
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiohybrid materials comprising an artificial peroxidase and differently shaped gold nanoparticles†
Emilia
Renzi‡
a,
Alessandra
Esposito‡
 a,
Linda
Leone
a,
Miriam
Chávez
b,
Teresa
Pineda
b,
Angela
Lombardi
*a and
Flavia
Nastri
*a
a,
Linda
Leone
a,
Miriam
Chávez
b,
Teresa
Pineda
b,
Angela
Lombardi
*a and
Flavia
Nastri
*a
aDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario Monte S. Angelo, via Cintia, Naples, 80126, Italy. E-mail: alombard@unina.it; flavia.nastri@unina.it
bDepartment of Physical Chemistry and Applied Thermodynamics, Institute of Chemistry for Energy and Environment, University of Cordoba, Campus Rabanales, Ed. Marie Curie, Córdoba, E-14014, Spain
First published on 3rd June 2024
Abstract
The immobilization of biocatalysts on inorganic supports allows the development of bio-nanohybrid materials with defined functional properties. Gold nanomaterials (AuNMs) are the main players in this field, due to their fascinating shape-dependent properties that account for their versatility. Even though incredible progress has been made in the preparation of AuNMs, few studies have been carried out to analyze the impact of particle morphology on the behavior of immobilized biocatalysts. Herein, the artificial peroxidase Fe(III)-Mimochrome VI*a (FeMC6*a) was conjugated to two different anisotropic gold nanomaterials, nanorods (AuNRs) and triangular nanoprisms (AuNTs), to investigate how the properties of the nanosupport can affect the functional behavior of FeMC6*a. The conjugation of FeMC6*a to AuNMs was performed by a click-chemistry approach, using FeMC6*a modified with pegylated aza-dibenzocyclooctyne (FeMC6*a-PEG4@DBCO), which was allowed to react with azide-functionalized AuNRs and AuNTs, synthesized from citrate-capped AuNMs. To this end, a literature protocol for depleting CTAB from AuNRs was herein reported for the first time to prepare citrate-AuNTs. The overall results suggest that the nanomaterial shape influences the nanoconjugate functional properties. Besides giving new insights into the effect of the surfaces on the artificial peroxidase properties, these results open up the way for creating novel nanostructures with potential applications in the field of sensing devices.
Introduction
Gold-based nanomaterials (AuNMs) have been widely applied as components of electrochemical and optical sensors by virtue of their enticing physico-chemical properties.1–6 The potential of AuNMs can be further expanded by merging them with biocatalysts, as the properties of the biomolecular components may endow nanomaterials with specific functions. Biocatalysts and AuNMs can both benefit from this interplay. The immobilization of enzymes on AuNMs represents a middle ground between heterogeneous (immobilized enzymes) and homogeneous (soluble free enzymes) catalysis.7–9 This strategy may provide a microenvironment suitable for increasing enzyme tolerance to pH and temperature, as well as stability in organic solvents.10–13 Further, the high surface-area-to-volume ratio of AuNMs allows high enzyme loading, thus influencing the specific activity of the functionalized nanomaterials. Other advantages for the biocatalyst include: (i) multiple mechanisms for enzyme attachment by virtue of tailor-made surface chemistry, (ii) high radii of curvature, which can improve the distances between enzymes and limit detrimental protein–protein interactions, (iii) biocompatibility, and (iv) quasi-homogeneous catalysis.14,15 On the AuNM side, their surface chemistry and unique electronic, magnetic and optical properties can be tuned by the presence of biocatalysts. Indeed, the localized surface plasmon resonance (LSPR) phenomenon is strongly dependent on the dielectric environment created by the AuNM coating shell.16–20 Thus, changes in the LSPR band induced by the surface binding of biocatalysts can be useful in optical biosensing.3,5 Thanks to this behavior, AuNMs play a crucial role in lateral flow assays,21–23 in which they act as transducers or bioreceptor immobilization platforms, giving an easy optical readout in the detection of a variety of target analytes.24–26 Further, the immobilization of redox-active enzymes on gold-based materials is of great interest in the field of electrochemical biosensors, as AuNMs may act as mediators and facilitate electron exchanges between the electrode and the biomolecular component.27–30 Within this topic, the introduction of shape anisotropy at the nanoscale has emerged as a potent way to access new properties and functionalities, enabling the exploration of complex nanomaterials across a wide range of applications.31 Differently shaped AuNMs, like rods (AuNRs), triangular nanoprisms (AuNTs), and branched multipods like nanostars, nanowires or nanocubes, have gained interest beyond spherical gold-nanoparticles (AuNPs). Due to their distinctive optical and electronic properties, these anisotropic AuNMs hold great potential as supporting materials for optical devices, biosensors and nanoconjugate assemblies.16,18,32–35 In particular, the organization and patterning of inorganic nanoparticles into surface-deposited well-ordered nanostructures are of increasing interest in the development of biosensing devices based on redox-active enzymes.28–30,36,37 In this scenario, the combination of the unique features of anisotropic AuNMs with artificial metalloenzymes (ArMs) may further broaden the array of functional nanomaterials useful for the assembly of chemical, optical, and electronic devices. Diverse and powerful strategies have been developed for the design of ArMs tailored ad hoc, endowed with a reduced size, and able to perform natural and unnatural reactions.38–44 Heme-enzymes have been widely used as a target for ArM design because of their versatility, which lies in their ability to tune heme properties toward different functions.45–49 In this field, Mimochromes (MCs), consisting of a metalloporphyrin embedded within two synthetic peptides, feature interesting properties.50–54 Apart from their considerable catalytic performances in solution,53,55 MCs can also be successfully anchored on gold nanosurfaces, while retaining structural and catalytic properties.56,57 The latest scaffold of the MCs' series, Mimochrome VI*a (MC6*a), is extremely versatile, accessing customized catalytic activities upon insertion of different metal ions into the porphyrin core (Fig. 1).58–61 The insertion of an iron ion affords FeMC6*a,62 which is provided with high catalytic versatility and enhanced performance in a simple miniaturized scaffold (Mw 3.5 kDa and radius of gyration ≈1 nm). Experimental data proved the ability of FeMC6*a to outclass natural and artificial biocatalysts in catalyzing diverse oxidation reactions.63–65 The repertoire of applications of FeMC6*a has been further widened through its interaction with nanomaterials, with the aim of preparing functional nanoconjugates.66,67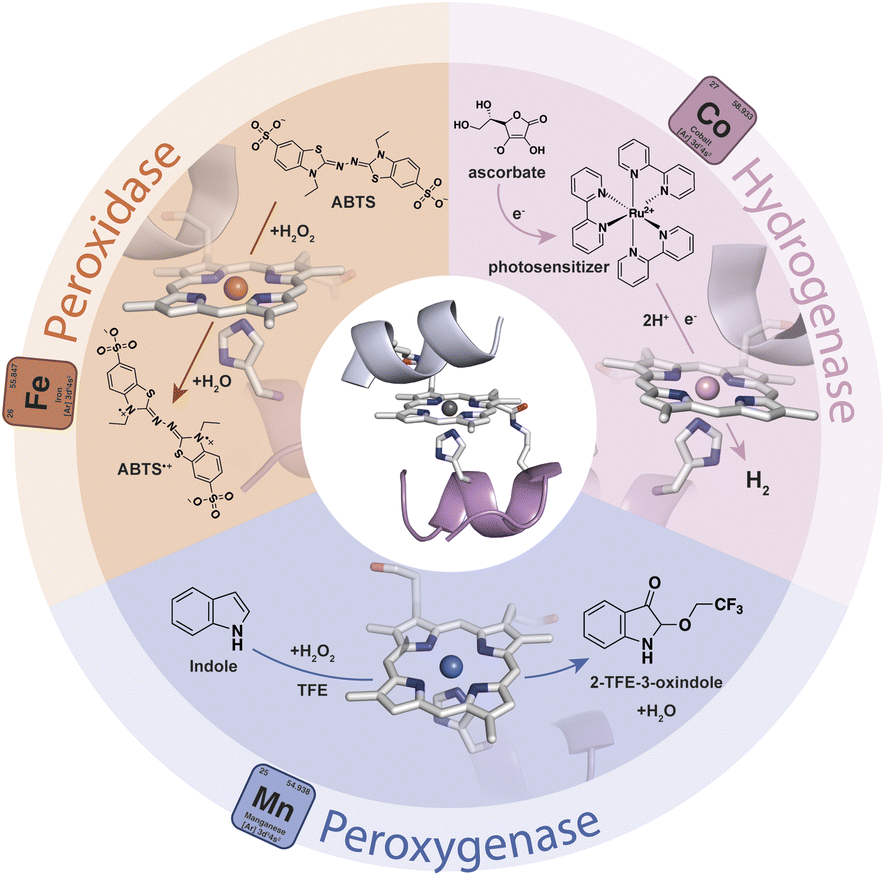 | ||
| Fig. 1 Schematic representation of the repertoire of the catalytic activity of MC6*a upon the insertion of different metal ions. | ||
In this study, we report the conjugation of FeMC6*a to differently shaped gold nanoparticles. Among the various gold nanomaterials available, we selected gold nanorods and triangular gold nanoprisms, as they can be easily produced using several well-developed and scalable methods.18,68–70 Further, as their surfaces can be manipulated and tuned with a variety of coating shells,18,35 they are optimal candidates for enzyme immobilization. This choice allows studying nanomaterials with distinct properties, specifically the sharp edges of nanoprisms and the smooth surfaces of nanorods. Our objective is to gain insights into how different morphologies influence the functional behavior of the miniaturized peroxidase. To this end, we first prepared AuNRs and AuNTs with exposed azide groups, to facilitate enzyme conjugation by click chemistry.71,72 Then, we analyzed the catalytic properties of the resulting nanoconjugates, as discussed here.
