation rate of composite hydrogels is not dependent on the SiP concentration (Fig. 3c). The bioavailability of the drugs loaded in the composite collagen hydrogels could be controlled by the degradation adjusted by the oligourethane crosslinking or by the swelling tailored by the silica particles. The present results suggest that the Dex release is related to the residual silica release during the swelling process. A higher concentration of Dex is released as long as a higher residual silica release is observed (Fig. 5). These results are congruent with the hydrolysis at neutral pH of silica particles.46 Thus, this delivery vehicle is capable of provide silica dissolution products that could have a synergic effect with therapeutic molecules to regulate cell response.
The observed direct relationship between the concentration of SiP and the Dex release efficiency is in line with the swelling behavior of composite hydrogels (Fig. 5). The swelling is result of the transport of water and mobile ions through the network, as well as of the absorption of water and the association, dissociation and binding of ions to the network chains.52,53 The present results suggest that the time taken to reach water uptake equilibrium dependent on the modification of collagen with oligourethane/silica. Moreover, at longer incubation times, the hydrolysis of silica and degradation of portions of uncrosslinked collagen could affect physicochemical parameters such as the fixed (bound to the network components) charge and the ionic strength of the solution; consequently affecting the swelling kinetic. An increment of the degree of water uptake with the incubation time appears to causes the hydrogels to increase the release of Dex. In addition, the swelling of the composite matrix favors the diffusion of drug-loaded silica particles outside the materials, as previously was proposed for other drug-release systems based on silica particles.54 From a physicochemical view point, the therapeutic can be adsorbed in the surface of mesoporous nanosilica forming both ionic interactions and hydrogen bonds.55,56 The “burst effect” observed in the release of Dex from collagen alone suggests that Dex does not have strong interactions with the collagen or that diffusivity in the collagen network is high (Fig. 5). On the other hand, the reduction of the burst release of Dex from composite collagen suggests that crosslinking with oligourethane and the addition of SiP induced stronger interactions with Dex or lower diffusivity. The dependence of the release efficacy of Dex with the SiP content suggests that the SiP in the composite hydrogels could be used in the control of the Dex release, presumably via the hydrogen bonds on SiP surface, as shown in Fig. 8. A higher concentration of SiP is related to higher surface area, which provided active sites for the drug adsorption.40 Moreover, the sustained Dex release can be used to increase its therapeutic effect while minimizing side-effects, as previously reported with a variety of polymeric carriers.57
 | ||
| Fig. 8 Scheme for the interactions between dexamethasone and components of the composite hydrogel. (a) Formation of urea linking between the primary collagen amine and the end-isocyanate of the oligourethane. (b) Hydrogen bond formed between Dex and silica–collagen. | ||
To have a first idea of the potential of the delivery system comprised of Dex and composite collagen for inducing activation of cells that secrete signaling molecules key in the control of inflammation and wound healing, the in vitro cell response was assessed. In this regard, the MTT results suggest that the metabolic activity of fibroblasts and macrophages is increased by the components of this novel Dex-releasing vehicle (Fig. 6). These results are consistent with the ability of Dex to active macrophages.58–60 This study also showed evidence to promote secretion of TGF-β1 cytokine by the vehicle releasing silica and Dex (Fig. 7a), which can be used in strategies to optimize the healing of chronic wounds. The CCL-2 secretion is not affected by the hybrid materials, which is expected due to the inflammatory action of this cytokine.59 The delivery of Dex from electrospun fibers materials has allowed the regulation of inflammatory related gene expression in Raw 264.7 macrophages,58 in turn, the sustained release of Dex from cyclodextrin/dextran hydrogels has induced the osteogenic differentiation of stem cells.61 To the best of the authors knowledge, to date a collagen-based material that has the ability to controlled release of Dex has not been reported. Thus, the biocomposites reported here represent a promising approach among strategies already available for modifying collagen biomaterials to effectively modulate the host responses. The in vivo response to this local delivery system of Dex and silica debris from composite collagen hydrogels is under study and will the subject of another paper.
