
Simultaneous detection of multiple viruses based on chemiluminescence and magnetic separation
Zeeshan
Ali†
ab,
Jiuhai
Wang†
a,
Yongjun
Tang
b,
Bin
Liu
c,
Nongyue
He
*ad and
Zhiyang
Li
*e
aState Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, P. R. China. E-mail: nyhe1958@163.com
bSchool of Applied Chemistry and Biotechnology, Shenzhen Polytechnic, Shenzhen 518055, P. R. China
cDepartment of Biomedical Engineering, School of Basic Medical Sciences, Nanjing Medical University, Nanjing 210029, China
dHunan Key Laboratory of Green Packaging and Application of Biological Nanotechnology, Hunan University of Technology, Zhuzhou 412007, P. R. China
eDepartment of Clinical Laboratory, the Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing 210008, P. R. China. E-mail: lizhiyangcn@qq.com
First published on 15th November 2016
Abstract
Nucleic acid testing (NAT) based methods are more sensitive and specific, and are preferred over enzyme immunoassays. Different NAT based protocols have been designed to detect multiple pathogens in order to reduce the inherent high cost for detection. However, these assays do not reliably detect a large number of pathogens at once. In this report, a DNA hybridization based chemiluminescence detection method has been proposed for reliable detection of multiple pathogens. The idea was practically demonstrated by carrying out simultaneous extraction and amplification of hepatitis B virus (HBV), hepatitis C virus (HCV), and human immunodeficiency virus (HIV-1 group M, herein referred to as HIV) through the one step multiplex reverse transcription polymerase chain reaction (RT-PCR). A detailed study was conducted to optimize the factors which could affect the chemiluminescence signal. The probes for HBV, HCV, and HIV demonstrated higher specificity by only capturing their respective target sequence. 10 viral copies per mL of serum were detected in the monoplex detection protocol for HBV, HCV, and HIV, respectively. However, when the sensitivity of each virus was analyzed in the presence of higher loads of other viruses in the multiplex detection assay, the assay finally detected 10 HBV copies, 10 HCV copies, and 100 HIV copies per microliter of serum. The use of silica modified magnetic nanoparticles during nucleic acid extraction and carboxyl coated magnetic nanoparticles during the chemiluminescence step can help modify this system into an automated platform for high throughput applications.
Introduction
Transmission of HBV, HCV, and HIV through blood transfusion can be markedly reduced through fast and reliable methods for pathogenic detection in blood donors. Despite huge efforts invested in improving immunoassays, their performance has been questioned during the window period, non-immunogenic and immune-variant forms of infection. Nucleic acid testing (NAT) based methods are more sensitive and specific, and are preferred over enzyme immunoassays. These assays directly target pathogen's genome as the detection strategy; however, the technical labor required for performing these tests and high detection costs are disadvantages for these assays.1 Multiplex PCRs have been designed to simultaneously detect various pathogens in one tube, to increase the detection capacity for the diagnostic assay and decrease the per pathogen detection cost. Different technical approaches have been adopted to develop multiplex assays, such as PCR, reverse transcription-polymerase chain reaction (RT-PCR),2,3 nucleic acid sequence-based amplification (NASBA),4 and transcription-mediated amplification (TMA).5,6Real time multiplex detection systems are most advanced and sensitive tools, and detection of amplified products in these devices is carried out using either fluorescent labeled probes or DNA binding fluorophores. However, the adsorption and fluorescence spectra for different fluorophores used in fluorescent labeled probe systems tend to interfere with one another, limiting the reliable detection of maximum four different pathogens.7 Moreover, the total amount of the amplified product is detected when using the DNA binding fluorophores like Sybr green, which limits their use.8,9
Oligonucleotide immobilization is an alternative approach for improving multiplex detection.10–13 Optical labels such as fluorescent tags,14,15 colorimetric nanoparticles16–18 and chemiluminescent (CL)19 labels are increasingly used in the development of DNA hybridization assays. CL and fluorescence based protocols are more practical approaches than others.20 However, the CL is preferred over fluorescence based detection owing to its high sensitivity, low background and simple instrumentation.21–23 Besides, surface modified magnetic particles have been extensively used for cell purification, nucleic acid isolation, drug delivery, immunoassays, and separation and enrichment of target molecules, because of their easy manipulation under an external magnetic field, which can be exploited when developing high throughput and automated detection platforms.24–28
Inspired by these advancements in nanotechnology, we combined the magnetic separation technology for nucleic acid purification with a chemiluminescence technique for post amplification detection of three lethal viruses. Besides the high sensitivity, the main advantage of our developed sensor is that reliable detection of even more than three different pathogens can be realized by optimizing few steps in our experimental protocol. Scheme 1 demonstrates the strategy which was followed during the experimentation. First, silica modified MNPs were used to extract viral (HBV, HCV, and HIV) nucleic acids. Then, the one step multiplex reverse transcription polymerase chain reaction (RT-PCR) was carried out to amplify the extracted viral nucleic acids. Different amino-modified probes for capturing HBV, HCV, and HIV target sequences were separately attached onto the surface of carboxyl coated MNPs. These probe modified MNPs were incubated with RT-PCR biotin labeled products in different tubes to capture their respective HBV, HCV or HIV sequences. Streptavidin-modified alkaline phosphatase (SA-AP) was allowed to bind with biotin. Finally, 3-(2′-spiroadamantane)-4-methoxy-4-(3′-phosphoryloxy)phenyl-1,2-dioxetane (AMPPD) was added, and chemiluminescence was recorded.