Experimental section
Synthesis of nanomaterials
Details about the synthetic procedures are reported in the ESI (see Sections S1 and S2).†Characterization of the nanomaterials
The samples for TEM analysis were prepared by loading a droplet (5 μL) of nanomaterial suspension on the surface of different carbon-coated copper grids (200 mesh). In detail, the sample was allowed to adsorb on the grid for 2 min, the excess nanomaterial was removed with filter paper and the grid was allowed to dry overnight before TEM measurements. In contrast, for the samples containing FeMC6*a, the grids were treated with UranyLess® staining solution to achieve contrast and visualize the protein shell around the gold core.73 To this end, the nanoconjugates were loaded on the grid as described above, and then the samples were treated with a droplet of UranyLess® solution (5 μL) for 2 min. After that, the excess of the staining solution was removed with filter paper and the grids were dried at room temperature overnight. At least 50 independent measurements were taken at different locations of the TEM grid. The mean particle size and standard deviation were determined from statistical measurements using ImageJ software.The ATR FT-IR spectra of CTAB-, PSS-, citrate-stabilized AuNRs/AuNTs and N3-exposing nanomaterials were acquired by loading a drop (4 μL) of the different concentrated colloidal solutions directly on a ZnSe crystal until dryness, to allow the formation of a thin film on the crystal. The spectra were recorded after the evaporation of the solvent (usually within 15–30 min). Baseline corrections for all spectra were made by means of the automatic baseline correction technique. Background bands, which were gathered under the same circumstances, were deducted from the sample spectra automatically. The spectra of the functionalized nanomaterials were compared to those of reference materials to further assess the nature of the gold coating shell. In particular, commercially available powders of CTAB, Na-PSS (Mw 70 kDa), Na3-citrate and lipoic acid (LA) were lyophilized prior to the use, whereas, for the synthesized azide-terminated derivative (LA@N3), 4 μL of a concentrated solution (44 mM) were used.
The ζ-potential of the prepared nanomaterials was assessed under the following conditions: (i) CTAB-AuNMs were stabilized in an aqueous solution of CTAB (2 mM); (ii) the values for PSS-AuNMs were evaluated after stage 3 of PSS/citrate treatment (AuNMs dispersed in aqueous solutions of Na-PSS Mw 70 kDa, 0.15% wt); (iii) the measurements using citrate-AuNMs were performed after the last step (stage 5) of PSS/citrate treatment (AuNMs dispersed in aqueous solutions of Na3-citrate, 5 mM); (iv) the measurements for N3-AuNRs/AuNTs, as well as for FeMC6*a-(PEG)4@AuNRs/AuNTs, were carried out with the samples in aqueous solutions at pH 9.
The samples for ICP-MS were mineralized as follows: each sample (250 μL) was treated with concentrated nitric and hydrochloric acids (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio). The reaction was carried out for 16 h at 90 °C. After the mineralization process, the samples were diluted with Milli-Q water (final volume 10.0 mL) and transferred into an ICP-MS vial for analysis. Metal concentrations were measured by performing the analysis in three replicates. The atomic gold concentrations of Au(III) in the stock solutions of citrate-AuNRs and citrate-AuNTs were estimated to be 445 and 300 μM, respectively. Prior to XPS measurements, the samples containing the differently functionalized nanomaterials were concentrated by centrifugation (7000 rpm, 30 min, and 25 °C). Then, a completely opaque layer was deposited on the surface of a quartz or ITO surface by drop-casting. The final size of the dry drop was ≈0.5 cm, which is an adequate size as the spot size employed was 2 mm. The spectrometer was calibrated assuming the binding energy (BE) of the Au 4f7/2 line at 84.0 eV. The standard deviation for the BE values was 0.2 eV. In the survey, low-resolution scans were run in the 0–1200 eV range (pass energy 60 eV), while higher resolution scans were recorded for the Au 4f, Br 3p, S 2p, N 1s, C 1s and O 1s regions. The analysis involved Shirley background subtraction, and spectral deconvolution was carried out by non-linear least-squares curve fitting adopting a Gaussian sum function, using CASA-XPS software.
:3 ratio). The reaction was carried out for 16 h at 90 °C. After the mineralization process, the samples were diluted with Milli-Q water (final volume 10.0 mL) and transferred into an ICP-MS vial for analysis. Metal concentrations were measured by performing the analysis in three replicates. The atomic gold concentrations of Au(III) in the stock solutions of citrate-AuNRs and citrate-AuNTs were estimated to be 445 and 300 μM, respectively. Prior to XPS measurements, the samples containing the differently functionalized nanomaterials were concentrated by centrifugation (7000 rpm, 30 min, and 25 °C). Then, a completely opaque layer was deposited on the surface of a quartz or ITO surface by drop-casting. The final size of the dry drop was ≈0.5 cm, which is an adequate size as the spot size employed was 2 mm. The spectrometer was calibrated assuming the binding energy (BE) of the Au 4f7/2 line at 84.0 eV. The standard deviation for the BE values was 0.2 eV. In the survey, low-resolution scans were run in the 0–1200 eV range (pass energy 60 eV), while higher resolution scans were recorded for the Au 4f, Br 3p, S 2p, N 1s, C 1s and O 1s regions. The analysis involved Shirley background subtraction, and spectral deconvolution was carried out by non-linear least-squares curve fitting adopting a Gaussian sum function, using CASA-XPS software.
Estimation of the maximum theoretical number of enzyme molecules covering the AuNM surface
To determine the maximum theoretical number (Nmax) of enzyme molecules that can be loaded on the different AuNM surfaces, the surface area of the AuNMs and the footprint area of a single FeMC6*a enzyme molecule were determined. AuNM average dimensions were taken from TEM data (see Table S1†). Considering the AuNR as a spherically capped cylinder, as already reported,74 the area was determined as AAuNR = 4πr2 + 2πr(l − 2r) where r is the radius of the rod (r = w/2) and l is its length (Fig. S1a†). For the AuNT, the total area was determined as AAuNT = 2Ab + AL considering it as a triangular nanoprism, in which Ab is the triangle base area Ab = (l × h)/2, with l and h corresponding to the edge length and the height of the equilateral triangle. AL is the lateral surface area AL = p × t, with p being the nanoprism perimeter (corresponding to 3 × l) and t being the triangular nanoprism thickness (Fig. S1b†). In this last formula, according to the synthetic method of Pelaz et al.68 used in this work, a value of 9 nm thickness was used. As previously reported,57 FeMC6*a is characterized by a cylindrical shape (dimensions of 1.6 nm in diameter and 2.6 nm in length). To take into account all possible randomly oriented cylinder molecules with respect to the AuNMs, the radius of gyration (RG)75 was used (RG = 0.9 nm).66 The footprint area AFeMC6*a was then calculated by considering the area of a circle of radius RG, according to eqn (1):| AFeMC6*a = π × RG2 | (1) |
According to the above consideration, the Nmax values were estimated for AuNRs and AuNTs as follows, respectively:
| Nmax FeMC6*a per AuNR = AAuNR/AFeMC6*a | (2) |
| Nmax FeMC6*a per AuNT = AAuNT/AFeMC6*a | (3) |
Results and discussion
Nanomaterials preparation
