
Local delivery of doxorubicin through supramolecular peptide amphiphile nanofiber gels†
Goksu
Cinar
,
Ayse
Ozdemir
,
Seren
Hamsici
,
Gokhan
Gunay
,
Aykutlu
Dana
,
Ayse B.
Tekinay
 * and
Mustafa O.
Guler
*
* and
Mustafa O.
Guler
*
Institute of Materials Science and Nanotechnology, National Nanotechnology Research Center (UNAM), Bilkent University, Ankara, 06800, Turkey. E-mail: atekinay@bilkent.edu.tr; moguler@unam.bilkent.edu.tr; Fax: +90 (312)266 4365; Tel: +90 (312)290 3552
First published on 7th November 2016
Abstract
Peptide amphiphiles (PAs) self-assemble into supramolecular nanofiber gels that provide a suitable environment for encapsulation of both hydrophobic and hydrophilic molecules. The PA gels have significant advantages for controlled delivery applications due to their high capacity to retain water, biocompatibility, and biodegradability. In this study, we demonstrate injectable supramolecular PA nanofiber gels for drug delivery applications. Doxorubicin (Dox), as a widely used chemotherapeutic drug for breast cancer treatment, was encapsulated within the PA gels prepared at different concentrations. Physical and chemical properties of the gels were characterized, and slow release of the Dox molecules through the supramolecular PA nanofiber gels was studied. In addition, the diffusion constants of the drug molecules within the PA nanofiber gels were estimated using fluorescence recovery after the photobleaching (FRAP) method. The PA nanofiber gels did not show any cytotoxicity and the encapsulation strategy enhanced the activity of drug molecules on cellular viability through prolonged release compared to direct administration under in vitro conditions. Moreover, the local in vivo injection of the Dox encapsulated PA nanofiber gels (Dox/PA) to the tumor site demonstrated the lowest tumor growth rate compared to the direct Dox injection and increased the apoptotic cells within the tumor tissue for local drug release through the PA nanofiber gels under in vivo conditions.
Introduction
Drug delivery systems have been developed to control drug release rates, maintain the drug concentration within therapeutic levels, improve the drug bioavailability and reduce potential side effects by lowering the therapeutic dosages necessary for treatment.1 A variety of organic/inorganic materials, devices or formulations have been used as delivery vehicles to develop effective therapeutic modalities.1,2 Advances in materials science and nanotechnology have provided a new generation of biodegradable and biocompatible delivery systems with considerable control over size, shape and functionality.2–5Self-assembly is a nature-inspired engineering tool that allows the construction of drug delivery systems with distinct chemical and physical properties.6 Polymers, synthetic or natural biomacromolecules, peptides, lipids or hybrid systems can be produced from relatively simple “building blocks” through multiple non-covalent interactions, hydrogen bonding and hydrophobic, aromatic, hydrophilic and electrostatic interactions. Although these interactions are individually weak and easily reversible compared to the covalent systems,7 the combined effect of multiple non-covalent forces is able to drive the self-assembly process and result in the formation of complex, adaptable and highly tunable structures as drug delivery architectures.8
Among self-assembled materials, peptide amphiphiles (PAs) consisting of an aliphatic alkyl tail and hydrophilic amino acids can form diverse nanostructures and supramolecular architectures in response to different factors such as the pH change,9 presence of oppositely charged molecules10,11 or electrolyte addition12 and enzyme activation.13 The PA assemblies provide structural organization for delivery of both hydrophobic and hydrophilic therapeutic moieties, which can be modulated through the design of building blocks.7,14–16 Moreover, targeted high efficacy delivery of small therapeutics can be achieved through functionalization of PA nanostructures with ligand-binding, cell-penetrating and internalization-associated peptide sequences.17,18 Biocompatibility and biodegradability of the PA assemblies through different proteases19–21 also enhance their utility as delivery architectures, especially for chemotherapeutics that show high cytotoxicity when administered directly to the blood stream. Previously, it was shown that bioresponsive anticancer drug delivery was sustained through self-assembled PA nanofiber gels containing matrix metalloproteinase-2 (MMP-2) cleavable amino acid sequences.22 In another study, noncovalent encapsulation of a hydrophobic anti-tumor drug into self-assembled PA nanofibers was shown both to enhance the aqueous solubility of the drug and to increase its efficiency against an in vivo breast cancer model.23
Local delivery of anticancer drugs using biocompatible scaffolds can be an alternative way to reduce systemic side effects on healthy tissues by improving their bioavailability at the target site.24–27 Self-assembled peptide nanofiber gels have been investigated extensively for controlled delivery of small molecules including proteins,28 antibodies29 and therapeutics,30–33 and are attractive candidates for local delivery of chemotherapeutics.22 The design of injectable peptide based gel systems has been presented in several studies.34–36 Recently, a β-hairpin peptide hydrogel developed as an injectable local drug delivery system was found to facilitate the continuous release of active chemotherapeutics for over a month.37
Doxorubicin (Dox) is an FDA approved and clinically used chemotherapeutic molecule for the treatment of various cancer types.38,39 Despite its widespread use, the intravenous injection of Dox may cause severe health problems in patients.40,41 In this study, we aimed to develop biocompatible, biodegradable and injectable PA nanofiber gels which provide both hydrophilic and hydrophobic nanofibrous environments for local delivery of drug molecules overcoming the limitations of Dox treatment. For this purpose, oppositely charged PA molecules were designed as the building blocks of self-supporting gels that are able to encapsulate drug molecules with 100% efficiency during sol–gel transition at pH 7.4 in water. The chemical and physical properties of nanofibrous PA networks including their secondary structure, concentration-dependent viscoelastic behaviors and injectability were studied using several characterization techniques. The biodegradability of the coassembled PA gels by different proteases was also tested under physiological conditions. Furthermore, the controlled release of Dox was investigated through in vitro bulk release experiments and fluorescence recovery after the photobleaching (FRAP) technique. Transport parameters, including diffusion coefficients and immobile fractions of drug molecules within the PA nanofiber gels, were estimated using a semi-empirical method. In addition, in vitro biocompatibility of the PA nanofiber gels and the effect of controlled drug release on cellular viability were studied to assess the anti-tumor efficiency of Dox encapsulated PA nanofiber gels. Finally, the potential of the PA nanofiber gel as a local drug delivery system was examined in a mouse model of breast cancer under in vivo conditions.
Experimental section
Materials
All protected amino acids, rink amide 4-methylbenzhydrylamine (MBHA) resin, Fmoc-Glu(OtBu)-Wang resin (100–200 mesh) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from NovaBiochem. Other chemicals including dichloromethane (DCM), dimethylformamide (DMF), acetonitrile, piperidine, acetic anhydride, N,N-diisopropylethylamine (DIAE), trifluoroacetic acid (TFA), uranyl acetate and doxorubicin hydrochloride were purchased from Fisher, Merck, Alfa Aesar or Sigma-Aldrich. Proteolytic enzymes (proteinase K from Tritirachium album and α-chymotrypsin) from bovine pancreas were purchased from VWR and Sigma, respectively. All chemicals and solvents used in this study were analytical grade.Peptide amphiphile synthesis and characterization
E3PA (Lauryl-VVAGEEE) and K3PA (Lauryl-VVAGKKK-Am) were synthesized according to a previously described solid phase 9-fluorenylmethoxycarbonyl (Fmoc) peptide synthesis method.11 Rink amide MBHA resin served as the solid support for K3PA synthesis while Fmoc-Glu(OtBu)-Wang resin was used as a solid support for E3PA synthesis. An Agilent 1200 series reverse-phase high performance liquid chromatograph (HPLC) equipped with an Agilent 6224 high resolution mass time-of-flight (TOF) mass spectrometer and an electrospray ionization (ESI) source was used for the characterization of the synthesized PAs (Fig. S1†). 1 mg mL−1 E3PA in water was analyzed at pH 7 using an Agilent Zorbax 300SB-C18, 3.5 μm (100 × 4.6 mm) column in a gradient of water (0.1% NH4OH) and acetonitrile (0.1% NH4OH) as the mobile phase, while an Agilent Zorbax Extend-C18, 3.5 μm 80A (100 × 4.6 mm) column was used for the analysis of 1 mg mL−1 K3PA at pH 7 in water in an optimized gradient of water (0.1% formic acid) and acetonitrile (0.1% formic acid). The mobile phase was optimized as 2% acetonitrile at the first 2 min, then gradually increased to 98% acetonitrile between 2 and 16 min, and finally returned to 2% acetonitrile for the last 2 min.Preparation of supramolecular PA nanofiber gels
E3PA and K3PA were separately dissolved in water, and the pH of the individual solutions was adjusted to 7.4 using 1 M NaOH. The negatively charged E3PA solution was mixed with the positively charged K3PA solution in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volumetric ratio to trigger the coassembly of PAs into supramolecular nanofiber gels through overall charge neutralization. The final PA concentrations within the coassembled gels were 3, 2, 1 and 0.5% (w/v), respectively.