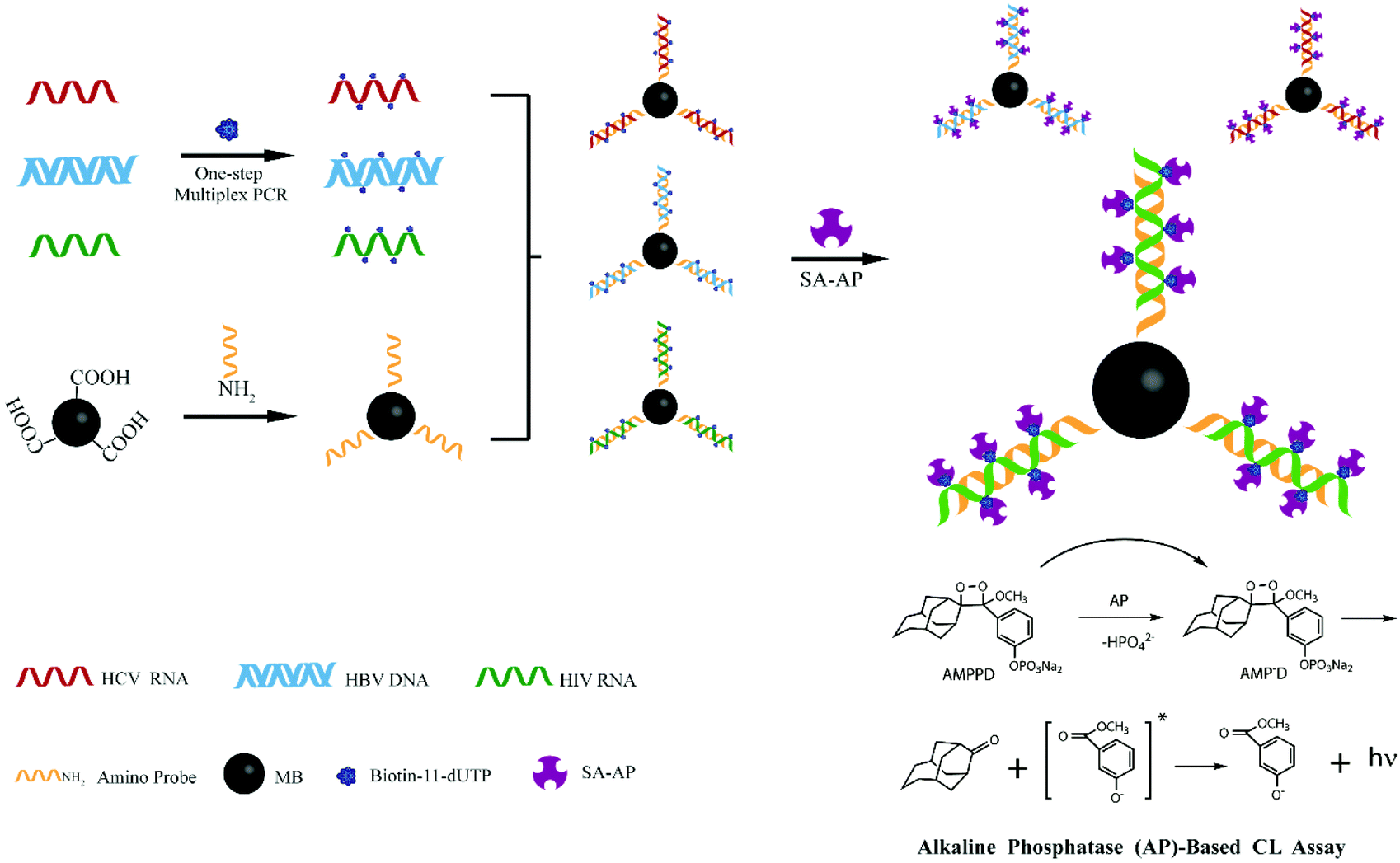 | ||
| Scheme 1 Illustration of one step multiplex RT-PCR and chemiluminescence detection of multiple viruses. | ||
Experimental
Materials
HBV, HCV, and HIV-1 group M (herein mentioned as HIV) infected serum samples were obtained from the Jiangsu Province Blood Center and the Second Affiliated Hospital of Nanjing Medical University.Guanidine hydrochloride (Gu·HCl), ethylenediaminetetraacetic acid (EDTA), Triton X-100, diethylpyrocarbonate (DEPC), magnesium chlorides (MgCl2), sodium dodecylsulfate (SDS), salt-sodium citrate (SSC), 2-(N-morpholino)ethanesulfonic acid (MES), N,N-dimethylformamide (DMF), bovine serum albumin (BSA), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), Tween 20, (3-aminopropyl)triethoxysilane (APTES), succinic anhydride (SA) and streptavidin-modified alkaline phosphatase (SA-AP) were acquired from the Sangon Company (China). 3-(2′-Spiroadamantane)-4-methoxy-4-(3′-phosphoryloxy)phenyl-1,2-dioxetane (AMPPD) was purchased from Biochem-ZX (China). Hybridization solution was bought from Sigma-Aldrich (USA). Carrier RNA was bought from Simgen Biological Reagent Development Co., Ltd (China). Oligonucleotides were synthesized and HPLC purified by Sangon Biotech (Shanghai) Co., Ltd. Reverse transcriptase, Taq DNA polymerase and other RT-PCR reagents were obtained from TaKaRa. Other unnamed reagents were domestic analytical reagents. DEPC treated water was used throughout the experiment.
Experimental procedures
HIV viral load determination and scalar dilutions
Positive serum samples in which HBV, HCV and HIV viral loads had been determined were provided by the Second Affiliated Hospital of Nanjing Medical University, Nanjing, China. The viral loads were quantified to be 1 × 108 copies per mL of serum for HBV, 3.4 × 107 copies per mL of serum for HCV and 5 × 106 copies per mL of serum for HIV. These known viral load serum samples (stocks) were diluted into HBV, HCV and HIV negative serum to achieve several scalar dilutions. HBV, HCV, and HIV positive samples in which viral loads were not determined were provided by the Jiangsu Blood Center, Nanjing, China.Viral nucleic acid (NA) extraction and purification from serum
The viral NA was extracted from serum samples using the following method. 100 μL of serum was mixed with 300 μL of lysis buffer containing 3 μg carrier RNA. The mixture was incubated at room temperature for 5 min. 300 μL of ethanol was blended with the lysate, then 0.5 mg silica coated MNPs were added, and NA adsorption was allowed. MNPs were separated after 3 minutes by applying an external magnetic field, followed by respective washing with washing buffer I and washing buffer II, to remove protein and salt contamination. Viral NA was eluted in 50 μL of TE buffer and stored at −80 °C until use. During the sensitivity experiment, the viral NA was eluted in 25 μL 2× PCR buffer.Oligonucleotides
Mega 6 alignment tools were used to align viral genotypes and conserved regions were selected to design primers. The oligonucleotide sequences were analyzed by Oligo 6.0 (Med Probe, Oslo, Norway) for their compatibility. The specificity of the primers was checked by using the NCBI blast function. The primers and probes used in this study are listed in Table 1.| Target | Oligonucleotide label | Oligonucleotide sequence (5′–3′) | Length (bp) |
|---|---|---|---|
| HBV (X gene) | FP | TGCACTTCGCTTCACCTCT | 119 |
| RP | CAAGGTCGGTCGTTGACATT | ||
| Probe | GGTGGGCGTTCACGGTGG-(15T)-NH2 | ||
| HCV (5′ UTR) | FP | GTATGAGTGTCGTGCAGCCT | 220 |
| RP | CACTCGCAAGCACCCTATCA | ||
| Probe | TTCCGCAGACCACTATGGCTCT-(15T)-NH2 | ||
| HIV (gag gene) | FP | AGGGGAAGTGACATAGCAGG | 174 |
| RP | TTCGCCCTTGTCTTATGTCCA | ||
| Probe | ATAGTAAGAATGTATAGCCC-(15T)-NH2 | ||
Reverse transcription-polymerase chain reaction
Multiplex RT-PCR was conducted using an ABI 9700 (Applied Biosystems, USA). The RT-PCR mixture contained the following: 7.5 μL 10× PCR buffer, 2.4 mM MgCl2, 0.2 mM dNTPs, 0.2 μM forward primer (each for HBV, HCV and HIV), 0.2 μM reverse primer (each for HBV, HCV, and HIV), 40 μM biotin-11-dUTP, 2 μL of template NA, 20 units AMV reverse transcriptase, 20 units ribonuclease inhibitor, 2.5 units hot start DNA polymerase and PCR grade water to the final volume of 50 μL. For sensitivity experiment, all the eluted NA was used in the RT-PCR.HBV, HCV, and HIV negative serum samples were also treated alike from extraction to RT-PCR and considered as blank controls during the RT-PCR and chemiluminescence detection steps. The RT-PCR thermocycling conditions were as follows; 50 °C for 15 min, 95 °C for 2 min, followed by 35 cycles of 95 °C for 20 s, 58 °C for 25 s and 72 °C for 30 s. A final extension step was continued at 72 °C for another 5 min.