Azide groups exposed on gold nanosurfaces have been widely applied to tightly immobilize enzymes on nanomaterials.76,77 We have previously demonstrated the feasibility of covalently linking FeMC6*a on spherical azido-exposing AuNPs,57,66 by means of strain-promoted alkyne-azide cycloaddition (SPAAC).71 To this end, FeMC6*a was derivatized with a pegylated aza-dibenzocyclooctyne moiety (DBCO),78 affording “clickable FeMC6*a-(PEG)4-DBCO”,66 and subsequently clicked onto monodisperse azido-functionalized AuNPs. Azido-exposing AuNPs were prepared by a ligand exchange reaction, upon treatment of citrate-capped AuNPs with a mixture of lipoic acid and its azide-terminated derivative (LA and LA@N3, see Fig. 2).57 In the present work, we tested the feasibility of applying this approach to azido-exposing gold nanorods (AuNRs) and triangular gold nanoprisms (AuNTs). To this end, the synthesis of azide-capped AuNRs and AuNTs would be needed. This functionalization was not a trivial task. Most of the methods applied to synthesize AuNRs and AuNTs involve the use of a cationic surfactant, as cetyltrimethylammonium bromide (CTAB), for its shape-directing power and stabilizing functions.79,80 However, as CTAB molecules arrange in an intimate double layer on gold nanosurfaces,4,79,81 their displacement from AuNMs by incoming ligands is a difficult process. In particular, given the anisotropic nature of the screened nanomaterials, the latter are characterized by different binding sites with distinct exchange reactivity. Vertex and edge sites show an increased propensity to ligand exchange with respect to the bulk surface, where strong CTAB absorption may hamper its displacement even by thiol-containing ligands.82 Indeed, under the examined conditions, the direct exchange of CTAB by the mixture of LA and LA@N3 in aqueous medium was unfeasible, and the aggregation of rods occurred prior to complete ligand exchange. This problem was faced by employing sodium polystyrene sulfonate (Na-PSS) in the ligand exchange protocol.83,84 This strategy was previously developed by Mehtala and co-workers84 for the synthesis of gold nanorods. To the best of our knowledge, it was herein demonstrated for the first time to be effective also for AuNTs. Fig. 2 summarizes the overall route employed.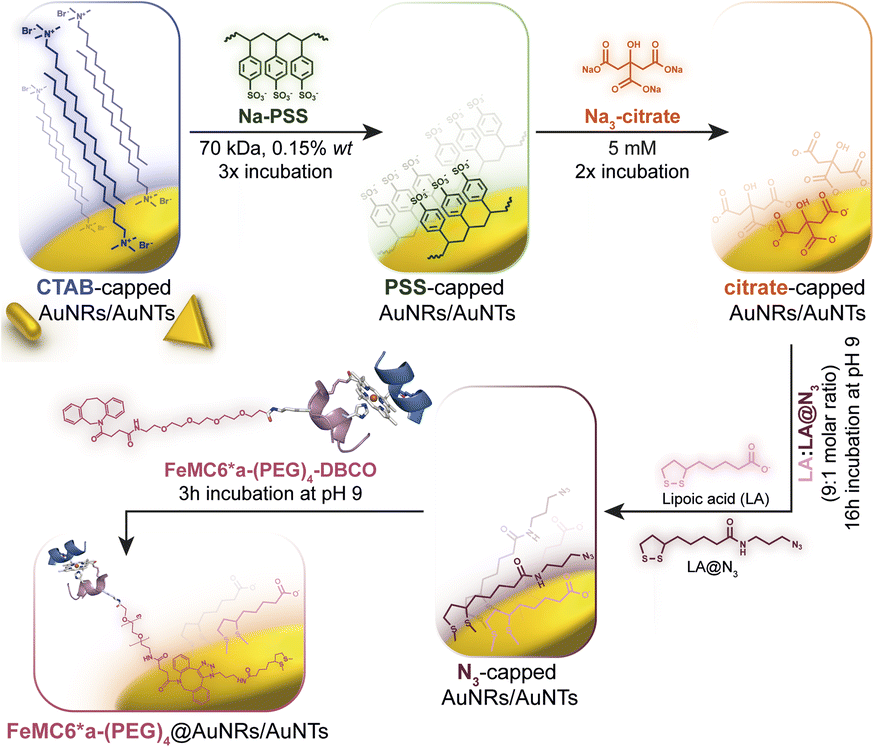 | ||
| Fig. 2 Route to prepare FeMC6*a-functionalized AuNRs and AuNTs, starting from CTAB-capped nanomaterials highlighting all the ligand exchange steps involved in the procedure. | ||
Synthesis of citrate-capped AuNMs
CTAB-capped AuNRs were prepared following an established procedure, namely the surfactant-directed seed-mediated growth method.79 Several centrifugation cycles were performed to remove any excess reagents and the recovered pellet was resuspended in a diluted aqueous CTAB solution (2 mM),85 to preserve colloidal stability (see Paragraph S2.1†).86,87 This procedure afforded a highly monodisperse CTAB-AuNR colloidal suspension, as highlighted by Vis-NIR and TEM analyses (Fig. S2a, b and Table S1†). CTAB-capped AuNTs were prepared by Au(III) reduction in the presence of Na2S2O3.68 This procedure afforded AuNTs of diverse sizes, along with differently shaped AuNMs (mostly nanoparticles), as evidenced by Vis–NIR and TEM analyses (Fig. S2c, d and Table S1†). To improve the purity and homogeneity of AuNTs, the depletion flocculation procedure, in the presence of a surfactant such as CTAB, was employed.88 At a critical CTAB concentration (0.167 M),88 the amount of micelles in solution was sufficient for allowing a separation of AuNTs of ≈100 nm from the reaction mixture. The sedimented AuNTs were redispersed in a minimal amount of water, and the surfactant concentration was decreased until 2 mM by centrifugation,85 leading to stable colloidal solutions of CTAB-capped AuNTs (details are outlined in Paragraph S2.2†). The analysis of the Vis-NIR spectra and TEM images of AuNTs, before and after the depletion procedure, demonstrates the effectiveness of the method (Fig. S2c–f†). To obtain citrate-capped AuNMs from the synthesized CTAB-AuNRs and CTAB-AuNTs, the method reported by Mehtala and co-workers84 was employed, which involved five stages of centrifugation and resuspension cycles. Within the first three cycles, CTAB-AuNMs were incubated with freshly prepared aqueous solutions of Na-PSS, for at least 1 h in each step, to allow the ligand exchange. As PSS is not a suitable stabilizer for CTAB-depleted AuNM solutions,89 two additional stages were performed, aimed at replacing PSS with citrate ions, using aqueous solutions of Na3-citrate (5 mM). The detailed procedure is reported in Paragraphs S2.3 and S2.4.† The progress of the various displacement cycles was followed by visible spectroscopy (Fig. S3†). The conversion of CTAB-AuNMs in PSS-AuNMs caused a shift of the plasmon resonance band for AuNRs (Fig. S3a†) and AuNTs (Fig. S3b†), consistent with literature data.84 Further, the absence of PSS UV bands was indicative of polyanion substitution by citrate ions (Fig. S4†).The effectiveness of the ligand exchange process was ascertained by X-ray photoelectron spectroscopy (XPS). Changes in the chemical composition of the AuNM coating shell were detected by analysing the signals associated with the binding energies (BEs in eV) of the different atomic species in the samples. The low-resolution survey spectra of CTAB-AuNRs and CTAB-AuNTs (Fig. S5†) show signals corresponding to Au 4f, C 1s, N 1s, Br (3p and 3d) and O 1s, as expected. CTAB exchange with Na-PSS led to the disappearance of Br and N signals, and the appearance of the S 2p signal, which almost completely vanished upon Na-PSS exchange by citrate ions. Additional information was gained by recording high-resolution spectra in the distinctive regions of the different elements. The surface chemical compositions of AuNMs, after each step of the ligand-exchange process, are listed in Table S2.† The spectrum of gold shows values that are consistent with the Au(0) oxidation state (Fig. S6†), for both AuNMs. The most informative elements in the context of the exchange procedure are bromine, nitrogen and sulfur. Fig. 3 shows a comparison of the representative XPS high-resolution Br 3p and N 1s spectra for AuNRs and AuNTs, for the intermediates obtained in the ligand-exchange procedure from CTAB to PSS. The high-resolution spectra of CTAB-AuNMs acquired at the BE of Br 3p feature a signal that can be deconvoluted into a doublet corresponding to Br 3p1/2 and Br 3p3/2 (Fig. 3a and e), confirming that the Br− ions cover the Au surface and interact with CTA+ for the formation of a bilayer. The high resolution N 1s spectra for CTAB-AuNMs (Fig. 3c and g) were fitted with just one peak, corresponding to the protonated amine of the surfactant head-group.90,91 Upon replacement of CTAB by PSS in both AuNRs and AuNTs, signals corresponding to Br 3p (Fig. 3b and f) and N 1s (Fig. 3d and h) were reduced to background noise. A similar trend has been reported in the literature for the displacement of CTAB by 11-mercaptoundecanoic and 16-mercaptohexanoic acids.85,92 In the final step of the procedure, the appearance of the signal corresponding to S 2p confirms the successful exchange of CTAB by PSS (Fig. 4a and c). This signal, which can be deconvoluted into a doublet related to S 2p3/2 and S 2p1/2,93 is lacking in the high-resolution spectra of citrate-AuNRs and citrate-AuNTs (Fig. 4b and d). High-resolution analysis of the C 1s region (Fig. 5) further revealed that PSS displacement by citrate is accompanied by a variation in the pattern of the peaks. Apart from hydrocarbon species (C–H/C–C; BE = 285.2 eV), signals related to C atoms bound to O atoms appeared at higher energies (C–OH/C–OR, BE = 286.8/286.6 eV, C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O, and BE = 288.6/288.5 eV) for citrate-AuNRs and citrate-AuNTs, respectively, as expected.
O)O, and BE = 288.6/288.5 eV) for citrate-AuNRs and citrate-AuNTs, respectively, as expected.
