BODIPY dyes functionalized with 2-(2-dimethylaminophenyl)ethanol moieties as selective OFF–ON fluorescent chemodosimeters for the nerve agent mimics DCNP and DFP†
Raúl Gotora,
Pablo Gaviñaa,
Luis E. Ochandob,
Katherine Chulvia,
Alejandro Lorentea,
Ramón Martínez-Máñezc and
Ana M. Costero*a
aCentro de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universidad de Valencia, Doctor Moliner 50 46100 Burjassot, Valencia, Spain. E-mail: Ana.Costero@uv.es; Fax: +34 963543831
bCentro de Reconocimiento Molecular y Desarrollo Tecnológico, Unidad Mixta Universidad de Valencia – Universidad Politécnica de Valencia, Departamento de Geología, Facultad de Ciencias Biológicas, Universidad de Valencia, Doctor Moliner 50, 46100 Burjassot, Valencia, Spain
cCentro de Reconocimiento Molecular y Desarrollo Tecnológico, Unidad Mixta Universidad Politécnica de Valencia – Universidad de Valencia Departamento de Química, Universidad Politécnica de Valencia Camino de Vera s/n, 46022, Valencia, Spain
First published on 12th March 2014
Abstract
Two OFF–ON fluorescent chemodosimeters based on a BODIPY core for the detection of nerve agent mimics have been synthesized. Their reactivity towards diethylcyanophosphonate (DCNP) and diisopropylfluorophosphate (DFP) has been tested in organic and aqueous phase. These chemodosimeters selectively detect the nerve agent mimics with good LODs. The chemodosimeters hold their sensing properties on solid supports, allowing the preparation of a hand held sensing kit. The sensing in solid–liquid phase has been demonstrated. The X-ray structure of compound 2 has been resolved.
Introduction
Sarin, Soman and Tabun are by far the most known chemical warfare agents among the nerve gases. Their extreme toxicity, together with their low cost and easy production makes these compounds a big concern for governments and civilians, especially in areas involved in war related conflicts. Chemically, nerve agents are organophosphorous derivatives with a good leaving group (see Scheme 1). These compounds irreversibly bind to a free serine residue of the acetylcholinesterase, inhibiting its enzymatic task.1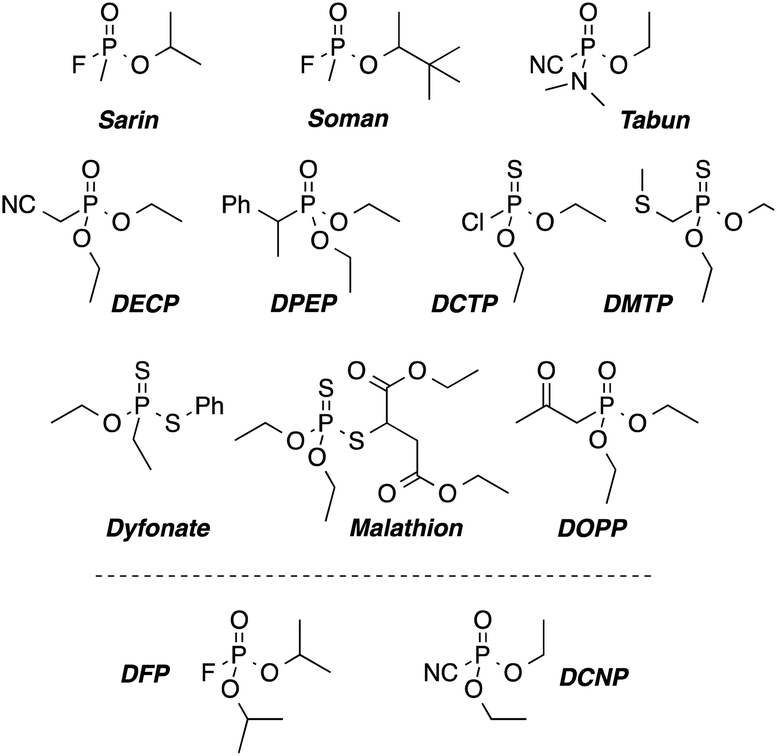 | ||
| Scheme 1 Chemical structures of nerve-agents Sarin, Soman, and Tabun, their simulants (DFP and DCNP) and some pesticides as potential interfering agents. | ||
The resulting over-accumulation of acetylcholine in the synaptic junctions of the nerves has immediate effects such as muscle relaxation hindering, “hyperventilation” and death in the worst cases.
Monitoring these agents has been achieved by means of different technologies, such as the use of ion mobility spectroscopy,2 biosensors,3 electrochemical methods,4 microcantilevers,5 photonic crystals,6 optical-fiber arrays,7 etc. but most of them show some limitations, such as low portability, high costs or the need of qualified personal to operate the devices.
In the last years, the use of colorimetric and/or fluorimetric chemosensors has gained importance. Colorimetric and/or fluorimetric chemosensors have proved to be a cost-effective method capable of performing quantitative analysis with widespread technologies, or even semiquantitative analysis with the “naked-eye”. Among the existing dosimeters for the nerve agent simulants, the transduction mechanism usually involves charge transfer (ICT) or photoinduced electron transfer (PET) processes, whereas the interaction with the analytes is performed by quaternization of nitrogen atoms,8 phosphorylation of hydroximate groups9 or displacement assays.10
Our research group has recently explored the use of 2-(2-(dimethylamino)phenyl)ethanol moieties11 in the selective detection of nervous gases. The detection mechanism starts with the phosphorylation of the hydroxyl group by the organophosphorous derivatives forming a phosphoester, which in a second step is displaced by the attack of the nitrogen atom to give a five membered ring quaternary ammonium salt.
On the other hand, borondipyrromethene (BODIPY) fluorophores have gained a huge attention due to their remarkable optic properties, such strong extinction coefficients, high quantum yields of fluorescence and good photostability.12 Herein we report the synthesis of two new fluorescent probes (1 and 2) based on 2-(2-dimethylaminophenyl)ethanol receptors connected to BODIPY fluorophores, and their use in the selective detection of the nerve agent simulants DCNP and DFP.
Results and discussion
The synthesized probes (1 and 2) are depicted in Scheme 2. It was expected that these chemodosimeters in polar solvents would have no fluorescence due to ICT processes involving the nonbonding electrons of the nitrogen atom.13,14 In the presence of nerve agents, the nitrogen atom should form the corresponding ammonium salt through a displacement reaction and thus an increase of the fluorescence was expected to occur.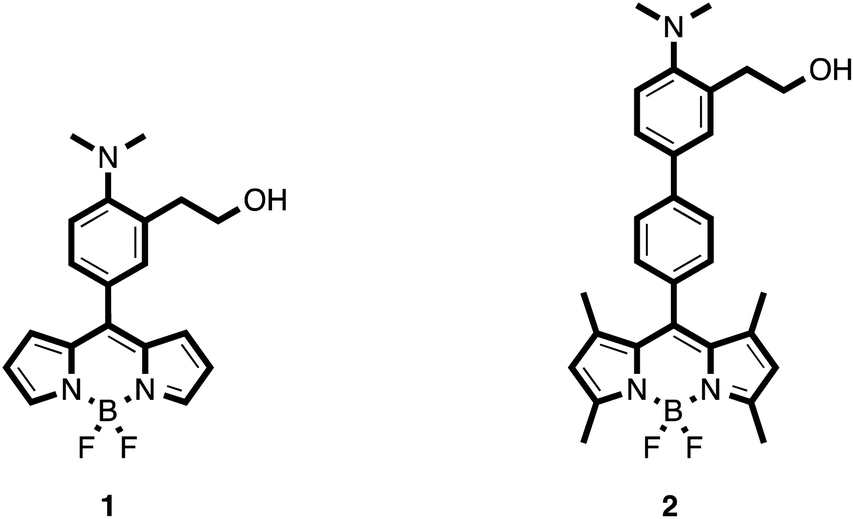 | ||
| Scheme 2 Chemical structures of the synthesized probes. | ||
Synthesis of the probes
BODIPY-based probe 1 was synthesized in 63% yield by means of a Liebeskind–Srögl coupling between the boronic acid derivative 3 and thiomethylbodipy 4 in the presence of Cu(I)-2-thienylcarboxylate (CuTC), trifurylphosphine (TFP) and a catalytic amount of Pd2(dba)314 (Scheme 3).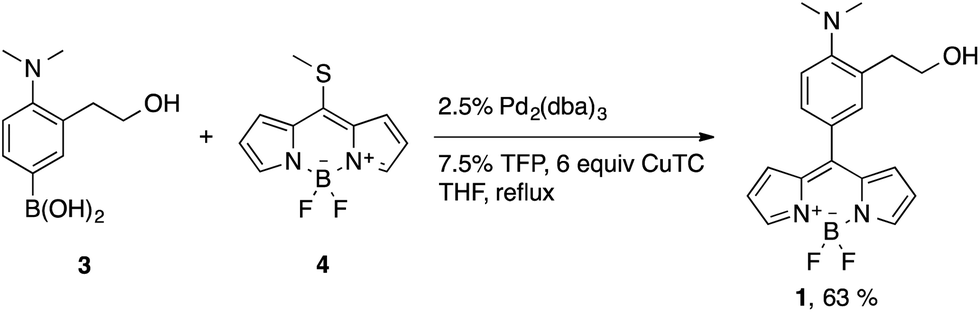 | ||
| Scheme 3 Coupling reaction to form probe 1. | ||
The synthesis of the boronic acid precursor 3 started with the reductive hydrogenation of the 2-(2-nitrophenyl)ethanol in the presence of formaldehyde to yield 2-(2-dimethylaminophenyl)ethanol 5, followed by an aromatic bromination with NBS catalyzed with ammonium acetate to form 6. Lithiation of this compound with two equivalents of BuLi followed by treatment with triethoxyborane and acidic water work-up gave rise to the building block 3 (Scheme 4).
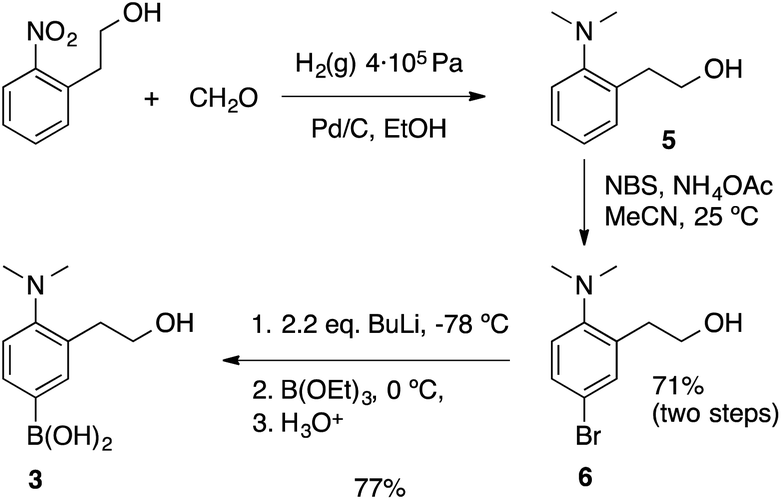 | ||
| Scheme 4 Synthetic procedures to form the boronic acid of sensing unit 3. | ||
On the other hand, the thiomethyl BODIPY derivative 4 was obtained by the condensation of pyrrole with thiophosgene, followed by methylation with MeI and reaction with BF3·Et2O following procedures described in the literature.15a The tetramethylated thiomethyl BODIPY analogous was also obtained by using 2,4-dimethylpyrrole instead of pyrrole. Unfortunately, the cross-coupling between 3 and this derivative was not successful, probably due to steric hindrance.
Probe 2 was synthesized by means of a Suzuki cross-coupling reaction between the boronic acid derivative 3 and the meso-(p-bromophenyl) BODIPY 7 in 86% yield (Scheme 5).