Preparation of surface modified MNPs
Fe3O4@SiO2 MNPs were prepared according to the previous studies.29–31 The surface modification of Fe3O4@SiO2 was successively carried out using APTES and SA (dissolved in DMF) to produce carboxyl groups on the particles.32,33 Briefly, 6 μL of APTES was dropped into 3 mL of ethanol containing 15 mg of MNPs and stirred at room temperature for 7 hours. The aminated MNPs were dispersed in 3 mL of DMF. The carboxylated MNPs were obtained by mixing and stirring equal volumes of aminated MNPs and SA (0.1 mM) at 20 °C for 24 h.The amino-modified probe was then attached to the carboxylated MNPs through covalent binding.34 Briefly, 30 μL of the amino-modified probe (5 μmol L−1) diluted with MES (25 mmol L−1, pH 6.0), 1.5 mg carboxyl-modified MNPs, 15 μL of EDC (5 mg mL−1) and 5 μL of MES were mixed together and incubated at 4 °C for 10 h. The resulting probe modified MNPs were washed with Tris-HCl (50 mmol L−1, pH 7.4) and stored in 150 μL of PBS buffer.
DNA hybridization
DNA hybridization was carried out according to our lab optimized protocol with some modifications.33 Briefly, 5 μL of probe modified MNPs were separated and supernatants were discarded. 5 μL of hybridization buffer, 10 μL of deionized water and 15 μL of a biotin labeled RT-PCR amplified segment were then mixed together with above separated MNPs. 15 μL of the blank RT-PCR control was also used as the blank control during chemiluminescence detection. The hybridization procedure was carried out under optimized conditions.Chemiluminescence detection
Chemiluminescence detection was carried out according to previous reports.35 A multi-function micro-hole plate reading machine Victor X3 (Perkin Elmer, USA) was used for detecting chemiluminescence. After hybridization, the MNPs were washed with 100 μL of each 2 × SSC, 0.1 × SSC and cleaned with 50 mM Tris, 0.15 M NaCl solutions. The MNPs were then suspended in 60 μL of blocking solution for 30 minutes. The blocking solution comprised of 0.25% BSA was dissolved in a cleaning solution. The MNPs were then incubated with 10 μL of SA-AP for 30 minutes. The MNPs were separated and washed thrice with 100 μL of cleaning buffer. 200 μL of 0.25 mM AMPPD, the substrate for AP, was finally added onto the MNPs and the chemiluminescence signal was recorded at 5 min intervals. The chemiluminescence intensity is highly time dependent even for the same sample. The reported results were the peak values obtained 50 minutes after the incubation of AMPPD.Data analysis
Each experiment was repeated 3 times. The data were expressed as mean ± SD (Standard Deviations). Student's t-test was used for comparing the data. SPSS 13.0 software was used for this purpose.Results and discussion
One step multiplex RT-PCR optimization to detect HBV, HCV and HIV genomes
We optimized the one step multiplex RT-PCR for the simultaneous detection of HBV, HCV and HIV. There were nine different genotypes of HBV.36 Similarly, the HCV and HIV had seven and nine different genotypes and subtypes, respectively.37,38 Specific primers were designed to target all the genotypes and sub-genotypes from these viruses. The HBV primers targeted the conserved region of the X gene in the HBV genome and successfully amplified the 119 bp segment (Fig. 1A), while the HIV primers amplified the 174 bp fragment of the gag gene in the HIV genome (Fig. 1B). For HCV, the sequence alignment results revealed that the 5′ untranslated region (5′ UTR) was well conserved and therefore primers were designed to target this region to amplify 220 bp fragments through one step RT-PCR (Fig. 1C).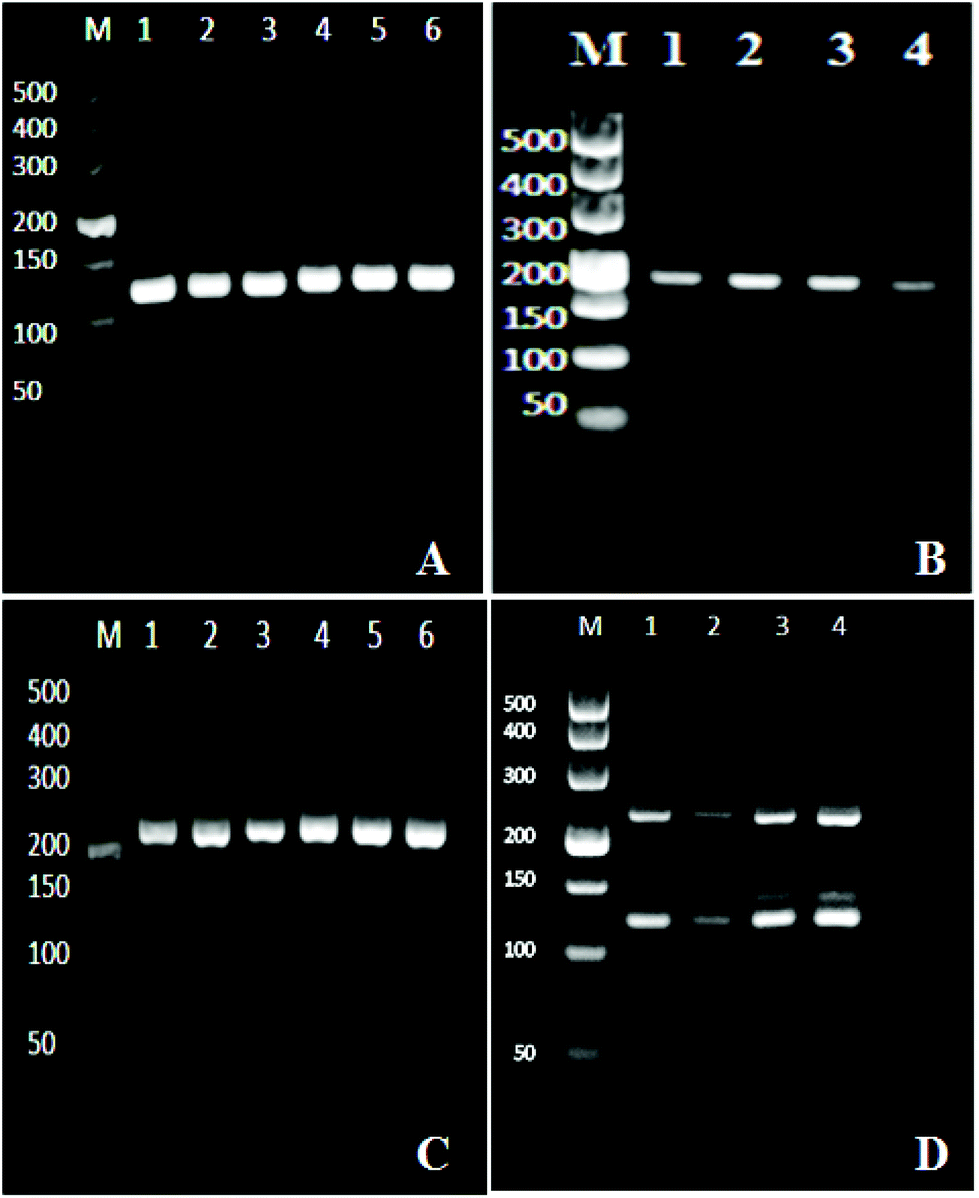 | ||
| Fig. 1 Electrophoresis results. Amplification of the HBV genome in monoplex PCR, the PCR product length was 119 bp (A). Monoplex RT-PCR for HIV amplification, the product length was 174 bp (B). Amplification of the HCV genome in monoplex RT-PCR, and the length of the amplified segment was 220 bp (C). The amplification of HBV, HCV, and HIV in one step multiplex RT-PCR (D). Clinical samples of an unknown viral load were tested. The band/bands in each lane in the pictures below show different samples. | ||
After the optimization and successful amplification of individual viruses through the monoplex reactions, one step multiplex RT-PCR was then performed by simultaneously extracting three viruses from one sample, followed by their amplification in one tube. The reaction components were adjusted to produce optimum conditions for the simultaneous amplification of three templates. Fig. 1D confirms the detection of the three viruses. The bands at the 119 bp, 174 bp, and 220 bp positions on the agarose gel showed the HBV, HCV, and HIV amplifications, respectively. The effect of non-specific amplification (visible Fig. 1D) was diminished by using the specific probe in the downstream chemiluminescence detection step.