:4 volumetric ratio to trigger the coassembly of PAs into supramolecular nanofiber gels through overall charge neutralization. The final PA concentrations within the coassembled gels were 3, 2, 1 and 0.5% (w/v), respectively.
Imaging of PA nanofibers and supramolecular gels
For the TEM imaging of individual PA nanofibers, 1% (w/v) PA gel was diluted by the addition of water, and the sample was cast onto a TEM grid. The sample was then stained with 2% (w/v) uranyl acetate, air-dried, and imaged using a FEI Tecnai G2 F30 TEM instrument. For SEM imaging, the coassembled PA nanofiber gels at 3, 2, 1 and 0.5% (w/v) concentrations were prepared at pH 7.4 and in water. The water content in the gels was replaced by a series of ethanol (40%, 60%, 80% and 100%) concentrations to preserve their nanofibrous architecture during critical point drying (CPD). The samples were coated with 6 nm Au/Pd and imaged using a FEI Quanta 200 FEG Scanning Electron Microscope.Zeta potential measurements
E3PA and K3PA solutions were prepared at 0.186 mM and 0.132 mM concentrations, respectively and the pH of the solutions was adjusted to 7.4 using 1 M NaOH. PA mixtures were prepared by continuously adding E3PA in 0.4 ml increments onto 10 ml of K3PA solution. After each addition of E3PA, the mixture was mixed and the zeta potential of the mixture was measured using a Malvern Nano-ZS ZetaSizer equipped with a titrator and pH meter.Secondary structure analysis using CD and FT-IR
For CD analysis, the coassembled PA gel was prepared according to the above protocol. The gel was then diluted to 0.272 mM prior to analysis. Individual solutions of E3PA and K3PA solutions were also prepared at the same concentrations as the control. A Jasco J-815 CD spectrophotometer was used for spectral analysis between 190 and 300 nm. For FTIR measurement, 1% (w/v) PA gel was formed on Petri dishes and instantly frozen in liquid nitrogen in order to preserve its architectural integrity. The frozen sample was kept at −80 °C overnight and then freeze-dried to remove its water content. 1 mg dried gel powder was mixed with 100 mg KBr to prepare KBr pellets for analysis. A Bruker VERTEX 70 FTIR spectrometer was used for the measurements between 300 and 4000 cm−1.Oscillatory rheology
Time sweep analysis of the coassembled PA nanofiber gels at 3, 2, 1 and 0.5% (w/v) concentrations was performed for 1 h at pH 7.4 at a constant angular frequency and a strain of 10 rad s−1 and 0.1%, respectively. The sample volume was determined as 250 μL with a 0.5 mm measuring distance from the stage. An Anton Paar MCR-301 Rheometer with a 25 mm PP25-SN17979 measuring device was used for the measurements. 1% (w/v) PA gel was also mixed within a syringe and injected through a 1/2 inch 26 gauge needle to the rheometer stage to determine the potential effect of injection on gel properties. The same parameters given above were also used for the analysis of the syringe-injected gel.Biodegradability of supramolecular PA nanofiber gels by different proteases
308 μL of 1% (w/v) PA gels were prepared in glass vials according to the gel preparation protocol explained above. Tare weights of the vials were also noted prior to sample preparation. The gels were then treated with 1 ml proteinase K or α-chymotrypsin solutions, prepared at a concentration of 1 mg ml−1 in 50 mM Tris buffer at pH 8. As a control group, Tris buffer without enzymes was also placed on the PA gels and the samples were weighed at the determined time intervals by removing the buffer solutions. After each measurement, freshly prepared buffer with or without the enzymes was replaced onto the gels. The experiment was conducted for 25 days, at which point the PA gels treated with proteinase K were found to have completely disappeared.Drug encapsulation and controlled release experiments
For drug encapsulation, E3PA and K3PA solutions at 3, 2, 1 and 0.5% (w/v) concentrations were prepared at pH 7.4 in water. A stock solution of Dox (833.3 μM) was also prepared in water. The stock drug solution initially was mixed with positively charged K3PA and then co-assembly was triggered by addition of the negatively charged E3PA at pH 7.4 in water at a 3:4 volumetric ratio. A final Dox concentration of 40 μM was encapsulated within 100 μL of PA gels prepared in small vials. After 2 h of incubation, 100 μL of water was added onto the PA gels, and Dox release was monitored at pre-determined time intervals by removing 1 μL of the sample for fluorescence-based concentration determination using a ThermoScientific Nanodrop 3300 spectrophotometer. After each measurement, the same amount of the water was added back to the gel samples. The measurements were replicated for 4 different groups for each gel concentration. λexcitation and λemission values of 565 and 630 nm were used for the measurements; and the maximum RFU value at 588 nm was recorded for fluorescence analysis.
FRAP measurements
SNOM confocal Raman microscopy was used for FRAP experiments at 200× magnification. The area was initially monitored by pre-bleach scanned images at low laser intensity, bleached at 100% laser intensity (40 mW) for 15 s, and subsequently followed by detection of the fluorescence recovery again at low intensity. Dox encapsulated PA gels at 3, 2, 1 and 0.5% (w/v) concentrations were prepared according to the protocol given above. Instead of small vials, the Dox encapsulated gels were prepared within closed Petri dishes to prevent drying. The final Dox concentration within the PA gels was 40 μM.In vitro studies
The 4T1 breast cancer cell line was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% antibiotics and 10% fetal bovine serum at 37 °C with 5% CO2 flow. The cellular viability and apoptotic behavior of 1% (w/v) PA gels (with or without 40 μM Dox prepared according to the procedures described above) were determined by flow cytometry.Before seeding the cells, all groups were treated with cell medium for 1 h, and 4T1 breast cancer cells (5 × 104 cells per well) were seeded onto 48-well culture plates. The cells were treated for 24 h and 48 h, and trypsin/collagenase (1:1 (v/v)) was then used to degrade the gels and to remove the cells from the peptide extract. Then, the degraded samples were strained with falcon strainers (40 μm pore size) and centrifuged at 4000 rpm for 6 min. The supernatants were removed and the remaining cells were washed with PBS to remove the cell medium. The cells were then centrifuged at 4000 rpm for 6 min, the supernatant was discarded, and the cell pellet was re-suspended using 100 μL of 1× annexin binding buffer. For the flow cytometry analysis, cells were labeled with Annexin V and propidium iodide and incubated at room temperature for 15 min. 400 μL of 1× annexin binding buffer was added to all samples immediately prior to flow cytometry analysis.
4T1 tumor model development and in vivo studies
Animals were maintained in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Diskapi Yildirim Beyazit Training and Research Hospital, Ankara, Turkey and approved by the Institutional Ethical Committee (IEC) of the Institutional Animal Care and Use Committee of the Diskapi Yildirim Beyazit Training and Research Hospital, Ankara, Turkey. A total of 40 female Balb/c mice (8–10 weeks old) from Adacell Laboratories (Ankara, Turkey) were used for the drug delivery experiments and histological studies. Animals were housed in a specific pathogen-free environment with 12 h light–dark cycles and ad libitum access to food and water. 4T1 cells were administered to mice in order to develop an orthotropic mammary carcinoma model. Just prior to the inoculation, 2 × 106 viable cells were suspended in 1 mL serum-free RPMI 1640 medium. Cells in 50 μL medium were subcutaneously injected into the second mammary fat pad on the right side of BALB/c mice using 1/2 inch 26 gauge needles. Tumor formation was observed 8 days after implantation. The tumor volume was calculated according to the following formula: V = 1/2(D × d2) where the tumor volume was defined as V, D is the measurement of the greatest longitudinal diameter of the tumor and d is the greatest transverse diameter of the tumor as measured with a digital caliper. When the tumor volume reached 100 mm3, forty mice were randomly divided into four groups and treated with Dox encapsulated 1% (w/v) PA gels or controls (n = 8): Dox/PA, only Dox, only PA and PBS. Samples were administered via subcutaneous injection adjacent to the tumor tissue at a dose equivalent of 10 mg Dox per kg body weight. The body weight (Fig. S7†) and tumor volume were monitored every 3 days. Animals were sacrificed at day 18.Histology and immunohistochemical staining
Tissues were fixed in 10% (v/v) formalin and embedded in paraffin. 5 μm sections were cut using a Leica microtome and stained with hematoxylin and eosin (H&E). Caspase-3 mediated apoptosis in tumor tissues of different treatment groups was evaluated. Slides were dried overnight at 60 °C before deparaffinization, and then rehydrated through a graded alcohol series. Sodium citrate buffer (pH 6.0) was used for heat-induced antigen retrieval. Sections were blocked in 1% Tris buffered saline (TBS) and 1% bovine serum albumin (BSA) solution for 2 h at room temperature (RT). Samples were then treated with the cleaved caspase-3 monoclonal antibody (1/300) (Asp175) (#9661 Cell Signaling Technology, USA) in TBS and BSA solution, and subsequently incubated at 4 °C overnight. Slides were rinsed with TBS and 0.025% Triton-X 100 solution following HRP conjugated goat-anti rabbit secondary antibody (1:500) incubation for 1 h at RT and then washed with TBS several times. Between each immunostaining step, slides were washed briefly in TBS buffer (pH 7.6). Controls were prepared by replacing the primary antibody with TBS buffer. The staining was developed by using diaminobenzidine (DAB) as the substrate. The sections were counterstained with hematoxylin. Bright-field microscopy was used to obtain the images.
Statistical analysis
All statistical analyses were carried out by one-way or two-way ANOVA, or an unpaired student's t-test, whichever applicable. A p-value of less than 0.05 was determined as statistically significant.Results and discussion
Design, synthesis and characterization of PA assemblies and supramolecular nanofiber gels
Two different PA molecules (E3PA and K3PA, Fig. 1a) were designed as building blocks of the drug delivery system and synthesized by the Fmoc solid phase peptide synthesis method11 (Fig. S1†). The PA molecules consist of an aliphatic tail, a β-sheet forming motif (–VVAG–) and oppositely charged amino acids to trigger coassembly through hydrophobic interactions, hydrogen bonding and electrostatic interactions at pH 7.4 in water. Particularly, the overall charge neutralization within the system, which was determined via zeta potential measurements of the PA mixtures (E3/K3PA) (Fig. 1b), enables the formation of the self-supporting supramolecular PA gel upon mixing the E3PA and K3PA solutions at a 3:4 molar ratio (Fig. 1c) above 0.5% (w/v) PA concentration. The nanofibrous structural organization within the PA gels prepared at different concentrations was analyzed via SEM imaging (Fig. 1d and S2†) and the individual coassembled PA nanofibers were observed by TEM (Fig. 1e).
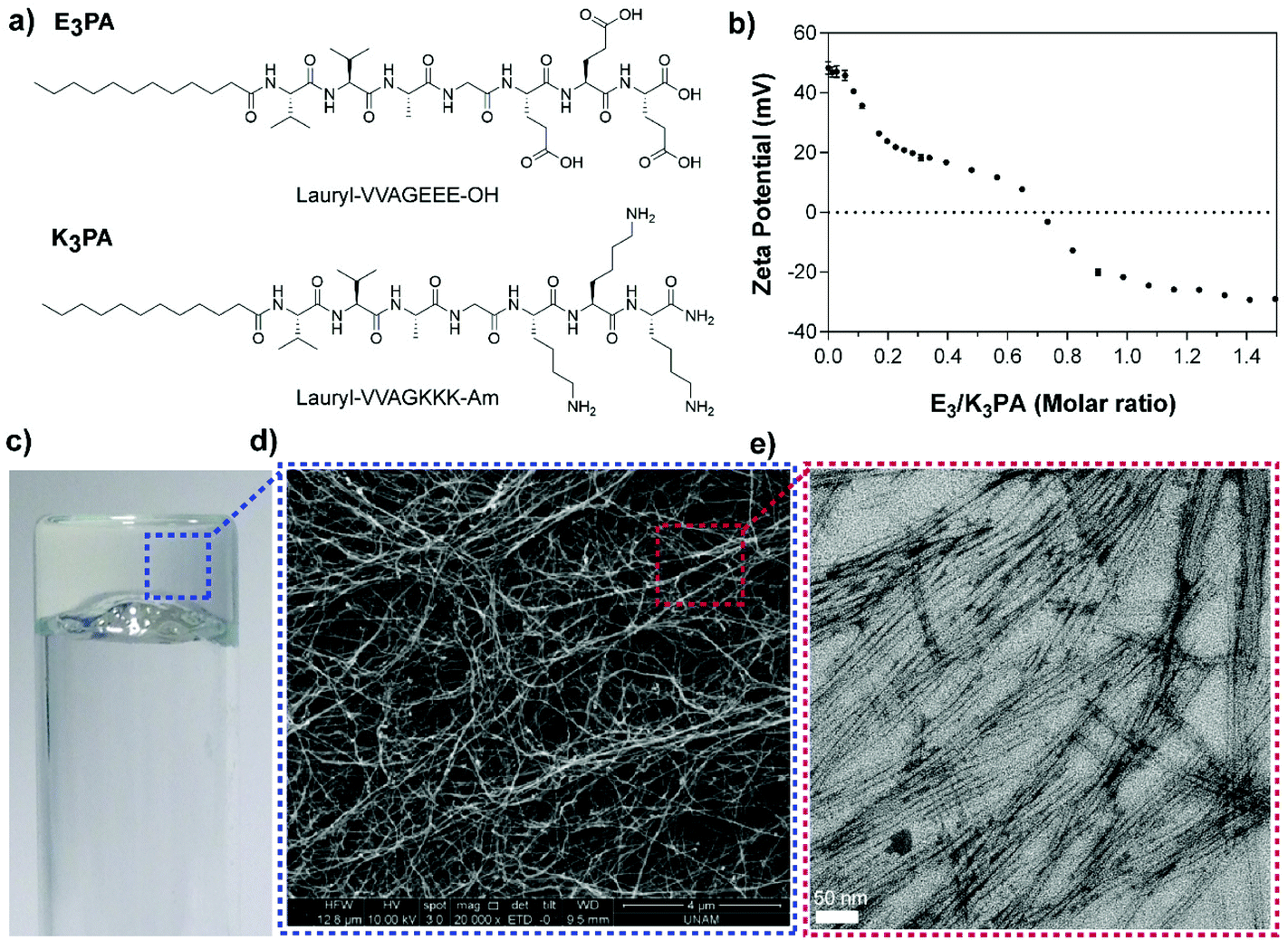 | ||
| Fig. 1 (a) Chemical representations of E3PA and K3PA molecules, (b) the zeta potential change of the E3PA upon addition of K3PA at pH 7.4 in water, (c) the supramolecular PA network showing self-supporting gel properties, and (d) SEM and (e) TEM images of the coassembled PA nanonetwork and nanofibers, respectively. | ||
The structural organization of the PA coassemblies was revealed by CD and FTIR measurements. The CD spectra of the E3/K3PA assemblies exhibited positive and negative peaks at around 190 and 220 nm respectively, which corresponded to the formation of twisted β-sheets within PA nanofibers (Fig. 2a). On the other hand, E3PA and K3PA solutions (which were used as control groups) did not show any aggregation and preserved their random coil organization at pH 7.4 in water (Fig. 2a). FTIR analysis of the PA assemblies also pointed the β-sheet organization of the coassembled nanofibers with the observed peaks at around 1632 cm−1 and 1672 cm−1 within the Amide I region42 complementary to the CD spectra (Fig. 2b and c).
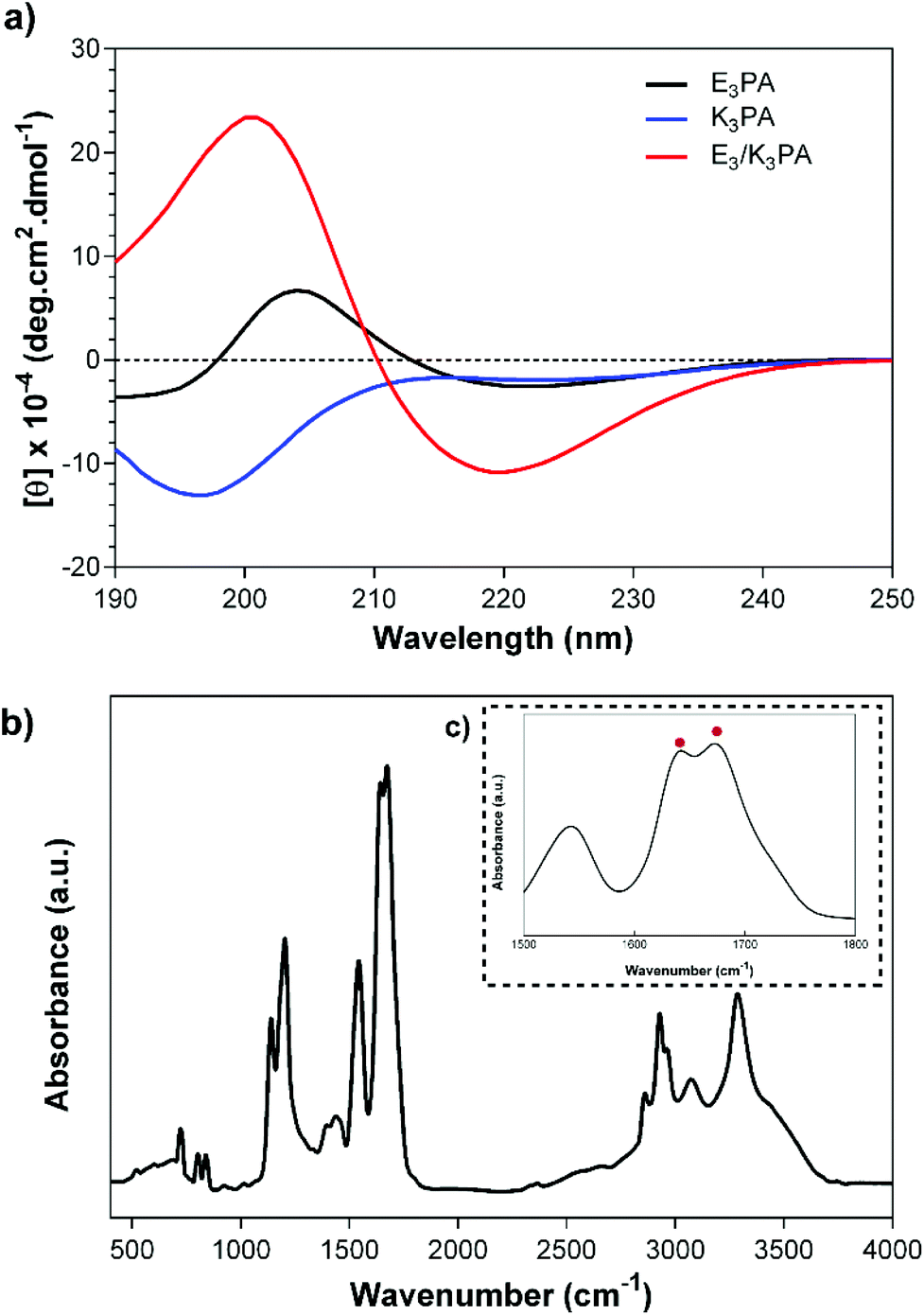 | ||
| Fig. 2 (a) CD spectra of E3PA, K3PA and E3/K3PA, and the FTIR analysis of the E3/K3PA assembly at pH 7.4 in water (b, c). | ||
The concentration dependent viscoelastic behavior of E3/K3PA assemblies was characterized using dynamic oscillatory rheology. Time sweep analysis of all groups revealed their gel-like behavior, as the storage modulus (G′) values of the samples were higher than their loss moduli (G′) (Fig. 3a). In addition, equilibrium storage and loss modulus values of the PA gels could be adjusted depending on the PA concentration within the coassembled gels (Fig. 3b). Although these architectures are formed through noncovalent interactions between PA building blocks, the viscoelasticity of the gel systems could reach up to 10 kPa and provided them with the mechanical stability required for their use as drug delivery systems in local soft tissue applications. In addition, the 1% (w/v) PA nanofiber gel preserved its mechanical stability upon its injection through a syringe (Fig. S3†).
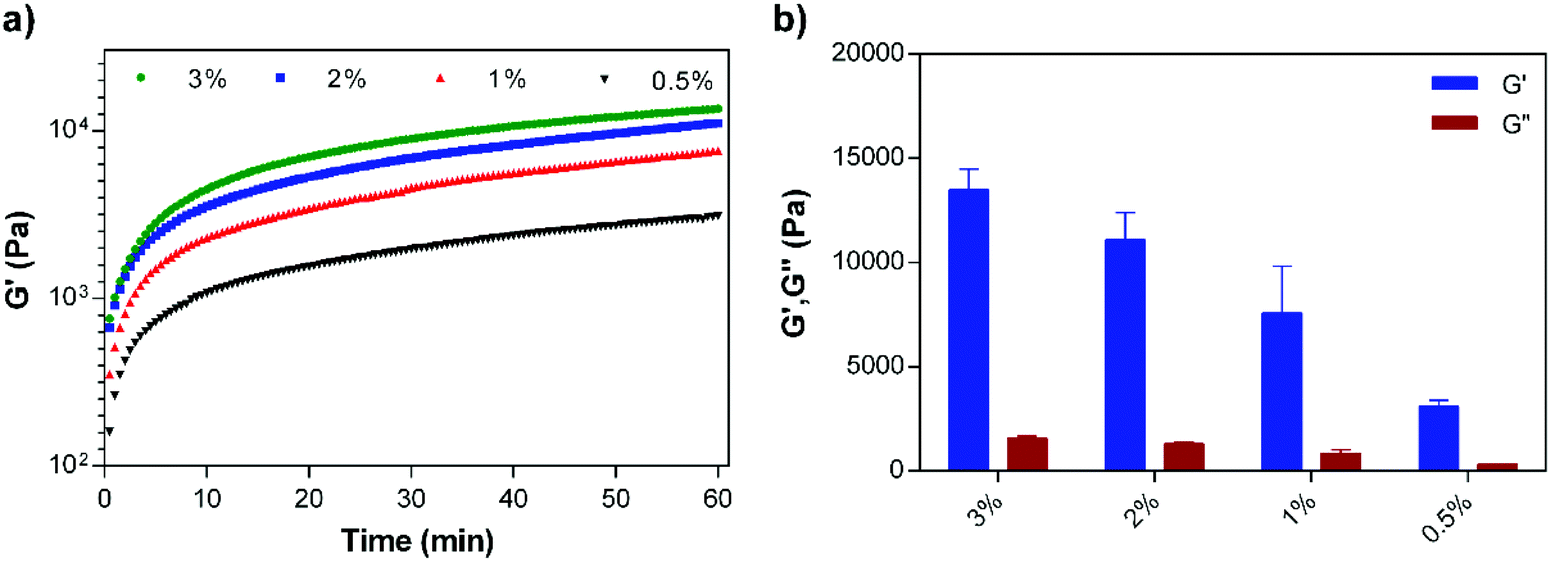 | ||
| Fig. 3 (a) Time sweep analysis of the supramolecular PA nanofiber gels prepared at different concentrations, and (b) equilibrium storage (G′) and loss moduli (G′′) of the PA gels. | ||
Biodegradability of supramolecular PA nanofiber gels
Proteolytic degradation profiles of 1% (w/v) coassembled PA gels were monitored based on the gel mass change over time (Fig. S4†). Two different serine proteases, proteinase K and α-chymotrypsin, were selected owing to their broad specificity for the cleavage of the peptide bonds, primarily from aromatic or hydrophobic residues.43 The gels were incubated with enzyme solutions prepared in Tris buffer at pH 7.4 or only the buffer as the control group. The solutions were removed and the remaining gel mass was weighed at determined time intervals (Fig. S4†). The PA gel was rapidly degraded by proteinase K and only 10% of the gel mass remained at day 25. On the other hand, the degradation of the PA gel by α-chymotrypsin was slower compared to proteinase K, and approximately 50% of the gel had remained at day 25 under the same concentrations. In the literature, α-chymotrypsin is generally utilized to cleave peptide bonds from aromatic residues.44 On the other hand, proteinase K exhibits activity against aliphatic amino acids in addition to hydrophobic and aromatic residues.45 This difference in the degradation behavior of the two enzymes could be related to the ability of proteinase K to easily cleave the aliphatic beta sheet forming motif (–VVAG–) that is present in the interspace between the hydrophobic aliphatic tail and hydrophilic amino acid regions of the PA molecules. Additionally, the control group (which is incubated only with the buffer solution) retains its weight for 25 days, suggesting that the coassembled system is stable for a considerable period of time in the absence of enzymatic degradation.Drug encapsulation and controlled drug release
To encapsulate the Dox molecules within the supramolecular PA nanofiber gels, the drug solution was initially mixed with positively charged K3PA and coassembly was subsequently triggered by addition of negatively charged E3PA at pH 7.4 (Fig. 4a). This approach enabled the 100% encapsulation and homogeneous dispersion of the drug molecules within PA gels. In addition, the nanofibrous architecture of the gels provided a suitable nanoporous environment for the controlled release of small molecules and to sustain the therapeutic level for local application of the delivery system in soft tissues (Fig. 4a).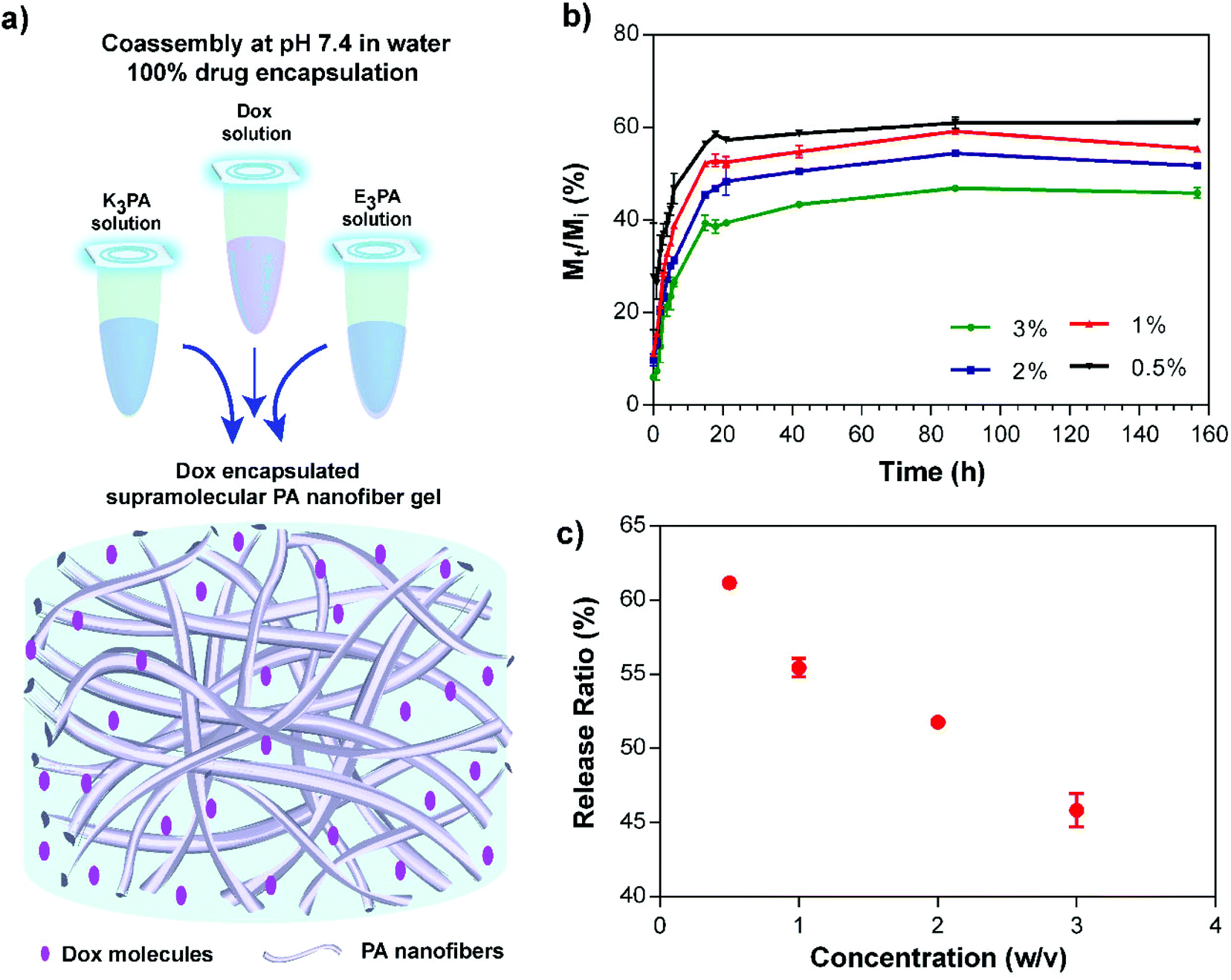 | ||
| Fig. 4 (a) Schematic representation of the drug encapsulation and coassembly of the oppositely charged PA molecules into supramolecular nanofiber gels, (b) controlled release profile of Dox through the PA gels prepared at different concentrations at pH 7.4 in water, and (c) the release ratios of the chemotherapeutic drug were modulated depending on the PA concentration within the gels. | ||
Drug release profiles depending on the PA concentration within the coassembled PA gels were monitored and quantified based on the concentration dependent fluorescence intensity calibration of the molecules. The sustained release of the drug molecules through the PA gels was observed for 48 h, and then in vitro release kinetics slowed down reaching an equilibrium after 156 h (Fig. 4b). Non-linear transport kinetics of all PA groups were consistent with non-Fickian release behavior, which may have resulted from the interactions of Dox with three different interfaces that are present in the PA system: (i) the hydrophobic alkyl domain, (ii) hydrophilic amino acids carrying both negative and positive charges, and (iii) water molecules entrapped within the system. Non-covalent interactions between Dox molecules and these interfaces may account for the non-linearity of the transport properties and prevent the complete release of the encapsulated drug molecules through the gel system. In addition, release kinetics and the amount of the released molecules under equilibrium conditions were modulated by the PA concentration (Fig. 4c) which has an effect on both the porosity of the network and the entanglement of nanofibers46 within the coassembled PA gel system. Furthermore, PA nanofiber gels preserved their structural integrity throughout the release experiment under given conditions for 156 h.
In addition to in vitro bulk release studies, the mobility of the drug molecules within coassembled PA gels was investigated by FRAP,46 in which fluorescent molecules are photobleached by using a high power laser source and the recovery of fluorescence intensity at the bleached area is monitored to estimate the transport parameters of the molecules. Dox encapsulated gels were prepared at different PA concentrations in an enclosed system to prevent drying throughout the experimental period. Fluorescence intensity changes were monitored after the photobleaching of drug molecules (Fig. 5a); and data analysis was performed using the program developed by Jönsson et al.47 Diffusion constants of Dox molecules within 3, 2, 1 and 0.5% (w/v) PA nanofiber gels were estimated by Hankel through transforming the resulting data (Fig. 5b and S5†). FRAP experiments suggest that the drug release and mobility of the molecules can be modulated and controlled depending on the PA concentration. In addition, immobile fractions of the Dox molecules within the gels were determined to be 0.56 ± 0.17, 0.26 ± 0.05, 0.19 ± 0.05 and 0.17 ± 0.02, respectively using the FRAP data. The increase in the immobile Dox molecule amount depending on the PA concentration within the gel systems also shows us the affinity of the PA network towards the drug molecules, which is consistent with the non-Fickian transport behavior that the gels exhibit. Although non-covalent interactions between PA nanofiber gels and drug molecules resulted in the entrapment of some portion of the molecules within the system, the biodegradability of the nanofibrous PA system could improve the availability of the immobile drug molecules via proteolytic degradation of the PA gel delivery system. On the other hand, 1% (w/v) PA concentration was determined for both in vivo and in vitro experiments considering the mechanical stability (Fig. 3), controlled release and transport properties of the PA nanofiber gels (Fig. 4 and 5).
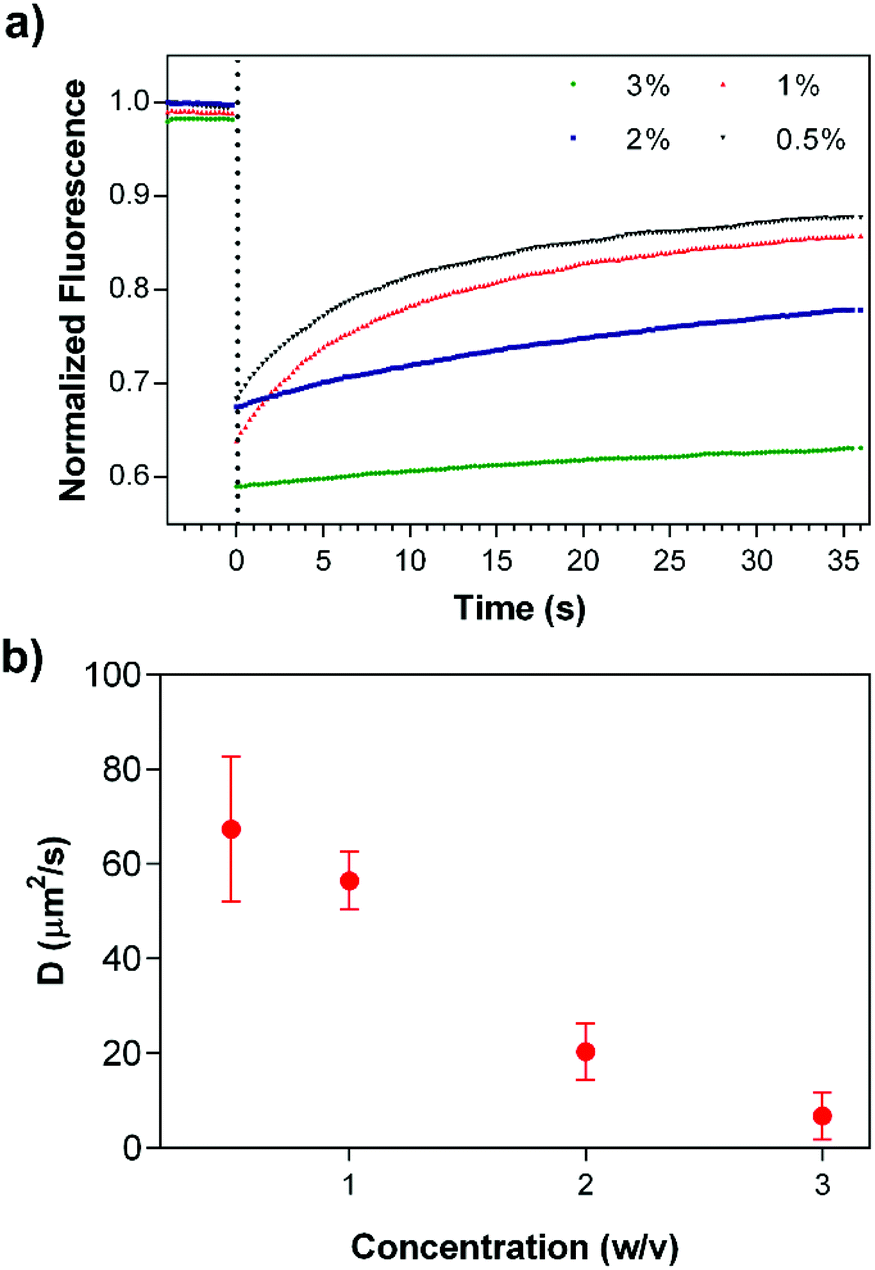 | ||
| Fig. 5 Mathematical fitting results of FRAP experiments: (a) the estimated diffusion constants, and (b) immobile fractions of the drug molecules encapsulated within the PA gels prepared at different concentrations. | ||
In vitro studies
To determine the biocompatibility of the delivery system and the effects of controlled drug release on cellular viability, breast cancer cells were cultured on the only PA gel, Dox encapsulated PA gel (Dox/PA) or uncoated tissue culture plate (TCP, control). The same amount of Dox encapsulated within the gels was also directly administered to the cell culture media as a positive control (only Dox) for 24 h and 48 h. The only PA gel did not exhibit any cytotoxicity compared to the TCP control (Fig. 6 and S6†). However, cellular viability had decreased at 24 h and 48 h for both only Dox and Dox/PA groups. Although no significant differences were present between these groups at 24 h, the Dox/PA group was more effective in inhibiting cellular growth at 48 h, potentially due to the sustained release of Dox molecules from the gel system. In addition, the PA nanofiber gel provides suitable protection for drug molecules and enhances their bioavailability and activity under in vitro conditions.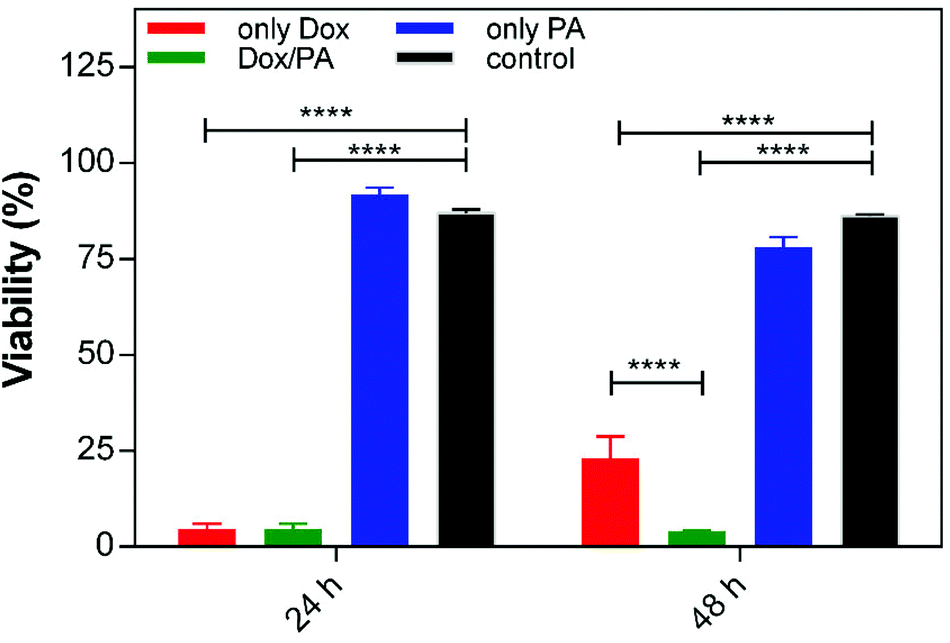 | ||
| Fig. 6 Cellular viability of (a) the control (TCP), (b) 1% (w/v) PA gel, (c) 1% (w/v) Dox/PA gel and (d) only Dox for 24 h and 48 h. | ||
In vivo applicability of supramolecular PA nanofiber gels for the treatment of solid tumors
After examining the biocompatibility of PA gels and controlled drug release dependent cellular viability under in vitro conditions, the applicability of the Dox/PA gels as a local drug delivery system was tested in vivo using a solid tumor model. For this purpose, highly aggressive and metastatic 4T1 breast cancer cells were subcutaneously injected to the mammary fat pad of Balb/c mice for tumor growth. When the tumor volume reached 100 mm3, identical amounts of the 1% (w/v) Dox/PA gel, only PA gel, Dox solution or PBS (as the control) were injected to the tumor site, and tumor sizes were measured every three days using an electronic caliper. The tumor growth rate was followed for 18 days (Fig. 7a), at which time the animals were sacrificed for histological analysis of tumors and other organs (Fig. S8†). The local delivery of Dox molecules by coassembled PA gels significantly limited tumor growth compared to the direct injection of the drug solution (Fig. 7b). On the other hand, PA gels without the drug encapsulation did not cause additional cytotoxicity compared to PBS control which indicated the in vivo biocompatibility of the coassembled PA nanofiber gels as a local delivery system.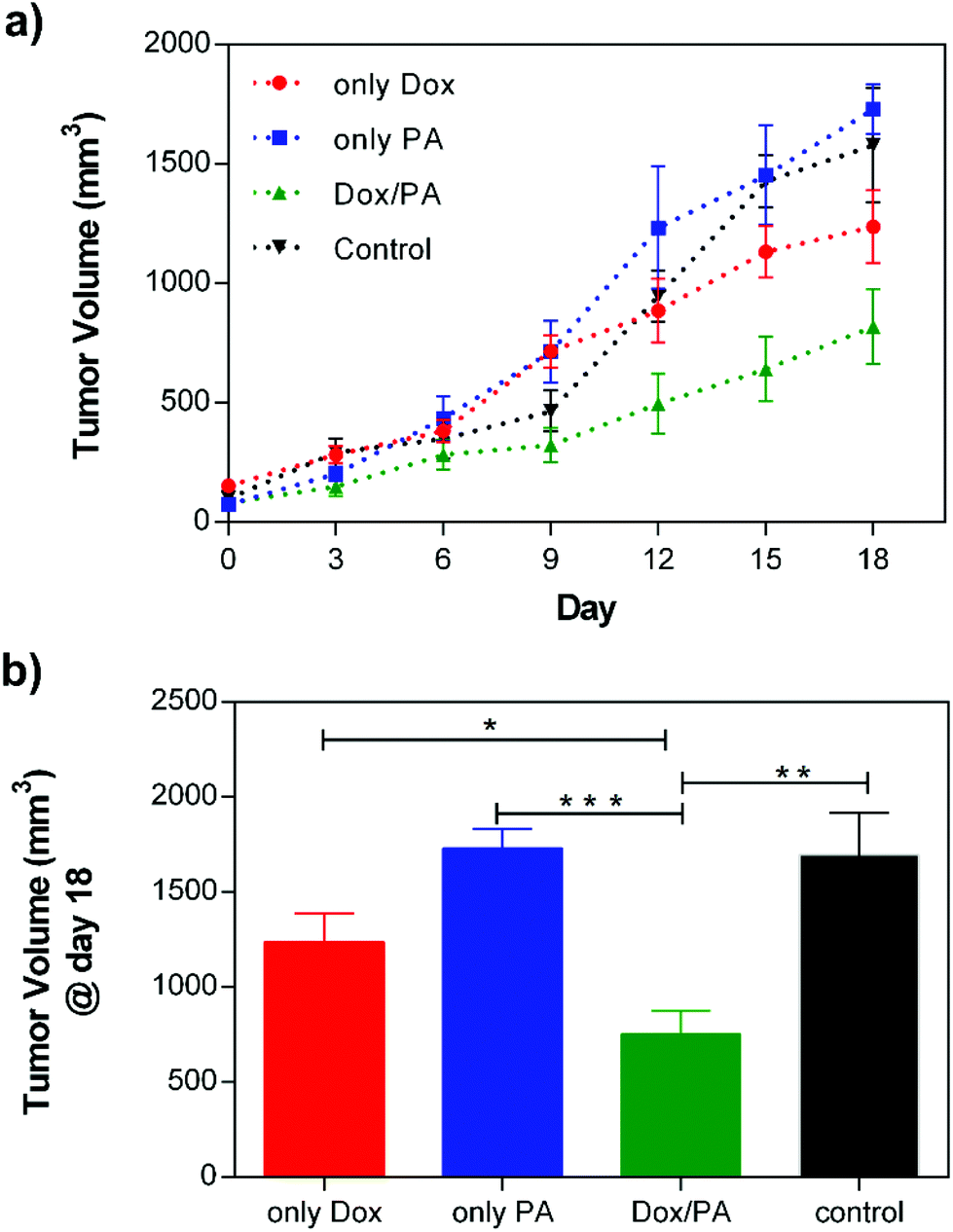 | ||
| Fig. 7 (a) Tumor growth of the only Dox, only PA, Dox/PA and non-treated control groups for 18 days, and (b) the final tumor volume (mm3) at day 18. | ||
Dox demonstrates its chemotherapeutic activity by inducing caspase-3 activation, which mediates apoptosis and DNA fragmentation through the p53 pathway.48 To reveal the effects of the administration of the Dox molecules via a controlled delivery system on the tumor tissue, immunostaining against caspase-3, which is an apoptotic cell marker, was performed on tissue sections. Cleaved caspase-3 positive cells were clearly observed in tumor tissues treated with Dox/PA. Analysis of tissue sections stained with caspase-3 indicated that the sustained release of the drug molecules through PA gels increased the number of apoptotic cells within the tumor tissue and facilitated the activity of the chemotherapeutic Dox molecules at the local site to a greater extent, compared to the local injection of the Dox molecules without a controlled delivery system (Fig. 8). Hematoxylin–eosin staining was also performed to observe the tissue morphology of the tumor, liver, kidney and spleen treated with the Dox/PA, and revealed the promoted activity and reduced side effects of the drug molecules compared to the healthy tissue controls (Fig. S8†).
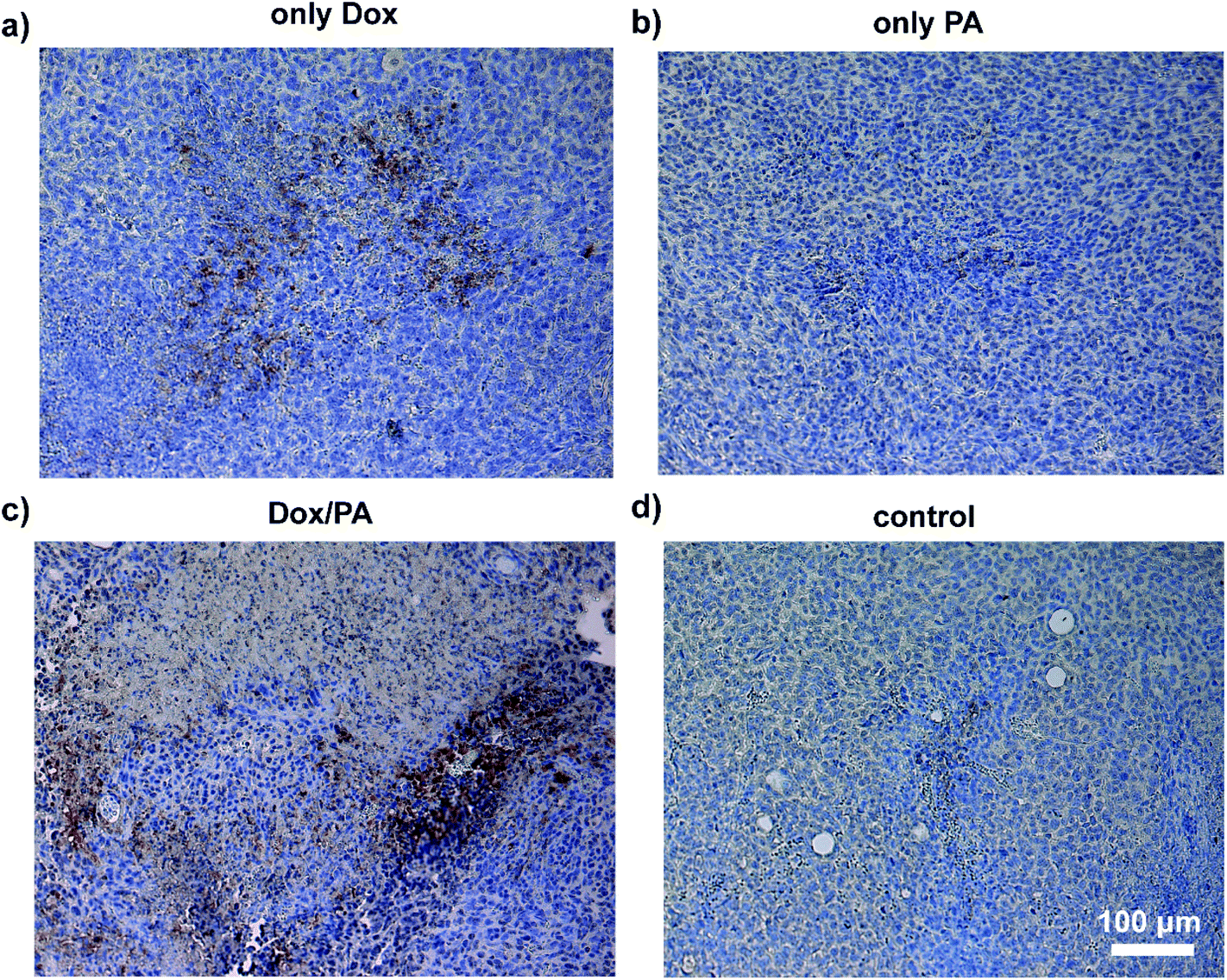 | ||
| Fig. 8 Caspase 3 immunohistochemical staining of paraffin-embedded sections of tumor tissues of (a) the only Dox, (b) only PA, (c) Dox/PA and (d) non-treated control groups at day 18. | ||
Conclusions
Advances in developing biocompatible and injectable drug delivery systems can facilitate the local chemotherapeutic applications for the treatment of soft-tissue cancer types. Supramolecular PA nanofiber gels are potential candidates for prolonged local delivery of chemotherapeutics under physiological conditions. In this study, we showed that the PA gels consisting of coassembled nanofibers with a β-sheet secondary structure organization could form with facile assembly properties at pH 7.4 in water through noncovalent interactions between oppositely charged PA molecules. In addition, controlled drug release characteristics of the PA nanofiber gels could be modulated depending on the PA concentration and noncovalent interactions between the PA network and drug molecules could be used to facilitate the affinity-controlled release of encapsulated drugs. Biocompatibility and biodegradability of the PA nanofiber gels were also other advantages of these architectures for drug delivery. The significant decrease in the tumor growth rate following the injection of Dox/PA gels in vivo compared to the other groups also showed the potential of supramolecular PA nanofiber gels for local chemotherapeutic applications in clinical settings.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
This work is partially supported by grants TUBITAK (213M406 and 113M900), TUBA-GEBIP, and FP7 Marie Curie IRG. G. C. is supported by the TUBITAK-BIDEB 2211-C PhD fellowship. We thank M. Guler for help in TEM imaging of PA nanostructures.References
- C. Alvarez-Lorenzo and A. Concheiro, Smart materials for drug delivery, 2013, vol. 1, pp. 1–32 Search PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS.
- D. Patra, S. Sengupta, W. Duan, H. Zhang, R. Pavlick and A. Sen, Nanoscale, 2013, 5, 1273–1283 RSC.
- G. Cinar, D. Mumcuoglu, A. B. Tekinay and M. O. Guler, Therapeutic Nanomaterials, 2016, p. 7 Search PubMed.
- G. Verma and P. Hassan, Phys. Chem. Chem. Phys., 2013, 15, 17016–17028 RSC.
- M. S. Ekiz, G. Cinar, M. A. Khalily and M. O. Guler, Nanotechnology, 2016, 27, 402002 CrossRef.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS.
- Y. Chen, H. X. Gan and Y. W. Tong, Macromolecules, 2015, 48, 2647–2653 CrossRef CAS.
- K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS.
- M. Goktas, G. Cinar, I. Orujalipoor, S. Ide, A. B. Tekinay and M. O. Guler, Biomacromolecules, 2015, 16, 1247–1258 CrossRef CAS.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS.
- S. Sundar, Y. Chen and Y. Tong, Curr. Med. Chem., 2014, 21, 2469–2479 CrossRef CAS.
- R. Mammadov, G. Cinar, N. Gunduz, M. Goktas, H. Kayhan, S. Tohumeken, A. E. Topal, I. Orujalipoor, T. Delibasi and A. Dana, Sci. Rep., 2015, 5, 16728 CrossRef CAS.
- S. Lu, Y. Ding, W. Cui, R. Pan, W. Xu and P. Chen, RSC Adv., 2016, 6, 86943–86946 RSC.
- D. Mumcuoglu, M. Sardan, T. Tekinay, M. O. Guler and A. B. Tekinay, Mol. Pharm., 2015, 12, 1584–1591 CrossRef CAS.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS.
- H. W. Jun, V. Yuwono, S. E. Paramonov and J. D. Hartgerink, Adv. Mater., 2005, 17, 2612–2617 CrossRef CAS.
- R. H. Zha, S. Sur and S. I. Stupp, Adv. Healthcare Mater., 2013, 2, 126–133 CrossRef CAS.
- A. Dehsorkhi, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 6665–6672 CrossRef CAS PubMed.
- J.-K. Kim, J. Anderson, H.-W. Jun, M. A. Repka and S. Jo, Mol. Pharm., 2009, 6, 978–985 CrossRef CAS.
- S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS PubMed.
- H. W. Seo, D. Y. Kwon, J. S. Kwon, L. M. Jin, B. Lee, J. H. Kim, B. H. Min and M. S. Kim, Biomaterials, 2013, 34, 2748–2757 CrossRef CAS PubMed.
- W. Wang, L. Deng, S. Xu, X. Zhao, N. Lv, G. Zhang, N. Gu, R. Hu, J. Zhang and J. Liu, J. Mater. Chem. B, 2013, 1, 552–563 RSC.
- G. Chang, T. Ci, L. Yu and J. Ding, J. Controlled Release, 2011, 156, 21–27 CrossRef CAS PubMed.
- J. Y. Lee, K. S. Kim, Y. M. Kang, E. S. Kim, S.-J. Hwang, H. B. Lee, B. H. Min, J. H. Kim and M. S. Kim, Int. J. Pharm., 2010, 392, 51–56 CrossRef CAS PubMed.
- S. Koutsopoulos, L. D. Unsworth, Y. Nagai and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef CAS PubMed.
- S. Koutsopoulos and S. Zhang, J. Controlled Release, 2012, 160, 451–458 CrossRef CAS PubMed.
- V. A. Kumar, S. Shi, B. K. Wang, I.-C. Li, A. A. Jalan, B. Sarkar, N. C. Wickremasinghe and J. D. Hartgerink, J. Am. Chem. Soc., 2015, 137, 4823–4830 CrossRef CAS PubMed.
- V. A. Kumar, N. L. Taylor, S. Shi, N. C. Wickremasinghe, R. N. D'Souza and J. D. Hartgerink, Biomaterials, 2015, 52, 71–78 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- I. C. Li, A. N. Moore and J. D. Hartgerink, Biomacromolecules, 2016, 17, 2087–2095 CrossRef CAS PubMed.
- X.-D. Xu, L. Liang, C.-S. Chen, B. Lu, N.-l. Wang, F.-G. Jiang, X.-Z. Zhang and R.-X. Zhuo, ACS Appl. Mater. Interfaces, 2010, 2, 2663–2671 CAS.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS PubMed.
- A. Baral, S. Roy, A. Dehsorkhi, I. W. Hamley, S. Mohapatra, S. Ghosh and A. Banerjee, Langmuir, 2014, 30, 929–936 CrossRef CAS PubMed.
- J. E. Sun, B. Stewart, A. Litan, S. J. Lee, J. P. Schneider, S. A. Langhans and D. J. Pochan, Biomater. Sci., 2016, 4, 839–848 RSC.
- R. B. Weiss, Semin. Oncol., 1992, 19, 670–686 CAS.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- J. L. Quiles, J. R. Huertas, M. Battino, J. Mataix and M. C. Ramírez-Tortosa, Toxicology, 2002, 180, 79–95 CrossRef CAS.
- G. Cinar, H. Ceylan, M. Urel, T. S. Erkal, E. Deniz Tekin, A. B. Tekinay, A. Dâna and M. O. Guler, Biomacromolecules, 2012, 13, 3377–3387 CrossRef CAS.
- J. J. Perona and C. S. Craik, Protein Sci., 1995, 4, 337–360 CrossRef CAS.
- W. K. Baumann, S. A. Bizzozero and H. Dutler, FEBS Lett., 1970, 8, 257–260 CrossRef CAS.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, FEBS J., 1974, 47, 91–97 CrossRef CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS.
- P. Jönsson, M. P. Jonsson, J. O. Tegenfeldt and F. Höök, Biophys. J., 2008, 95, 5334–5348 CrossRef.
- S. Wang, E. A. Konorev, S. Kotamraju, J. Joseph, S. Kalivendi and B. Kalyanaraman, J. Biol. Chem., 2004, 279, 25535–25543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6bm00656f |
| This journal is © The Royal Society of Chemistry 2017 |