
Unveiling the effects of ions in the electric double layer on the carbon dioxide reduction reaction
Fenglou
Ni
,
Kangkang
Jia
,
Yangyang
Chen
,
Yunzhou
Wen
and
Sisi
He
 *
*
School of Science, Harbin Institute of Technology (Shenzhen), University Town, Shenzhen, Guangdong 518055, China. E-mail: hesisi@hit.edu.cn
First published on 26th May 2023
Abstract
As a potential approach to minimize carbon emissions, the electrocatalytic transfer of carbon dioxide (CO2) to valuable chemicals has attracted great attention. Since such a process takes place at the liquid/solid interface, the electrolyte plays a vital role in determining the catalytic performance. Particularly, the cations including alkali metal cations and organic cations, and/or anions of the electrolyte can greatly change the activity and selectivity of the CO2 reduction reaction (CO2RR). Here, we review the development of the electric double layer and recent advances in the understanding of cation effects and anion effects on their electrocatalytic mechanisms and performance. Insights into the behavior of cations and anions in the electric double layer and their various mechanistic impacts on the electrochemical reduction of CO2 are of critical importance in optimizing reaction conditions and designing reactors for the efficient conversion of CO2. Finally, several challenges and outlook for better understanding and utilizing the electric double layer in the enhancement of the CO2RR are proposed, and we hope that this review can provide guidance for the design of more efficient CO2RR electrocatalysts.
 Sisi He | Sisi He is an Associate Professor at the Harbin Institute of Technology (Shenzhen) in China. She received her B.E. (2011), M.S. (2014) and PhD (2017) from Northeastern University, Tianjin University and Fudan University, respectively, and then worked as a postdoctoral fellow in Okinawa Institute of Science and Technology Graduate University, Japan and MacMaster University, Canada during 2018–2021. Her current interests focus on electrochemistry-related research areas including flexible sensor/energy storage and conversion platforms and electrocatalytic syntheses. |
1. Introduction
The electrochemical reduction of carbon dioxide into valuable chemical commodities has significant potential to achieve the carbon-neutral goal while at the same time making full use of intermittent and renewable energy sources.1–3 The development of catalysts, as a key factor in the CO2RR, has been progressing rapidly and tremendously in recent years.4–7 Many strategies, such as adjusting the morphology,8 chemical state,9,10 surface facet,11 and coordination structure12 and introducing different elements,13,14 have been applied to the design of highly active catalysts. Thus, near-unity faradaic efficiencies (FEs) for CO2 reduction to carbon monoxide (CO) and formate have been achieved on various catalysts, including metal, single-atom material, and molecular catalysts.15–22 The FEs for CO2 reduction to multi-carbon products (C2+) are now above 70% on copper.23–29 Besides, the electrodes and reactors also play significant roles in the CO2RR.30,31 Generally, there are two kinds of reactors, a conventional H-type reactor and a gas diffusion electrode (GDE) based flow-cell reactor. The H-type reactor has been widely used in fundamental research on the CO2RR, as the dissolved CO2 concentration in the electrolyte mainly determines the current density (less than 100 mA cm−2 in most cases). But, using the GDEs in flow cell reactors, CO2 is able to directly reach the catalyst in the gas phase, greatly improving the mass transport of CO2 and thus achieving higher current density (more than 1 A cm−2).25,26 Meanwhile, the high local alkalinity generated by high current densities can suppress the activity of the hydrogen evolution reaction (HER),28 which further enhances the selectivity for the CO2RR.27,32While seeing the advances in designing CO2RR catalysts and reactors, one may find that some major issues, e.g., high overpotentials33 and carbonate (CO32−) formation,34,35 seem challenging to be addressed solely by the optimization of catalysts and reactors. Indeed, today's CO2RR reactors suffer from large cell voltages above 3 V and huge additional energy costs due to the overpotential and CO32− formation issues.27,36 With more and more attention paid to the catalyst–electrolyte interface of the CO2RR, mainly the electric double layer (EDL), the interplay between the local reaction microenvironment and the catalytic performance emerges as a new direction progressing the CO2RR towards practical applications.37–39
Therefore, this review aims to briefly summarize the progress of the research on the role of an EDL in the CO2RR. An EDL consists of catalyst surfaces, ions, and important intermediates and reactants, determining the selectivity and activity. We first narrate the classic theory of EDLs and their development history, then focus on outlining the effects of EDLs on the CO2RR with the research toolkits for studying EDLs which were divided into two types, i.e., cation effects and anion effects. Finally, we end this review by discussing the remaining challenges in understanding and utilizing EDLs to enhance CO2RR performance.
2. The theory of EDLs
An EDL is a structure existing at a charged interface consisting of a fluid and an object (e.g., a solid, a gas bubble, and a liquid droplet) exposed to the fluid.40 In electrochemistry, the EDL can be understood in a simplified model having two parallel layers: one is the charged electrode surface and the other consists of counterions accumulated at a distance of a few nanometers to the surface.41 This model is similar to a parallel-plate capacitor first proposed by Helmholtz.42 The two plates, corresponding to the charged electrode and the accumulated counterions layer, define the Helmholtz layer. Within it, the differential capacitance is a constant, and the potential decreases linearly (Fig. 1a).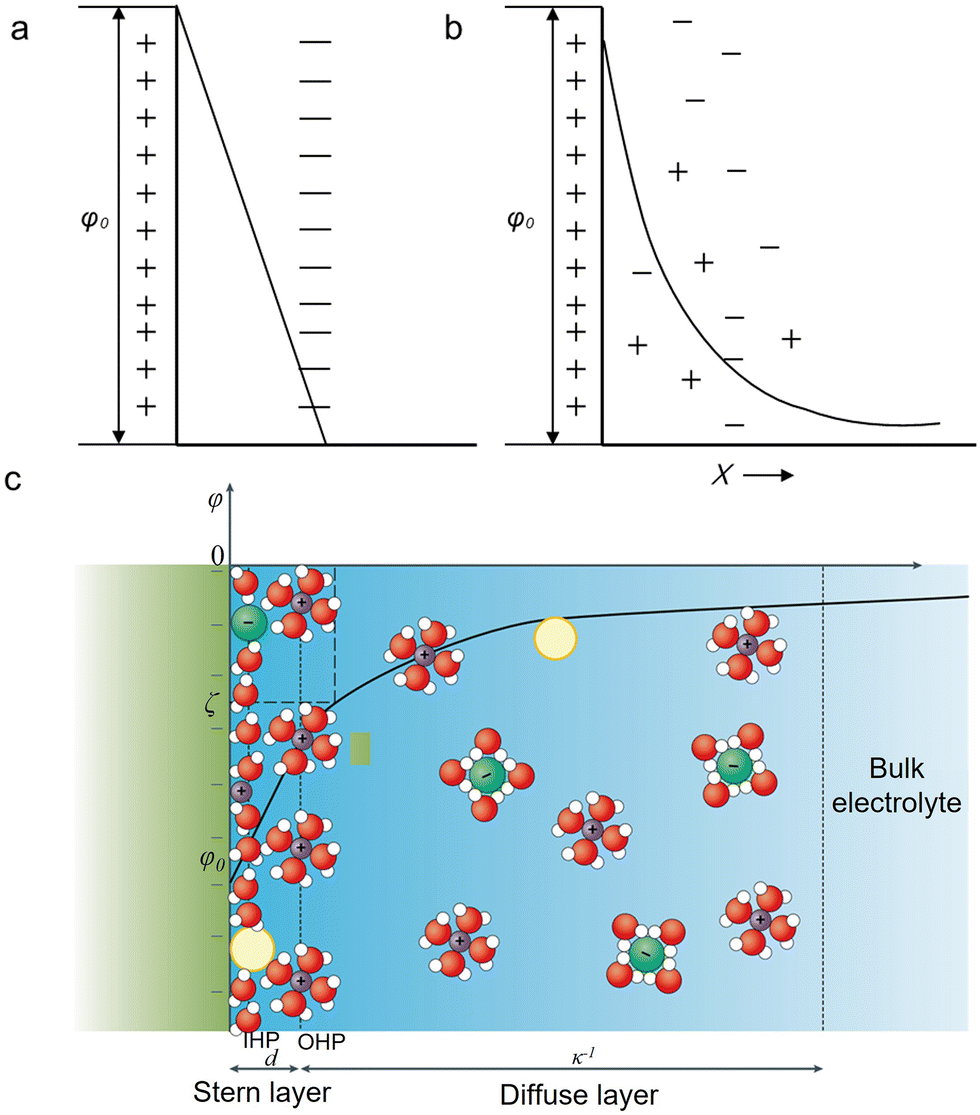 | ||
| Fig. 1 Schematic description of different EDL models at a negatively charged electrode. (a) The Helmholtz model. (b) The Gouy-Chapman model. (c) The Gouy-Chapman-Stern model.40 The yellow, green, purple, red, and white spheres represent neutral species, the anions, the cations, oxygen and hydrogen. Reproduced from ref. 40 with permission from the Springer Nature, copyright 2021. | ||
However, as observed by Gouy and Chapman, the capacitance of an EDL is positively proportional to the applied potential and the ionic concentration.43,44 Thereby, the “Gouy-Chapman model” was proposed. This model describes that the equilibrium distribution of ions as a function of the distance to the surface follows the Maxwell–Boltzmann statistics. That is, the concentration of the counterions will progressively reduce to the bulk concentration at a certain distance. As a result, the interfacial potential exponentially decays away from the surface (Fig. 1b).
Combining the Helmholtz model and the Gouy-Chapman model, the Gouy-Chapman-Stern (GCS) model was obtained.45 The GCS model proposed by Stern regarded ions as point charges. Later, this model was further modified by considering the solvation shells of ions and their specific adsorption on the electrode surface. Thus, this classical description of the EDL consists of three different regions (Fig. 1c): the inner Helmholtz plane (IHP),46 the outer Helmholtz plane (OHP), and the diffusion layer. The IHP passes through the centers of the specifically adsorbed species. The OHP passes through the centers of solvated counterions (hydrated cations in the CO2RR) accumulated at a distance closest to the surface. Finally, the diffusion layer refers to the region beyond the OHP. Within this EDL structure, the electrode surface, ions, and electrically neutral species play essential roles in determining the activity and selectivity for electrochemical processes.47,48 For the CO2RR, as documented in the literature, cations and anions strongly affect the catalytic performance.38,49–51 Thus, in the next section, we will elaborate on how the above factors tune the CO2RR performance.
3. Mechanisms of cation effects
In 1969, Paik et al. found that cations impacted the CO2RR kinetics and the current density increased in the order Li+ < Na+ < (Et)4N+ at a given potential on the mercury electrodes.52 Later, Hori et al. observed that increasing the cation size (crystal ionic size) from Li+ to Cs+ could enhance the selectivity of C2+ products on Cu electrodes.53 Since then, lots of research has been conducted to further deeply investigate the effect of monovalent alkali cations, including Li+, Na+, K+, Rb+ and Cs+, on the CO2RR. Moreover, multivalent cations54 and organic cations55 are also used to improve the activity and selectivity, and explore the mechanisms of cation effects. To date, several main hypotheses, including (1) the specific adsorption of cations,53 (2) the hydrolysis of hydrated cations,56 (3) the interactions between the intermediates and the electric field or cations,57,58 (4) the suppression of the HER,59 and (5) other effects including multivalent or organic cations have been proposed to interpret the above disparity. With a better understanding by combining the density functional theory (DFT) calculations and advanced characterization methods, the evolution of these assumptions is still ongoing.3.1. Specific adsorption of cations
Murata and Hori first proposed that the potential change at the OHP led to the differences in selectivity and activity among different cations.53,60 Such a potential change is highly related to the hydration extent of cations, which further affects the local pH. Smaller cations (e.g., Li+) are strongly hydrated, thus hindering their specific adsorption on the electrode surface. In contrast, larger cations (e.g., K+ and Cs+) with a lower hydration number are able to adsorb more easily on the surface. As the specific adsorption of cations gave rise to a positive shift of the OHP potential, the electrode potential became more positive for larger cations at a constant current density.61 Meanwhile, the concentration of hydrogen anions (H+) decreased in the electrolytes containing larger cations, resulting in higher pH values at the OHP and thereby increasing the C2/C1 product ratio. Besides, specifically adsorbed larger cations have also been theoretically proven to promote the C–C coupling through stabilizing the *CHO intermediates.62 More recently, similar cation effects were also observed in the CO2RR on silver GDEs, where an enhancement of CO FE was achieved with increasing cation size.63 This improvement was also rationalized by the interplay between the hydration level of cations and the extent of cations’ special adsorption. Furthermore, surface-enhanced infrared absorption spectroscopy (SEIRAS) was experimentally conducted to probe the asymmetric CH3 deformation band of tetramethylammonium (methyl4N+) by Vincent et al. and they observed the specific adsorption of alkali cations on Au electrodes.64 Compared with the pure methyl4N+ electrolyte, a negative-going band at ∼1842 cm−1 appeared after the addition of cations, which is due to the displacement of specifically adsorbed methyl4N+ by cations. Meanwhile, through integrating the area of the above band, the surface coverage of specifically adsorbed cations increased in the order Li+ < Na+ < K+ < Cs+, indicating that cations with softer hydration shells are more likely to specifically adsorb on the surface.However, Strmcnik et al. and Milles et al. argued that the specific adsorption of hydrated cations failed to account for the above-observed impact, based on the DFT calculations.65,66 The equilibrium potentials for the specific adsorption of alkali cations are more negative than −2.4 V vs. the normal hydrogen electrode (NHE) on Au, suggesting that the specific adsorption of cations may not occur at commonly applied potentials of the CO2RR.
So far, whether cations adsorb specifically or electrostatically below the potential of zero charge is still ambiguous in the CO2RR. But most of the recent studies on the CO2RR tended to accept that cations accumulate near the electrode surface via non-covalent interactions rather than specific adsorption.55–58
3.2. Hydrolysis of hydrated cations
Recently, another hypothesis about cation effects in the CO2RR was proposed by Singh and co-workers, attributing this to the buffering capacity of the hydrolysis of hydrated cations near the cathode.56| [M+(H2O)n] + H2O ⇌ [MOH(H2O)n−1] + (H3O)+ |
The pKa of the above reaction at the electrode surface is
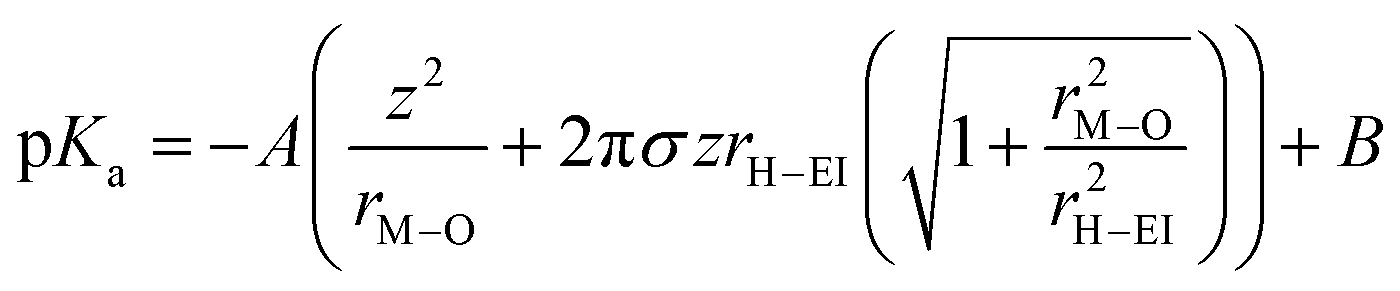
In light of the above equation, the calculated pKa values for different hydrated cations were comparable in the bulk electrolytes, but decreased near the electrodes in the sequence of Li+ (11.64) > Na+ (10.26) > K+ (7.95) > Rb+ (6.97) > Cs+ (4.31) at −1 V versus the reversible hydrogen electrode (RHE). Thus, Cs+ displayed a stronger buffering ability, offering more protons to counteract the increasingly local hydroxide anions (OH−) and thereby maintaining the local pH close to that in the bulk (pH = 7) in comparison with the case of Li+.67 This neutral microenvironment impeded the reaction of CO2 molecules with OH− to produce bicarbonate (HCO3−) or CO32− anions, thus enhancing the local concentration of CO2 available for the reduction (Fig. 2a). Clearly, according to the calculation results, this pronounced effect promoted the FE of CO on Ag cathodes from 55.8% in Li + to 90.2% in Cs+. Meanwhile, such a noticeable improvement also occurred in the C2/C1 selectivity ratio over Cu electrodes.
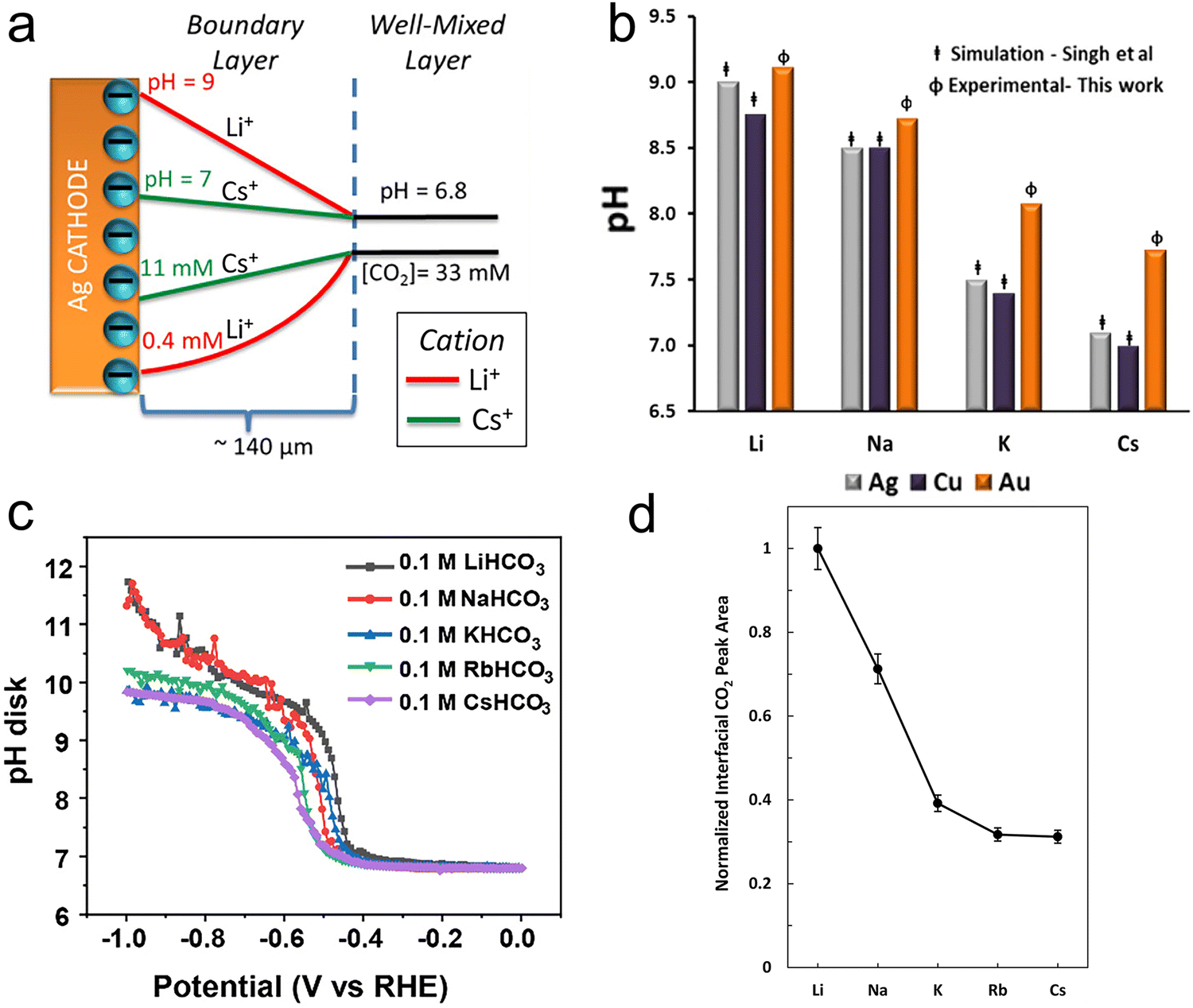 | ||
| Fig. 2 (a) Profiles of pH and CO2 concentrations as a result of the cations’ pKa values on Ag electrodes.56 Reproduced from ref. 56 with permission from the American Chemical Society, copyright 2016. (b) Steady-state pH values at the interface in CO2-saturated 0.05 M carbonate solutions at −1.0 V vs. RHE on Au electrodes in comparison with the results from Singh et al.'s calculations.68 Reproduced from ref. 68 with permission from the American Chemical Society, copyright 2017. (c) The interfacial pH values under various potentials in 0.1 M CO2-saturated electrolytes with different cations.70 Reproduced from ref. 70 with permission from the Royal Society of Chemistry, copyright 2023. (d) The interfacial CO2 concentration was determined by measuring the areas of the dissolved CO2 band in different cations and then normalizing the band areas with that of Li+.71 Reproduced from ref. 71 with permission from the American Chemical Society, copyright 2020. | ||
However, this theoretical buffering capability of cations was overestimated, when compared with the experimental results. Cuesta et al. noted that the pH at the electrode surface could be probed using SEIRAS.68 As the concentration equilibrium between CO2 and HCO3− species is related to the concentration of H+, the pH change at the interface during the CO2RR can be determined by the ratio of the integrated intensity between the CO2 and HCO3− bands. As shown in Fig. 2b, while the local pH decreased with increasing cation size, the experimental pH values for larger cations were much higher than those obtained using DFT calculations. Moreover, similar results were obtained on a rotating ring-disc electrode as well.68–70 Liu et al. developed a considerably sensitive pH sensor by using the redox couple of 4-hydroxylaminothiophenol (4-HATP)/4-nitrosothiophenol (4-NSTP), and combined this sensor with a rotating ring-disc electrode system to directly estimate the interfacial pH of gold electrodes.70 As illustrated in Fig. 2c, the interfacial pH decreases with increasing the cation size from Li+ to Cs+. However, the trend and the cations’ buffering ability are both less pronounced compared to the calculated theoretical results. On the other hand, since the peak potential for the oxidation of CO on the Pt ring electrode is sensitive to the local pH, and shifts by −86 ± 2 mV pH−1, such a potential change can quantitatively describe deviations in the local pH among cations.69 The local basicity followed the trend Li+ > Na+ > K+ > Cs+ under the same CO2RR conditions in Singh et al.'s work, but the difference in local pH values between Li+ and Cs+ electrolytes was only 0.28 at a current density of 5 mA cm−2, and 0.74 even at 15 mA cm−2. Such a small difference in pH values between Li+ and Cs+ may not be considered the reason for the activity improvement.
Instead of boosting the interfacial CO2 concentration by increasing the cation size according to previous theoretical calculations, Xu et al. found the opposite trend by using attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS).71 As displayed in Fig. 2d, while the integrated area of the dissolved CO2 band decreased by 69% from Li+ to Cs+, the partial current density of CO increased more than 10 times. This suggests that the local CO2 concentration mainly depended on the consumption rate, specifically the reaction rate, rather than on the buffering capacity of hydrated cations. Thus, a lower local CO2 concentration was obtained in electrolytes containing larger cations, since they accelerated the rate of the CO2RR.
3.3. Interactions between intermediates and the electric field or cations
The above two hypotheses do not consider the effect of an electric field on the energetics of the uncharged species which have a large dipole moment and/or polarizability. As the electric field over 0.1 V Å−1 could vary the energies of molecular orbitals of adsorbates, the electrostatic forces would markedly alter the adsorption free energies of intermediates and the activation barrier, thus changing the activity and selectivity for electrochemical reactions.72 Through introducing an explicit model of the electrochemical interface, Nørskov et al. investigated the effect of the electric field resulting from hydrated cations and their corresponding image charges at the interface in the CO2RR on Ag(111) electrodes.57 With K+, an applied electric field of −1 V Å−1 significantly lowers the free energy of the intermediate *CO2 and thus stabilizes this key intermediate. On Cu electrodes, the surface intermediates with strong dipole moments (e.g., *CO2, *CO, *OCCO) could also be stabilized by the cation-induced electrostatic field.73,74Furthermore, the interfacial field strength varies with different cations, which impacts the extent of the stabilization effect on the dipolar and polarizable intermediates and further gives rise to pronounced differences between cations.73 Increasing the cation size leads to higher electric field strength, which can greatly stabilize the intermediates. Besides, as the intermediate *CO has a larger dipole moment than *CHO, the key intermediate for forming methane, larger cations are more inclined to enhance the formation rates of C2 products than C1 products on Cu electrodes (Fig. 3a). Perez-Gallent et al. also noted that the cation-induced field could dramatically stabilize the C2 intermediates with respect to C1 species in the CO reduction reaction, resulting in an increasing trend of the selectivity towards ethylene with the increase of the cation size.75 To interpret the above changes, the enhancement of the electric field intensity was attributed to the accumulation of more cations at the OHP, determined by a few factors, including (i) the driving force for cations,73 (ii) the repulsion effect,76 and (iii) the effect of volume exclusion.77
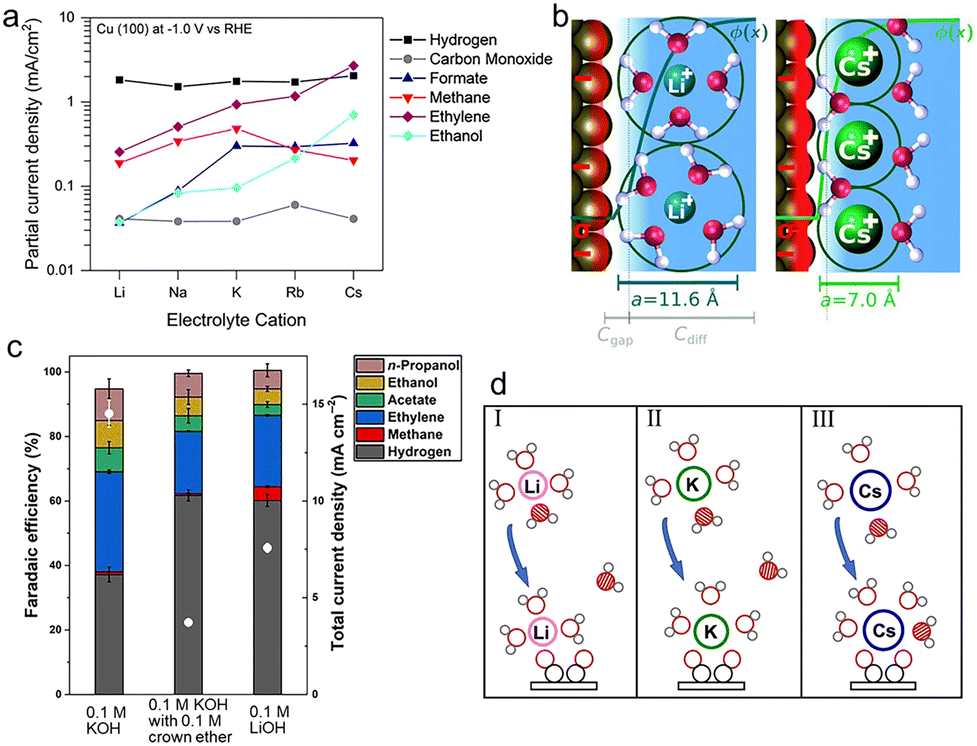 | ||
| Fig. 3 (a) Partial current densities for each product of CO2 reduction on Cu(100) at −1.0 V vs. RHE as a function of the electrolyte cation.73 Reproduced from ref. 73 with permission from the American Chemical Society, copyright 2017. (b) The repulsion between solvated cations at the OHP reduces the local concentrations of cations, the EDL field, and the surface charge density (σ, depicted by the red region). The diameters for solvated Li+ and Cs+ are 11.6 Å and 7.0 Å, respectively.76 Reproduced from ref. 76 with permission from the Royal Society of Chemistry, copyright 2019. (c) FEs for each product and total current densities (white dots) obtained on Cu electrodes at −0.7 V vs. RHE in 0.1 M potassium hydroxide (KOH) with and without 0.1 M crown ether, and 0.1 M lithium hydroxide (LiOH), respectively.82 Reproduced from ref. 82 with permission from the Science Advances is the American Association for the Advancement of Science, copyright 2020. (d) Schematic diagrams of the coordination of hydrated cations Li+(I), K+(II), and Cs+(III) with water and the intermediate *OCCO during the CO coupling process.85 Reproduced from ref. 85 with permission from the American Chemical Society, copyright 2021. | ||
In terms of the driving force, it would be enhanced by increasing the cation size, thus improving the movement of cations to be at the OHP from the bulk electrolyte and leading to a higher concentration of larger cations near the electrode. Especially, the driving force for Cs+ is more favorable by ∼1 eV compared to that for Li+.73 This result can be experimentally proved by analyzing the peak frequency of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretch band of surface-adsorbed CO using time-resolved and surface-sensitive infrared spectroscopy.78 With increasing cation size, the CO stretch frequency shifted to lower energies, suggesting that surface-adsorbed CO molecules experienced a stronger interfacial field.
O stretch band of surface-adsorbed CO using time-resolved and surface-sensitive infrared spectroscopy.78 With increasing cation size, the CO stretch frequency shifted to lower energies, suggesting that surface-adsorbed CO molecules experienced a stronger interfacial field.
As for the repulsion effect, Ringe et al. noted that larger cations with a smaller hydrated cation radius displayed smaller repulsion and therefore became more concentrated at the OHP by combining the size-modified Poisson–Boltzmann theory and ab initio simulation (Fig. 3b).76 The effective radii of hydrated cations follow the order Li+ (5.8 Å) > Na+ (5.2 Å) > K+ (4.1 Å) > Rb+ (3.9 Å) > Cs+ (3.5 Å),79,80 indicating that the highest concentration of Cs+ is formed close to the electrode. Moreover, due to the volume exclusion, the migration of hydrated cations to the OHP would reach the steric limit with increasing applied potentials, further forming a condensed layer.77 The higher concentration of larger cations at the OHP was observed with a thinner layer, which was also in favor of the mass transport of CO2 to the electrode surface.
According to the GCS model, the variation of the electric field strength among different cations can be experimentally quantified by measuring the Stark tuning rate, which is dependent on the effective size of hydrated cations.81,82 The Stark tuning rate increases from 29 to 39 cm−1 V−1 from Li+ to K+, and stabilizes at 39 to 41 cm−1 V−1 from K+ to Cs+. Compared with Li+, the relatively larger Stark tuning rates correspond to higher field intensity, as well as smaller effective sizes of hydrated cations following the order Li+(H2O)x > Na+(H2O)x > K+(H2O)x ∼ Rb+(H2O)x ∼ Cs+(H2O)x. Moreover, smaller cations like Li+ have a thicker hydration shell with more than one layer of water molecules, still effectively impacting the reactions at the interface.
Besides the above-mentioned intermediate-field medium-range interactions, the short-range interactions between intermediates and hydrated cations may have a similar stabilization effect as well. For instance, the FE for C2+ products increased from Li+ to K+ and kept primarily unchanged from K+ to Cs+, which was in line with the trend of electric field strength.82 However, the current density increased monotonically with the cation size. Due to the observed similar peak positions and lineshapes of adsorbed CO with K+ and Cs+ from the ATR-SEIRAS spectra, these cations showed comparable interactions with the adsorbed CO. Furthermore, chelating K+ with the crown ether would result in the same Stark tuning rate as that of hydrated Li+, demonstrating that the chelated K+ and hydrated Li+ had similar field strength and effective sizes. The comparable field strength resulted in a similar product distribution, but the current density for Li+ was 2 times higher than that for the chelated K+ (Fig. 3c). The above results manifested that the reaction rate was directly related to the nature and structure of the cation. The differences between larger cations could be due to the increasingly looser association with the hydration shell. Furthermore, at the same pH, increasing the concentration of Na+ from 0.1 to 1.0 M dramatically promoted the formation of C2+ products without changing the field strength.83 This improvement demonstrates that the nature of cations plays a significant role in tuning the reactivity and selectivity as well.
Such an effect of the nature of cations was further proved using theoretical calculations. By integrating the ab initio molecular dynamics simulations with the slow-growth sampling method, Qin et al. observed that the presence of K+ cations facilitated the rate-determining CO2 activation step by the coordination interactions.84 Through coordinating with CO2 molecules to form a cation–CO2 complex, cations favor the stabilization of the *CO2− intermediates by ∼0.5 eV than the solvation of water molecules, hence initiating the CO2RR.58 The average CO2 coordination numbers obtained from ab initio molecular dynamics simulations are 0.1 ± 0.3, 0.9 ± 0.2, 0.9 ± 0.4, 1.3 ± 0.5 for Li+, Na+, K+ and Cs+, respectively. Due to its hard solvation shell, Li+ poorly coordinates with CO2. In contrast, larger cations with a softer hydration shell can not only strongly interact with CO2, but also become more concentrated at the OHP, accounting for the activity trend Cs+ > K+ > Na+ > Li+ in the CO2RR. Moreover, the decrease of the O–C–O angle from a linear 180° to below 140° caused by the cation–CO2 complex, and the enhancement of the electron transfer from the interface to CO2 might also promote the stabilization effect. Another cation–OCCO complex has also been found to improve the conversion of CO2 to C2 products.85 As displayed in Fig. 3d, compared to Li+, larger cations K+ and Cs+ would get coordinated with two oxygen atoms in the intermediates *OCCO simultaneously, which made the cation–intermediate complex more stable.
3.4. Suppression of the HER
As a major competing reaction during the CO2RR, the HER significantly affects the selectivity of main products.86 Thus, the suppression of the HER is vital to improve the conversion of CO2. In the alkaline and neutral electrolytes, high OH− concentrations at the interface greatly suppress the formation of H2.28,87 However, OH− reacts with CO2 to form CO32−, drastically reducing the local concentration of CO2 and its utilization efficiency.88–90 Such an issue could be avoided by using acidic electrolytes, while the HER can be suppressed by the addition of cations.91,92 For instance, nearly 100% FE of hydrogen was obtained on Au electrodes in CO2-saturated 0.1 M perchloric acid (HClO4) solutions (pH = 1), while an 80% FE of CO was achieved in CO2-saturated 0.1 M potassium perchlorate (KClO4) solutions (pH = 3).93 Moreover, the efficient reduction of CO2 on Cu electrodes was reported in mixed electrolytes containing 1 M phosphoric acid (H3PO4) and 3 M potassium chloride (KCl).92 More recently, single-pass CO2 utilization efficiencies of 85% for CO2 to CO at 600 mA cm−2 and 60% for CO2 to C2+ at 500 mA cm−2 under acidic conditions, respectively, were achieved, which greatly reduced the CO2 loss.94,95 However, the mechanism of the cation suppression effect is still unclear.The HER consists of two pathways, the reduction of protons (2H+ + 2e− → H2) and the reduction of water (2H2O + 2e− → H2 + 2OH−).96–98 As the proton reduction has an earlier onset potential than water reduction, the water reduction will become the major reaction by replacing the proton reduction under more negative potentials.99 Gu et al. noticed that cations could suppress the proton reduction under the potentials more positive than the onset potential for the water reduction.59 In pure trifluoromethanesulfonic acid (HOTf), no limiting current density of the proton reduction was observed. In contrast, with the addition of 0.4 M potassium trifluoromethanesulfonate (KOTf), a plateau of limiting current density appeared at −0.6 V vs. the standard hydrogen electrode (SHE), which was due to the mass transport limitation of hydronium. Thus, in comparison with the K+-free medium, hydrated cations at the OHP impeded the migration of hydronium ions toward the electrode, thereby decreasing the concentration of hydronium ions near the electrode. Through a further quantitative study, the identity of cations hardly impacted the plateau current density.100 Meanwhile, increasing the concentration of cations, especially when it is higher than that of H+, would substantially suppress the migration rate and the diffusion rate of H+, and further restrain the HER.
Furthermore, the cations also highly affect the surface coverage of protons, further the activity of the HER. Xu et al. noticed that the proton adsorption was energetically unfavored in K+ compared to that in Na+, owing to the higher orbital sensitivity and a stronger preference in the binding of K+ ions.101 Thus, the H2 FE largely decreased from 73% (Na+) to 24% (K+). Additionally, due to the competitive adsorption of cations, increasing the concentration of cations could markedly decline the local concentration of protons near electrodes.102 Such competition could suppress the HER and offer more local interactions to stabilize the intermediates *OCOH, ultimately improving the efficient conversion of CO2.
3.5. Other effects (multivalent cations and organic cations)
Apart from alkali cations, multivalent cations and ionic surfactants can also promote the CO2RR, according to recent studies.54,103 As multivalent cations having larger surface charges could improve the interfacial electric field strength, the cations including Ba2+ and La3+ displayed up to 2 orders of magnitude higher activity for the production of CO than Cs+.76 Such an acceleration effect was also observed on the rate of the CO2RR over the Cu–Sn–Pb alloy electrodes.104 The reaction rate in the electrolyte containing La3+ was 2 times higher than that in the case of Na+ at a lower overpotential (−0.65 V vs. Ag/AgCl).To explicitly unravel the effect of multivalent cations, more dilute electrolytes (2 mM Mn+= Li+, Cs+, Be2+, Mg2+, Ca2+, Ba2+, Al3+, Nd3+, and Ce3+; bulk pH = 3) were used to minimize the possible deposition including oxides and hydroxides. As shown in Fig. 4a, Monteiro et al. found that the cation acidity, determining the accumulation of cations at the OHP and water dissociation kinetics, affected the activities of the HER and CO2RR.54 On the one hand, as the activation barrier of water dissociation decreased with cation acidity, the weakly hydrated trivalent cations greatly increased the reaction rate of water reduction. Meanwhile, the softly hydrated cations including Ba2+ and Nd3+ could increasingly accumulate at the OHP and further accelerate the water reduction, due to the minimal repulsion. As for the CO2RR, the short-range interactions generated by coordination between cations and the *CO2− species could stabilize this intermediate, hence promoting the CO2 reduction. Therefore, at low potentials where the proton reduction reaction mainly takes place, more CO can be produced with increasing cation acidity, since the proton reduction is independent of the cation identity. In contrast, at high potentials, the activity for CO increased in the order Ca2+ < Li+ < Ba2+ < Cs+, as acidic cations extremely promoted the water reduction. In consequence, the interplay between the specific cation-intermediate interactions, the concentration of cations at the OHP, and the activity for water reduction should be taken into account for the investigation of the cation effect.
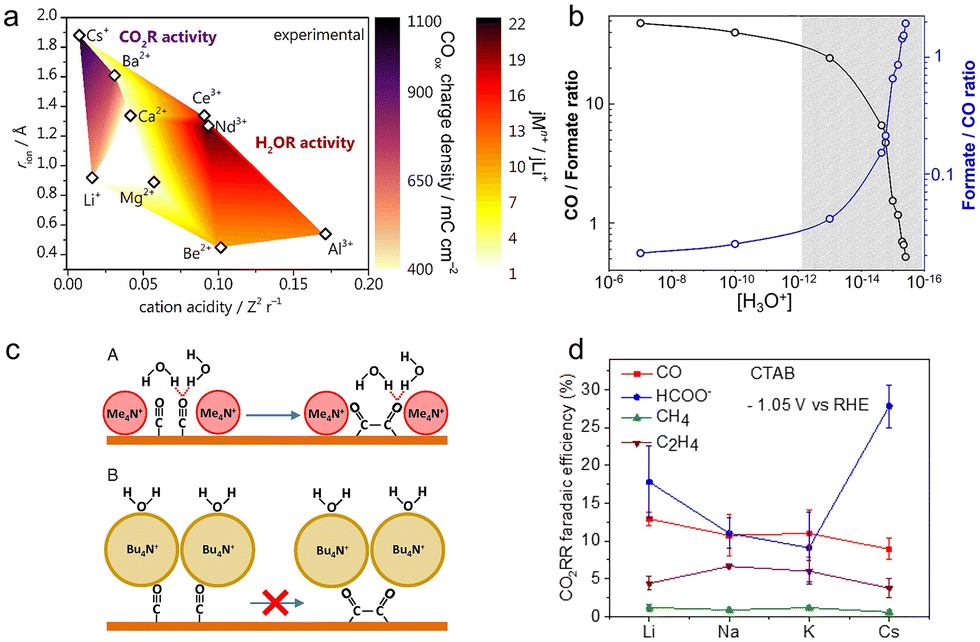 | ||
| Fig. 4 (a) The color map of the CO2RR activity (purple shades) and HER activity (red shades) at high potentials as a function of cation acidity and cationic radius.54 Reproduced from ref. 54 with permission from the American Chemical Society, copyright 2022. (b) The CO/formate ratio and formate/CO ratio at different concentrations of hydronium.106 Reproduced from ref. 106 with permission from the American Chemical Society, copyright 2018. (c) The schematic of the CO coupling mechanism in the presence of methyl4N+ and butyl4N+.103 Reproduced from ref. 103 with permission from the Proceedings of the National Academy of Sciences of the United States of America, copyright 2019. (d) FEs for main products in the presence of CTAB and different cations at −1.05 V vs. RHE.112 Reproduced from ref. 112 with permission from the American Chemical Society, copyright 2020. | ||
In contrast to the above promotion effect, Bhargava et al. noticed that multivalent cations hindered the reaction rate and selectivity for CO production.105 Compared to monovalent cations, multivalent cations would impede the CO2 adsorption on the catalyst surface by blocking the active sites with in situ generated deposits including hydroxide, oxide, and carbonate. But this research was conducted in the electrolytes with a high concentration of cations (3 M) under high current densities, which led to a higher local pH value. This may be a possible reason for the reverse trend.
Hydronium within the double layer can switch product selectivity on Ag electrodes by altering the activation barrier for the hydrogenation of the intermediate *CO2−.106 Compared to the formation of *COOH, the intermediate for producing CO, the activation energy barrier for forming *CHOO, and the intermediate for producing formate significantly dropped in the absence of hydronium. In the experiment, drastically reducing the local concentration of hydronium could hugely contribute to the switch of the reaction pathway from CO2 to formate, achieving ∼60% FE of formate in 11 M KOH (Fig. 4b).
Specially, for the reduction of CO2 to ethylene, the change in its selectivity correlates with the hydrogen bonding between the interfacial water and the CO dimer, rather than the strength of the cation-induced electric field.103 Under the experimental conditions, the field strength had a negligible effect on the adsorption energy of CO on Cu, although the sizes of quaternary alkyl ammonium cations follow the order methyl4N+ < ethyl4N+ < propyl4N+ < butyl4N+. Using SEIRAS, a sharp band was observed for the electrolytes containing methyl4N+ and ethyl4N+, which was due to the hydrogen bonding of water to the terminal oxygen of adsorbed CO. Such interactions tended to stabilize the CO dimer and facilitate the formation of ethylene. But, due to the larger size and more hydrophobic nature, propyl4N+ and butyl4N+ effectively displaced water molecules from the interface. This displacement further led to the disruption of the interaction between the adsorbed CO and water, thus hindering CO dimerization (Fig. 4c). This difference can account for the production of ethylene only in methyl4N+- and ethyl4N+-containing electrolytes.
Introducing the organic cation of cetyltrimethylammonium bromide (CTAB) to the electrolytes can increase the rate of the CO2RR and suppress the HER.108 With the presence of CTAB in 0.1 M sodium bicarbonate (NaHCO3), the FE of H2 approximately dropped by half, while the formate selectivity reaches almost 50% at −0.6 V vs. RHE.109 Besides, increasing the concentration and chain length of CTAB could further improve the selectivity of CO and formate, and suppress the HER. According to the electrochemical impedance spectroscopy (EIS) study, the double layer capacitance value was lower in the presence of CTAB, which was attributed to the displacement of hydronium and hydrated Na+ cations109 and the formation of an ordered structure of CTAB enhancing the electron transfer at the interface.110 This ordered surfactant assembly would repel isolated water near the interface and hinder the tendency of hydrogen atoms of local water molecules to approach the interface.110 Combined with the depletion of the local proton source, both factors significantly inhibited the HER. At the same time, the CTAB at the interface disturbed the binding configuration of CO, shifting the CO band to a lower frequency, further increasing the CO surface coverage, and thus enhancing the activity of the CO2RR.111
The presence of CTAB disfavored the formation of C2 products but improved the formation of formate.112 In Fig. 4d, the activity and selectivity for the CO2RR were virtually independent of the identity of cations (Li+, Na+ and K+) with CTAB. But an unexpected enhancement in formate selectivity was observed in the presence of Cs+ and CTAB, while the selectivity for ethylene significantly dropped compared to the CTAB-free Cs+ electrolytes. Due to its hydrophobicity, the CTAB inhibited the C–C bond formation by disturbing the hydrogen bonding, reducing the formation of C2+ products. Meanwhile, Cs+ with a smaller hydrated size formed a more compact layer, thereby more easily displacing partial CTAB than other cations, leading to a higher formate FE.
4. Mechanisms of the anion effect
According to the buffer capacity, anions can be divided into two types: buffering anions (HCO3−, CO32−, and H2PO4−) and non-buffering anions (Cl−, Br−, I−, ClO4−, SO42−, and OH−). Buffering anions can modulate the local pH by acting as a proton source, thus affecting the selectivity of H2 and methane (CH4) whose rate-determining steps (RDSs) involve the participation of protons.107,113–116 Compared with non-buffering anions, the local pH would not change dramatically in buffering electrolytes (HCO3− or H2PO4−), preferentially facilitating the production of CH4 and H2.117 Particularly, the phosphate anions showed negligible activity towards C2+ products and largely favored the formation of H2 (FE >70%) and CH4. Besides, increasing the concentration of HCO3− from 0.05 to 0.2 M could enhance partial current densities of H2 and CH4. Resasco et al. ascribed the above enhancement to the pKa of the buffering anions.118 The equilibrium constant for the deprotonation of HCO3− is 104 times higher than that for the deprotonation of H2O, proving that HCO3− could supply its own protons to the electrode surface. Furthermore, upon decreasing the pKa of buffering anions, the reaction rates for producing H2 and CH4 increases in the sequence HCO3− (pKa = 10.33) < H3BO3 (pKa = 9.23) < H2PO4− (pKa = 7.21). Thus, this substantially low pKa of phosphate anions may account for the high selectivity of H2 as well.In buffering anions, HCO3− also acts as a carbon source.119 Through analyzing the CO product from the isotopically labelled 13CO2 reduction reaction in the NaH12CO3 electrolytes, 89% of the CO product was 12CO (Fig. 5a). Moreover, surface adsorbed CO2 molecules were also from the HCO3− anions at the start of the reaction, observed by the real-time ATR-SEIRAS.120 In light of the above results, it is clear that the vast majority of adsorbed CO2 molecules are from HCO3− rather than the freely dissolved CO2 molecules. Meanwhile, such a rapid equilibrium between HCO3− and dissolved CO2 molecules increased the effective concentration of CO2 near the electrode surface, hence boosting the rate of CO production (Fig. 5b). Moreover, Shan et al. noted that HCO3− could improve the stability of the intermediate *OCO− on the surface, hence facilitating the formation of this intermediate and further formate.121
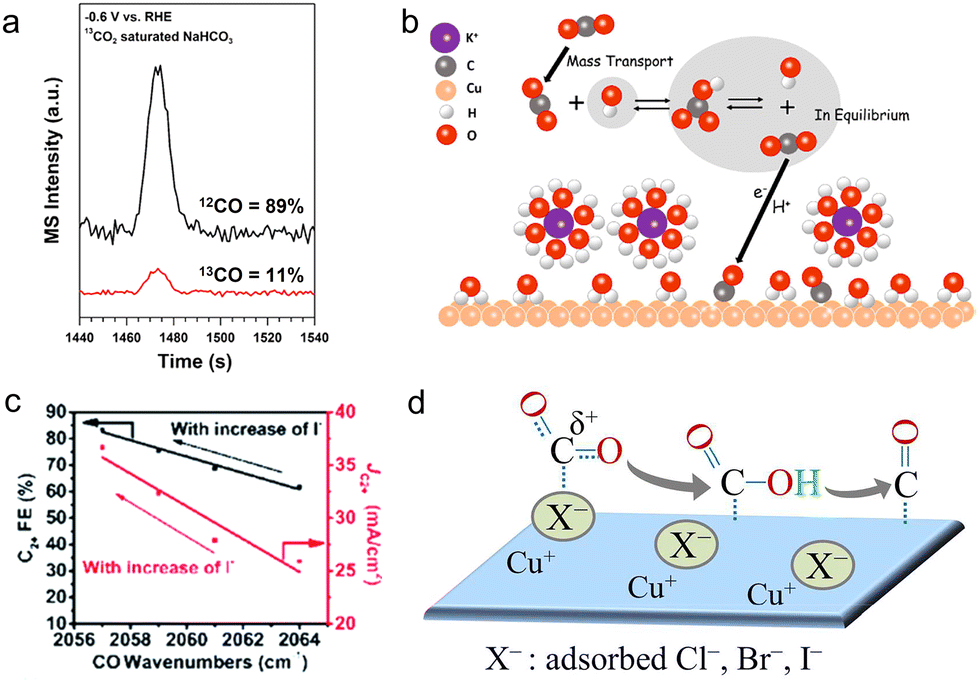 | ||
| Fig. 5 (a) The mass spectra of the CO product under −0.6 V vs. RHE in the 13CO2 saturated NaH12CO3 electrolyte.119 Reproduced from ref. 119 with permission from the American Chemical Society, copyright 2017. (b) The schematic of the proposed mechanism for providing CO2 molecules from HCO3−.120 Reproduced from ref. 120 with permission from the American Chemical Society, copyright 2017. (c) Variation of FE and current density of C2+ with CO wavenumbers as the concentration of I− varies.126 Reproduced from ref. 126 with permission from the Royal Society of Chemistry, copyright 2022. (d) Illustration of the improvement in the adsorption of CO2 and the stabilization of the intermediate *CO in the presence of halides.123 Reproduced from ref. 123 with permission from the American Chemical Society, copyright 2017. | ||
In terms of non-buffering anions, the formed higher local pH gave better selectivity to ethylene (C2H4) (>45%) and total C2+ products (>65%). Especially for halide anions of Cl−, Br− and I−, they could specifically adsorb on the surface of catalysts, changing surface morphologies and surface charges during the CO2RR.122–124 According to the DFT calculations, the specific halide adsorption is increasingly favorable in the order Cl− < Br− < I− on a well-defined Cu surface.122 In the meantime, the enhanced adsorption ability of halides will affect the negative charge transfer of adsorbed halides to the Cu surface. Hence, compared with Cl− and Br−, a more negative charge of I− could be readily transferred to the Cu surface, modifying the local electronic environment.125 This further improves the interactions between Cu and the intermediates *CO2 and *CO, thus promoting the protonation of *CO and further forming CH4. Consequently, the presence of I− impedes the CO production and enhances the CH4 selectivity 6 times higher compared with the halide-free (potassium bicarbonate (KHCO3)) electrolyte. Besides, the specific adsorption of halide anions may lead to the transfer of their negative charges to the intermediates *CO, giving rise to the stronger adsorption of *CO and further faster C–C coupling kinetics. This was demonstrated by the decrease of the wavenumbers of CO with increasing concentration of halide anions using in situ ATR-SEIRAS, and a higher C2+ FE of 84.5% was achieved (Fig. 5c).126
Different from the above results, Dunfeng et al. noted that the addition of halides had a minor effect on the selectivity of all products, but mainly increased the reaction rates and positively shifted the onset potentials for forming C2 products, following the trend no halide < Cl− < Br− < I− in the mixed electrolytes containing 0.1 M KHCO3 and 0.3 M KX (X = Cl, Br, and I).123 Such improvement may result from the effect of halides on the common step for the generation of all products. As shown in Fig. 5d, the specifically adsorbed halide formed a covalent X−–C bond with a local CO2 molecule by donating its partial charge to the carbon atom. This further changed the linear form of CO2 to the bent species, thereby facilitating the adsorption of CO2 and stabilizing the intermediate *COOH. Moreover, while the specific adsorption of halides led to the nanostructuring of the Cu surface, especially I−, the observed similar current densities among halides demonstrated that the surface morphology differences could not account for the change in activity.
Unlike the above effects of halides on the CO2RR in mixed solutions, the FE of C2 products could be greatly enhanced in the halide-only electrolytes with the sequence of ClO4− < Cl− < Br− < I−.127 The ethylene and ethanol FEs increased from 31% and 7% (ClO4−) to 50% and 16% (I−), and the associated current densities increased five and seven times, respectively. By using linear sweep voltammetry, a higher population of *CO adsorbed on the surface was observed in the presence of I−, thus promoting the C–C coupling to C2 products. A similar effect was also found on the zinc (Zn) electrodes in the CO2RR.128 In comparison with the FE of CO (∼13%) in the KHCO3 solution, a four times higher CO selectivity (∼48%) was achieved in the KCl electrolyte.
Hydroxide anions also favor the reduction of CO2 to C2+ products and a high ethylene FE of 70% at −0.55 V vs. RHE was obtained on the GDE.28 According to the DFT calculations, the OH− tended to lower the binding energy of the intermediate *CO. Meanwhile, by increasing the charge imbalance between carbon atoms in the adsorbed intermediate *OCCO, the OH− could greatly stabilize this intermediate. Thus, the combined effects could reduce the energy barrier for the *CO coupling, significantly increasing the formation of C2+ products, especially ethylene.
5. Conclusions and outlook
In this review, we presented the development of an EDL model and discussed the effects of different species in the electrolytes on the properties of the EDL, and thus on the activity and selectivity for the CO2RR. The solvated cations, adsorbed at the interface, could stabilize the intermediates including *CO2 and *OCCO through coordination. Meanwhile, the accumulation of cations at the OHP would enhance the interfacial electric field, favoring the stabilization of intermediates with larger dipole moments. Besides, the pKa values varying with the sizes of cations affect the local pH and the concentration of CO2, and further product selectivity. In addition, the effects of different anions on the activity and selectivity for the CO2RR were also discussed.Apart from the investigation on the mechanisms of the cation effect, researchers have developed different strategies to leverage this effect. Based on the DFT calculations, the adsorbed K+ can lower the thermodynamic energy barrier for the conversion of CO2 to CO on Au electrodes.129 Sargent et al. managed to significantly improve the concentration of K+ near the electrode by using the Au nanoneedles that produce high local electric fields, resulting in an order of magnitude improvement in performance compared to other Au electrodes with different morphologies. Besides, in the acidic systems, the perfluorosulfonic acid ionomer has also been used to concentrate the K+ on the catalyst surface, which could increase the C2+ selectivity by the electrostatic interactions of K+ with specific adsorbates.92 At a current density of 1.2 A cm−2, 61% of the CO2RR selectivity including 40% FE of C2+ products was achieved. On the other hand, because of having a compact EDL, K+ has also been introduced to improve the electron transfer from the electrode to carbamate and the adsorption energy of reactants, further promoting the conversion of CO2 captured by amine-based chemical solvents.130
On the other hand, in situ spectroscopies including SEIRAS and surface-enhanced Raman spectroscopy (SERS) have often been employed to explore the reaction interface and mechanisms of the CO2RR.131–133 Both in situ studies aim at identifying the identity and configuration of intermediates during the CO2RR, and their evolution as a function of electrode potential, electrolyte composition and other parameters. Hence, this will vastly assist researchers in investigating the reaction pathways and the effect of reaction environments (especially ions in the EDL). In our review, SEIRAS can be used to probe the local pH and observe a dynamic equilibrium between bicarbonate and dissolved CO2, and investigate the interaction between cations and the adsorbed intermediate *CO. However, for other intermediates, it is considerably hard to detect and confirm their identity, owing to their short residual time and low coverage on the surface. To address this issue, combining SEIRAS and online mass spectroscopy or other instruments which are capable of identifying products will be a possible and challenging strategy. Developing stabilizers and trapping reagents for stabilizing or capturing intermediates might be another way to further understand the mechanism.
Despite the development of in situ spectroscopies, there are still plenty of issues that need to be addressed. In most instances, the catalyst surface is more complicated to study experimentally than theoretically. Such complexity will lead to changes in the local cation distribution and the interfacial electric field and its strength. It is not clear to what extent the catalyst surface will severely affect the cation distribution and the field. Furthermore, the degree of cation hydration and the impact of cations on the structure of water molecules in the hydration shell or the interfacial water molecules and thus on the interactions between intermediates and cations/water molecules warrant further investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work acknowledges the funding support from the National Natural Science Foundation of China (52103300), Guangdong Basic and Applied Basic Research Foundation (2023A1515010572), and the Shenzhen Science and Technology Program (JCYJ20210324132806017 and GXWD20220811163904001).References
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Recent progress in the electrochemical conversion and utilization of CO2, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Recent Advances in Inorganic Heterogeneous Electrocatalysts for Reduction of Carbon Dioxide, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- T. Zheng, K. Jiang and H. Wang, Recent Advances in Electrochemical CO2-to-CO Conversion on Heterogeneous Catalysts, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Recent Progresses in Electrochemical Carbon Dioxide Reduction on Copper-Based Catalysts toward Multicarbon Products, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- M.-G. Kim, J. Park, Y. Choi, H. C. Song, S.-H. Kim, K.-M. Bang, H. C. Ham, N.-K. Kim, D. H. Won, B. K. Min, S. J. Yoo and W. Kim, CuIr Nanoparticles for Electrochemical Reduction of CO2 to t-BuOH, Adv. Energy Mater., 2023, 2300749 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- J. L. Wang, H. Y. Tan, Y. P. Zhu, H. Chu and H. M. Chen, Linking the Dynamic Chemical State of Catalysts with the Product Profile of Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS PubMed.
- Z. Z. Wu, F. Y. Gao and M. R. Gao, Regulating the oxidation state of nanomaterials for electrocatalytic CO2 reduction, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- W. J. Luo, X. W. Nie, M. J. Janik and A. Asthagiri, Facet Dependence of CO2 Reduction Paths on Cu Electrodes, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- R. Dorakhan, I. Grigioni, B.-H. Lee, P. Ou, J. Abed, C. O’Brien, A. Sedighian Rasouli, M. Plodinec, R. K. Miao, E. Shirzadi, J. Wicks, S. Park, G. Lee, J. Zhang, D. Sinton and E. H. Sargent, A silver–copper oxide catalyst for acetate electrosynthesis from carbon monoxide, Nat. Synth., 2023, 2, 448–457 CrossRef.
- X. Wang, P. F. Ou, A. Ozden, S. F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. G. de Arquer, R. K. Miao, C. P. O'Brien, Z. Y. Wang, J. Abed, A. S. Rasouli, M. J. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag-Ru-Cu catalyst, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, Size-Dependent Electrocatalytic Reduction of CO2 over Pd Nanoparticles, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, The p-Orbital Delocalization of Main-Group Metals to Boost CO2 Electroreduction, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- Y. Zhou, R. Zhou, X. Zhu, N. Han, B. Song, T. Liu, G. Hu, Y. Li, J. Lu and Y. Li, Mesoporous PdAg Nanospheres for Stable Electrochemical CO2 Reduction to Formate, Adv. Mater., 2020, 32, 2000992 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- X. Y. Cao, L. L. Zhao, B. R. Wulan, D. X. Tan, Q. W. Chen, J. Z. Ma and J. T. Zhang, Atomic Bridging Structure of Nickel-Nitrogen-Carbon for Highly Efficient Electrocatalytic Reduction of CO2, Angew. Chem., Int. Ed., 2022, 61, e2021139 Search PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- H. Huo, J. Wang, Q. Fan, Y. Hu and J. Yang, Cu-MOFs Derived Porous Cu Nanoribbons with Strengthened Electric Field for Selective CO2 Electroreduction to C2+ Fuels, Adv. Energy Mater., 2021, 11, 2102447 CrossRef CAS.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Efficient Electrocatalytic CO2 Reduction to CO2 Alcohols at Defect-Site-Rich Cu Surface, Joule, 2021, 5, 429–440 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Electrolytic CO2 Reduction in a Flow Cell, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Continuous Carbon Dioxide Electroreduction to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Unlocking direct CO2 electrolysis to C3 products via electrolyte supersaturation, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- M. Duarte, B. De Mot, J. Hereijgers and T. Breugelmans, Electrochemical Reduction of CO2: Effect of Convective CO2 Supply in Gas Diffusion Electrodes, ChemElectroChem, 2019, 6, 5596–5602 CrossRef CAS.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, Insights into the Low Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an Alkaline Flow Electrolyzer, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Catalyst–electrolyte interface chemistry for electrochemical CO2 reduction, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- J. Chen and L. Wang, Effects of the Catalyst Dynamic Changes and Influence of the Reaction Environment on the Performance of Electrochemical CO2 Reduction, Adv. Mater., 2022, 34, 2103900 CrossRef CAS PubMed.
- S.-J. Shin, D. H. Kim, G. Bae, S. Ringe, H. Choi, H.-K. Lim, C. H. Choi and H. Kim, On the importance of the electric double layer structure in aqueous electrocatalysis, Nat. Commun., 2022, 13, 174 CrossRef CAS PubMed.
- G. Gonella, E. H. G. Backus, Y. Nagata, D. J. Bonthuis, P. Loche, A. Schlaich, R. R. Netz, A. Kühnle, I. T. McCrum, M. T. M. Koper, M. Wolf, B. Winter, G. Meijer, R. K. Campen and M. Bonn, Water at charged interfaces, Nat. Rev. Chem., 2021, 5, 466–485 CrossRef CAS PubMed.
- L. R. F. Allen and J. Bard, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2000 Search PubMed.
- H. Helmholtz, Ueber einige Gesetze der Vertheilung elektrischer Ströme in körperlichen Leitern mit Anwendung auf die thierisch-elektrischen Versuche, Ann. Phys., 1853, 165, 211–233 CrossRef.
- M. Gouy, Sur la constitution de la charge électrique à la surface d'un électrolyte, J. Phys. Theor. Appl., 1910, 9, 457–468 CrossRef.
- D. L. Chapman, LI. A contribution to the theory of electrocapillarity, London Edinburgh Philos. Mag. J. Sci., 1913, 25, 475–481 CrossRef.
- O. Stern, Zur Theorie Der Elektrolytischen Doppelschicht, Z. Elektrochem. Angew. Phys. Chem., 1924, 30, 508–516 CAS.
- D. C. Grahame, The Electrical Double Layer and the Theory of Electrocapillarity, Chem. Rev., 1947, 41, 441–501 CrossRef CAS PubMed.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, How cations affect the electric double layer and the rates and selectivity of electrocatalytic processes, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed.
- M. Dunwell, Y. Yan and B. Xu, Understanding the influence of the electrochemical double-layer on heterogeneous electrochemical reactions, Curr. Opin. Chem. Eng., 2018, 20, 151–158 CrossRef.
- M. Moura de Salles Pupo and R. Kortlever, Electrolyte Effects on the Electrochemical Reduction of CO2, ChemPhysChem, 2019, 20, 2926–2935 CrossRef CAS PubMed.
- M. König, J. Vaes, E. Klemm and D. Pant, Solvents and Supporting Electrolytes in the Electrocatalytic Reduction of CO2, iScience, 2019, 19, 135–160 CrossRef PubMed.
- S. J. Shin, H. Choi, S. Ringe, D. H. Won, H. S. Oh, D. H. Kim, T. Lee, D. H. Nam, H. Kim and C. H. Choi, A unifying mechanism for cation effect modulating C1 and C2 productions from CO2 electroreduction, Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed.
- W. Paik, T. N. Andersen and H. Eyring, Kinetic studies of the electrolytic reduction of carbon dioxide on the mercury electrode, Electrochim. Acta, 1969, 14, 1217–1232 CrossRef CAS.
- A. Murata and Y. Hori, Product Selectivity Affected by Cationic Species in Electrochemical Reduction of CO2 and CO at a Cu Electrode, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, The Role of Cation Acidity on the Competition between Hydrogen Evolution and CO2 Reduction on Gold Electrodes, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- S. Banerjee, X. Han and V. S. Thoi, Modulating the Electrode–Electrolyte Interface with Cationic Surfactants in Carbon Dioxide Reduction, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager, III and A. T. Bell, Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, Electric Field Effects in Electrochemical CO2 Reduction, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- Y. Hori and S. Suzuki, Electrolytic Reduction of Carbon Dioxide at Mercury Electrode in Aqueous Solution, Bull. Chem. Soc. Jpn., 1982, 55, 660–665 CrossRef CAS.
- A. N. Frumkin, Influence of cation adsorption on the kinetics of electrode processes, Trans. Faraday Soc., 1959, 55, 156–167 RSC.
- S. A. Akhade, I. T. McCrum and M. J. Janik, The Impact of Specifically Adsorbed Ions on the Copper-Catalyzed Electroreduction of CO2, J. Electrochem. Soc., 2016, 163, F477 CrossRef CAS.
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, Effect of Cations on the Electrochemical Conversion of CO2 to CO, J. Electrochem. Soc., 2013, 160, F69 CrossRef CAS.
- V. J. Ovalle, Y. S. Hsu, N. Agrawal, M. J. Janik and M. M. Waegele, Correlating hydration free energy and specific adsorption of alkali metal cations during CO2 electroreduction on Au, Nat. Catal., 2022, 5, 624–632 CrossRef CAS.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum, Nat. Chem., 2009, 1, 466–472 CrossRef CAS PubMed.
- J. N. Mills, I. T. McCrum and M. J. Janik, Alkali cation specific adsorption onto fcc(111) transition metal electrodes, Phys. Chem. Chem. Phys., 2014, 16, 13699–13707 RSC.
- M. R. Singh, E. L. Clark and A. T. Bell, Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC.
- O. Ayemoba and A. Cuesta, Spectroscopic Evidence of Size-Dependent Buffering of Interfacial pH by Cation Hydrolysis during CO2 Electroreduction, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed.
- F. Zhang and A. C. Co, Direct Evidence of Local pH Change and the Role of Alkali Cation during CO2 Electroreduction in Aqueous Media, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- X. Liu, M. C. O. Monteiro and M. T. M. Koper, Interfacial pH measurements during CO2 reduction on gold using a rotating ring-disk electrode, Phys. Chem. Chem. Phys., 2023, 25, 2897–2906 RSC.
- A. S. Malkani, J. Anibal and B. Xu, Cation Effect on Interfacial CO2 Concentration in the Electrochemical CO2 Reduction Reaction, ACS Catal., 2020, 10, 14871–14876 CrossRef CAS.
- F. Che, J. T. Gray, S. Ha, N. Kruse, S. L. Scott and J.-S. McEwen, Elucidating the Roles of Electric Fields in Catalysis: A Perspective, ACS Catal., 2018, 8, 5153–5174 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- H. Y. Yu, S. E. Weitzner, J. B. Varley, B. C. Wood and S. A. Akhade, Surface Engineering of Copper Catalyst through CO* Adsorbate, J. Phys. Chem. C, 2023, 127, 1789–1797 CrossRef CAS.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Structure- and Potential-Dependent Cation Effects on CO Reduction at Copper Single-Crystal Electrodes, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Correction: Understanding cation effects in electrochemical CO2 reduction, Energy Environ. Sci., 2019, 12, 3609–3610 RSC.
- D. Bohra, J. H. Chaudhry, T. Burdyny, E. A. Pidko and W. A. Smith, Modeling the electrical double layer to understand the reaction environment in a CO2 electrocatalytic system, Energy Environ. Sci., 2019, 12, 3380–3389 RSC.
- C. M. Gunathunge, V. J. Ovalle and M. M. Waegele, Probing promoting effects of alkali cations on the reduction of CO at the aqueous electrolyte/copper interface, Phys. Chem. Chem. Phys., 2017, 19, 30166–30172 RSC.
- M. Nakamura, Y. Nakajima, K. Kato, O. Sakata and N. Hoshi, Surface Oxidation of Au(111) Electrode in Alkaline Media Studied by Using X-ray Diffraction and Infrared Spectroscopy: Effect of Alkali Metal Cation on the Alcohol Oxidation Reactions, J. Phys. Chem. C, 2015, 119, 23586–23591 CrossRef CAS.
- C. A. Lucas, P. Thompson, Y. Gründer and N. M. Markovic, The structure of the electrochemical double layer: Ag(111) in alkaline electrolyte, Electrochem. Commun., 2011, 13, 1205–1208 CrossRef CAS.
- D. K. Lambert, Vibrational Stark effect of adsorbates at electrochemical interfaces, Electrochim. Acta, 1996, 41, 623–630 CrossRef CAS.
- A. S. Malkani, J. Li, N. J. Oliveira, M. He, X. Chang, B. Xu and Q. Lu, Understanding the electric and nonelectric field components of the cation effect on the electrochemical CO reduction reaction, Sci. Adv., 2020, 6, eabd2569 CrossRef CAS PubMed.
- J. Li, D. Wu, A. S. Malkani, X. Chang, M.-J. Cheng, B. Xu and Q. Lu, Hydroxide Is Not a Promoter of C2+ Product Formation in the Electrochemical Reduction of CO on Copper, Angew. Chem., Int. Ed., 2020, 59, 4464–4469 CrossRef CAS PubMed.
- X. P. Qin, T. Vegge and H. A. Hansen, Cation-Coordinated Inner-Sphere CO2 Electroreduction at Au-Water Interfaces, J. Am. Chem. Soc., 2023, 145, 1897–1905 CrossRef CAS PubMed.
- H. Liu, J. Liu and B. Yang, Promotional Role of a Cation Intermediate Complex in C2 Formation from Electrochemical Reduction of CO2 over Cu, ACS Catal., 2021, 11, 12336–12343 CrossRef CAS.
- M. Ma, K. Djanashvili and W. A. Smith, Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- X. Y. Chen, J. F. Chen, N. M. Alghoraibi, D. A. Henckel, R. X. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- C. Chen, Y. Li and P. Yang, Address the “alkalinity problem” in CO2 electrolysis with catalyst design and translation, Joule, 2021, 5, 737–742 CrossRef.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Efficiency and selectivity of CO2 reduction to CO on gold gas diffusion electrodes in acidic media, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, CO2 electrolysis to multicarbon products in strong acid, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- S. Ringe, C. G. Morales-Guio, L. D. Chen, M. Fields, T. F. Jaramillo, C. Hahn and K. Chan, Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on Gold, Nat. Commun., 2020, 11, 33 CrossRef CAS PubMed.
- Y. Xie, P. F. Ou, X. Wang, Z. Y. Xu, Y. C. Li, Z. Y. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. H. Wang, T. X. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, High carbon utilization in CO2 reduction to multi-carbon products in acidic media, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over a Ni–N–C catalyst in a membrane electrode assembly electrolyzer, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, Suppression of Hydrogen Evolution in Acidic Electrolytes by Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- H. G. Qin, F. Z. Li, Y. F. Du, L. F. Yang, H. Wang, Y. Y. Bai, M. Lin and J. Gu, Quantitative Understanding of Cation Effects on the Electrochemical Reduction of CO2 and H+ in Acidic Solution, ACS Catal., 2023, 13, 916–926 CrossRef CAS.
- Z. Y. Xu, M. Z. Sun, Z. S. Zhang, Y. Xie, H. S. Hou, X. B. Ji, T. F. Liu, B. L. Huang and Y. Wang, Steering the Selectivity of Electrochemical CO2 Reduction in Acidic Media, ChemCatChem, 2022, 14, e202200052 CAS.
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, Engineering the Local Microenvironment over Bi Nanosheets for Highly Selective Electrocatalytic Conversion of CO2 to HCOOH in Strong Acid, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- J. Li, X. Li, C. M. Gunathunge and M. M. Waegele, Hydrogen bonding steers the product selectivity of electrocatalytic CO reduction, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9220–9229 CrossRef CAS PubMed.
- A. Schizodimou and G. Kyriacou, Acceleration of the reduction of carbon dioxide in the presence of multivalent cations, Electrochim. Acta, 2012, 78, 171–176 CrossRef CAS.
- S. S. Bhargava, E. R. Cofell, P. Chumble, D. Azmoodeh, S. Someshwar and P. J. A. Kenis, Exploring multivalent cations-based electrolytes for CO2 electroreduction, Electrochim. Acta, 2021, 394, 139055 CrossRef CAS.
- A. Seifitokaldani, C. M. Gabardo, T. Burdyny, C.-T. Dinh, J. P. Edwards, M. G. Kibria, O. S. Bushuyev, S. O. Kelley, D. Sinton and E. H. Sargent, Hydronium-Induced Switching between CO2 Electroreduction Pathways, J. Am. Chem. Soc., 2018, 140, 3833–3837 CrossRef CAS PubMed.
- A. Bagger, L. Arnarson, M. H. Hansen, E. Spohr and J. Rossmeisl, Electrochemical CO Reduction: A Property of the Electrochemical Interface, J. Am. Chem. Soc., 2019, 141, 1506–1514 CrossRef CAS PubMed.
- F. Quan, M. Xiong, F. Jia and L. Zhang, Efficient electroreduction of CO2 on bulk silver electrode in aqueous solution via the inhibition of hydrogen evolution, Appl. Surf. Sci., 2017, 399, 48–54 CrossRef CAS.
- S. Banerjee, X. Han and V. S. Thoi, Modulating the Electrode-Electrolyte Interface with Cationic Surfactants in Carbon Dioxide Reduction, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- W. X. Ge, Y. X. Chen, Y. Fan, Y. H. Zhu, H. L. Liu, L. Song, Z. Liu, C. Lian, H. L. Jiang and C. Z. Li, Dynamically Formed Surfactant Assembly at the Electrified Electrode-Electrolyte Interface Boosting CO2 Electroreduction, J. Am. Chem. Soc., 2022, 144, 6613–6622 CrossRef CAS PubMed.
- Z.-Q. Zhang, S. Banerjee, V. S. Thoi and A. Shoji Hall, Reorganization of Interfacial Water by an Amphiphilic Cationic Surfactant Promotes CO2 Reduction, J. Phys. Chem. Lett., 2020, 11, 5457–5463 CrossRef CAS PubMed.
- S. Banerjee, Z. Q. Zhang, A. S. Hall and V. S. Thoi, Surfactant Perturbation of Cation Interactions at the Electrode-Electrolyte Interface in Carbon Dioxide Reduction, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- M. N. Jackson, O. Jung, H. C. Lamotte and Y. Surendranath, Donor-Dependent Promotion of Interfacial Proton-Coupled Electron Transfer in Aqueous Electrocatalysis, ACS Catal., 2019, 9, 3737–3743 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- M. Schreier, Y. Yoon, M. N. Jackson and Y. Surendranath, Competition between H and CO for Active Sites Governs Copper-Mediated Electrosynthesis of Hydrocarbon Fuels, Angew. Chem., Int. Ed., 2018, 57, 10221–10225 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. T. Bell, Effects of Anion Identity and Concentration on Electrochemical Reduction of CO2, ChemElectroChem, 2018, 5, 1064–1072 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO2 Electrochemical Reduction Reaction on Cu Surfaces, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- W. Y. Shan, R. Liu, H. C. Zhao and J. F. Liu, Bicarbonate Rebalances the *COOH/*OCO- Dual Pathways in CO2 Electrocatalytic Reduction: In Situ Surface-Enhanced Raman Spectroscopic Evidence, J. Phys. Chem. Lett., 2022, 13, 7296–7305 CrossRef CAS PubMed.
- I. T. McCrum, S. A. Akhade and M. J. Janik, Electrochemical specific adsorption of halides on Cu 111, 100, and 211: A Density Functional Theory study, Electrochim. Acta, 2015, 173, 302–309 CrossRef CAS.
- D. Gao, F. Scholten and B. Roldan Cuenya, Improved CO2 Electroreduction Performance on Plasma-Activated Cu Catalysts via Electrolyte Design: Halide Effect, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Electrocatalytic Production of C3-C4 Compounds by Conversion of CO2 on a Chloride-Induced Bi-Phasic Cu2O-Cu Catalyst, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, Tuning the Catalytic Activity and Selectivity of Cu for CO2 Electroreduction in the Presence of Halides, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- T. H. Yuan, T. Wang, G. Zhang, W. Y. Deng, D. F. Cheng, H. Gao, J. Zhao, J. Yu, P. Zhang and J. L. Gong, The effect of specific adsorption of halide ions on electrochemical CO2 reduction, Chem. Sci., 2022, 13, 8117–8123 RSC.
- Y. Huang, C. W. Ong and B. S. Yeo, Effects of Electrolyte Anions on the Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper (100) and (111) Surfaces, ChemSusChem, 2018, 11, 3299–3306 CrossRef CAS PubMed.
- M. Zhao, H. Tang, Q. Yang, Y. Gu, H. Zhu, S. Yan and Z. Zou, Inhibiting Hydrogen Evolution using a Chloride Adlayer for Efficient Electrochemical CO2 Reduction on Zn Electrodes, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Electrochemical upgrade of CO2 from amine capture solution, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- Y. W. Choi, H. Mistry and B. Roldan Cuenya, New insights into working nanostructured electrocatalysts through operando spectroscopy and microscopy, Curr. Opin. Electrochem., 2017, 1, 95–103 CrossRef CAS.
- A. D. Handoko, F. X. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- S. Q. Zhu, T. H. Li, W. B. Cai and M. H. Shao, CO2 Electrochemical Reduction As Probed through Infrared Spectroscopy, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
| This journal is © the Partner Organisations 2023 |