Optimization of factors for chemiluminescence detection of HBV
The HBV probe was first modified onto the surface of the carboxyl coated MNPs and then employed to hybridize with the 119 bp HBV fragment. The chemiluminescence signal directly depends on the efficiency of the probe to capture its target sequence;39 therefore, different factors which might affect probe–target hybridization were optimized. The optimized hybridization temperature, probe concentration, hybridization time, and amount of probe-MNPs were determined to be 45 °C, 5 μM, 30 minutes, and 50 μg, respectively (Fig. 2).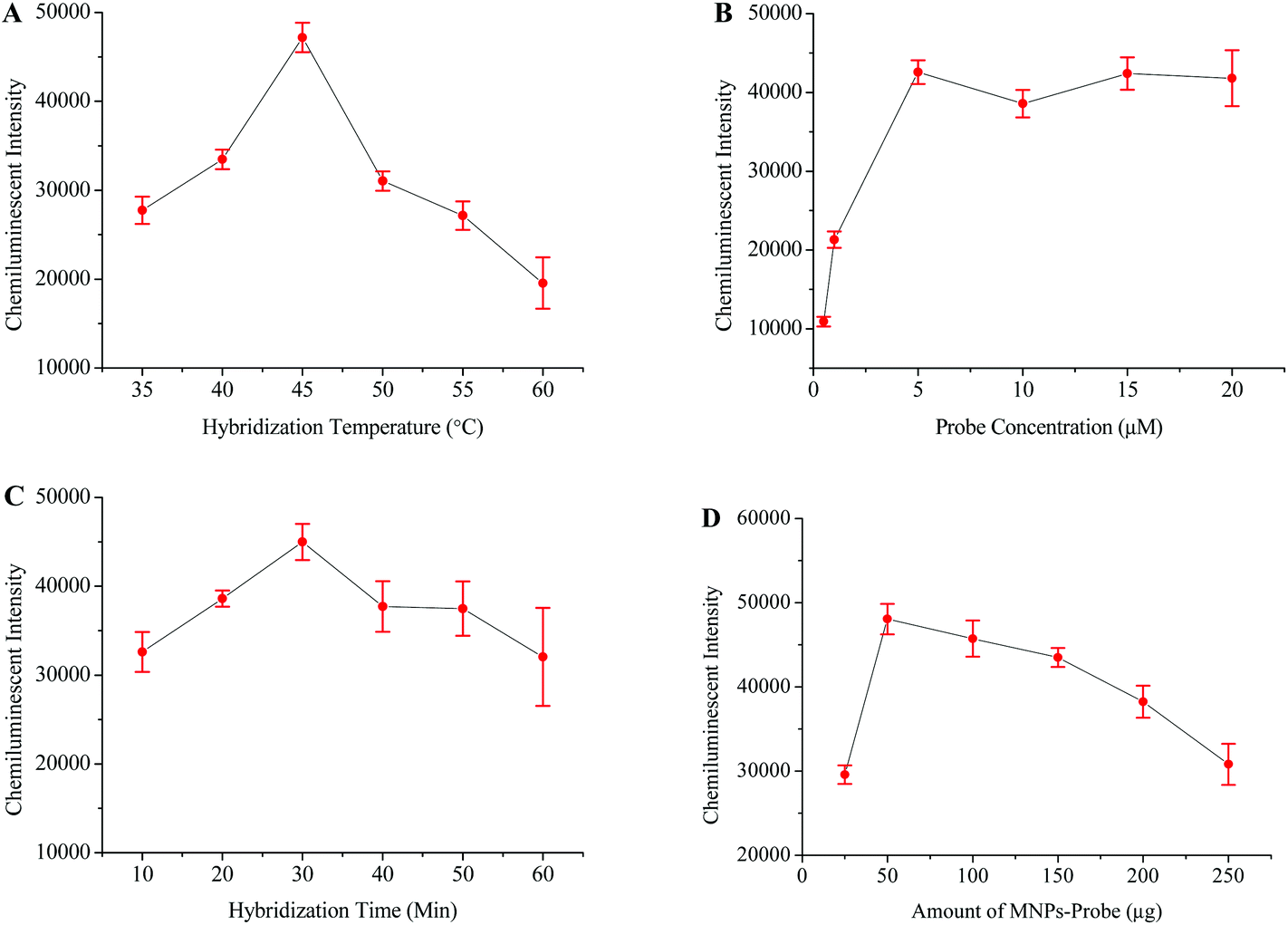 | ||
| Fig. 2 Optimization of different factors for achieving maximum HBV detection signals. Relationship between the chemiluminescence intensity and hybridization temperature (A), probe concentration (B), hybridization time (C) and concentration of the MNP-probe (D). | ||
The optimized protocol was followed to check the specificity and sensitivity of this method. Apart from the HBV samples, the HCV and HIV positive samples were tested in specificity analysis. HBV negative serum was used as a negative control while distilled water replaced the PCR products to serve as the blank control. Fig. 5A shows that the intensity of the chemiluminescence signal was very poor when the HCV, HIV and blank controls were tested. A strong signal was however recorded when the HBV positive sample was tested.
In order to determine the sensitivity for this method, the HBV sample of a known viral load was diluted into HBV negative serum to obtain different scalar dilutions. Fig. 5B shows that this method successfully detected 10 viral copies per ml of serum. The clinical samples were used throughout the development and optimization of this method, which proved the reliability of this method.
