Preparation of open and closed forms of the lvt network with Cu(II) complexes of structurally related 1,2-diazole ligands†
Chris S. Hawes‡
* and
Paul E. Kruger*
MacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Chemistry, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. E-mail: paul.kruger@canterbury.ac.nz
First published on 24th March 2014
Abstract
Reported here are the synthesis and structural characterisation of two topologically related coordination polymers: [Cu(L1)(CH3CONH2)], 1, and [Cu(L2)(NMP)]·0.5NMP·0.5H2O, 2, (NMP = N-methyl-pyrrolidinone) prepared from 1H-indazole-3-carboxylic acid, H2L1, and 1H-pyrazole-3-carboxylic acid, H2L2, respectively. Both 1 and 2 crystallise in the tetragonal space group I41/a and form 3-dimensional coordination polymers described by the lvt topology, in which [Cu2L2] dimers form four-connected nodes linked through carboxylate groups. Within 1 the presence of a Cu-bound acetamide molecule (formed in situ by the solvothermal hydration of acetonitrile) propagates hydrogen bonding between dimer units that, combined with the steric bulk of the indazole fragment, yield a closed (non-porous) framework. In 2, however, Cu-bound NMP, which does not participate in hydrogen bonding, coupled with the less bulky nature of the pyrazole-backbone, gives rise to an open framework of an otherwise isostructural network.
Introduction
The study of coordination polymers (CPs) and metal organic frameworks (MOFs) is at the forefront of modern inorganic chemistry due to the potential such species have in various applications ranging from magnetic and luminescent materials, through catalysis and sensing, to gas separation and storage.1 While many important advances have been made in the synthesis and physical characterisation of these framework materials, a key area of investigation remains the synthesis of ligands containing novel donor groups or additional backbone functionality.2 By developing new ligands some of the critical limitations recognized in CPs and MOFs, such as hydrolytic and structural instability common for zinc cluster-based MOFs, may be addressed.3 In this regard the use of nitrogen heterocyclic ligand scaffolds provides an excellent base from which to explore the synthesis of new framework materials, as azolate-based CPs and MOFs in particular have been shown to be resistant to hydrolysis and more structurally resilient than their related carboxylate analogues.4Recently we reported several nitrogen heterocyclic and carboxylic acid ligand based coordination polymers,5 including the first example of an indazole-carboxylate coordination polymer, featuring 1H-indazole-5-carboxylic acid.6 The fused ring back-bone of indazole, a heterocyclic ligand only rarely employed in coordination chemistry,7 offers an opportunity through functionalisation at the 3, 4, 5, 6, or 7 ring positions, or combinations thereof, to give ligand geometries not readily available in more commonly used heterocyclic ligand systems.
Herein we report the synthesis of two structurally related 3D coordination polymers prepared from the iso-functional diazole-carboxylate ligands: 1H-indazole-3-carboxylic acid, H2L1, and 1H-pyrazole-3-carboxylic acid, H2L2 (Fig. 1). This contribution reports rare examples of coordination polymers formed from either H2L1 or H2L2. To date, the only reported structurally-characterised metal complexes of L1 consist of discrete species,8 whilst of the few known metal complexes of L2,9 only one polymeric material has been reported, a polymeric alkyltin material.10 Whilst the dearth of L1 complexes in the literature might be rationalised by its somewhat obscure nature, it is surprising to note that L2 has been similarly under-explored in coordination chemistry. This is especially surprising when contrasted to the related compound 1H-pyrazole-3,5-dicarboxylic acid, which has been used extensively in the synthesis of coordination polymers.11 We address this oversight here.
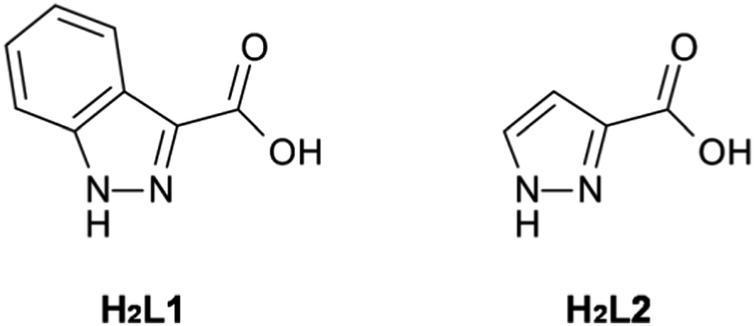 | ||
| Fig. 1 Ligands of interest within this study; H2L1 and H2L2. | ||
Experimental
Materials and methods
All reagents and starting materials were used as received from standard suppliers and were of reagent grade or higher. The ligands H2L1 (ref. 12) and H2L2 (ref. 13) were prepared according to published procedures. Infrared spectra were recorded on a Perkin Elmer Spectrum One FTIR instrument operating in diffuse reflectance sampling mode with samples diluted in powdered KBr in the range 400–4000 cm−1. Melting points were recorded on an electrothermal melting point apparatus and are uncorrected. Thermogravimetric analysis was performed on an AlphaTech Q600 SDT DSC/TGA instrument where samples were heated on alumina crucibles under nitrogen flow of 100 mL min−1 and were heated at a rate of 1 °C min−1 to 500 °C. Elemental analysis for C, H and N were carried out at Campbell Micro Analytical Laboratories, Otago University, Dunedin. Phase purity of crystalline materials was confirmed by indexing unit cell collections from a representative sampling of crystals from each batch.X-ray crystallography
Single crystal data and experimental details for 1 and 2 are presented in Table 1, and hydrogen bonding parameters are presented in Table S1 (ESI†). X-ray crystallographic data collection and refinement was carried out with either an Oxford-Agilent SuperNova instrument with focused micro-source Cu Kα (λ = 1.5418 Å) radiation and ATLAS CCD area detector (1) or a Bruker APEX-II instrument (2), using graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation. Both structures were solved using direct methods with SHELXS14 and refined on F2 using all data by full matrix least-squares procedures with SHELXL-97 (ref. 15) within OLEX-2.16 Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in calculated positions with isotropic displacement parameters 1.2 times the isotropic equivalent of their carrier atoms, except H16A and H16B of structure 1, which were manually located from residual Fourier peaks and modelled with appropriate bond length restraints and Uiso dependences. The functions minimized were ∑w(Fo2 − Fc2), with w = [σ2(Fo2) + aP2 + bP]−1, where P = [max(Fo)2 + 2Fc2]/3.| Compound reference | 1 | 2 |
|---|---|---|
| Chemical formula | C10H9CuN3O3 | C9H11CuN3O3 |
| Formula mass | 282.74 | 272.75 |
| Crystal system | Tetragonal | Tetragonal |
| a/Å | 16.6545 (16) | 18.0584 (8) |
| b/Å | 16.6545 (16) | 18.0584 (8) |
| c/Å | 16.3362 (18) | 18.3413 (11) |
| α/° | 90.00 | 90.00 |
| β/° | 90.00 | 90.00 |
| γ/° | 90.00 | 90.00 |
| Unit cell volume/Å3 | 4531.2 (8) | 5981.2 (5) |
| Temperature/K | 113 (2) | 113 (2) |
| Radiation source | Cu Kα | Mo Kα |
| Space group | I41/a | I41/a |
| No. of formula units per unit cell, Z | 16 | 16 |
| No. of reflections measured | 9990 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 629 629 |
| No. of independent reflections | 1998 | 2647 |
| No. of reflections observed (I > 2σ(I)) | 1320 | 2497 |
| Rint | 0.0853 | 0.0626 |
| Final R1 values (I > 2σ(I)) | 0.0455 | 0.0651 |
| Final wR(F2) values (I > 2σ(I)) | 0.0931 | 0.1589 |
| Final R1 values (all data) | 0.0876 | 0.0702 |
| Final wR(F2) values (all data) | 0.1097 | 0.1620 |
Synthesis of [Cu(L1)(CH3CONH2)], 1
H2L1 (10 mg; 0.062 mmol) was added to Cu(NO3)2·2.5H2O (15 mg; 0.062 mmol) in 5 mL of acetonitrile within a 23 mL Parr instruments Teflon-lined acid digestion bomb, which was sealed and heated to 120 °C, allowed to dwell at this temperature for 36 hours, and then cooled at 5 °C h−1 to room temperature. The product was isolated as a pure crystalline phase consisting of green octahedral blocks. Yield 12.1 mg, 69%; mp: >300 °C; found: C, 42.4; H, 3.16; N, 14.9%; C10H8N3O3Cu requires: C, 42.5; H, 3.21; N, 14.9%;![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) max/cm−1 (KBr): 3397 s, 3153 m, 1667 s, 1580 s, 1470 m, 1343 m, 1195 m, 1149 w, 1091 w, 1003 w, 958 w, 846 s, 799 m, 734 m, 650 m, 562 m.
max/cm−1 (KBr): 3397 s, 3153 m, 1667 s, 1580 s, 1470 m, 1343 m, 1195 m, 1149 w, 1091 w, 1003 w, 958 w, 846 s, 799 m, 734 m, 650 m, 562 m.
Synthesis of [Cu(L2)(NMP)]·0.5NMP·0.5H2O, 2
H2L2 (200 mg; 1.8 mmol) was added to Cu(NO3)2·2.5H2O (220 mg; 0.9 mmol) in 20 mL of N-methylpyrrolidone containing 2 mL of water. The mixture was heated to 50 °C and stirred briefly to ensure dissolution of the starting materials. The mixture was then filtered, and the filtrate left to stand in a calcium chloride desiccator for a period of several weeks, giving the product as large blue blocks which were isolated by filtration. Yield 125 mg, 13%. mp: >300 °C; found: C, 41.9; H, 4.87; N, 14.6%; {C9H11N3O3Cu·0.5(H2O)·0.5(C5H9NO)} requires: C, 41.7; H, 5.02; N, 14.8%.max/cm−1 (KBr): 3519 m, 2873 s, 1668 s, 1582 m, 1524 m, 1385 m, 1302 s, 1145 s, 1114 m, 1073 w, 986 w, 823 s, 773 s, 632 m.
Results and discussion
Synthesis and X-ray crystal structure of 1
The ligand H2L1 was reacted in acetonitrile solution with one equivalent of Cu(NO3)2·2.5H2O under solvothermal conditions whereupon green octahedral-shaped crystals of 1 were isolated directly following cooling. The crystals of 1 were analysed by single crystal X-ray diffraction and the data obtained were solved and refined in the tetragonal space group I41/a, Table 1. The asymmetric unit of 1 contains a single 5-coordinate Cu(II) ion with distorted square pyramidal geometry (τ5 = 0.43),17 and a doubly deprotonated molecule of L1 that coordinates through all four hetero-atoms to the equatorial positions of symmetry-equivalent Cu(II) centres. This arrangement gives rise to an [M2L2] dimer which is further linked to adjacent dimers via additional Cu–O bonds, Fig. 2.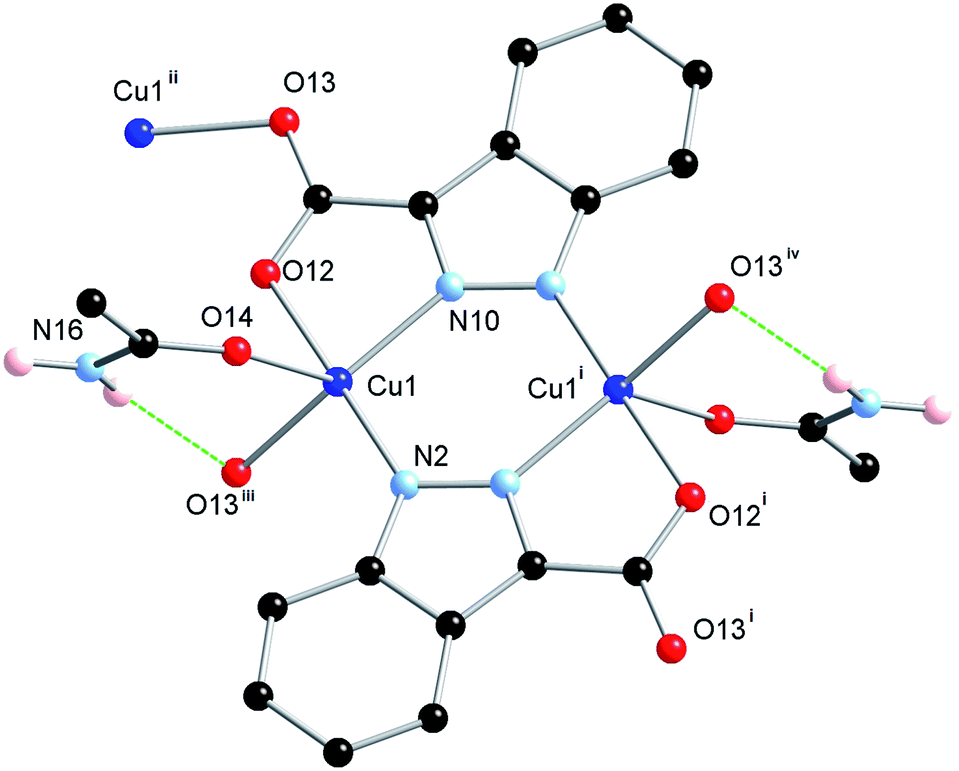 | ||
| Fig. 2 Molecular structure and atomic numbering scheme for 1. Hydrogen atoms not involved in hydrogen bonding omitted for clarity. Symmetry operations used to generate equivalent atoms: (i) 1/2 − X, 1/2 − Y, 3/2 − Z; (ii) 1/4 + Y, 1/4 − X, 5/4 − Z; (iii) 3/4 − Y, −1/4 + X, 7/4 − Z; (iv) −1/4 + Y, 3/4 − X, −1/4 + Z. | ||
The apical coordination site of the square pyramidal Cu(II) ion is occupied by the terminal atom of a trigonally disposed species consisting of four-atoms. Charge balance requires a neutral species and consideration of the electron density and hydrogen bond donor–acceptor interactions is consistent with an acetamide molecule coordinating through the oxygen atom. This assignment was supported by IR spectroscopy (max 1667 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and microanalyses which rule out other possible identities, such as acetic acid, for the coordinating species. We have shown previously that acetate can form via the hydrolysis of acetonitrile in the presence of Cu(II) under solvothermal conditions and it is most likely that acetamide is the precursor to this species.18 Indeed, 1 could also be prepared in similar yield by adding acetamide to the initial reaction mixture.
O stretch) and microanalyses which rule out other possible identities, such as acetic acid, for the coordinating species. We have shown previously that acetate can form via the hydrolysis of acetonitrile in the presence of Cu(II) under solvothermal conditions and it is most likely that acetamide is the precursor to this species.18 Indeed, 1 could also be prepared in similar yield by adding acetamide to the initial reaction mixture.
The extended structure of 1 constitutes a three-dimensional polymer, where the copper dimers bridged by the indazole nitrogen atoms are further linked by coordination through the non-chelating oxygen atom of the carboxylate group. Topologically, the most sensible description of the network can be arrived at by considering the [Cu2(L1)2] dimer as a single node, which is linked to four adjacent dimers by the Cu1–O13 bonds. When the network is simplified in this manner, the structure can be described by the (42·84) lvt network, Fig. 3. Despite the apparent presence of channels within the network, no significant void space is observed within 1. In reality, these channels are filled by both the steric bulk of the indazole rings, and the coordinating acetamide molecules. The acetamide molecule within the asymmetric unit forms two hydrogen bonds, both originating from the NH2 group; one hydrogen atom points towards the nearby coordinating carboxylate oxygen atom, to form a six-membered hydrogen bonding ring, while the other interacts with the coordinating acetamide oxygen atom on an adjacent dimer, to propagate a hydrogen bonding chain parallel to the c unit cell axis.
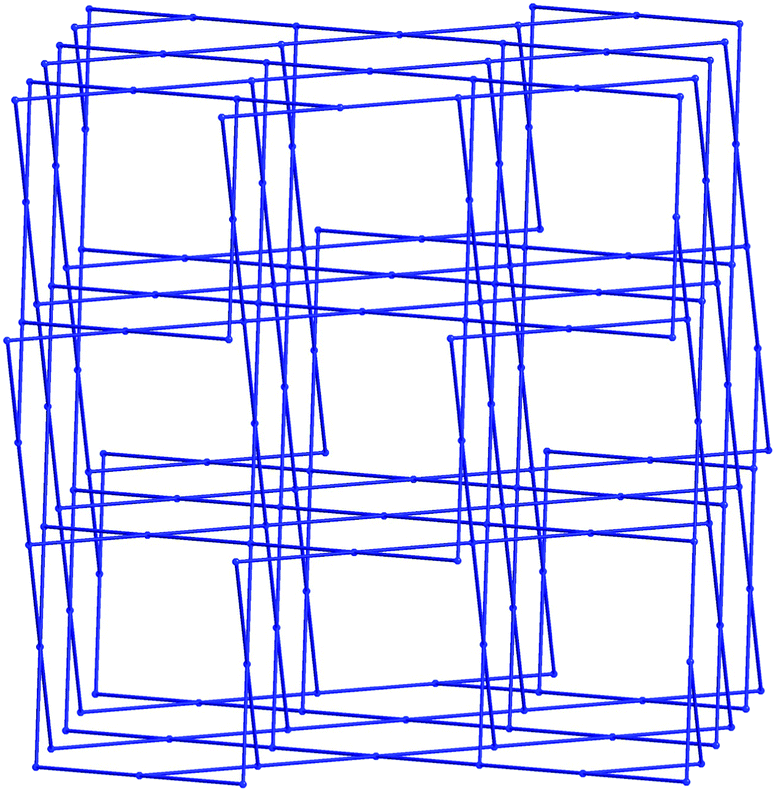 | ||
| Fig. 3 Topological representation of the lvt network in 1 with [Cu2L2] units selected as nodes. | ||
Thermogravimetric analysis of 1 shows 20% mass loss with onset at 180 °C and centred at 250 °C, consistent with the loss of acetamide (calculated 21%). A small plateau is observed in the TGA trace from ca. 250–280 °C; however, on heating crystals of 1 to this temperature a loss of single crystallinity was observed, suggesting a thermally activated single crystalline phase is not accessible from this starting material.
Synthesis and X-ray crystal structure of 2
Considering the homology between H2L1 and H2L2 we expected an isostructural framework to that found in 1 to form, although the less bulky nature of H2L2 would potentially allow the formation of voids. Subjecting H2L2 to the exact reaction conditions used to generate 1 failed to yield any identifiable products, most likely due to the low stability of 5-unsubstituted pyrazoles under harsh solvothermal conditions. Instead, H2L2 was combined with Cu(NO3)2·2.5H2O in a variety of solvents under more mild conditions. It was found that carrying out the reaction in N-methylpyrrolidinone (NMP) produced a blue micro-crystalline solid on standing. Furthermore, addition of water to the mixture caused the crystals to redissolve. As such, the reaction was carried out in a 9:1 mixture of NMP and water, and 2 slowly crystallised by allowing the solution to stand in a CaCl2 desiccator for several weeks. Smaller crystals could also be obtained by using a 20:1 NMP–H2O mixture and allowing the mixture to stand in a sealed vial. The blue crystals obtained were subjected to single crystal X-ray diffraction, and the data obtained were solved and refined in the tetragonal space group I41/a, Table 1. Notably, the crystal structure was solved in the same space group as 1, although with a considerably larger unit cell with a volume of 5981.2(5) Å3 (cf. 4531.2(8) Å3 for 1).
The structure model revealed an equivalent coordination mode for the L2 ligand as was the case for L1, in which each heteroatom coordinates to the Cu(II) ion, firstly with the formation of a [Cu2(L2)2] dimer, which is then linked into a three-dimensional network by bridging through the second carboxylate oxygen atom. The structures of the dimer units in 1 and 2 are superimposable (Fig. S1†), however, the geometry of coordination to the linking carboxylate oxygen atom shows a slight deviation. While the Cu(II) centre in 1 showed distortion towards trigonal bipyramidal geometry (τ5 = 0.43), the Cu(II) centre geometry in 2 is essentially square pyramidal with τ5 = 0.09. This leads to a greater distance between the nodes in 2 (7.8606(2) Å vs. 7.1660(4) Å for 1), and gives rise to the larger unit cell volume. The coordination sphere of the Cu(II) ion is completed by an NMP molecule in the axial position, with Cu(1)–O(10) distance 2.337(4) Å, significantly longer than the equivalent Cu(1)–O(14) bond in 1 of 2.268(3) Å, as would be expected for a truly square planar coordination geometry, and consistent with the weaker nature of NMP as an oxygen donor ligand when compared to acetamide. A small amount of crystallographic disorder was detectable on the NMP molecule, visualised as a two fold rotation about the coordinating oxygen atom; however, as the minor contributor displayed less than 20% occupancy, and was further disordered by a slight plane-tilting motion, only the major contributor was modelled. The structure of 2 is shown in Fig. 4.
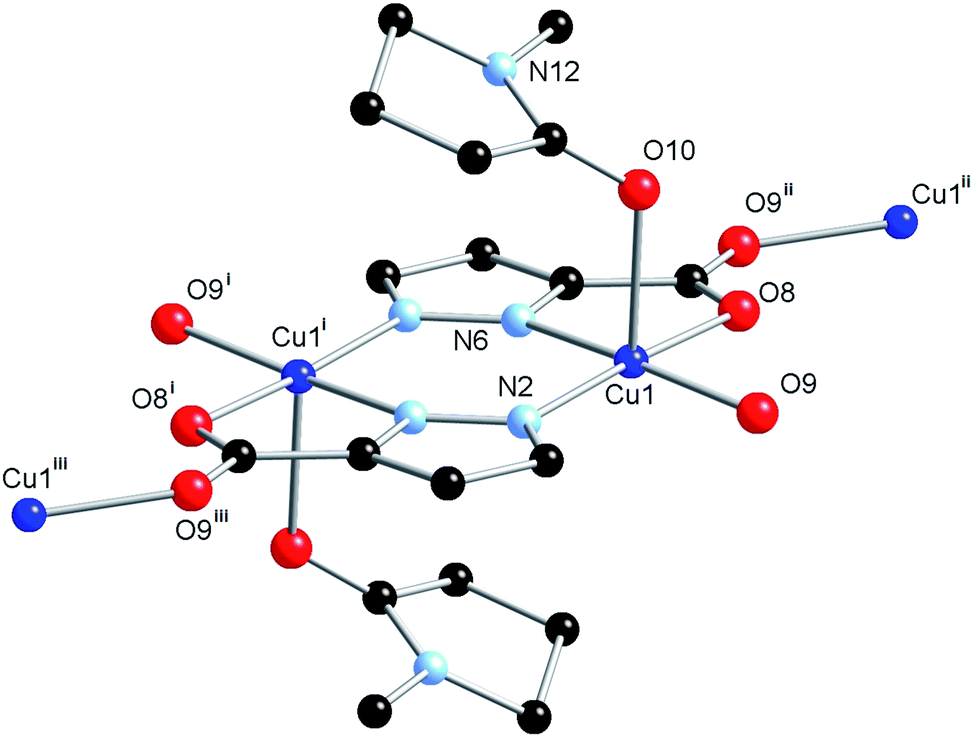 | ||
| Fig. 4 Structure of compound 2 with unique heteroatom labelling scheme. Hydrogen atoms omitted for clarity. Symmetry operations used to generate equivalent atoms: (i) 1/2 − X, 3/2 − Y, 1/2 − Z; (ii) −3/4 + Y, 3/4 − X, 3/4 − Z; (iii) 5/4 − Y, 3/4 + X, −1/4 + Z. | ||
As would be expected due to the structural similarities with complex 1, the extended network of 2 can be described by the lvt topology by making the same assignment of nodes to the centroid of the pyrazole-bridged copper dimer. Interestingly, while the topological descriptions are identical, the physical structures of 1 and 2 are substantially different. While 1 comprised a densely packed network with no appreciable void space, 2 defines a more open structure, where channels are observed running parallel to the a and equivalent b unit cell axes, with interatomic dimensions ca. 9 Å, (Fig. 5) while no void space is observed directly parallel to the unique c axis. When the structures of 1 and 2 are compared, this discrepancy becomes intuitive as when the additional carbon atoms comprising the indazole ring are considered separately, a channel structure akin to 2 but occupied by the indazole backbone is observed, Fig. 6.
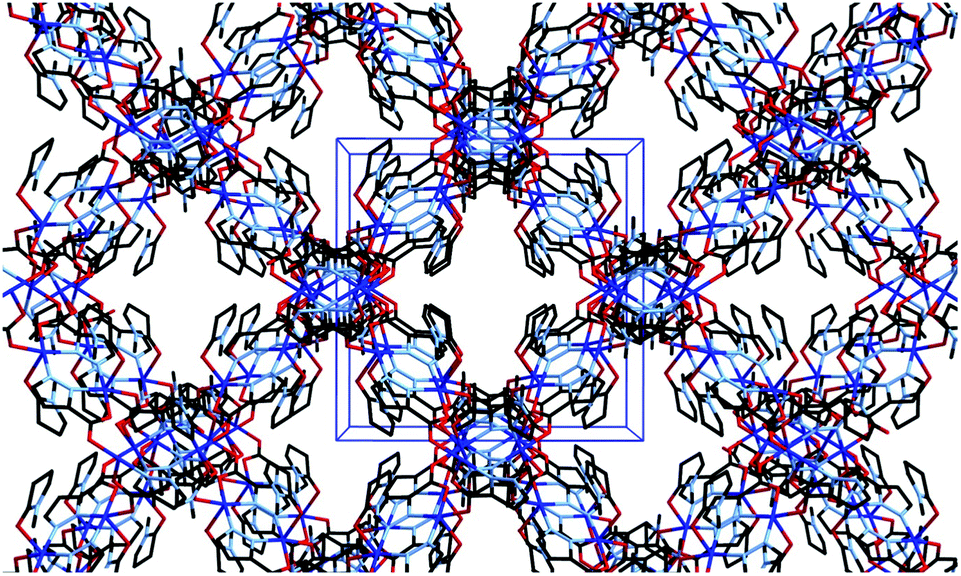 | ||
| Fig. 5 Channel structure of 2 viewed parallel to the a axis, showing unit cell dimensions in blue. Hydrogen atoms and channel solvate omitted for clarity. | ||
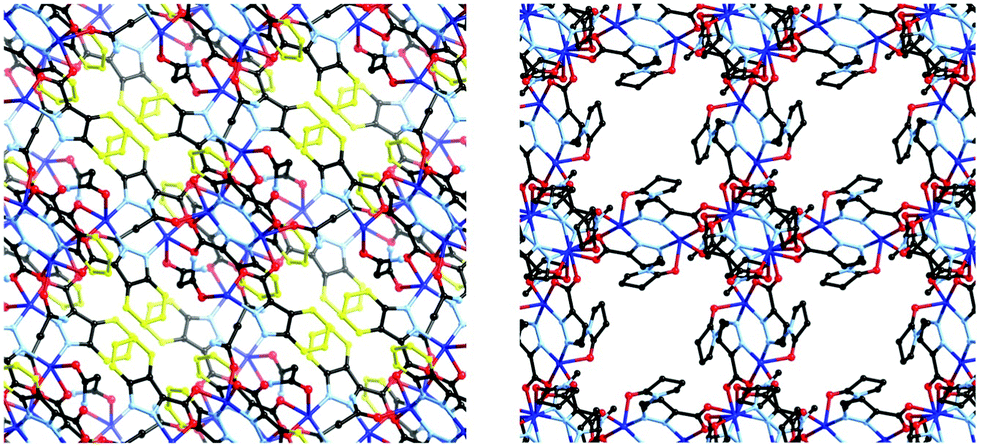 | ||
| Fig. 6 Comparison of 1 (left) and 2 (right), viewed along equivalent vectors. The additional atoms comprising the phenyl ring of the indazole fragment of L1 are highlighted in yellow. | ||
The diffuse electron density within the channels in 2 was unable to be satisfactorily modelled, and as such the SQUEEZE19 routine was applied to the crystallographic data, which suggested a void content of 1230 electrons per unit cell, equivalent to 153 electrons per [Cu2L2] unit, occupying 36% of the unit cell volume. These data are consistent with a void occupancy of approximately 3 non-coordinating molecules of NMP per dimer, suggesting a total volatile mass percentage of 59%, including the coordinating solvent molecules. Thermogravimetric analysis is in approximate agreement with this value, showing a total of 52% mass loss in two overlapping steps by 150 °C, with onset at room temperature, followed by a brief plateau, a further 6% mass loss centred at 210 °C, and a rapid decomposition centred at 310 °C. Elemental analysis confirmed the partial loss of lattice solvent on prolonged standing in air, suggesting a molecular formula for a dry sample of [Cu(L2)(NMP)]·0.5(NMP)·0.5(H2O). Although the narrow plateau region in the TGA trace of the as-synthesised compound discouraged attempts to directly desolvate compound 2, the material was observed to retain single crystallinity on soaking in diethyl ether, chloroform or acetonitrile; however, the crystals disintegrated instantly on removal from any of these more volatile solvents, preventing the collection of a guest-exchanged crystal structure. Furthermore, attempts to measure gas sorption on activated samples of 2 failed to observe any significant uptake, most likely due to framework collapse brought on during the desolvation activation procedure. The material was observed to lose crystallinity on soaking in methanol, preventing supercritical CO2 activation due to the lack of a suitable intermediate fluid.
Conclusions
We have demonstrated the reproducibility of formation of the lvt network in the case of 3-carboxy-1,2-diazoles in the presence of Cu(II). This retention of coordination functionality on altering the nature of the ligand system has allowed exploration into the effect of the steric bulk present on the indazole framework in comparison to that of the smaller pyrazole ring system, whilst also demonstrating the amenability of two different synthetic methods, including the in situ formation of the acetamide co-ligand.Coordination polymers built upon indazole ligands are almost unknown within the literature; 1 is an especially rare additional example, and the synthesis and structural characterisation of 2 represents only the second reported example of a coordination polymer featuring the 1H-pyrazole-3-carboxylic acid ligand. This is somewhat surprising considering the wealth of examples of coordination polymers incorporating the 1H-pyrazole-3,5-dicarboxylic acid manifold and is most probably due to the lower stability associated with 5-unsubstituted pyrazoles. Work in our laboratory is currently continuing to employ the indazole framework in the preparation of coordination polymer networks, and results to this end will be reported in due course.
Acknowledgements
The authors gratefully acknowledge the University of Canterbury (Scholarship to CSH) and the Royal Society of New Zealand Marsden Fund for financial support. The authors thank Assoc. Prof. Brendan Abrahams and Dr Keith White (University of Melbourne) for carrying out gas sorption tests on compounds 2, and Dr Anthea Lees (UC) for helpful discussions.Notes and references
- Metal–Organic Frameworks: Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH, Weinheim, 2011 RSC; S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC Publishing, Cambridge, 2009 RSC; L. R. MacGillivray, Metal–Organic Frameworks Design and Application, John Wiley and Sons, Hoboken, 2010 RSC; C. J. Kepert, in Porous Materials, ed. D. W. Bruce, D. O'Hare and R. I. Walton, John Wiley and Sons, Chichester, 2010 RSC; D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC; A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed; M. Schroder, Functional Metalorganic Frameworks: Gas Storage, Separation and Catalysis, Top. Curr. Chem., 2010, 293, 1–262 Search PubMed.
- P. Usov, T. Keene and D. D'Alessandro, Aust. J. Chem., 2013, 66, 429–435 CrossRef CAS; D. H. Hong and M. P. Suh, Chem. Commun., 2012, 48, 9168–9170 RSC; E. J. Mensforth, M. R. Hill and S. R. Batten, Inorg. Chim. Acta, 2013, 403, 9–24 CrossRef PubMed; A. D. Burrows, D. J. Kelly, M. I. H. Mohideen, M. F. Mahon, V. M. Pop and C. Richardson, CrystEngComm, 2011, 13, 1676–1682 RSC.
- L. Bellarosa, J. M. Castillo, T. Vlugt, S. Calero and N. Lopez, Chem. - Eur. J., 2012, 18, 12260–12266 CrossRef CAS PubMed.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC; J.-P. Zhang and S. Kitagawa, J. Am. Chem. Soc., 2008, 130, 907–917 CrossRef CAS PubMed; C. Pettinari, A. Tâbâcaru, I. Boldog, K. V. Domasevitch, S. Galli and N. Masciocchi, Inorg. Chem., 2012, 51, 5235–5245 CrossRef PubMed; S.-Q. Bai, D. J. Young and T. S. A. Hor, Chem. –Asian J., 2011, 6, 292–304 CrossRef PubMed; W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef PubMed.
- C. S. Hawes, C. M. Fitchett, S. R. Batten and P. E. Kruger, Inorg. Chim. Acta, 2012, 389, 112–117 CrossRef CAS PubMed; N. R. Kelly, S. Goetz, S. R. Batten and P. E. Kruger, CrystEngComm, 2008, 10, 1018–1026 RSC; N. R. Kelly, S. Goetz, C. S. Hawes and P. E. Kruger, Inorg. Chim. Acta, 2013, 403, 102–109 CrossRef PubMed; P. E. Kruger, Chimia, 2013, 67, 403–410 CrossRef PubMed.
- C. S. Hawes, R. Babarao, M. R. Hill, K. F. White, B. F. Abrahams and P. E. Kruger, Chem. Commun., 2012, 48, 11558–11560 RSC.
- R. Jothibasyu and H. V. Huynh, Chem. Commun., 2010, 46, 2986–2988 RSC; R. Pettinari, C. Pettinari, F. Marchetti, R. Gobetto, C. Nervi, M. R. Chierotti, E. J. Chan, B. W. Skelton and A. H. White, Inorg. Chem., 2010, 49, 11205–11215 CrossRef CAS PubMed; C. Janiak, S. Temizdemir, S. Dechert, W. Deck, F. Girgsdies, J. Heinze, M. J. Kolm, T. G. Scharmann and O. M. Zipffel, Eur. J. Inorg. Chem., 2000, 1229–1241 Search PubMed; Z. G. Gu, Y. F. Xu, L. C. Kang, Y. Z. Li, J. L. Zuo and X. Z. You, Inorg. Chem., 2009, 48, 5073–5080 CrossRef PubMed; J. C. Cuevas, J. Demendoza, P. Prados, F. Hernandezcano and C. Focesfoces, J. Chem. Soc., Chem. Commun., 1986, 1641–1642 RSC; R. Pritchard, C. A. Kilner and M. A. Halcrow, Tetrahedron Lett., 2009, 50, 2484–2486 CrossRef PubMed.
- B. Machura, A. Swietlicka, M. Wolff and R. Kruszynski, Polyhedron, 2010, 29, 2061–2069 CrossRef CAS PubMed; B. Machura, M. Wolff, D. Tabak, J. A. Schachner and N. C. Mosch-Zanetti, Eur. J. Inorg. Chem., 2012, 3764–3773 CrossRef PubMed.
- S.-Y. Zhang, Y. Li and W. Li, Inorg. Chim. Acta, 2009, 362, 2247–2252 CrossRef CAS PubMed; T. Hajra, J. K. Bera and V. Chandrasekhar, Aust. J. Chem., 2011, 64, 561–566 Search PubMed.
- V. Chandresekhar, R. Thirumoorthi, R. K. Metre and B. Mahanti, J. Organomet. Chem., 2011, 696, 600–606 CrossRef PubMed.
- P. King, R. Clérac, C. E. Anson, C. Coulon and A. K. Powell, Inorg. Chem., 2003, 42, 3492–3500 CrossRef CAS PubMed; J. H. Yang, S. L. Zheng, X. L. Yu and X. M. Chen, Cryst. Growth Des., 2004, 4, 831–836 Search PubMed; L. Pan, X. Y. Huang, J. Li, Y. G. Wu and N. W. Zheng, Angew. Chem., Int. Ed., 2000, 39, 527–530 CrossRef; L. Pan, N. Ching, X. Y. Huang and J. Li, Chem. –Eur. J., 2001, 7, 4431–4437 CrossRef; C. W. Hahn, P. G. Rasmussen and J. C. Bayon, Inorg. Chem., 1992, 31, 1963–1965 CrossRef; X.-H. Zhou, Y.-H. Peng, X.-D. Du, C.-F. Wang, J.-L. Zuo and X.-Z. You, Cryst. Growth Des., 2009, 9, 1028–1035 Search PubMed; L. Pan, T. Frydel, M. B. Sander, X. Huang and J. Li, Inorg. Chem., 2001, 40, 1271–1283 CrossRef PubMed.
- H. S. Lowrie, J. Med. Chem., 1966, 9, 664–669 CrossRef CAS; R. Stolle and W. Becker, Chem. Ber., 1924, 57, 1123–1124 CrossRef PubMed.
- N. Fatin-Rouge, E. Toth, D. Perret, R. H. Backer, A. E. Merbach and J.-C. G. Bünzli, J. Am. Chem. Soc., 2000, 122, 10810–10820 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement, University of Gottingen, 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildrea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Vanrijn and G. C. Verschor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- C. S. Hawes and P. E. Kruger, Aust. J. Chem., 2013, 66, 401–408 CrossRef CAS.
- P. Vandersluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables of bond lengths, angles and hydrogen bonding parameters for 1 and 2; thermogravimetric analysis plots for 1 and 2, overlay diagram for repeat unit of 1 and 2. CCDC 983017 and 983018. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra02147a |
| ‡ Current address: School of Chemistry, Monash University, Clayton, Victoria 3800, Australia. |
| This journal is © The Royal Society of Chemistry 2014 |