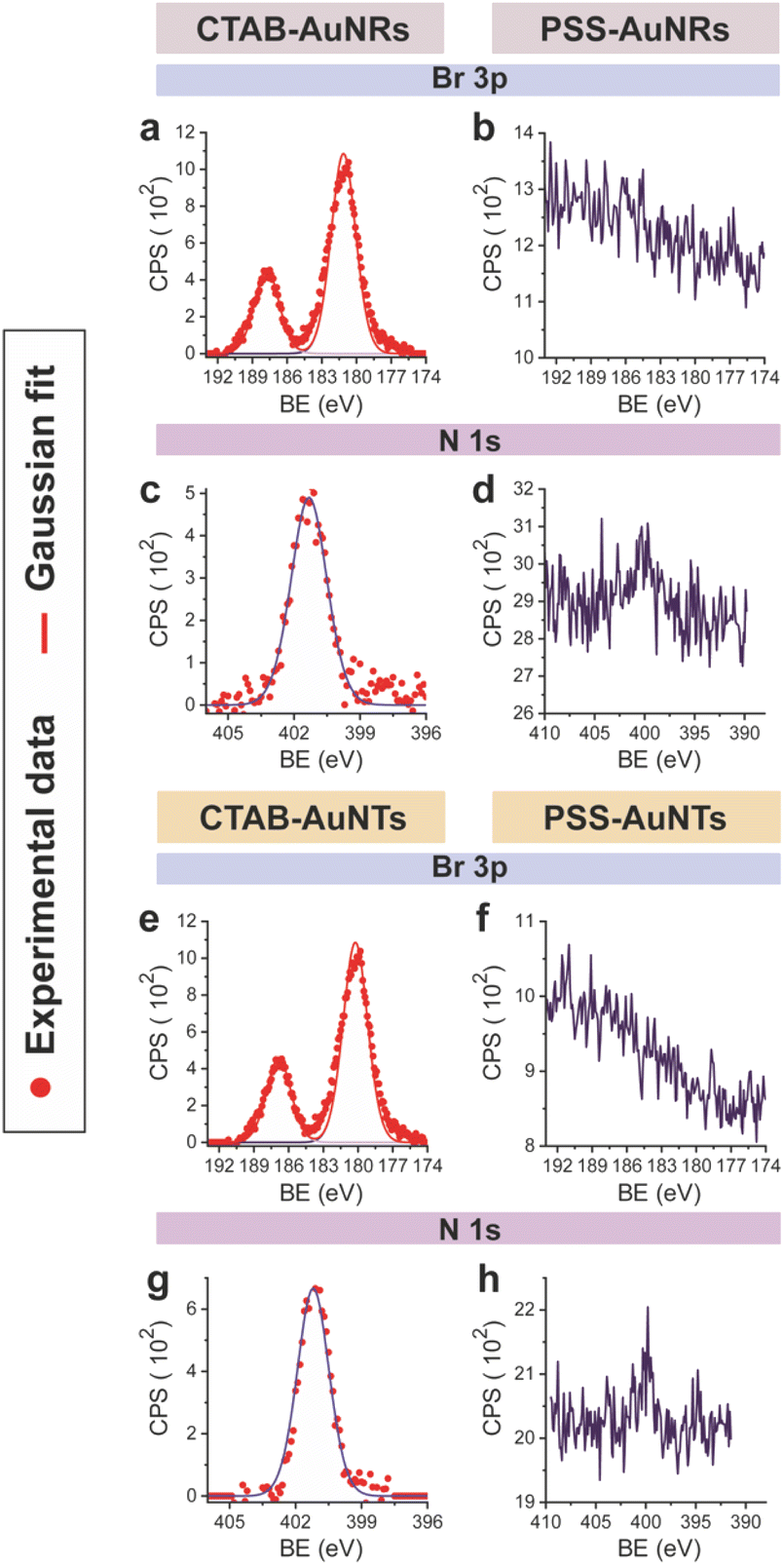 | ||
| Fig. 3 XPS high-resolution spectra in the Br 3p region for: (a) CTAB-AuNRs, (b) PSS-AuNRs, (e) CTAB-AuNTs and (f) PSS-AuNTs. In the N 1s region for: (c) CTAB-AuNRs, (d) PSS-AuNRs, (g) CTAB-AuNTs and (h) PSS-AuNTs. | ||
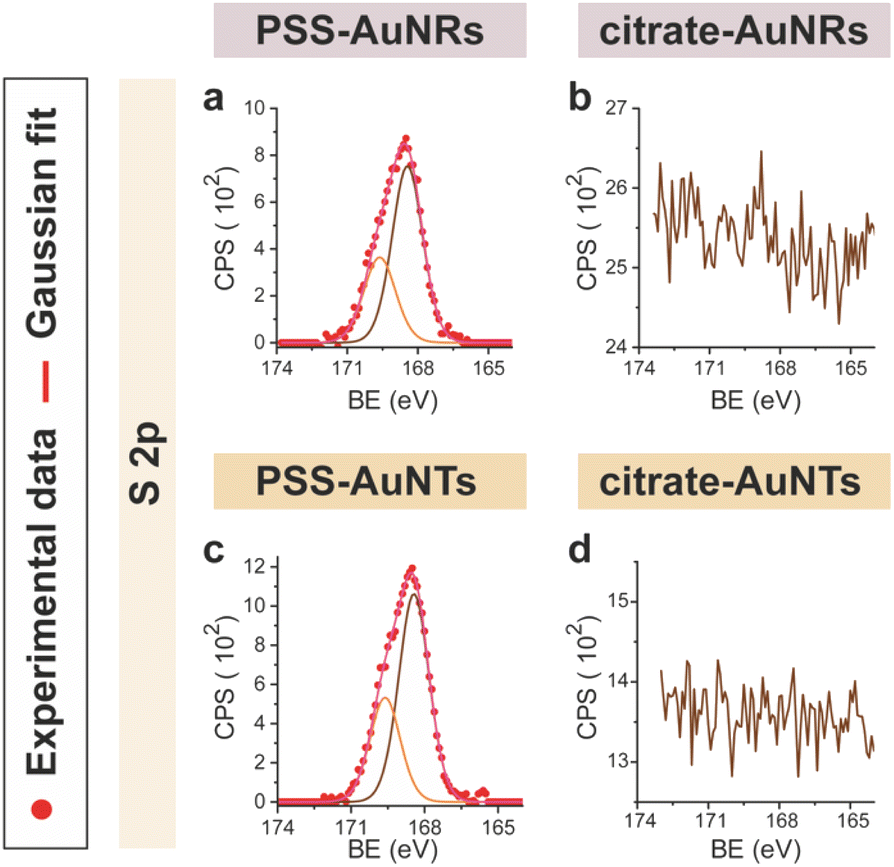 | ||
| Fig. 4 XPS high-resolution spectra in the S 2p region for: (a) PSS-AuNRs, (b) citrate-AuNRs, (c) PSS-AuNTs, (d) citrate-AuNTs. | ||
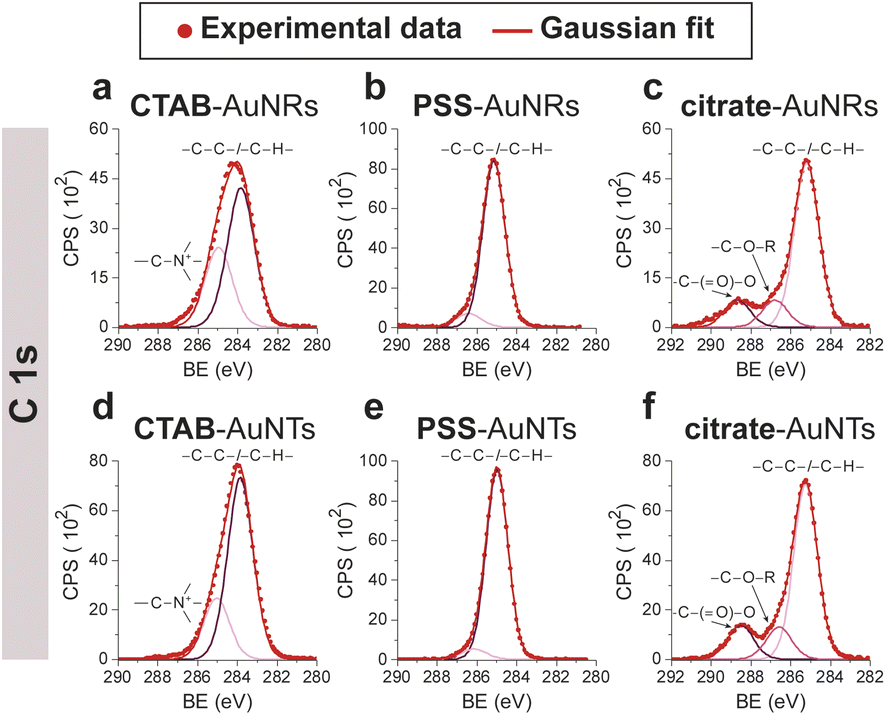 | ||
| Fig. 5 XPS high-resolution spectra in the C 1s region for: (a) CTAB-AuNRs, (b) PSS-AuNRs, (c) citrate-AuNRs, (d) CTAB-AuNTs, (e) PSS-AuNTs, (f) citrate-AuNTs. | ||
ζ-Potential measurements further evidenced the ligand exchange processes. As expected, AuNMs capped with CTAB show a positive ζ-potential value, whereas negative values were observed for PSS- or citrate-capped AuNMs. Table S3† reports the ζ-potential values measured for all the prepared nanomaterials. Notably, when moving from CTAB-AuNRs/AuNTs to citrate-AuNRs/AuNTs no significant changes in the size distribution were observed (Fig. S3c† for citrate-AuNRs and S3d† for citrate-AuNTs, and Table S1†), thus highlighting that the ligand exchange protocol does not affect either the morphology or the size of the nanomaterials.
Synthesis and characterization of FeMC6*a-labeled gold nanoconjugates
Azide-exposing AuNRs and AuNTs were prepared and purified by a ligand exchange reaction from citrate-capped precursors, using a mixture containing LA and LA@N3 (Fig. 1, details in Paragraph S2.6†).66 As expected, the ligand exchange caused a shift of the SPR bands for both AuNMs. Morphological analysis and statistical measurements demonstrated that the displacement of citrate with an alkanethiol mixture affected neither the integrity of the nanomaterials nor their size distributions (Fig. 6a and c). Indeed, no significant changes in the dimensions of the gold core were observed (Fig. 6b for N3-AuNRs: average length of 49 ± 3 nm, width of 17.5 ± 0.9 nm, aspect ratio of 2.7 ± 0.2 and Fig. 6d for N3-AuNTs: average edge length of 101 ± 19 nm). The success of the ligand exchange was also ascertained by ATR FT-IR spectroscopy (Fig. S7d and h†), as evidenced by the presence of an intense signal at 2100 cm−1 relatable to azide stretching,76 which was absent in the spectra of the precursors (Fig. S8†). Further, because of the presence of lipoate anions on the nanosurfaces, N3-AuNRs/AuNTs are characterized by a negative ζ-potential value (−36.6 ± 0.9 mV and −33 ± 2 mV respectively), as previously observed for PSS- or citrate-capped AuNMs (see Table S3†). Next, the artificial catalyst FeMC6*a-(PEG)4-DBCO was conjugated to the target nanomaterials via in situ SPAAC reaction,71 by adding a solution of the enzyme to N3-AuNR or N3-AuNT aqueous solutions under vigorous stirring (see Paragraph S2.7†). The mixtures were allowed to react for 4 h, until no further shifts in plasmon resonance bands were spectroscopically detected. Purification from the excess of unbound enzyme was performed through several centrifugation cycles. After each cycle, the supernatant containing the unbound enzyme was discarded and the pellet, containing the nanoconjugates, was redispersed in an equal volume of an aqueous solution of NaOH (pH 9), to preserve the colloidal solution stability. As already performed for the conjugation of FeMC6*a to AuNPs,57,66 after the first cycle, the pellet was resuspended in an aqueous solution of NaOH at pH 9 with 50% (v/v) 2,2,2-trifluoroethanol (TFE). Thanks to the high solvation properties of TFE and solubility of FeMC6*a in this solvent, this washing procedure allows to get rid of the enzyme excess, which could be adsorbed on the AuNM surface. The conjugation of FeMC6*a to AuNMs was ascertained by UV-visible spectroscopy. Fig. 7a and c report the superposition of the visible spectra of azide-AuNMs and the corresponding FeMC6*a-labeled nanoconjugates. A slight shift in the SPRB of both FeMC6*a-(PEG)4@AuNMs is observed, and the absence of significant band broadening suggests that, upon FeMC6*a coating, AuNMs remained well dispersed without any aggregation in the colloidal suspension. Finally, the weak absorption band around 398 nm, consistent with the heme Soret absorption band, provides further evidence of the successful catalyst conjugation to AuNMs. The morphology of AuNMs upon enzyme conjugation was assessed by TEM analysis. Fig. 7b and d show the TEM images of FeMC6*a-(PEG)4@AuNRs and FeMC6*a-(PEG)4@AuNTs, respectively. Upon negative staining, in both samples the presence of a white halo is indicative of FeMC6*a loading on gold nanosurfaces. The average number of FeMC6*a molecules loaded on AuNMs was determined by dividing the concentration of FeMC6*a by the concentration of AuNMs in the nanoconjugate samples: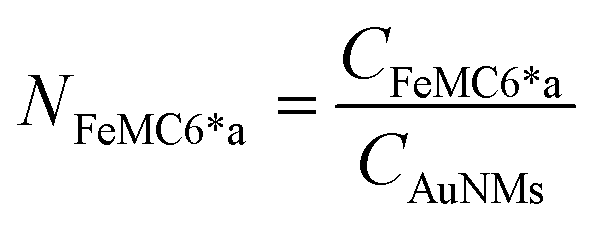 | (4) |
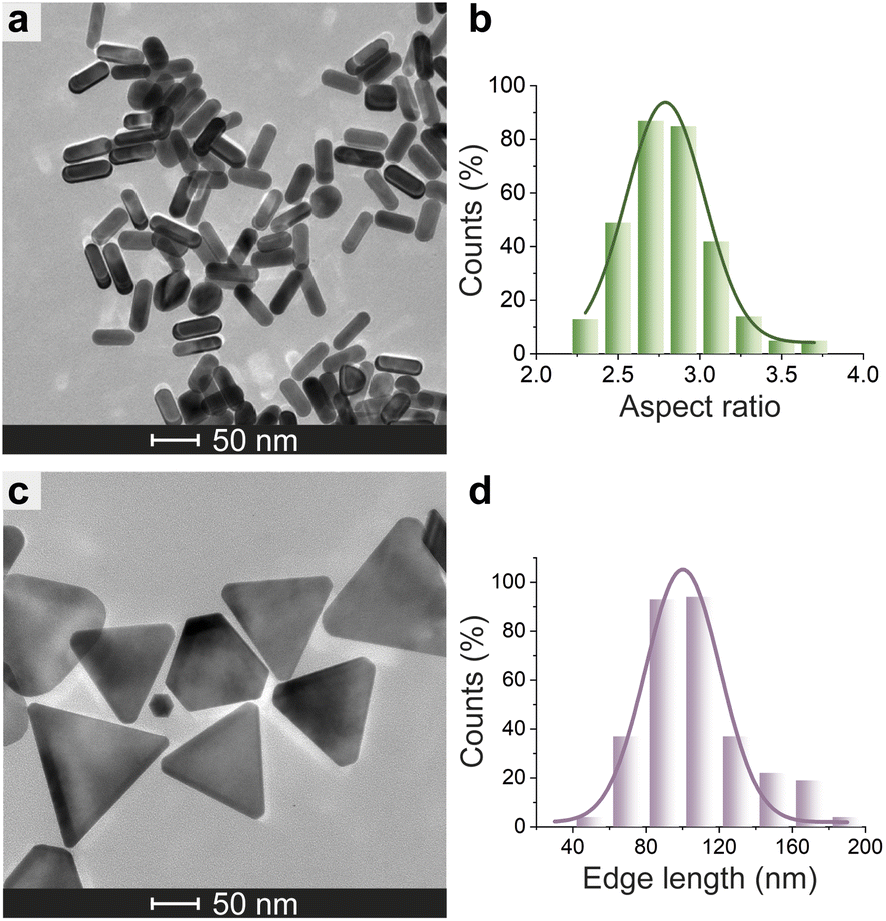 | ||
| Fig. 6 Morphological analysis of N3-AuNMs. TEM micrograph of N3-AuNRs (a) and N3-AuNTs (c). Histogram of the aspect ratio of N3-AuNRs (b) and edge length of N3-AuNTs (d) with a Gaussian fit. | ||
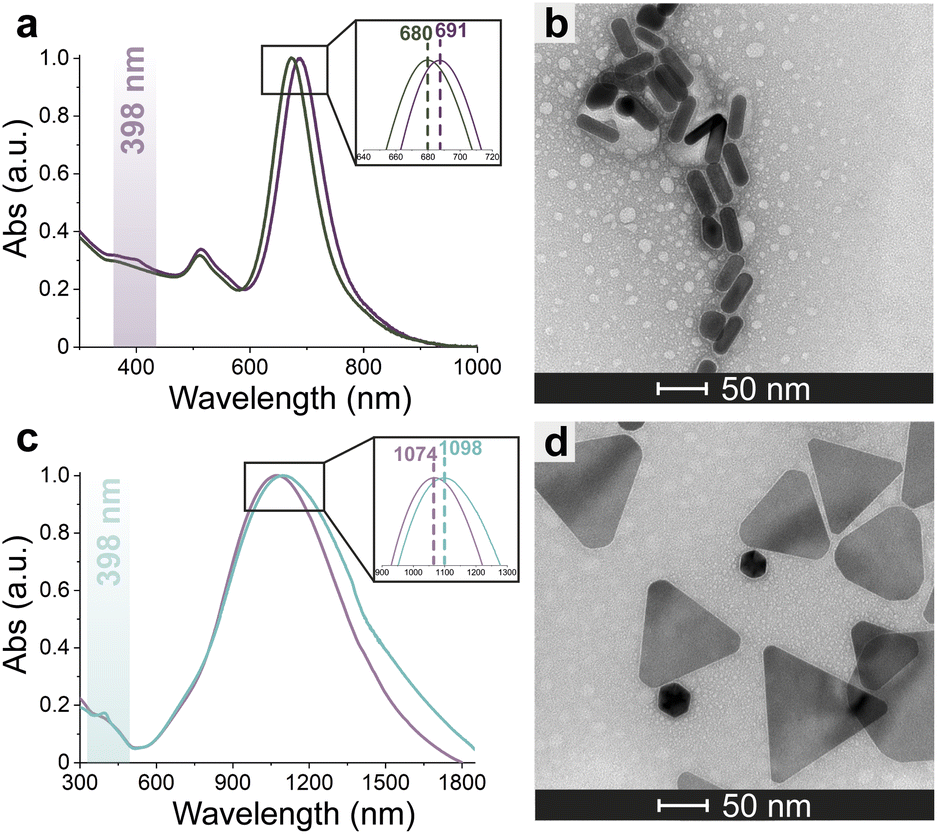 | ||
| Fig. 7 Conjugation of FeMC6*a-(PEG)4-DBCO to N3-AuNMs. Superimposition of the normalized adsorption spectra of N3-AuNMs and FeMC6*a-(PEG)4@AuNMs (a and c). TEM images of FeMC6*a-(PEG)4@AuNMs acquired upon negative staining (b and d). | ||
The concentration of the nanomaterial in the nanoconjugate sample was determined by first quantifying the total amount of gold by ICP-MS analysis. Next, the concentrations of AuNRs and AuNTs were determined by calculating the average number of gold atoms per AuNR and AuNT (through geometrical considerations; see Paragraph S2.5†).
Enzyme concentration was determined by quantifying the heme moiety in FeMC6*a-(PEG)4@AuNR and FeMC6*a-(PEG)4@AuNT samples (see Paragraph S3.4 and Fig. S9†), using the cyanide method, following a procedure reported in the literature94 and already used by us.67 Sample treatment with potassium cyanide promotes the disruption of the gold nanomaterials, thus allowing the spectroscopic measurement of heme concentration as a heme-cyanide complex. Using this approach, a loading of 1900 and 13300 NFeMC6*a molecules per AuNRs and AuNTs was obtained, respectively (Table S4†). The maximum theoretical number of enzyme molecules, Nmax, that can be loaded on each AuNR and AuNT was estimated by geometrical consideration, as reported in the literature.57,74,95 On the basis of the size of the enzyme footprint area and the AuNM available total area, the maximum theoretical number of FeMC6*a molecules per AuNM was estimated to be as follows: for AuNRs Nmax ≅ 1270 and for AuNTs Nmax ≅ 4320 (eqn (2) and (3)). The comparison of the experimentally determined loading with the theoretical Nmax suggests the formation of an enzyme monolayer around each AuNR. For AuNTs, the experimentally determined loading value largely deviates from the theoretical Nmax value (13300 vs. 4320). This enzyme overloading for AuNTs may be attributed to FeMC6*a multilayer formation. However, the shape of AuNMs could also play an additional role in affecting enzyme loading on the surface. Indeed, it is reported that loading density increases with surface curvature.96 Consequently, it is reasonable to assume that sharp edges allow for a higher enzyme loading than planar or soft-curved surfaces.