Post-amplification chemiluminescence detection of HCV
The intensity of the chemiluminescence signal is directly proportional to the efficiency of probe–template hybridization, and hybridization temperature is considered as an important factor which greatly influences the hybridization efficiency.40 Therefore, the hybridization temperature, probe concentration, hybridization time and amount of probe-MNPs were also optimized in order to generate maximum signals (Fig. 3).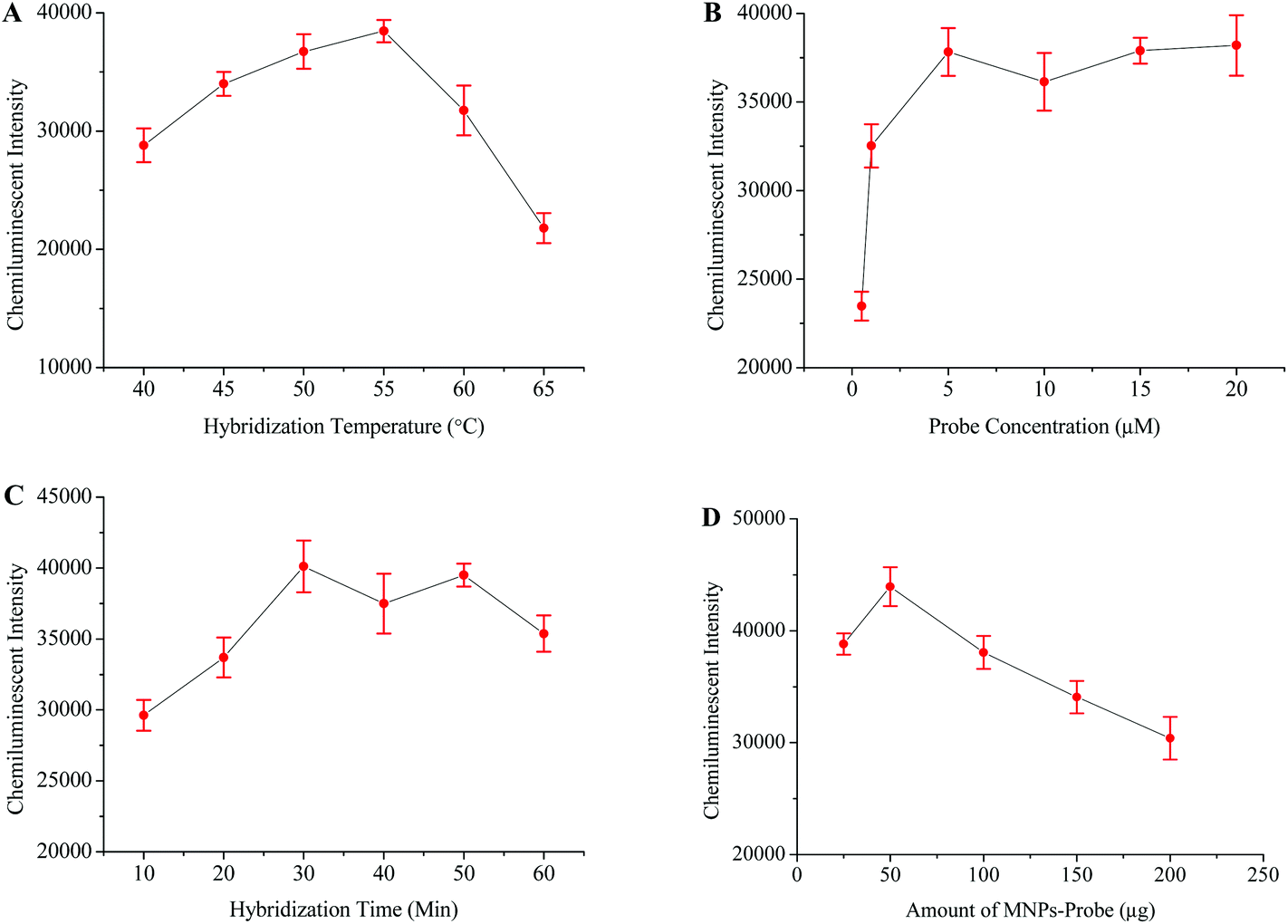 | ||
| Fig. 3 Optimization of different factors for obtaining maximum HCV detection signals. The relationship between the chemiluminescence intensity and hybridization temperature (A), probe concentration (B), hybridization time (C), and concentration of the MNP-probe (D). | ||
HBV and HIV positive serum samples were used to evaluate the specificity of this HCV detection protocol. The HCV negative sample and distilled water were respectively used as negative and blank controls in this experiment. The results from this part of experiment verified that the primers and probes specifically targeted the HCV genome (Fig. 5C). To examine the sensitivity of this method, several scalar dilutions were then made by diluting the HCV positive sample of a known viral load. The results in Fig. 5D confirm the excellent sensitivity of this method with a detection limit of 10 viral copies per mL of serum.
Chemiluminescence based post-amplification detection of HIV
The HIV genome was amplified through one step RT-PCR (Fig. 1B). The post-amplification detection was then carried out by using the magnetic separation and chemiluminescence technique. Initially, the HIV probes were modified onto the surface of the MNPs-COOH, and different parameters which influence probe–target hybridization were then studied in detail. Fig. 4 shows that the best hybridization temperature, probe concentration, and hybridization time were 35 °C, 5 μM, and 30 minutes, respectively.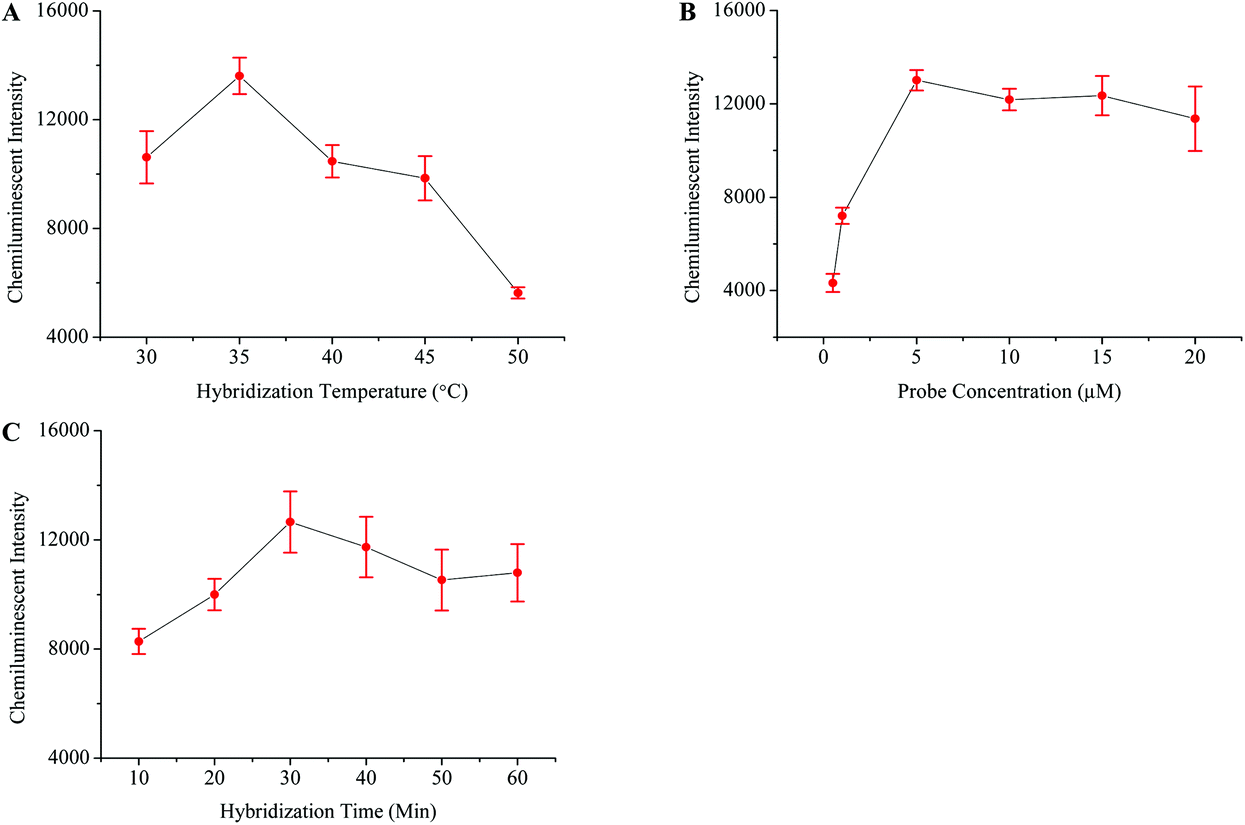 | ||
| Fig. 4 Optimization of different factors for achieving maximum HIV detection signals. The relationship between the chemiluminescence intensity and hybridization temperature (A), probe concentration (B), and hybridization time (C). | ||
The specificity of this method was examined by testing the HBV and HCV samples. Negative and blank controls were also included in this experiment, which consisted of HIV negative serum and distilled water instead of RT-PCR products, respectively. Fig. 5E demonstrates that this method unequivocally detected the HIV positive sample while other viral samples produced a very weak signal similar to that of control samples. The sensitivity of this assay was detected by diluting the HIV positive sample of a known viral load. The results in Fig. 5F showed that the detection limit of this method was 10 viral copies per ml of serum.
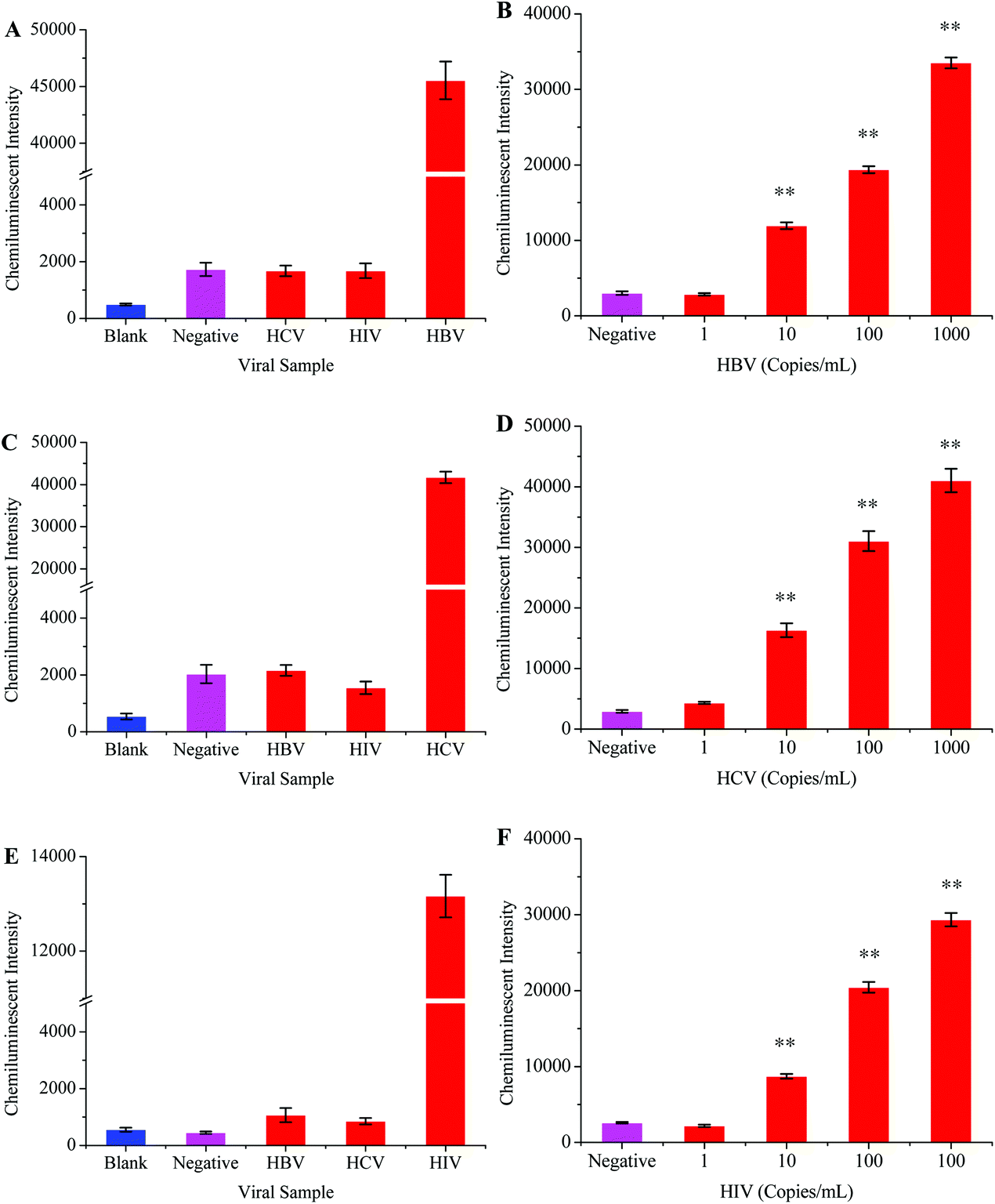 | ||
| Fig. 5 The specificity and sensitivity analyses of chemiluminescence based assays. The specificity analyses for HBV, HCV, and HIV detection are shown in A, C, and E, respectively (clinical samples with an unknown viral load were used during specificity assay). The limit of HBV, HCV, and HIV detection for their respective assays is shown in B, D, and F, respectively. **, P < 0.01 versus control. | ||
Evaluating the specificity and sensitivity of the multiplex detection assay
One step multiplex RT-PCR (herein referred to as multiplex RT-PCR) was designed to achieve higher detection sensitivity. The multiplex RT-PCR products were allowed to hybridize separately with HBV, HCV, and HIV probes. The probes captured their target sequences, and a downstream detection procedure was followed according to the protocol given in Experimental procedures. Scheme 1 shows the post-amplification chemiluminescence detection of multiple viruses.The multiplex RT-PCR successfully amplified all the three viral templates and produced 119 bp HBV, 174 bp HIV and 220 bp HCV fragments. Besides, the non-specific amplification also occurred, producing few undesired DNA fragments. We designed specificity tests in order to confirm that the probes only captured their respective target sequences from the multiplex RT-PCR products. Primarily, the HBV, HCV, and HIV were separately amplified through the monoplex PCRs, followed by their simultaneous amplification in one tube through the multiplex RT-PCR. A negative sample (negative for all the three viruses) was also processed in the multiplex RT-PCR, and this sample was termed the blank control. The HBV probe was then incubated with all the PCR products, followed by the detection procedure to generate the CL signal. The results in Fig. 6A showed that the signal intensity was high when the HBV probe was incubated with HBV monoplex PCR and multiplex RT-PCR products, while the blank control produced a very weak signal. As expected, poor signals were recorded when the HBV probe was allowed to capture both the HCV and HIV monoplex PCR products. This inferred that the specificity of the HBV probe was excellent as it hybridized only with the 119 bp HBV sequence which was co-amplified with the HCV and HIV in multiplex RT-PCR. Similar experiments were conducted to evaluate the specificity of the HCV and HIV. The results in Fig. 6B and C also confirmed the high specificity demonstrated by the HCV and HIV probes, respectively.