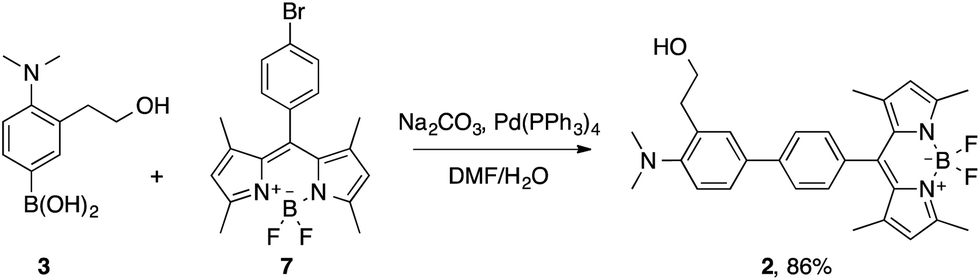 | ||
| Scheme 5 Synthesis of probe 2. | ||
Compound 7 was prepared in a 19% yield by the condensation of 2,4-dimethylpyrrole with p-bromobenzaldehyde, followed by aromatization with DDQ and reaction with BF3·Et2O.15b
X-ray structure of compound 2
Single dark red crystals of compound 2 were obtained by slow evaporation of acetonitrile solutions. X-ray diffraction analysis of several batches revealed a monoclinic P21/c crystal system, with two molecules in the asymmetric unit.In general, the bond lengths and angles within the BODIPY core exhibit the same geometric parameters as those examples reported in the literature.16 Thus, the boron atom has the typical bond lengths (B–N 1.548 Å and B–F 1.394 Å) and the N1–B1–N2 and F1–B1–F2 angles indicate a tetrahedral BF2N2 configuration.17 Moreover, the dihedral angle between the pyrrole rings is 2.16° indicating a high level of planarity of the BODIPY core. By contrast, the phenyl group at the meso position is almost perpendicular to the indacene plane with a dihedral angle of 81.53°. The two aromatic rings in the biphenyl moiety are also not coplanar (dihedral angle of 22.16°) following the trend observed in other related compounds14 (Fig. 1). To our surprise, the nitrogen atom of the dimethylamino group shows a remarkable pyramidalization with average C19–N3–C19′ bond angles of 112.8°. The Car–N3 bond length is 1.429 Å indicating that there is no conjugation between the nitrogen lone pair and the π-system of the aromatic ring. Besides, the methyl groups of the dimethylaniline moiety are facing away from the ortho alkyl substituent, observation that has been previously spotted in other o-substituted anilines.18
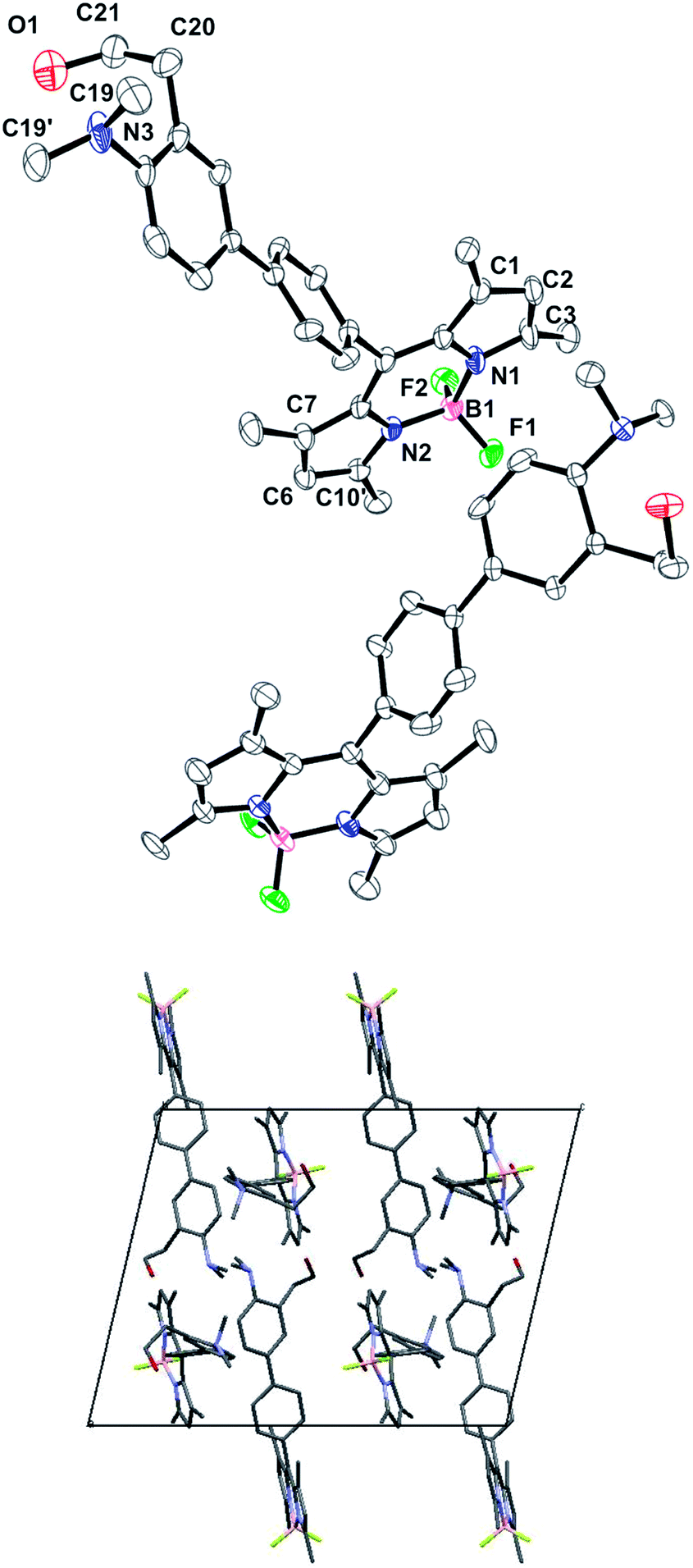 | ||
| Fig. 1 (Top) Asymmetric unit of compound 2. Thermal ellipsoids indicate the atom positions at probability levels of 35%. (Bottom) Molecular packing in the unit cell of the crystal structure along the b axis. Hydrogen atoms have been omitted for clarity. | ||
The 2-ethanol moiety shows a high grade of disorder in one of the molecules of the asymmetric unit, with three different positions for the oxygen atom, one of them at the 50% of occupation and the other two at 25% each.
Spectroscopic properties
The spectroscopic properties of compounds 1 and 2 were evaluated in acetonitrile solution (10−6 M). Compound 1 shows an absorption band at 495 nm (ε = 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1) whereas compound 2, in addition to the main absorption band at 498 nm (ε = 91400 M−1 cm−1), shows two small bands at 240 and 290 nm, which can be attributed to the biphenyl moiety attached at meso position. The fluorescence emission spectra of these compounds in acetonitrile show weak emission bands at λem = 515 nm (λexc = 470 nm) for 1 and λem = 507 nm (λexc = 480 nm) for 2. The fluorescence quantum yields (Φf) of the free species in acetonitrile are rather low (<0.02 for 2 and <0.001 for 1).
600 M−1 cm−1) whereas compound 2, in addition to the main absorption band at 498 nm (ε = 91400 M−1 cm−1), shows two small bands at 240 and 290 nm, which can be attributed to the biphenyl moiety attached at meso position. The fluorescence emission spectra of these compounds in acetonitrile show weak emission bands at λem = 515 nm (λexc = 470 nm) for 1 and λem = 507 nm (λexc = 480 nm) for 2. The fluorescence quantum yields (Φf) of the free species in acetonitrile are rather low (<0.02 for 2 and <0.001 for 1).
The strong fluorescence quenching observed for these compounds presumably resides in an intramolecular charge transfer (ICT) from the donor dimethylaniline moiety to the acceptor BODIPY core. In fact, it is known for similar compounds that the initial locally excited state (LE), after solvent reorganization, leads to a ICT state which is highly stabilized in polar solvents such acetonitrile.13 BODIPYs showing an ICT processes use to have low quantum yields due to the non-radiative deactivation nature of the ICT processes. Moreover, in the case of 1, the existence of a twisted intramolecular charge transfer (TICT) could be considered, as it has previously been observed in related compounds.14
Reducing the dimethylaniline electron-donating power (i.e.: protonation or quaternization), would result in the cancellation of the ICT processes with the concomitant increase in the lifetime of the LE state thereby restoring the fluorescence emission.13
In fact, we could observe that after protonation with HClO4, compound 2 restored its fluorescence to emission quantum yields of Φf = 0.446, while the quantum yield of 1 + H+ increased to Φf = 0.014 (see Table 1).
Detection experiments in solution
As a first approach, detection studies were carried in liquid phase using acetonitrile solutions (10−6 M) of compounds 1 and 2. Both probes showed small changes in the UV spectrum after addition of different aliquots of DCNP or DFP. Only compound 2 showed a new UV band at 260 nm along with a decrease of the bands at 240 and 290 nm. This change on the UV region can be ascribed to a change in the dihedral angle of the biphenyl moiety19,20 (see Fig. 2 for compound 2 and Fig. S-1† for compound 1). On the other hand, the addition of DCNP or DFP to acetonitrile solutions of 1 and 2 immediately gave rise to a large enhancement of the fluorescence emission.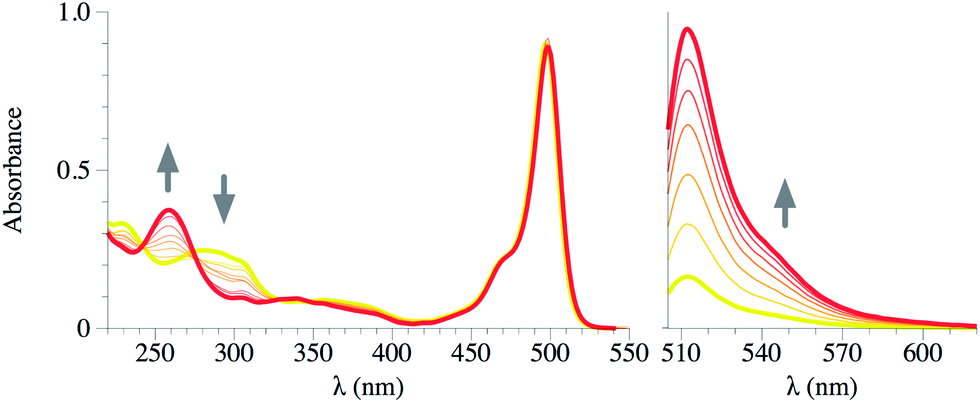 | ||
| Fig. 2 UV-vis (left) and emission (right, λexc = 470 nm) spectra of compound 2 in MeCN (1 × 10−6 M for emission, 1 × 10−5 M for UV-vis) titrated with DCNP (from 100 μM to 20 mM). Yellow: free ligand, red: ligand + DCNP 20 mM. | ||
The side products from the nerve agents decomposition by hydrolysis or other mechanisms (i.e.: formation of the pyrophosphate) include the formation of acids. Thus, in order to demonstrate that the previously described changes were due to the reaction with the simulant and not to a mere protonation process, buffered solutions of the probes were used in the detection experiments.
Thus, 10−6 M solutions of sensors 1 and 2 in water–MeCN 3:1, buffered with 0.1 M MES pH 5.5 were also studied.11 Addition of DCNP or DFP in the range of 100 μM to 20 mM also gave rise to a large enhancement of the corresponding emission bands.
The nature of the resulting product of the reaction of 1 or 2 with the simulants was studied by the synthesis of the expected product through a different pathway. Thus, cyclization reaction of 2 was carried out with one equivalent of tosyl chloride in acetonitrile in the presence of K2CO3 (Scheme 6).
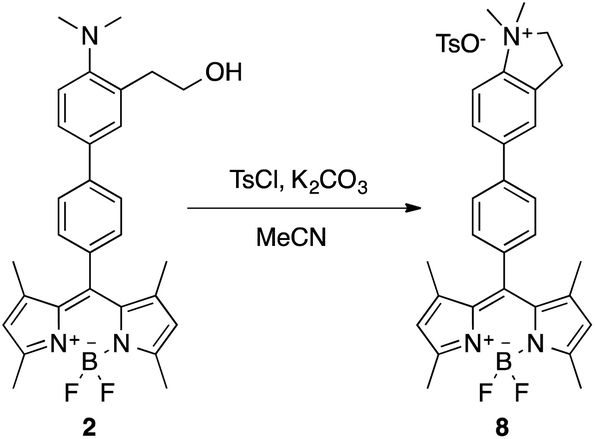 | ||
| Scheme 6 Cyclization reaction of 2 in the presence of tosyl chloride to form compound 8. | ||
The isolated product 8 was characterized by NMR and HRMS, and its UV-vis and emission spectrum (λabs = 499 nm, λem = 509 nm, Φf = 0.435, τ1 = 424 ps, τ2 = 2289 ps with relative amplitudes of a1 = 14% and a2 = 86%) perfectly matched with that resulting of 2 + DCNP (see ESI, Fig. S-17†).
Kinetic studies
The kinetic of the reaction between compounds 1 and 2 and DCNP and DFP in buffered water–MeCN (3:1 v/v) solutions (pH 5.5, MES 0.1 M) was studied. For each solution, a battery of experiments was carried out using different amounts of simulant. The simulant was always in an excess to consider pseudo first-order conditions.
The intensity of the emission band was continuously monitored (520 nm and 510 nm for 1 and 2 respectively). The obtained data allowed us to determine the observed rate constants at different simulant concentrations (kobs). Plotting kobs vs. the concentration of the simulant, let us evaluate the constant rates (k) and the corresponding half-life time (t1/2 = ln2/k) for the reaction of DCNP and DFP with 1 and 2. Table 2 summarizes the results obtained. Moreover, as an example, Fig. 3 shows the increment in the emission intensity at 510 nm for probe 2 in the presence of 6, 12, 18, 24 mM of DCNP in buffered water–acetonitrile 3:1 v/v mixtures (for compound 1 see ESI, Fig. S-18†).
:1 v/v mixture
| Probe | kobs | k (10−3 s−1) | t1/2 (s) | |||
|---|---|---|---|---|---|---|
| 1 | 0.116 | 0.173 | 0.235 | 0.286 | 9.53 | 72 |
| 2 | 0.122 | 0.156 | 0.198 | 0.221 | 5.68 | 122 |
| DCNP (mM) | 6 | 12 | 18 | 24 | ||
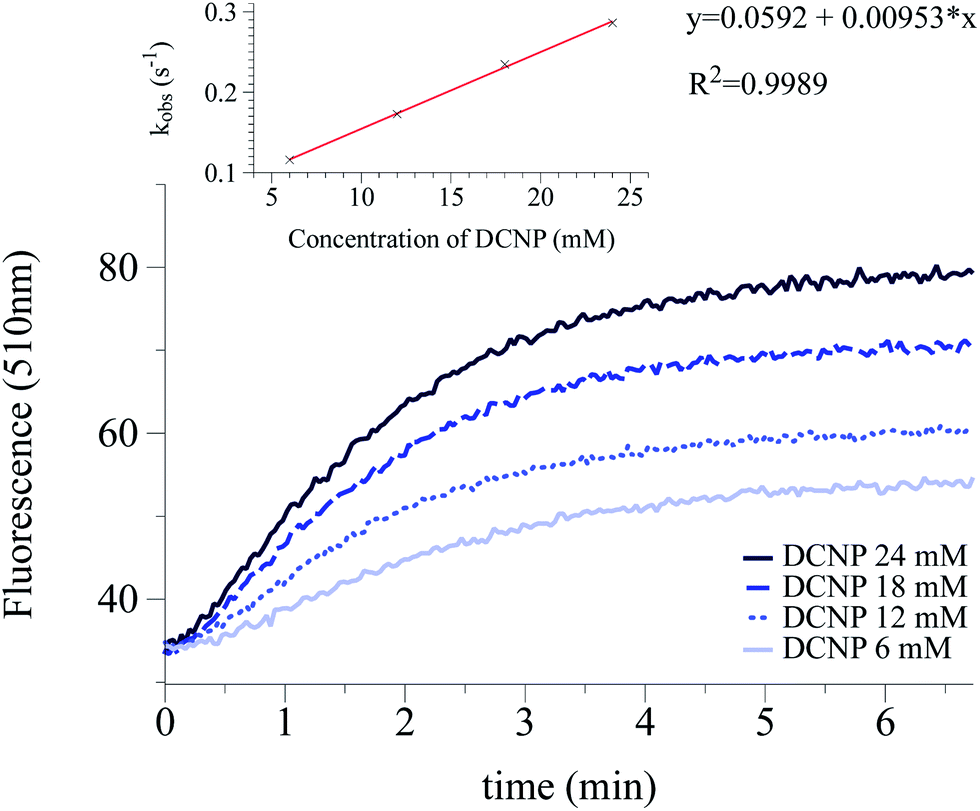 | ||
| Fig. 3 Kinetic profile of the emission intensity at 510 nm of probe 2 after the addition of 6, 12, 18 and 24 mM DCNP. Probe 2 is 10−6 M in pH = 5.5 buffered water–acetonitrile 3:1 v/v mixture. The inset shows the correlation between kobs and concentration of DCNP. | ||
Limits of detection and interferents
Detection limits for compounds 1 and 2 are summarized in Table 3. For this determination, increasing amounts of the simulants were added to solutions containing probes 1 or 2. The emission values for these determinations were measured two minutes after the addition of the simulant. The limit of detection was considered to be reached at concentrations of simulant that fulfilled the condition:| Is = Iblank + 3 × σblank |
:1 v/v) solutions (pH 5.5, MES 0.1 M)
| (ppm) | DCNP | DFP | DCNP (water–MeCN) |
|---|---|---|---|
| 1 | 5 | 3 | 54 |
| 2 | 7 | 8 | 4 |
Additionally, the reactivity of probes 1 and 2 towards other organophosphorous compounds (OP) and 4,4′-DDE, or 4,4′-DDD (see Scheme 1) was studied in buffered aqueous solutions (water–acetonitrile 3:1 v/v pH = 5.5 MES 0.1 M). Thus, 100 ppm of the interfering species were added to the probe solutions. In general, no increase of fluorescence emission was observed indicating that no reaction occurred in the presence of potential interferents that may be present in common environments (see Fig. 4).
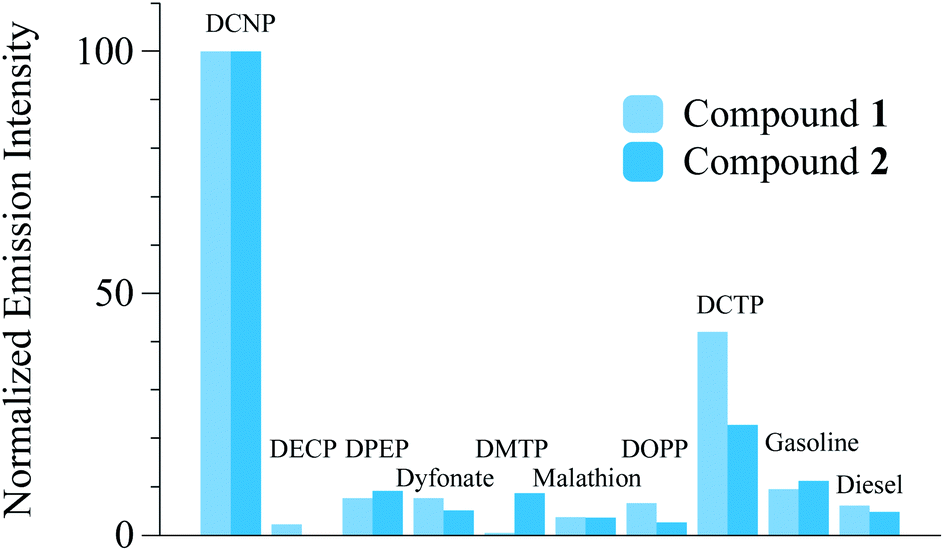 | ||
| Fig. 4 Relative (to DCNP) emission intensity of compounds 1 and 2 (measured at 520 and 510 nm respectively) with 1000 ppmv of DCNP, OP compounds, gasoline and diesel. | ||
Moreover, DCNP or DFP were added to the water–acetonitrile 3:1 v/v solutions of 1 and 2, containing the tested interferents and, as expected, the detection event still was observed, indicating that these species do not ruin the sensing capabilities of the probes.
Sensing experiments over supporting material
In order to assets the possibility of using these chemosensors as on-field sensing devices, we decided to fabricate solid polymer strips doped with the probes 1 and 2. The strips, which had no initial fluorescence, were dipped in distilled water contaminated with DCNP (from 2 to 2000 ppm). Clear enhancement of the fluorescence was observed by the naked eye from 20 to 2000 ppm for 2 (see Fig. 5).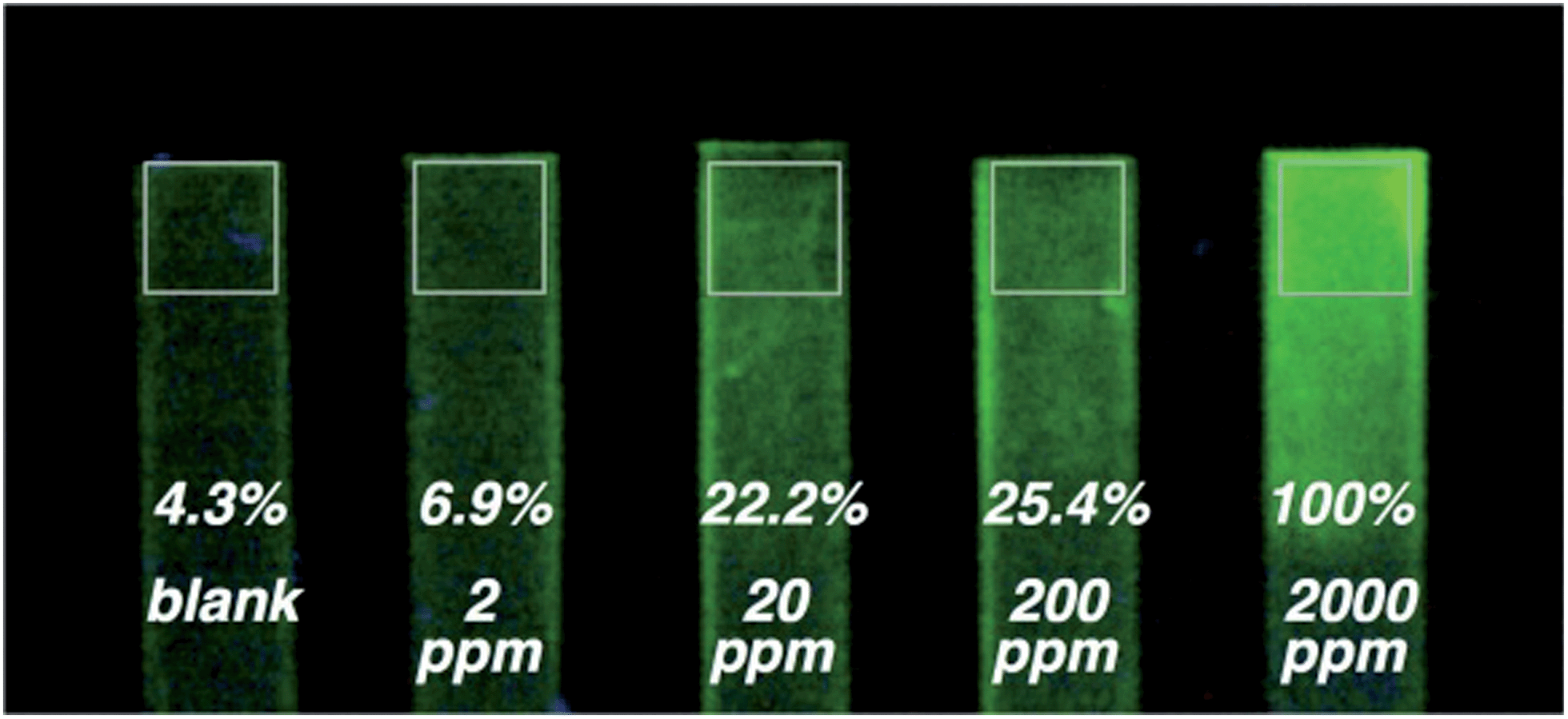 | ||
| Fig. 5 Hydrogel coated polyethylene strips. Hydrogel was previously doped with compound 2. The strips were dipped in distilled water contaminated with (from the left to the right): 0, 2, 20, 200, and 2000 ppmv of DCNP. Values indicate the relative (to the rightmost strip) luminance of the measured squared areas. Measurement was carried out by meaning the CIE luminance of all the pixels inside the squared area with a custom-made image processing software. (Fluorescent changes are more easily appreciated on the real samples rather than on the paper). | ||
Additionally, tap water was also used as solvent in the tests showing that this complicated matrix has no influence in the sensing properties of the system. After the test was performed, washing the strips with distilled water did not quench the fluorescence, indicating that the irreversible cyclization had occurred.
Gas phase sensing
As a test experiment, we hang the strips in the center of a round bottom flask, containing an air atmosphere. Then, this atmosphere was contaminated with 5 ppm of DCNP. In the period of 5 minutes, the strip was removed from the system, and a clear increase of the fluorescence emission could be observed under a hand held UV254nm lamp. With this simple procedure, we demonstrated the possibility of detection of nerve agent simulants in the gas phase. At the moment, this part of the investigation is still in progress.Experimental
General methods
All the synthetic manipulations were performed in a dry argon atmosphere using standard techniques. Compound 4 and 7 were prepared following the procedures described in the literature.15a,b Tetrahydrofuran was distilled over Na prior to use. The other materials were purchased and used as received. Silica gel 60 F254 (Merck) plates were used for TLC. 1H and 13C NMR spectra were recorded using a Bruker DRX-500 spectrometer (500 MHz for 1H and 126 MHz for 13C) and a Bruker Avance 400 MHz (400 MHz for 1H and 100 MHz for 13C) with the deuterated solvent as the lock and residual solvent as the internal reference. HRMS were recorded using a Shimadzu QP5050A. Absorption spectra were recorded with a Shimadzu UV-2101PC spectrophotometer. Fluorescence spectra were carried out in a Varian Cary Eclipse fluorimeter.Crystal structure data collection and refinement
Intensity data were collected at 120(2) K with an Oxford Diffraction S Ultra diffractometer equipped with a Mo co-mounted (λ = 0.71073 Å) enhance X-ray sapphire CCD detector. Structures were solved by direct methods and refined by full-matrix least-squares using SHELX-97.20,21 Molecular graphics of the structures (Fig. 1) were produced with Mercury and Ortep.22 Solution, refinement, geometrical calculations and graphics were performed with the WinGX package.†23Experimental procedures
:6 as eluent to yield 10.61 g (71%) of 6 as brownish oil. Rf = 0.77 (EtOAc–hexane 6:4). 1H NMR (500 MHz, CDCl3) δ 7.24 (dd, J = 8.5, 2.4 Hz, 1H), 7.21 (d, J = 2.3 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 3.75 (t, J = 5.7 Hz, 2H), 2.87 (t, J = 5.7 Hz, 2H), 2.59 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 151.36, 138.22, 133.64, 130.35, 121.77, 117.61, 77.00, 63.86, 44.81, 35.61. HR-MS: calcd for C10H14BrNO [M + H]+: 244.0337, found: 244.0341.:1 mixture of EtOAc–hexane was slowly added in order to precipitate the boronic acid 3. The compound was isolated by centrifugation (0.96 g, 77%). Rf = 0.59 (EtOAc–MeOH 9:1). 1H NMR (500 MHz, DMSO-d6) δ 7.81 (br. s, 2H), 7.62 (d, J = 1.2 Hz, 1H), 7.59 (dd, J = 8.0, 1.2 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 4.70 (s, 1H), 3.64 (t, J = 7.5 Hz, 2H), 2.82 (t, J = 7.5 Hz, 2H), 2.65 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ 136.69, 133.44, 132.29, 118.49, 62.20, 45.16, 34.72. HR-MS: calcd for C10H17BNO3 [M + H]+: 210.1296, found: 210.1301.Tetrakis(triphenylphosphine)palladium(0) (13.7 mg, 12 μmol) was added, and the mixture was degassed for 5 more minutes. Then, the mixture was heated to 100 °C and allowed to react for 1 h. After this time, TLC plate revealed quantitative consumption of starting materials. Solvents were then evaporated and the mixture was dissolved in dichloromethane. The organic solvent was washed twice with water, and the aqueous layer was extracted with dichloromethane. The combined organic layers were washed with NaCl (sat.) and dried with MgSO4. After solvent evaporation, the remaining mixture was purified by silica column chromatography using EtOAc–hexane 7:3 as eluent to yield 99 mg (86%) of an orange solid. Rf = 0.4 (EtOAc–hexane 4:6). 1H NMR (500 MHz, CD2Cl2) δ 7.78 (d, J = 8.2 Hz, 2H), 7.60 (dd, J = 8.2, 2.4 Hz, 1H), 7.56 (d, J = 2.3 Hz, 1H), 7.39 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.3 Hz, 1H), 6.08 (s, 2H), 3.91 (t, J = 5.8 Hz, 2H), 3.11 (t, J = 5.8 Hz, 2H), 2.78 (s, 6H), 2.57 (s, 6H), 1.52 (s, 6H). 13C NMR (125 MHz, CD2Cl2) δ 155.35, 152.50, 143.38, 141.88, 141.21, 136.42, 136.10, 133.46, 131.44, 129.52, 128.49, 127.31, 125.93, 121.12, 120.40, 64.05, 44.76, 36.06, 14.33. HR-MS: calcd for C29H33BN3OF2 [M + H]+: 488.2679, found: 488.2685.
Conclusions
Two OFF–ON BODIPY-based probes have been prepared and their utility in the detection of nerve agent simulant (DCNP and CFP) has been demonstrated. The dimethylamino groups present in compounds 1 and 2 inhibit the typical fluorescence of the BODIPY core probably due to an ICT mechanism. In the presence of the target species, the fluorescence of the system is restored through the quaternization of the dimethylamino nitrogen atom. The intensity of the fluorescence is stronger in compound 2 than in 1. Registered LOD were in the 50 ppm, and in the case of 1 with DFP, went down to 3 ppm. In addition, selectivity towards different potential interferents present in civilian or military settings has been demonstrated. The properties of the probes are still present in solid supported kits which are able to detect the studied simulant not only in solution but also in gas phase. The achieved LOD are even lower when these solid supports are used.Acknowledgements
We thank the Spanish Government and the European FEDER funds (project MAT2012-38429-C04-02 and 01). SCSIE and ICMOL (Universidad de Valencia) are gratefully acknowledged for all the equipment employed. R.G. is grateful to the Spanish Ministry of Education for its FPU grant. Dr Knut Rurack is gratefully acknowledged for instrumental support and fruitful discussion.Notes and references
- S. M. Somani, Chemical Warfare Agents, Academic Press, San Diego, 1992 Search PubMed.
- E. Steiner, S. J. Klopsch, W. A. English, B. H. Clowers and H. H. Hill, Anal. Chem., 2005, 77, 4792 CrossRef PubMed.
- (a) L. M. Eubanks, T. J. Dickerson and K. D. Janda, Chem. Soc. Rev., 2007, 36, 458 RSC; (b) J. J. Gooding, Anal. Chim. Acta, 2006, 559, 137 CrossRef CAS; (c) M. T. McBride, S. Gammon, M. Pitesky, T. W. O'Brien, T. Smith, J. Aldrich, R. G. Langlois, B. Colston and K. S. Venkateswaran, Anal. Chem., 2003, 75, 1924 CrossRef CAS PubMed; (d) A. J. Russell, J. A. Berberich, G. F. Drevon and R. R. Koepsel, Annu. Rev. Biomed. Eng., 2003, 5, 1 CrossRef CAS PubMed; (e) H. Wang, J. Wang, D. Choi, Z. Tang, H. Wu and Y. Lin, Biosens. Bioelectron., 2009, 24, 2377 CrossRef CAS PubMed; (f) A. Mulchandani, I. Kaneva and W. Chen, Anal. Chem., 1998, 70, 5042 CrossRef CAS PubMed; (g) S. Munchaldani, P. Mulchandani, I. Kaneva and W. Chen, Anal. Chem., 1998, 70, 4140 CrossRef.
- (a) M. A. K. Khan, Y.-T. Long, G. Schatte and H.-B. Kraatz, Anal. Chem., 2007, 79, 2877 CrossRef CAS PubMed; (b) O. V. Shulga and C. Palmer, Anal. Bioanal. Chem., 2006, 385, 1116 CrossRef CAS PubMed; (c) J.-C. Chen, J.-L. Shih, C.-H. Liu, M.-Y. Kuo and J.-M. Zen, Anal. Chem., 2006, 78, 3752 CrossRef CAS PubMed; (d) G. Liu and Y. Lin, Anal. Chem., 2006, 78, 835 CrossRef CAS PubMed; (e) K. A. Joshi, M. Prouza, M. Kum, J. Wang, J. Tang, R. Haddon, W. Chen and A. Mulchandani, Anal. Chem., 2006, 78, 331 CrossRef CAS PubMed; (f) G. Liu and Y. Lin, Anal. Chem., 2005, 77, 5894 CrossRef CAS PubMed; (g) J. Wang, L. Chen, S. Mulchandani, P. Mulchandani and W. Chen, Electroanalysis, 1999, 11, 866 CrossRef CAS.
- (a) G. Zuo, X. Li, P. Li, T. Yang, Y. Wang, Z. Chen and S. Feng, Anal. Chim. Acta, 2006, 580, 123 CrossRef CAS PubMed; (b) C. Karnati, H. Du, H.-F. Ji, X. Xu, Y. Lvov, A. Mulchandani, P. Mulchandani and W. Chen, Biosens. Bioelectron., 2007, 22, 2636 CrossRef CAS PubMed; (c) Q. Zhao, Q. Zhu, W. Y. Shih and W.-H. Shih, Sens. Actuators, B, 2006, 117, 74 CrossRef CAS.
- (a) W. He, Z. Liu, X. Du, Y. Jiang and D. Xiao, Talanta, 2008, 76, 698 CrossRef CAS PubMed; (b) J. P. Walker, K. W. Kimble and S. A. Asher, Anal. Bioanal. Chem., 2007, 389, 2115 CrossRef CAS PubMed; (c) J. P. Walker and S. A. Asher, Anal. Chem., 2005, 77, 1596 CrossRef CAS PubMed.
- M. J. Aernecke and D. R. Walt, Sens. Actuators, B, 2009, 142, 464 CrossRef CAS.
- (a) K. A. Van Houten, D. C. Heath and R. S. Pilato, J. Am. Chem. Soc., 1998, 120, 12359 CrossRef CAS; (b) S.-W. Zhang and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 340 CrossRef PubMed; (c) T. J. Dale and J. Rebek, Jr, J. Am. Chem. Soc., 2006, 128, 4500 CrossRef CAS PubMed; (d) A. M. Costero, S. Gil, M. Parra, P. M. E. Mancini, R. Marínez-Máñez, F. Sancenón and S. Royo, Chem. Commun., 2008, 6002 RSC.
- K. J. Wallace, J. Morey, V. M. Lynch and E. V. Anslyn, New J. Chem., 2005, 29, 1469 RSC.
- D. Knapton, M. Burnworth, S. J. Rowan and C. Weder, Angew. Chem., Int. Ed., 2006, 45, 5825 CrossRef CAS PubMed.
- A. M. Costero, M. Parra, S. Gil, R. Gotor, P. M. E. Mancini, R. Martínez-Máñez, F. Sancenón and S. Royo, Chem.–Asian J., 2010, 5, 1573 CrossRef CAS PubMed.
- (a) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed. Engl., 2008, 47, 1184 CrossRef CAS PubMed; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed; (c) R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496 RSC.
- (a) M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. A, 1998, 102, 10211 CrossRef CAS; (b) W. Qin, M. Baruah, M. Van der Auweraer, F. C. De Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371 CrossRef CAS PubMed.
- E. Lager, J. Liu, A. Aguilar-Aguilar, B. Y. Tang and E. Pena-Cabrera, J. Org. Chem., 2009, 74, 2053 CrossRef CAS PubMed.
- (a) E. Pena-Cabrera, A. Aguilar-Aguilar, M. Gonzalez-Dominguez, E. Lager, R. Zamudio-Vazquez, J. Godoy-Vargas and F. Villanueva-Garcia, Org. Lett., 2007, 9, 3985 CrossRef CAS PubMed; (b) A. Nierth, A. Y. Kobitski, G. U. Nienhaus and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 2646–2654 CrossRef CAS PubMed.
- (a) J.-S. Lu, S.-B. Ko, N. R. Walters and S. Wang, Org. Lett., 2012, 14, 5660 CrossRef CAS PubMed; (b) H. Lu, Q. Wang, L. Gai, Z. Li, Y. Deng, X. Xiao, G. Lai and Z. Shen, Chem.–Eur. J., 2012, 18, 7852 CrossRef CAS PubMed.
- Z. Shen, H. Röhr, K. Rurack, H. Uno, M. Spieles, B. Schulz, G. Reck and N. Ono, Chem.–Eur. J., 2004, 10, 4853 CrossRef CAS PubMed.
- (a) J.-Y. Shin, B. O. Patrick and D. Dolphin, Tetrahedron Lett., 2008, 49, 5515 CrossRef CAS; (b) I. G. Voigt-Martin, Z. X. Zhang, D. H. Yan, A. Yakimanski, R. Matschiner, P. Krämer, C. Grania, D. Schollmeyer, R. Wortmann and N. Detzer, Colloid Polym. Sci., 1997, 275, 18 CrossRef CAS.
- A. M. Costero, J. Sanchis, S. Gil, V. Sanz and J. A. G. Williams, J. Mat. Chem., 2005, 15, 2848 RSC.
- G. M. Sheldrick, SHELX97, Programs for Crystal Structure Analysis (Release 97–2), University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Scheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112 CrossRef PubMed.
- (a) C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS; (b) L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- C. A. Osorio-Martínez, A. Urías-Benavides, C. F. A. Gómez-Durán, J. Bañuelos, I. Esnal, I. L. Arbeloa and E. Peña-Cabrera, J. Org. Chem., 2012, 77, 5434 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 982038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra00710g |
| This journal is © The Royal Society of Chemistry 2014 |