5. Conclusions
An efficient dexamethasone-release system based on hydrogels comprised of collagen, oligourethane and silica was reported. The oligourethane crosslinking regulated the viscoelastic properties and degradation of the biocomposites, while the incorporation of colloidal silica accelerated the collagen polymerization modifying the structural parameters of the collagenous network. The incorporation and release of dexamethasone as well as the matrix swelling were controlled by the presence of the mesoporous silica particles in the composite collagen hydrogels. Consequently, the metabolic activity and TGF-β1 secretion of macrophages were stimulated by the local release of dexamethasone.Acknowledgements
We thank to Maria C. Lona by assistance with cells viability assays and Prof. Jorge Delgado by support for the use of a rheometer in the UG' soft matter laboratory. The authors are grateful for the financial support from the Consejo Nacional de Ciencia y Tecnología (grant PDCAPN 1310-2015, México), and the Universidad de Guanajuato (grant 1146/2016-17).References
- G. S. Alvarez, C. Hélary, A. M. Mebert, X. Wang, T. Coradin and M. F. Desimone, J. Mater. Chem. B, 2014, 2, 4660 RSC.
- D. Wallace, Adv. Drug Delivery Rev., 2003, 55, 1631–1649 CrossRef CAS PubMed.
- M. R. Blatchley and S. Gerecht, Biomed. Mater., 2015, 10, 034001 CrossRef PubMed.
- C. Caramella, B. Conti, T. Modena, F. Ferrari, M. C. Bonferoni, I. Genta, S. Rossi, M. L. Torre, G. Sandri, M. Sorrenti, L. Catenacci, R. Dorati and G. Tripodo, J. Drug Delivery Sci. Technol., 2016, 32, 206–228 CrossRef CAS.
- S. Hinderer, S. L. Layland and K. Schenke-Layland, Adv. Drug Delivery Rev., 2016, 97, 260–269 CrossRef CAS PubMed.
- T. D. Sargeant, A. P. Desai, A. S. Banerjee, A. Agawu and J. B. Stopek, Acta Biomater., 2012, 8, 124–132 CrossRef CAS PubMed.
- D. J. Munoz-Pinto, A. C. Jimenez-Vergara, T. P. Gharat and M. S. Hahn, Biomaterials, 2015, 40, 32–42 CrossRef CAS PubMed.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, J. Mater. Chem. B, 2014, 2, 5319 RSC.
- J. A. Claudio-Rizo, B. Mendoza-Novelo, J. Delgado, L. E. Castellano and J. L. Mata-Mata, Biomed. Mater., 2016, 11, 035016 CrossRef PubMed.
- D. Choi, J. Heo, J. H. Park, Y. Jo, H. Jeong, M. Chang, J. Choi and J. Hong, J. Ind. Eng. Chem., 2016, 36, 326–333 CrossRef CAS.
- H. Teles, T. Vermonden, G. Eggink, W. E. Hennink and F. A. de Wolf, J. Controlled Release, 2010, 147, 298–303 CrossRef CAS PubMed.
- G. T. Tihan, I. Rău, R. G. Zgârian and M. V. Ghica, C. R. Chim., 2016, 19, 390–394 CrossRef.
- S. Heinemann, T. Coradin and M. F. Desimone, Biomater. Sci., 2013, 1, 688 RSC.
- M. F. Desimone, C. Helary, I. B. Rietveld, I. Bataille, G. Mosser, M. M. Giraud-Guille, J. Livage and T. Coradin, Acta Biomater., 2010, 6, 3998–4004 CrossRef CAS PubMed.
- X. Wang, C. Helary and T. Coradin, ACS Appl. Mater. Interfaces, 2015, 7, 2503–2511 CAS.
- S. D. Patil, F. Papadmitrakopoulos and D. J. Burgess, J. Controlled Release, 2007, 117, 68–79 CrossRef CAS PubMed.
- S. Bae, H. J. Lee, J. S. Lee and K. Webb, Biomacromolecules, 2015, 16, 2757–2765 CrossRef CAS PubMed.
- C. Z. Liang, H. Li, Y. Q. Tao, L. H. Peng, J. Q. Gao, J. J. Wu, F. C. Li, J. M. Hua and Q. X. Chen, Acta Biomater., 2013, 9, 9423–9433 CrossRef CAS PubMed.
- X. Du, X. Li, L. Xiong, X. Zhang, F. Kleitz and S. Z. Qiao, Biomaterials, 2016, 91, 90–127 CrossRef CAS PubMed.
- G. Arakeri and P. A. Brennan, Med. Hypotheses, 2012, 78, 682–686 CrossRef CAS PubMed.
- D. Yu, C. Sun, Z. Zheng, X. Wang, D. Chen, H. Wu, X. Wang and F. Shi, Int. J. Pharm., 2016, 503, 229–237 CrossRef CAS PubMed.
- I. Galeska, T. Kim, S. D Patil, U. Bhardwaj, D. Chatttopadhyay, F. Papadimitrakopoulos and D. J. Burgess, AAPS J., 2005, 7, E231–E240 CrossRef CAS PubMed.
- H. Jung, J. S. Park, J. Yeom, N. Selvapalam, K. M. Park, K. Oh, J. A. Yang, K. H. Park, S. K. Hahn and K. Kim, Biomacromolecules, 2014, 15, 707–714 CrossRef CAS PubMed.
- M. D. Moya-Ortega, C. Alvarez-Lorenzo, H. H. Sigurdsson, A. Concheiro and T. Loftsson, Carbohydr. Polym., 2010, 80, 900–907 CrossRef CAS.
- C. Gao, Y. Cai, X. Kong, G. Han and J. Yao, Mater. Lett., 2013, 93, 312–315 CrossRef CAS.
- R. Sánchez-Sánchez, E. Martínez-Arredondo, V. Martínez-López, Y. Melgarejo-Ramírez, A. Brena-Molina, H. Lugo-Martínez, R. Gómez-García, D. Garciadiego-Cázares, P. Silva-Bermúdez, E. Márquez-Gutiérrez, C. Ibarra and C. Velasquillo, J. Nanomater., 2016, 2016, 1–8 CrossRef.
- J. A. Claudio-Rizo, M. Rangel-Argote, P. U. Muñoz-González, L. E. Castellano, J. Delgado, G. Gonzalez-García, J. L. Mata-Mata and B. Mendoza-Novelo, J. Mater. Chem. B, 2016, 4, 6497–6509 RSC.
- B. Mendoza-Novelo, M. C. Lona-Ramos, G. González-García, L. E. Castellano, J. Delgado, P. Cuellar-Mata, J. M. Flores-Moreno, J. Vargas, J. A. Gutiérrez, E. E. Ávila and J. L. Mata-Mata, RSC Adv., 2014, 4, 63457–63465 RSC.
- B. Mendoza-Novelo, G. González-García, J. L. Mata-Mata, L. E. Castellano, P. Cuéllar-Mata and E. E. Ávila, Mater. Lett., 2013, 106, 369–372 CrossRef CAS.
- C. Dima, L. Patrascu, A. Cantaragiu, P. Alexea and S. Dima, Food Chem., 2016, 195, 39–48 CrossRef CAS PubMed.
- J. Zhu and L. J. Kaufman, Biophys. J., 2014, 106, 1822–1831 CrossRef CAS PubMed.
- E. M. Carr and J. Hermans, Macromolecules, 1978, 11, 46–50 CrossRef PubMed.
- Y. Hayashido, K. Shirasuna, T. Sugiura, M. Nakashima and T. Matsuya, Cancer Lett., 1996, 108, 81–86 CrossRef CAS PubMed.
- Z. Ruszczak and W. Friess, Adv. Drug Delivery Rev., 2003, 55, 1679–1698 CrossRef CAS PubMed.
- A. M. S. Costa and J. F. Mano, Eur. Polym. J., 2015, 72, 344–364 CrossRef CAS.
- M. de Wild, W. Pomp and G. H. Koenderink, Biophys. J., 2013, 105, 200–210 CrossRef CAS PubMed.
- R. Hartwell, V. Leung, C. Chavez-Munoz, L. Nabai, H. Yang, F. Ko and A. Ghahary, Acta Biomater., 2011, 7, 3060–3069 CrossRef CAS PubMed.
- T. Hickey, D. Kreutzer, D. J. Burgess and F. Moussy, Biomater. Sci., 2002, 23, 1649–1656 CrossRef CAS.
- K. Wang, X. Xu, Y. Wang, X. Yan, G. Guo, M. Huang, F. Luo, X. Zhao, Y. Wei and Z. Qian, Int. J. Pharm., 2010, 389, 130–138 CrossRef CAS PubMed.
- L. W. Norton, E. Tegnell, S. S. Toporek and W. M. Reichert, Biomaterials, 2005, 26, 3285–3297 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- G. Cevc and G. Blume, Biochim. Biophys. Acta, 2004, 1663, 61–73 CrossRef CAS PubMed.
- S. Zhu, Z. Gu, S. Xiong, Y. An, Y. Liu, T. Yin, J. Yu and Y. Hu, RSC Adv., 2016, 6, 66180–66190 RSC.
- Z. Liu and P. Yao, Polym. Chem., 2014, 5, 1072–1081 RSC.
- S. Heinemann, C. Heinemann, R. Bernhardt, A. Reinstorf, B. Nies, M. Meyer, H. Worch and T. Hanke, Acta Biomater., 2009, 5, 1979–1990 CrossRef CAS PubMed.
- M. F. Desimone, C. Helary, S. Quignard, I. B. Rietveld, I. Bataille, G. J. Copello, G. Mosser, M. M. Giraud-Guille, J. Livage, A. Meddahi-Pelle and T. Coradin, ACS Appl. Mater. Interfaces, 2011, 3, 3831–3838 CAS.
- H. S. Yu, E. J. Lee, S. J. Seo, J. C. Knowles and H. W. Kim, J. Biomater. Appl., 2015, 30, 338–350 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- S. Perumal, S. K. Ramadass, A. Gopinath, B. Madhan, G. Shanmugam, J. Rajadas and A. B. Mandal, J. Mech. Behav. Biomed. Mater., 2015, 52, 131–138 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- F. Zhao, D. Yao, R. Guo, L. Deng, A. Dong and J. Zhang, Nanomaterials, 2015, 5, 2054–2130 CrossRef CAS.
- A. D. Drozdov, J. de Claville-Christiansen and C. G. Sanporean, Int. J. Solids Struct., 2016, 87, 11–25 CrossRef CAS.
- S. K. De, N. R. Aluru, B. Johnson, W. C. Crone, D. J. Beebe and J. Moore, J. Microelectromech. Syst., 2002, 11, 544–555 CrossRef CAS.
- E. Haartman, D. Lindberg, N. Prabhakar and J. M. Rosenholm, Eur. J. Pharm. Sci., 2016, 95, 17–27 CrossRef PubMed.
- M. Prokopowicz, K. Czarnobaj, A. Szewczyk and W. Sawicki, Mater. Sci. Eng., C, 2016, 60, 7–18 CrossRef CAS PubMed.
- A. Salis, L. Medda, F. Cugia and M. Monduzzi, Colloids Surf., B, 2016, 137, 77–90 CrossRef CAS PubMed.
- J. Urbanska, A. Karewicz and M. Nowakowska, Life Sci., 2014, 96, 1–6 CrossRef CAS PubMed.
- Y. F. Li, M. Rubert, Y. Yu, F. Besenbacher and M. Chen, RSC Adv., 2015, 5, 34166–34172 RSC.
- G. D. Keeler, J. M. Durdik and J. A. Stenken, Acta Biomater., 2015, 12, 11–20 CrossRef CAS PubMed.
- W. Wang, S. Lin, Y. Xiao, Y. Huang, Y. Tan, L. Cai and X. Li, Life Sci., 2008, 82, 190–204 CrossRef CAS PubMed.
- A. C. Lima, A. M. Puga, J. F. Mano, A. Concheiro and C. Alvarez-Lorenzo, J. Mater. Chem. B, 2014, 2, 4943–4956 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25989h |
| This journal is © The Royal Society of Chemistry 2017 |