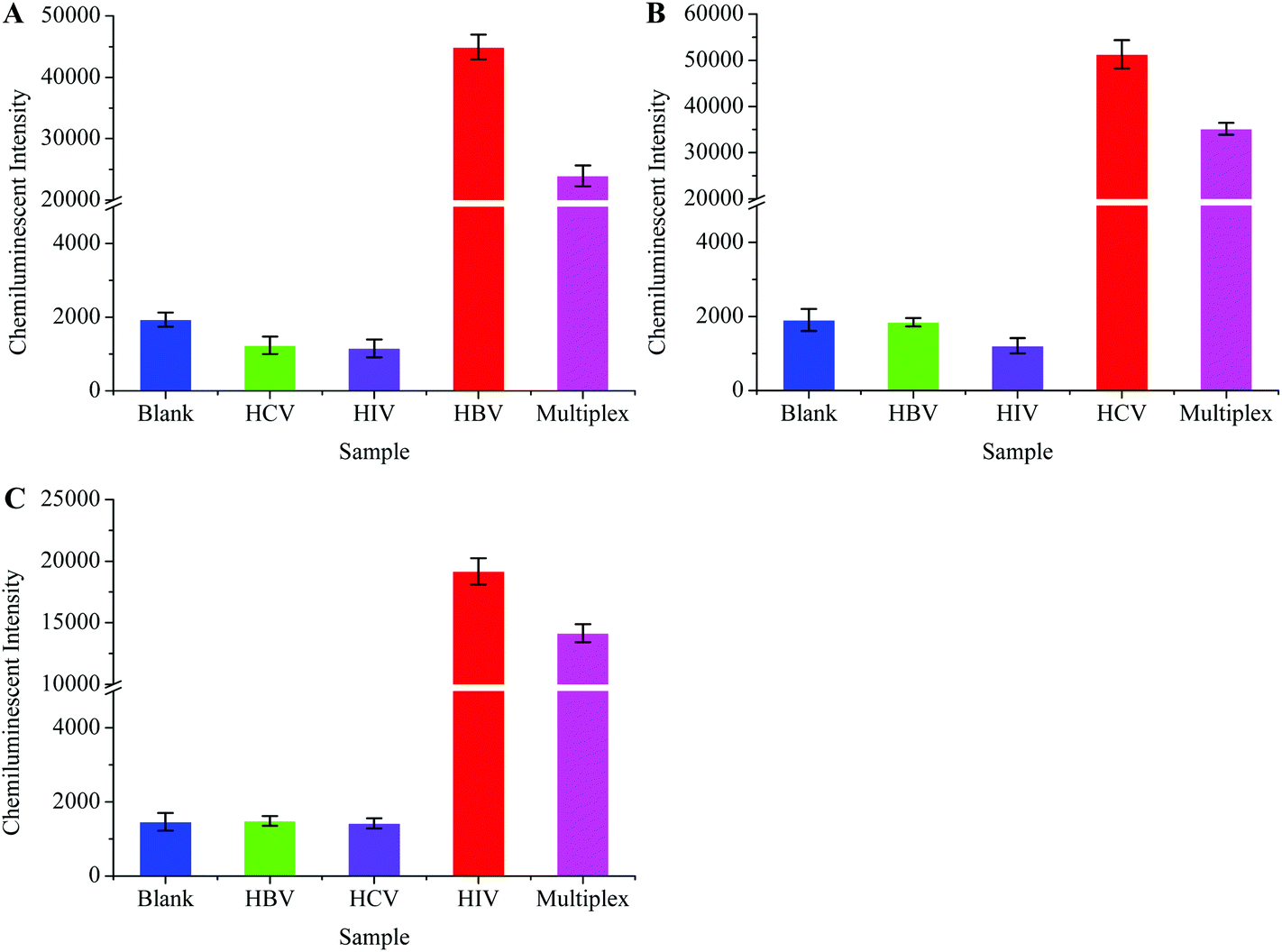 | ||
| Fig. 6 Specificity of virus specific probes for capturing their targeted sequences in one-step multiplex RT-PCR products during the chemiluminescence detection step. The HBV probe only captured the HBV target sequence (A), and the HCV probe specifically captured the HCV target sequence (B), while the HIV probe hybridized specifically with the HIV target segment (C). Clinical samples with an unknown viral load were used during the specificity assay. | ||
Preferential amplification is the main problem for multiplex PCR because some loci are amplified more efficiently than others.41 Any detection system that is based on multiplex PCR amplification should be carefully tested before it is used in clinical laboratories as preferential amplification may lead to false negative results for some pathogens. We therefore thoroughly studied this problem in our experiment. Different scalar dilutions with different viral loads were made as shown in Table 2. These samples were then tested to evaluate the preferential amplification and detection limit for our method. Fig. 7A, B, and C show the detection sensitivity for HBV, HCV, and HIV, respectively. These results showed that the HBV and HCV were equally amplified in the multiplex RT-PCR even when the viral loads for other viruses were comparatively high, and the detection limit for these two viruses was 10 viral copies per mL of serum.
 | ||
| Fig. 7 Analytical sensitivity of chemiluminescence and magnetic separation based multiplex detection assay. The sensitivity for HBV (A), HCV (B), and HIV (C) was determined in different ratios of standard serum dilutions as shown in Table 2. **, P < 0.01 versus control. | ||
| Sample ID | HBV (copies per mL) | HCV (copies per mL) | HIV (copies per mL) |
|---|---|---|---|
| A1 | 10 | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
100000 |
| A2 | 100 | 100000 |
100000 |
| A3 | 1000 | 100000 |
100000 |
| A4 | 10000 |
100000 |
100000 |
| B1 | 100000 |
10 | 100000 |
| B2 | 100000 |
100 | 100000 |
| B3 | 100000 |
1000 | 100000 |
| B4 | 100000 |
10000 |
100000 |
| C1 | 100000 |
100000 |
10 |
| C2 | 100000 |
100000 |
100 |
| C3 | 100000 |
100000 |
1000 |
| C4 | 100000 |
100000 |
10000 |
However, the phenomenon of preferential amplification was noticed in the case of HIV and the amplification efficiency for this virus diminished to some extent when it was co-amplified with HBV and HCV in the multiplex RT-PCR. The lower limit of HIV detection in the context of multiplex detection was 100 copies per mL of serum. The HIV viral load between 2 × 102–105 copies per mL has been observed during the window period.42 Thus, the sensitivity of this method is sufficient enough to be used in clinical laboratories.
In summary, the HBV, HCV and HIV were simultaneously extracted in this study from a single sample and amplified in the one step multiplex reverse transcription polymerase chain reaction (RT-PCR), with biotin-11-dUTP being incorporated into the amplified products during the RT-PCR. The probe coated MNPs were then used to capture the virus specific RT-PCR products, and SA-AP was added to bind with the biotin modified RT-PCR products, followed by several washings to remove non-specifically bound SA-AP. Finally, the chemiluminescence signal was generated after incubating the MNPs with AMPPD.
Conclusions
We have proposed a novel method for simultaneous detection of multiple viruses. This method has demonstrated high efficiency and suitability for processing clinical samples. Besides, a magnetic separation technique was used throughout the experiment, from nucleic acid extraction to chemiluminescence detection. Owing to this successful use of the MNPs, this assay can be modified into a high throughput automated set up for the simultaneous detection of multiple infections, which will further reduce the detection cost per pathogen. The use of silica modified magnetic nanoparticles during nucleic acid extraction and carboxyl coated magnetic nanoparticles during the chemiluminescence step can help modify this system into an automated platform for high throughput applications. This assay provides an alternative for analyzing clinical samples in blood screening units.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the 863 Program (2012AA022703), the National Key Special Science Program (2013ZX10004103-002), the National Natural Science Foundation of China (61271056, 61401217 and 61527806), the Clinical Science and Technology Special Projects in Jiangsu Province (BL2014094), the China Postdoctoral Science Foundation funded project (2014M551491), the Jiangsu Planned Projects for Postdoctoral Research Funds (1302007A and 1301093C), the Science and Technology Development Projects in Nanjing (201503017), the Natural Science Foundation of Jiangsu Province (BK20140900), and the open project of State Key Laboratory of Bioelectronics (2014HX12). Scientific Research Fund of Hunan Provincial Education Department (14B043), and Shenzhen Basic Research Projects (JCYJ 20140901141243646) GDPRSFS (2012).References
- M. El Ekiaby, N. Lelie and J. P. Allain, Biologicals, 2010, 38, 59–64 CrossRef PubMed.
- L. Zhou, R. Gong, X. Lu, Y. Zhang and J. Tang, Jpn. J. Infect. Dis., 2015, 68, 481–487 CrossRef CAS PubMed.
- Y. Peng, L. E. Tellier and J. S. Temenoff, Biomater. Sci., 2016, 4, 1371–1380 RSC.
- P. V. Gorelkin, A. S. Erofeev, G. A. Kiselev, D. V. Kolesov, E. V. Dubrovin and I. V. Yaminsky, Analyst, 2015, 140, 6131–6137 RSC.
- T. Zako and M. Maeda, Biomater. Sci., 2014, 2, 951–955 RSC.
- D. Candotti, A. Richetin, B. Cant, J. Temple, C. Sims, I. Reeves, J. A. Barbara and J. P. Allain, Transfusion, 2003, 43, 215–225 CrossRef CAS PubMed.
- T. Matsubara, M. Ujie, T. Yamamoto, M. Akahori, Y. Einaga and T. Sato, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8981–8984 CrossRef CAS PubMed.
- T. B. Morrison, J. J. Weis and C. T. Wittwer, Biotechniques, 1998, 24, 954–958 CAS.
- D. Gibellini, F. Gardini, F. Vitone, P. Schiavone, G. Furlini and M. C. Re, Mol. Cell. Probes, 2006, 20, 223–229 CrossRef CAS PubMed.
- J. P. Defoort, M. Martin, B. Casano, S. Prato, C. Camilla and V. Fert, J. Clin. Microbiol., 2000, 38, 1066–1071 CAS.
- F. Ye, M. S. Li, J. D. Taylor, Q. Nguyen, H. M. Colton, W. M. Casey, M. Wagner, M. P. Weiner and J. Chen, Hum. Mutat., 2001, 17, 305–316 CrossRef CAS PubMed.
- D. Wang, L. Coscoy, M. Zylberberg, P. C. Avila, H. A. Boushey, D. Ganem and J. L. DeRisi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15687–15692 CrossRef CAS PubMed.
- G. Mitterer and W. M. Schmidt, Methods Mol. Biol., 2006, 345, 37–51 CAS.
- R. K. Thapa, J. Y. Choi, B. Gupta, T. Ramasamy, B. K. Poudel, S. K. Ku, Y. S. Youn, H. G. Choi, C. S. Yong and J. O. Kim, Biomater. Sci., 2016, 4, 1340–1350 RSC.
- W. Liao, W. Li, T. Zhang, M. Kirberger, J. Liu, P. Wang, W. Chen and Y. Wang, Biomater. Sci., 2016, 4, 1051–1061 RSC.
- Q. Wang, R. D. Li, B. C. Yin and B. C. Ye, Analyst, 2015, 140, 6306–6312 RSC.
- M. Varshney and Y. Li, Biosens. Bioelectron., 2007, 22, 2408–2414 CrossRef CAS PubMed.
- A. Muangchuen, P. Chaumpluk, A. Suriyasomboon and S. Ekgasit, Sensors, 2014, 14, 14472–14487 CrossRef PubMed.
- I. Bronstein and P. McGrath, Nature, 1989, 338, 599–600 CrossRef CAS PubMed.
- J. Wang, P. Lu, J. Yan, Y. Zhang, L. Huang, Z. Ali, B. Liu, Z. Li and N. He, J. Biomed. Nanotechnol., 2016, 12, 710–716 CrossRef CAS PubMed.
- M. Luo, X. Chen, G. Zhou, X. Xiang, L. Chen, X. Ji and Z. He, Chem. Commun., 2012, 48, 1126–1128 RSC.
- F. J. Lara, D. Airado-Rodriguez, D. Moreno-Gonzalez, J. F. Huertas-Perez and A. M. Garcia-Campana, Anal. Chim. Acta, 2016, 913, 22–40 CrossRef CAS PubMed.
- K. Nakashima, R. Ikeda and M. Wada, Anal. Sci., 2009, 25, 21–31 CrossRef CAS PubMed.
- M. Fuentes, C. Mateo, A. Rodriguez, M. Casqueiro, J. C. Tercero, H. H. Riese, R. Fernandez-Lafuente and J. M. Guisan, Biosens. Bioelectron., 2006, 21, 1574–1580 CrossRef CAS PubMed.
- J. H. Min, M. K. Woo, H. Y. Yoon, J. W. Jang, J. H. Wu, C. S. Lim and Y. K. Kim, Anal. Biochem., 2014, 447, 114–118 CrossRef CAS PubMed.
- S. Berensmeier, Appl. Microbiol. Biotechnol., 2006, 73, 495–504 CrossRef CAS PubMed.
- R. D. Peterson, W. Chen, B. T. Cunningham and J. E. Andrade, Biosens. Bioelectron., 2015, 74, 815–822 CrossRef CAS PubMed.
- Y. Gao, J. Pallister, F. Lapierre, G. Crameri, L. F. Wang and Y. Zhu, J. Virol. Methods, 2015, 222, 170–177 CrossRef CAS PubMed.
- B. Zeshan, A. Sadaf and N. H. Othman, J. Nanosci. Nanotechnol., 2016, 16, 6622–6633 CrossRef CAS.
- B. Zhou, Z. Li, H. Yang and N. He, J. Biomed. Nanotechnol., 2014, 10, 2865–2890 CrossRef CAS PubMed.
- C. Ma, C. Li, N. He, F. Wang, N. Ma, L. Zhang, Z. Lu, Z. Ali, Z. Xi, X. Li, G. Liang, H. Liu, Y. Deng, L. Xu and Z. Wang, J. Biomed. Nanotechnol., 2012, 8, 1000–1005 CrossRef CAS PubMed.
- H. Yang, Y. Guo, S. Li, G. Lan, Q. Jiang, X. Yang, J. Fan, Z. Ali, Y. Tang, X. Mou, H. Liu, M. A. Shah, S. Jin, H. Jiang and Z. Li, J. Nanosci. Nanotechnol., 2014, 14, 3337–3342 CrossRef CAS PubMed.
- Y. K. Jung, T. W. Kim, C. Jung, D. Y. Cho and H. G. Park, Small, 2008, 4, 1778–1784 CrossRef CAS PubMed.
- N. He, F. Wang, C. Ma, C. Li, X. Zeng, Y. Deng, L. Zhang and Z. Li, J. Biomed. Nanotechnol., 2013, 9, 267–273 CrossRef CAS PubMed.
- Y. Tang, J. Zou, C. Ma, Z. Ali, Z. Li, X. Li, N. Ma, X. Mou, Y. Deng, L. Zhang, K. Li, G. Lu, H. Yang and N. He, Theranostics, 2013, 3, 85–92 CrossRef CAS PubMed.
- B. S. Guirgis, R. O. Abbas and H. M. Azzazy, Int. J. Infect. Dis., 2010, 14, e941–e953 CrossRef CAS PubMed.
- R. Gill, R. Polsky and I. Willner, Small, 2006, 2, 1037–1041 CrossRef CAS PubMed.
- J. Hemelaar, E. Gouws, P. D. Ghys and S. Osmanov, W.-U. N. f. H. Isolation and Characterisation, AIDS, 2011, 25, 679–689 CrossRef PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- J. Wang, Z. Ali, N. Wang, W. Liang, H. Liu, F. Li, H. Yang, L. He, L. Nie and N. He, Sci. China: Chem., 2015, 58, 1774–1778 CrossRef CAS.
- P. Markoulatos, N. Siafakas and M. Moncany, J. Clin. Lab. Anal., 2002, 16, 47–51 CrossRef CAS PubMed.
- G. Albertoni, M. J. Castelo Girao and N. Schor, Int. J. Infect. Dis., 2014, 25, 145–149 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |