
Anode-selective coating of titanium(iv) oxide (TiO2) using electrophoretic sulfone-containing click polyester
Abstract
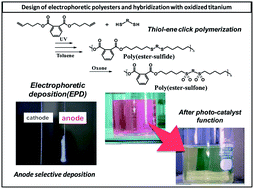
We synthesized, at room temperature, aliphatic (1–4) and aromatic (5–8) poly(ester-sulfide)s, via a thiol–ene click polymerization of bis(pentenyl)methylsuccinate (BPMSA) and bis(pentenyl)phthalate (BPPh), respectively, with several dithiols including 1,2-ethanedithiol (EtDt), 1,3-propanedithiol (PrDt), 1,4-butanedithiol (BuDt), and 1,5-pentanedithiol (PeDt) (Mn = 0.7–5.2 × 104, Mw/Mn = 1.6–2.6). Subsequent Oxone oxidation of poly(BPPh-alt-BuDt) (7), as an example, led to the corresponding poly(ester-sulfone) 7′ (Mn = 30 000, Mw/Mn = 1.8). We then prepared a composite of 7′ and TiO2 by using electrophoretic deposition (EPD). The TiO2/poly(ester-sulfone) 7′ composite was selectively deposited onto a stainless-steel anode. The electrode's morphology was confirmed by scanning electron microscopy. We characterized the relationship between the structure of the composite-coated electrode and the zeta potential of an N,N-dimethylformamide/alcohol suspension of poly(ester-sulfone) 7′. The measured values, −(33.27–18.3) mV, indicate that the composite had indeed been selectively deposited on the anode. Notably, from the suspension with the negative potential of −33.27 mV, the thickest composite film was obtained. Furthermore, photo-catalytic activity occurred on the surface of the composite when immersed in aqueous rhodamine-B for 1 month.
 Please wait while we load your content...
Please wait while we load your content...