Catalytic activity and conformational properties of FeMC6*a on differently shaped AuNMs
In order to analyze whether FeMC6*a retains its catalytic behavior once immobilized on differently shaped AuNMs, the peroxidase activity of FeMC6*a-(PEG)4@AuNRs and FeMC6*a-(PEG)4@AuNTs nanoconjugates was screened, using ABTS as a substrate and H2O2 as oxidizing agent, under the optimal experimental conditions (50 mM phosphate buffer at pH 6.5 with 50% TFE v/v) previously determined for the freely diffusing FeMC6*a and for the FeMC6*a-PEG4@AuNPs conjugate.57,62,66 Control assays were first performed on the supernatant of the last washing/centrifugation step of the synthetic procedure for obtaining FeMC6*a-(PEG)4@AuNRs/AuNTs. Only background autoxidation activity was detected in the solution (data not shown), thus excluding the presence of unbound freely diffusing enzyme molecules in the purified nanomaterials. The kinetic parameters for both nanoconjugates were determined by varying H2O2 concentration using fixed concentrations of ABTS, and vice versa (H2O2 0–250 mM range, ABTS 5 mM; ABTS 0–5 mM range, and H2O2 100 mM), using an amount of FeMC6*a-(PEG)4@AuNM solution corresponding to a fixed enzyme concentration (20 nM FeMC6*a-(PEG)4@AuNRs and 25 nM FeMC6*a-(PEG)4@AuNTs). The initial rates (v0) of ABTS oxidation were plotted against ABTS and H2O2 concentrations for both nanoconjugates as reported in Fig. 8. Data fitting, using a two-substrate Michaelis–Menten equation, allowed the determination of the catalytic parameters (Table 1).57,66 Both FeMC6*a-(PEG)4@AuNRs and FeMC6*a-(PEG)4@AuNTs were catalytically active toward ABTS oxidation (see Table 1). However, as already observed for FeMC6*a-(PEG)4@AuNPs,66 FeMC6*a-(PEG)4@AuNRs and FeMC6*a-(PEG)4@AuNTs showed lower catalytic performances than the freely diffusing enzyme. In detail, all the AuNMs show similar kcat values, smaller than the freely diffusing enzyme. A slight increase in the apparent Km value for ABTS is observed, following the order FeMC6*a-(PEG)4@AuNPs < FeMC6*a-(PEG)4@AuNRs < FeMC6*a-(PEG)4@AuNTs. On the basis of these data, the catalytic efficiency for ABTS conversion by FeMC6*a decreases when moving from the spherical gold nanoparticles (AuNPs) to the triangular nanoprisms (AuNTs).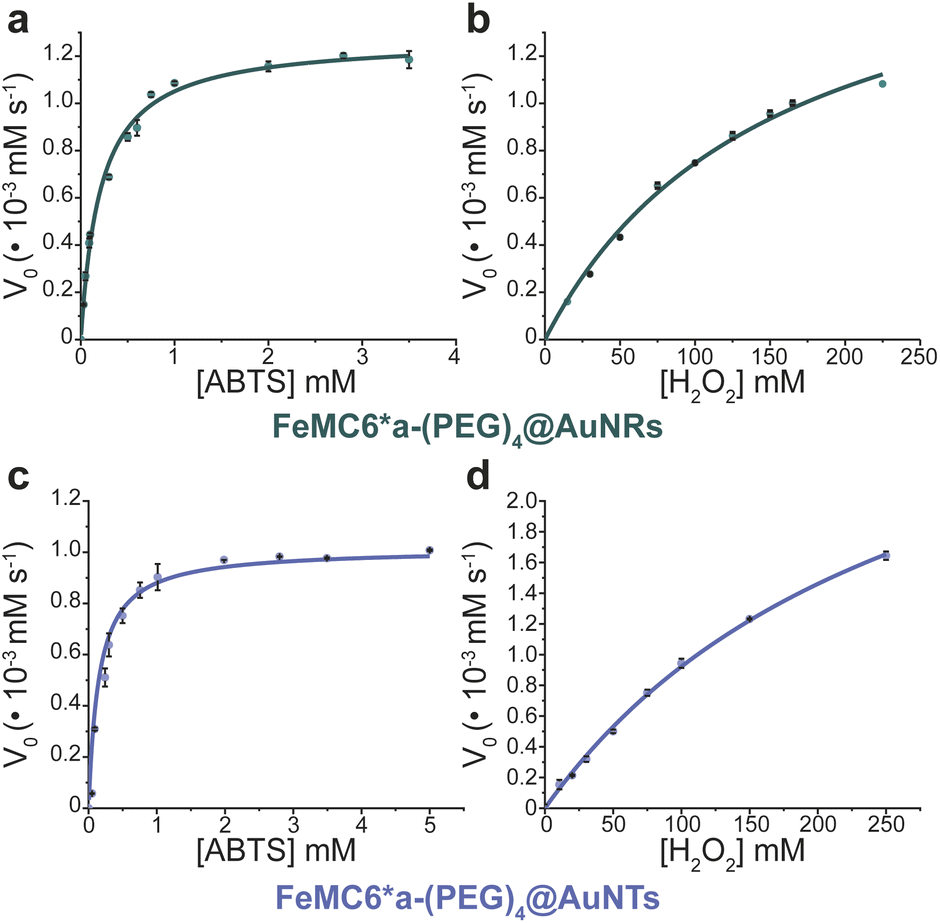 | ||
| Fig. 8 Peroxidase activity of FeMC6*a-(PEG)4@AuNMs. Initial rate dependence vs. ABTS concentration (a and c) and H2O2 concentration (b and d). | ||
| Catalyst | K ABTSm (10−1 mM) |
|
k cat (102 s−1) | k cat/KABTSm (103 mM−1 s−1) |
|
|---|---|---|---|---|---|
| FeMC6*a62 | 0.9 ± 0.1 | 4.4 ± 0.5 | 58 ± 3 | 64 ± 8 | 13 ± 2 |
| FeMC6*a-(PEG)4@AuNPs66 | 1.93 ± 0.05 | 2.16 ± 0.04 | 1.10 ± 0.03 | 0.570 ± 0.001 | 0.509 ± 0.003 |
| FeMC6*a-(PEG)4@AuNRs | 3.3 ± 0.2 | 1.6 ± 0.1 | 1.00 ± 0.02 | 0.30 ± 0.02 | 0.62 ± 0.04 |
| FeMC6*a-(PEG)4@AuNTs | 8.2 ± 0.8 | 3.3 ± 0.1 | 1.63 ± 0.04 | 0.20 ± 0.02 | 0.50 ± 0.01 |
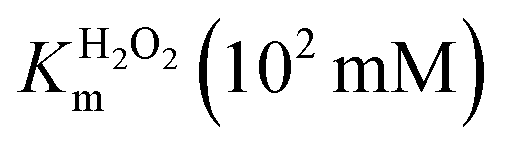
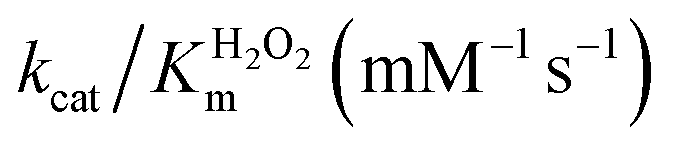
These results indicate that FeMC6*a catalytic activity is preserved upon immobilization of the artificial enzyme on different AuNMs, but also suggest that it is modulated by the shape of the gold nanomaterials. As for FeMC6*a, the catalytic performance are strongly related to the peptide helical folding around the porphyrin moiety,62 and the CD spectra of FeMC6*a immobilized on the differently shaped AuNMs were recorded (Fig. 9). The CD spectrum of FeMC6*a-(PEG)4@AuNRs (Fig. 9 orange line) displays two minima around 222 and 210 nm, typical of helical conformation, with a slight decrease in the helical content with respect to the freely diffusing FeMC6*a (Fig. 9 dark red line, molar ellipticity at 222 nm [θ]222 = −11370 deg cm2 dmol−1 res−1 and [θ]222 = −15500 deg cm2 dmol−1 res−1 respectively). A similar behavior was observed for FeMC6*a once immobilized on spherical AuNPs (FeMC6*a-(PEG)4@AuNPs [θ]222 = −11830 deg cm2 dmol−1 res−1).66 Conversely, a significant decrease in the helical content was observed upon FeMC6*a immobilization on AuNTs. The FeMC6*a-(PEG)4@AuNTs conjugate (Fig. 9, green line) shows a very low value of [θ]222 ([θ]222 = −4890 deg cm2 dmol−1 res−1) and spectral features suggesting helical aggregation, as the [θ]ratio value (the ratio of ellipticity at 222 nm to that at the shorter wavelength minimum) was found to be >1.0. The analysis of these results suggests that the shape of the AuNMs strongly influences the FeMC6*a conformation. Several investigations have been performed to correlate the effect of the size and shape of AuNMs on the protein conformation, structures and activity.36,97,98 By analyzing the lysozyme structure upon adsorption on silica nanoparticles of various diameters, Vertegel et al.98 demonstrated that the surface curvature had a strong influence on the protein structure.
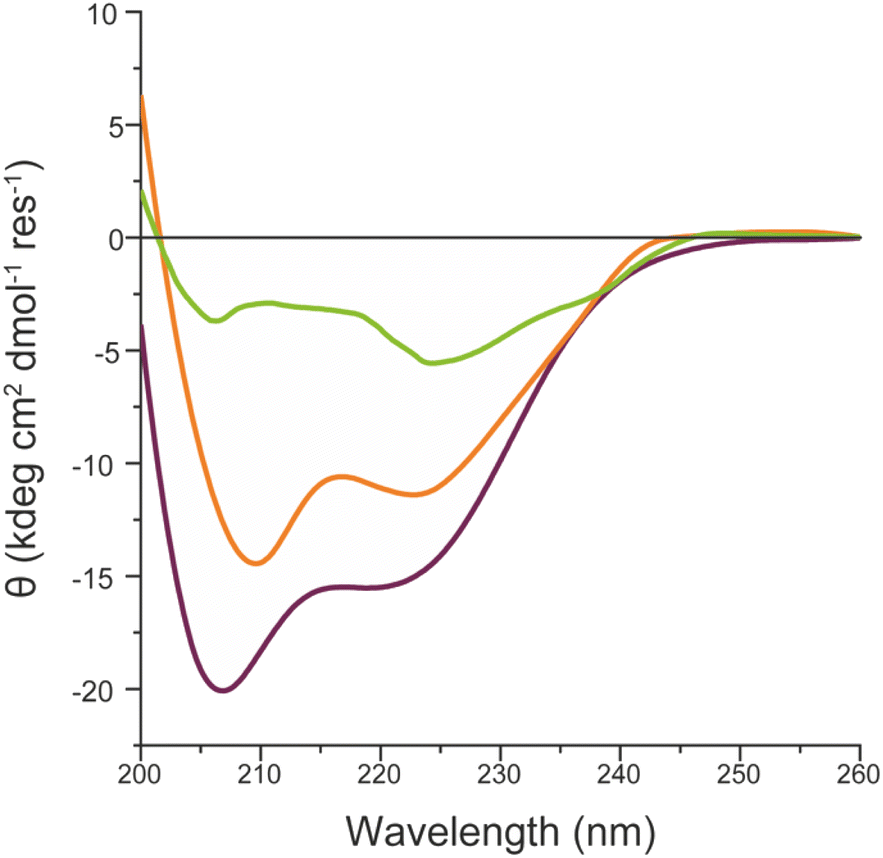 | ||
| Fig. 9 Superimposition of the CD spectra of FeMC6*a-(PEG)4@AuNRs (orange line), FeMC6*a-(PEG)4@AuNTs (green line) and FeMC6*a (dark red line), in 10 mM phosphate buffer (pH 6.5) with 50% TFE (v/v). | ||
A more native-like structure was found for protein adsorbed on smaller nanoparticles as their higher curvature causes fewer protein surface interactions and in turn fewer changes in the structure. Hence, the curvature of the nanomaterial used to immobilize FeMC6*a may have a significant impact on the enzyme activity. Indeed, with AuNTs being flatter than AuNRs, it can be hypothesized that major crowding and aggregation of the enzyme molecules may occur on the AuNT surface with respect to the more curved AuNRs, thus lowering the catalytic efficiency. Actually, the comparison of the experimental and theoretical values of Nmax strongly suggests major crowding and multilayer formation when FeMC6*a is immobilized on the AuNT material, with respect to AuNRs.
Conclusions
In this paper, we demonstrated the feasibility of using an artificial heme-peroxidase, FeMC6*a, as a “clickable” enzyme for the surface decoration of differently shaped gold-based nanomaterials. Starting from our previous studies on spherical nanoparticles, we moved toward anisotropic nanomaterials, with the aim of exploiting the effect of the AuNM shape on immobilized mini-enzyme performance. Azide-capped AuNRs and AuNTs were successfully prepared and used for FeMC6*a immobilization, affording two nanoconjugates that retain the intrinsic peroxidase behavior of the artificial enzyme. The overall results suggest that the nanomaterial shape influences the nanoconjugate functional properties, with curved AuNMs, i.e., AuNPs and AuNRs, showing better catalytic performance. The outcomes of these studies provide new insights into the effect of the surfaces on the artificial heme peroxidase catalytic activity and open up the way for the construction of novel nanostructures for potential applications in the field of sensing devices. In this context, several studies have reported the successful applications of anisotropic gold nanomaterials in protein electrochemistry, as they appeared to be suitable host matrices for modulating the direct electron transfer between redox-active proteins and a variety of electrodes.28–30 To this end, we are currently investigating the redox and electrocatalytic response towards H2O2 of FeMC6*a, when immobilized on differently shaped AuNMs or adsorbed on glassy carbon electrodes, to better clarify the relationship between AuNM anisotropy and enzyme activity.Author contributions
E. R. and A. E.: investigation, formal analysis, and writing original draft. L. L.: formal analysis, methodology, and validation. M. C.: investigation and formal analysis. T. P.: supervision and conceptualization. A. L. and F. N.: supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Rocco Di Girolamo and Antonella Giarra for their help in collecting TEM images. The authors wish to thank Gabriele Soriano for preliminary data collection. A. E. thanks the Italian Ministry of University and Research (MUR) for being awarded a PhD position (PONR&I 2014–2020, CUP: E65F21003750003). L. L. is grateful to the European Research Council (ERC) for being granted a research fellow position in the frame of the “BioDisOrder” project (UGOV 000005_HORIZON2020_ERC_2018_BioDisOrder, CUP: E64I19002960006). The work described in this paper was supported by Italian MUR, PRIN 2020, Project SEA-WAVE 2020BKK3W9, [CUP E69J22001140005]. The authors finally acknowledge the Italian MUR program “Dipartimenti di Eccellenza 2023–2027” for the projects arCHIMede [CUP E63C22003710006].References
- H. Hassan, P. Sharma, M. R. Hasan, S. Singh, D. Thakur and J. Narang, Mater. Sci. Energy Technol., 2022, 5, 375–390 CAS.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- J. Cao, T. Sun and K. T. V. Grattan, Sens. Actuators, B, 2014, 195, 332–351 CrossRef CAS.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- J.-H. Lee, H.-Y. Cho, H. K. Choi, J.-Y. Lee and J.-W. Choi, Int. J. Mol. Sci., 2018, 19, 2021 CrossRef CAS PubMed.
- L. Qin, G. Zeng, C. Lai, D. Huang, P. Xu, C. Zhang, M. Cheng, X. Liu, S. Liu, B. Li and H. Yi, Coord. Chem. Rev., 2018, 359, 1–31 CrossRef CAS.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- A. A. Yaqoob, H. Ahmad, T. Parveen, A. Ahmad, M. Oves, I. M. I. Ismail, H. A. Qari, K. Umar and M. N. Mohamad Ibrahim, Front. Chem., 2020, 8, 341 CrossRef CAS PubMed.
- R. M. Williams, S. Chen, R. E. Langenbacher, T. V. Galassi, J. D. Harvey, P. V. Jena, J. Budhathoki-Uprety, M. Luo and D. A. Heller, Nat. Chem. Biol., 2021, 17, 129–137 CrossRef CAS PubMed.
- J. M. Bolivar and B. Nidetzky, Molecules, 2019, 24, 3460 CrossRef CAS PubMed.
- G. A. Ellis, S. A. Díaz and I. L. Medintz, Curr. Opin. Biotechnol., 2021, 71, 77–90 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- L. Ren, H. Ji, K. Heuzé, B. Faure, E. Genin, P. Rousselot Pailley and T. Tron, Colloids Surf., B, 2021, 206, 111963 CrossRef CAS PubMed.
- M. Chen, G. Zeng, P. Xu, C. Lai and L. Tang, Trends Biochem. Sci., 2017, 42, 914–930 CrossRef CAS PubMed.
- M. Bilal and H. M. N. Iqbal, Coord. Chem. Rev., 2019, 388, 1–23 CrossRef CAS.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415 RSC.
- A. S. Stender, G. Wang, W. Sun and N. Fang, ACS Nano, 2010, 4, 7667–7675 CrossRef CAS PubMed.
- N. D. Burrows, W. Lin, J. G. Hinman, J. M. Dennison, A. M. Vartanian, N. S. Abadeer, E. M. Grzincic, L. M. Jacob, J. Li and C. J. Murphy, Langmuir, 2016, 32, 9905–9921 CrossRef CAS PubMed.
- A. C. Templeton, J. J. Pietron, R. W. Murray and P. Mulvaney, J. Phys. Chem. B, 2000, 104, 564–570 CrossRef CAS.
- S. K. Ghosh, S. Nath, S. Kundu, K. Esumi and T. Pal, J. Phys. Chem. B, 2004, 108, 13963–13971 CrossRef CAS.
- C. Parolo, A. Sena-Torralba, J. F. Bergua, E. Calucho, C. Fuentes-Chust, L. Hu, L. Rivas, R. Álvarez-Diduk, E. P. Nguyen, S. Cinti, D. Quesada-González and A. Merkoçi, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed.
- C. Parolo and A. Merkoçi, Chem. Soc. Rev., 2012, 42, 450–457 RSC.
- J. H. Soh, H.-M. Chan and J. Y. Ying, Nano Today, 2020, 30, 100831 CrossRef CAS.
- Y. Gupta and A. S. Ghrera, Arch. Microbiol., 2021, 203, 3767–3784 CrossRef CAS PubMed.
- A. M. López_Marzo, J. Pons, D. A. Blake and A. Merkoçi, Biosens. Bioelectron., 2013, 47, 190–198 CrossRef PubMed.
- Y. Panraksa, A. Apilux, S. Jampasa, S. Puthong, C. S. Henry, S. Rengpipat and O. Chailapakul, Sens. Actuators, B, 2021, 329, 129241 CrossRef CAS.
- G. Siciliano, A. Alsadig, M. S. Chiriacò, A. Turco, A. Foscarini, F. Ferrara, G. Gigli and E. Primiceri, Talanta, 2024, 268, 125280 CrossRef CAS PubMed.
- M. Chávez, Á. Fernandez-Merino, R. del Caño, G. Sánchez-Obrero, R. Madueño, M. Blázquez and T. Pineda, Biosensors, 2023, 13, 467 CrossRef PubMed.
- P. Bollella, Y. Hibino, P. Conejo-Valverde, J. Soto-Cruz, J. Bergueiro, M. Calderón, O. Rojas-Carrillo, K. Kano and L. Gorton, Anal. Bioanal. Chem., 2019, 411, 7645–7657 CrossRef CAS PubMed.
- Y. Niu, J. Liu, W. Chen, C. Yin, W. Weng, X. Li, X. Wang, G. Li and W. Sun, Anal. Methods, 2018, 10, 5297–5304 RSC.
- A. K. Pearce, T. R. Wilks, M. C. Arno and R. K. O'Reilly, Nat. Rev. Chem, 2021, 5, 21–45 CrossRef CAS PubMed.
- I. Hammami, N. M. Alabdallah, A. A. jomaa and M. kamoun, J. King Saud Univ., Sci., 2021, 33, 101560 CrossRef.
- L. Yang, Z. Zhou, J. Song and X. Chen, Chem. Soc. Rev., 2019, 48, 5140–5176 RSC.
- E. Martinsson, M. M. Shahjamali, N. Large, N. Zaraee, Y. Zhou, G. C. Schatz, C. A. Mirkin and D. Aili, Small, 2016, 12, 330–342 CrossRef CAS PubMed.
- K. K. Bharadwaj, B. Rabha, S. Pati, T. Sarkar, B. K. Choudhury, A. Barman, D. Bhattacharjya, A. Srivastava, D. Baishya, H. A. Edinur, Z. Abdul Kari and N. H. Mohd Noor, Molecules, 2021, 26, 6389 CrossRef CAS PubMed.
- H. Liu, Y. Tian and Z. Deng, Langmuir, 2007, 23, 9487–9494 CrossRef CAS PubMed.
- S. Komathi, A. I. Gopalan, S.-K. Kim, G. S. Anand and K.-P. Lee, Electrochim. Acta, 2013, 92, 71–78 CrossRef CAS.
- A. Lombardi, F. Pirro, O. Maglio, M. Chino and W. F. DeGrado, Acc. Chem. Res., 2019, 52, 1148–1159 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- M. J. Chalkley, S. I. Mann and W. F. DeGrado, Nat. Rev. Chem, 2021, 6, 31–50 CrossRef PubMed.
- K. J. Koebke, T. B. J. Pinter, W. C. Pitts and V. L. Pecoraro, Chem. Rev., 2022, 122, 12046–12109 CrossRef CAS PubMed.
- J.-P. Mahy, F. Avenier, W. Ghattas, R. Ricoux and M. Salmain, Enzymes for Solving Humankind's Problems: Natural and Artificial Systems in Health, Agriculture, Environment and Energy, ed. J. J. G. Moura, I. Moura and L. B. Maia, Springer International Publishing, Cham, 2021, pp. 363–411 Search PubMed.
- L. Leone, G. Sgueglia, S. La Gatta, M. Chino, F. Nastri and A. Lombardi, Int. J. Mol. Sci., 2023, 24, 8605 CrossRef CAS PubMed.
- L. Yin, H. Yuan, C. Liu, B. He, S.-Q. Gao, G.-B. Wen, X. Tan and Y.-W. Lin, ACS Catal., 2018, 8, 9619–9624 CrossRef CAS.
- C. Liu, J. Xu, S.-Q. Gao, B. He, C.-W. Wei, X.-J. Wang, Z. Wang and Y.-W. Lin, RSC Adv., 2018, 8, 33325–33330 RSC.
- R. Stenner, J. W. Steventon, A. Seddon and J. L. R. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1419–1428 CrossRef CAS PubMed.
- A. D'Souza and S. Bhattacharjya, Biochemistry, 2021, 60, 431–439 CrossRef PubMed.
- K. Oohora and T. Hayashi, Dalton Trans., 2021, 50, 1940–1949 RSC.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- M. Chino, L. Leone, G. Zambrano, F. Pirro, D. D'Alonzo, V. Firpo, D. Aref, L. Lista, O. Maglio, F. Nastri and A. Lombardi, Biopolymers, 2018, 109, e23107 CrossRef PubMed.
- F. Nastri, D. D'Alonzo, L. Leone, G. Zambrano, V. Pavone and A. Lombardi, Trends Biochem. Sci., 2019, 44, 1022–1040 CrossRef CAS PubMed.
- L. Leone, M. Chino, F. Nastri, O. Maglio, V. Pavone and A. Lombardi, Biotechnol. Appl. Biochem., 2020, 67, 495–515 CrossRef CAS PubMed.
- M. Chino, S. La Gatta, L. Leone, M. De Fenza, A. Lombardi, V. Pavone and O. Maglio, Int. J. Mol. Sci., 2023, 24, 11070 CrossRef CAS PubMed.
- R. Vitale, L. Lista, C. Cerrone, G. Caserta, M. Chino, O. Maglio, F. Nastri, V. Pavone and A. Lombardi, Org. Biomol. Chem., 2015, 13, 4859–4868 RSC.
- A. Ranieri, S. Monari, M. Sola, M. Borsari, G. Battistuzzi, P. Ringhieri, F. Nastri, V. Pavone and A. Lombardi, Langmuir, 2010, 26, 17831–17835 CrossRef CAS PubMed.
- G. Zambrano, E. Ruggiero, A. Malafronte, M. Chino, O. Maglio, V. Pavone, F. Nastri and A. Lombardi, Int. J. Mol. Sci., 2018, 19, 2896 CrossRef PubMed.
- L. Leone, D. D'Alonzo, V. Balland, G. Zambrano, M. Chino, F. Nastri, O. Maglio, V. Pavone and A. Lombardi, Front. Chem., 2018, 6, 590 CrossRef CAS PubMed.
- L. Leone, D. D'Alonzo, O. Maglio, V. Pavone, F. Nastri and A. Lombardi, ACS Catal., 2021, 11, 9407–9417 CrossRef CAS.
- V. Firpo, J. M. Le, V. Pavone, A. Lombardi and K. L. Bren, Chem. Sci., 2018, 9, 8582–8589 RSC.
- E. H. Edwards, J. M. Le, A. A. Salamatian, N. L. Peluso, L. Leone, A. Lombardi and K. L. Bren, J. Inorg. Biochem., 2022, 230, 111753 CrossRef CAS PubMed.
- G. Caserta, M. Chino, V. Firpo, G. Zambrano, L. Leone, D. D'Alonzo, F. Nastri, O. Maglio, V. Pavone and A. Lombardi, ChemBioChem, 2018, 19, 1823–1826 CrossRef CAS PubMed.
- G. Zambrano, F. Nastri, V. Pavone, A. Lombardi and M. Chino, Sensors, 2020, 20, 3793 CrossRef CAS PubMed.
- G. Zambrano, A. Sekretareva, D. D'Alonzo, L. Leone, V. Pavone, A. Lombardi and F. Nastri, RSC Adv., 2022, 12, 12947–12956 RSC.
- D. D'Alonzo, M. De Fenza, V. Pavone, A. Lombardi and F. Nastri, Int. J. Mol. Sci., 2023, 24, 8058 CrossRef PubMed.
- G. Zambrano, M. Chino, E. Renzi, R. Di Girolamo, O. Maglio, V. Pavone, A. Lombardi and F. Nastri, Biotechnol. Appl. Biochem., 2020, 67, 549–562 CrossRef CAS PubMed.
- E. Renzi, A. Piper, F. Nastri, A. Merkoçi and A. Lombardi, Small, 2023, 2207949 CrossRef CAS PubMed.
- B. Pelaz, V. Grazu, A. Ibarra, C. Magen, P. del Pino and J. M. de la Fuente, Langmuir, 2012, 28, 8965–8970 CrossRef CAS PubMed.
- R. Yamada, R. Kimura and S. Kuwahara, RSC Adv., 2023, 13, 32143–32149 RSC.
- X. Yu, Z. Wang, H. Cui, X. Wu, W. Chai, J. Wei, Y. Chen and Z. Zhang, Molecules, 2022, 27, 8766 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- S. De Carlo and J. R. Harris, Micron, 2011, 42, 117–131 CrossRef CAS PubMed.
- Y. Wang, K. van Asdonk and P. Zijlstra, Langmuir, 2019, 35, 13356–13363 CrossRef CAS PubMed.
- M. Yu. Lobanov, N. S. Bogatyreva and O. V. Galzitskaya, Mol. Biol., 2008, 42, 623–628 CrossRef CAS.
- M.-X. Zhang, B.-H. Huang, X.-Y. Sun and D.-W. Pang, Langmuir, 2010, 26, 10171–10176 CrossRef CAS PubMed.
- N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- D. Nepal, K. Park and R. A. Vaia, Small, 2012, 8, 1013–1020 CrossRef CAS PubMed.
- H.-M. Gao, H. Liu, H.-J. Qian, G.-S. Jiao and Z.-Y. Lu, Phys. Chem. Chem. Phys., 2018, 20, 1381–1394 RSC.
- A. P. Leonov, J. Zheng, J. D. Clogston, S. T. Stern, A. K. Patri and A. Wei, ACS Nano, 2008, 2, 2481–2488 CrossRef CAS PubMed.
- J. G. Mehtala, D. Y. Zemlyanov, J. P. Max, N. Kadasala, S. Zhao and A. Wei, Langmuir, 2014, 30, 13727–13730 CrossRef CAS PubMed.
- R. del Caño, J. M. Gisbert-González, J. González-Rodríguez, G. Sánchez-Obrero, R. Madueño, M. Blázquez and T. Pineda, Nanoscale, 2020, 12, 658–668 RSC.
- B. C. Rostro-Kohanloo, L. R. Bickford, C. M. Payne, E. S. Day, L. J. E. Anderson, M. Zhong, S. Lee, K. M. Mayer, T. Zal, L. Adam, C. P. N. Dinney, R. A. Drezek, J. L. West and J. H. Hafner, Nanotechnology, 2009, 20, 434005 CrossRef PubMed.
- S. Lee, L. J. E. Anderson, C. M. Payne and J. H. Hafner, Langmuir, 2011, 27, 14748–14756 CrossRef CAS PubMed.
- F. Zhang, J. Zhu, H.-Q. An, J.-J. Li and J.-W. Zhao, J. Mater. Chem. C, 2016, 4, 568–580 RSC.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- S. D. Techane, L. J. Gamble and D. G. Castner, Biointerphases, 2011, 6, 98–104 CrossRef CAS PubMed.
- R. Ahmad, L. Boubekeur-Lecaque, M. Nguyen, S. Lau-Truong, A. Lamouri, P. Decorse, A. Galtayries, J. Pinson, N. Felidj and C. Mangeney, J. Phys. Chem. C, 2014, 118, 19098–19105 CrossRef CAS.
- J. Cao, E. K. Galbraith, T. Sun and K. T. V. Grattan, Sens. Actuators, B, 2012, 169, 360–367 CrossRef CAS.
- Y. Joseph, I. Besnard, M. Rosenberger, B. Guse, H.-G. Nothofer, J. M. Wessels, U. Wild, A. Knop-Gericke, D. Su, R. Schlögl, A. Yasuda and T. Vossmeyer, J. Phys. Chem. B, 2003, 107, 7406–7413 CrossRef CAS.
- A. Onoda, Y. Ueya, T. Sakamoto, T. Uematsu and T. Hayashi, Chem. Commun., 2010, 46, 9107 RSC.
- Y. Wang, R. Jonkute, H. Lindmark, J. D. Keighron and A.-S. Cans, Langmuir, 2020, 36, 37–46 CrossRef CAS PubMed.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS PubMed.
- M.-E. Aubin-Tam and K. Hamad-Schifferli, Biomed. Mater., 2008, 3, 034001 CrossRef PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, UV-vis-NIR spectra, TEM images, size histograms, ATR FTIR, and XPS. See DOI: https://doi.org/10.1039/d4na00344f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |