
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilencing of proinflammatory NF-κB and inhibition of herpes simplex virus (HSV) replication by ultrasmall gold nanoparticles (2 nm) conjugated with small-interfering RNA†
Natalie
Wolff‡
a,
Sebastian
Kollenda‡
a,
Kai
Klein
a,
Kateryna
Loza
a,
Marc
Heggen
b,
Leonie
Brochhagen
c,
Oliver
Witzke
c,
Adalbert
Krawczyk
c,
Ingrid
Hilger
 d and
Matthias
Epple
*a
d and
Matthias
Epple
*a
aInorganic Chemistry and Centre for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, 45117 Essen, Germany. E-mail: matthias.epple@uni-due.de
bErnst-Ruska Centre for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany
cDepartment of Infectious Diseases, West German Centre of Infectious Diseases, University Hospital Essen, University of Duisburg-Essen, Hufelandstr. 55, 45147 Essen, Germany
dDepartment of Experimental Radiology, Institute of Diagnostic and Interventional Radiology, Jena University Hospital, Friedrich Schiller University Jena, Am Klinikum 1, 07740 Jena, Germany
First published on 5th September 2022
Abstract
Azide-terminated ultrasmall gold nanoparticles (2 nm gold core) were covalently functionalized with alkyne-terminated small-interfering siRNA duplexes by copper-catalyzed azide–alkyne cycloaddition (CuAAC; click chemistry). The nanoparticle core was visualized by transmission electron microscopy. The number of attached siRNA molecules per nanoparticle was determined by a combination of atomic absorption spectroscopy (AAS; for gold) and UV-Vis spectroscopy (for siRNA). Each nanoparticle carried between 6 and 10 siRNA duplex molecules which corresponds to a weight ratio of siRNA to gold of about 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Different kinds of siRNA were conjugated to the nanoparticles, depending on the gene to be silenced. In general, the nanoparticles were readily taken up by cells and highly efficient in gene silencing, in contrast to free siRNA. This was demonstrated in HeLa-eGFP cells (silencing of eGFP) and in LPS-stimulated macrophages (silencing of NF-κB). Furthermore, we demonstrated that nanoparticles carrying antiviral siRNA potently inhibited the replication of Herpes simplex virus 2 (HSV-2) in vitro. This highlights the strong potential of siRNA-functionalized ultrasmall gold nanoparticles in a broad spectrum of applications, including gene silencing and treatment of viral infections, combined with a minimal dose of gold.
:1. Different kinds of siRNA were conjugated to the nanoparticles, depending on the gene to be silenced. In general, the nanoparticles were readily taken up by cells and highly efficient in gene silencing, in contrast to free siRNA. This was demonstrated in HeLa-eGFP cells (silencing of eGFP) and in LPS-stimulated macrophages (silencing of NF-κB). Furthermore, we demonstrated that nanoparticles carrying antiviral siRNA potently inhibited the replication of Herpes simplex virus 2 (HSV-2) in vitro. This highlights the strong potential of siRNA-functionalized ultrasmall gold nanoparticles in a broad spectrum of applications, including gene silencing and treatment of viral infections, combined with a minimal dose of gold.
Introduction
Gold nanoparticles have a long history in biomedicine for drug delivery and imaging.1–15 This is due to their comparatively easy synthesis, the absence of toxic effects, and their chemical inertness. They can be surface-functionalized by thiol- or phosphane-ligands to carry drugs, dyes, or other molecules.2,5,7,9,11,13,14,16–20 The surface functionalization of gold nanoparticles with nucleic acids has been pioneered by Mirkin, Alivisatos, Parak and others.21–25 DNA on the surface of nanoparticles also permits the construction of three-dimensional networks of nanoparticles that carry complementary DNA strands.26–28 If small-interfering RNA (siRNA) is attached to 13 nm gold nanoparticles by ligand exchange, they can be used for gene silencing after cellular uptake.29–32 RNA interference (RNAi) is a potent approach in regulating gene expression during the treatment of diseases.33 In particular, exogenous small interfering RNAs (siRNAs) delivered into cells initiate the degradation of complementary mRNA molecules by their own cellular machinery.34 In contrast, dissolved (“naked”) siRNA is rapidly degraded in the presence of RNAses and not taken up by cells due to its negative charge.35Ultrasmall nanoparticles with a diameter of 1 to 2 nm are on the borderline between molecular atom-sharp clusters and “classical” plasmonic nanoparticles (typically 10 to 15 nm in diameter).36–43 They have a much higher specific surface area than bigger nanoparticles which permits a higher dose of attached molecules per mass of administered gold. They also do not quench the fluorescence of attached molecules.44 The common ligand exchange reactions for thiolated DNA make it difficult to control the number of DNA molecules.24 However, gold nanoparticles functionalized with siRNAs were prepared by Yamankurt et al. by ligand exchange:29 Citrate-stabilized gold nanoparticles with a diameter of 13 nm from a modified Turkevich synthesis45 were coated by thiolated siRNAs via ligand exchange in dispersion and denoted as “siRNA spherical nucleic acids”.29
Here, we demonstrate how click chemistry can be used for a covalent attachment of alkyne-terminated siRNA duplex molecules to the surface of azide-terminated ultrasmall gold nanoparticles.46 This method permits the attachment of alkynylated compounds like fluorescent dyes,46,47 molecular tweezers,48 or aggregation-induced emission molecules (AIE)49 to the particle surface. Ultrasmall gold nanoparticles are taken up by endocytosis, but they appear to use different endocytosis pathways. If one is blocked, another one opens as we have recently shown.50 This is consistent with earlier results on nanoparticles and quantum dots of similar size as reported by Yang et al.51 and Carrillo-Carrion et al.52 and different from the behavior of larger nanoparticles.41 Ultrasmall nanoparticles have the advantage of even penetrating cell nuclei in an in vitro 3D brain spheroid model.53 Thus, ultrasmall nanoparticles represent an interesting agent for drugs that are to be transported across the blood–brain barrier.53,54 They are usually non-toxic to cells41 due to the inert gold–sulfur bond.55 This is in line with general studies on the absence of toxicity of conventional gold nanoparticles (10–15 nm)56–58 It shall be noted that cytotoxic effects have been discussed for very small gold nanoparticles and gold clusters in eukaryotic cells,59–61 bacteria,62 and zebrafish.63 However, there are other studies (including our previous results) where it has been shown that ultrasmall gold nanoparticles are not cytotoxic.2,43,50,54,64–67 The attached ligands appear to play an important role for the cytotoxicity, especially if they can leave the gold surface.57 In any case, the particles prepared here were not cytotoxic. So far, there are no data on the fate of the nanoparticles after cellular uptake, but an eventual excretion is likely.52,57,68 Due to the noble character of gold, a dissolution inside the cell or the body can be excluded.57
We show that gold nanoparticles are advantageous as siRNA delivery vehicles into cells because they promote siRNA uptake and silence the expression of specific genes. This is demonstrated in a model system for gene silencing (HeLa-eGFP cells) and in two examples of clinical relevance where they suppress inflammation and viral infection: the silencing of NF-κB in macrophages and the inhibition of viral replication in Herpes Simplex Virus (HSV)-infected cells.
Results and discussion
For the copper-catalyzed click reaction with different siRNA-alkyne sequences, Au-GSH nanoparticles were prepared with a modified Brust-Schiffrin synthesis.69 Azide groups were introduced by substituting the amine group in the stabilizing ligand glutathione (GSH) to Au-N3 nanoparticles with an azide-transfer reagent.46 Au-GSH and Au-N3 nanoparticles were analyzed with UV-Vis-spectroscopy and disc centrifugal sedimentation (DCS). Dynamic light scattering (DLS) was not possible because the particles were too small to scatter the light, in agreement with earlier reports on Au-GSH and Au-N3 nanoparticles.46 UV-Vis spectroscopy was used to verify the successful synthesis of ultrasmall nanoparticles and the absence of larger nanoparticles. There was no typical surface plasmon resonance (SPR) band between 500 and 520 nm that would have indicated larger gold nanoparticles.47 The hydrodynamic diameter of Au-GSH and Au-N3 nanoparticles was determined by DCS with less than 2 nm. The ligand shell influenced the particle size by DCS, thereby underestimating the actual particle size.70 NMR spectroscopy which can be applied to ultrasmall nanoparticles71–76 was used to identify and quantify GSH and N3 bound to the ultrasmall nanoparticles.46 Alkyne-terminated duplex siRNA was then clicked to the Au-N3 nanoparticles. Fig. 1 shows the full reaction scheme.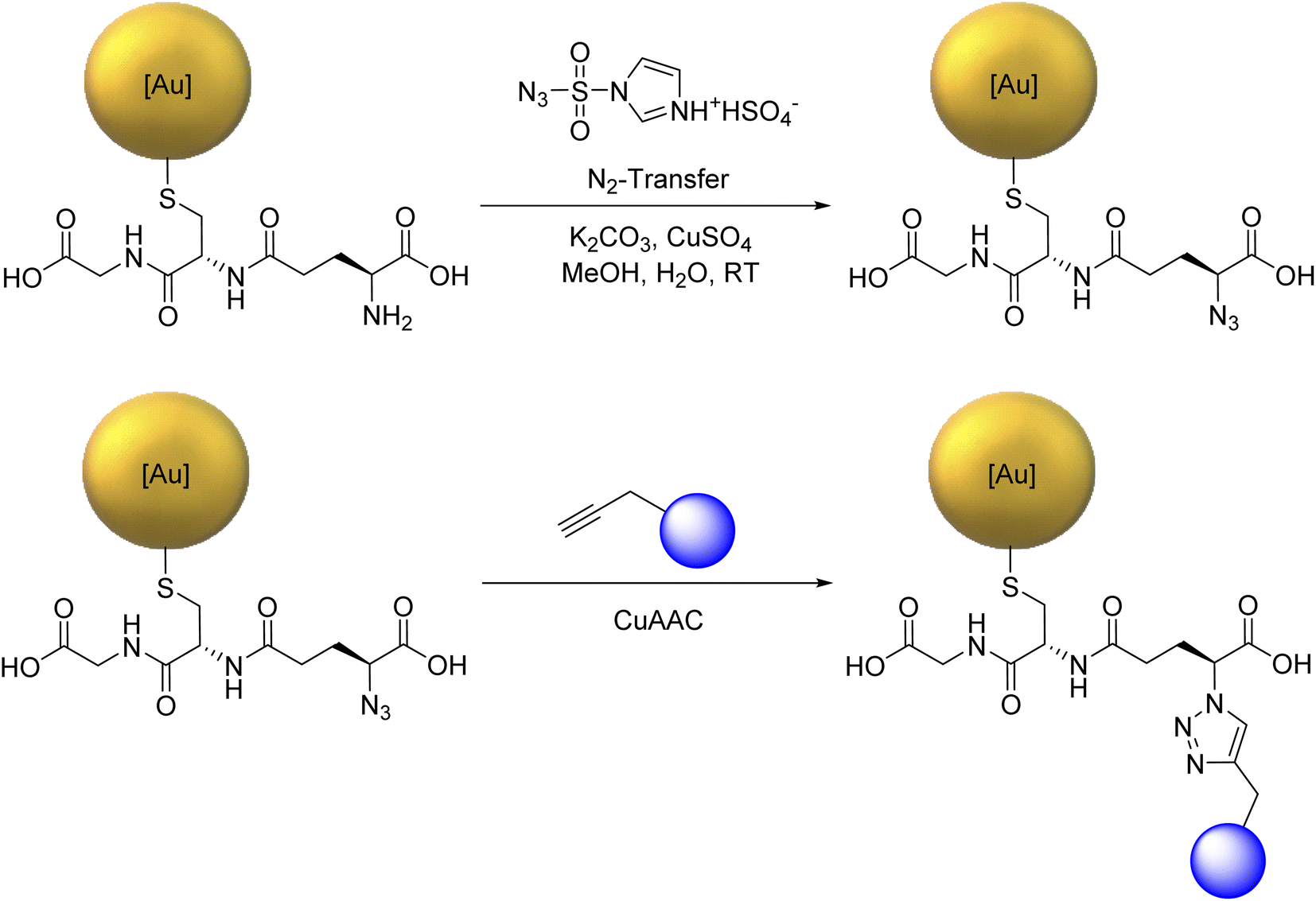 | ||
| Fig. 1 Reaction scheme for the attachment of alkyne-terminated siRNA (depicted as blue balls). | ||
We have used functional duplex siRNA as well as Cy5-labelled duplex siRNA. The labelling with Cy5 was important to quantify the number of siRNA molecules on each nanoparticle and to follow the nanoparticles inside a cell by fluorescence spectroscopy. The gold nanoparticles that carry only siRNA are only very weakly fluorescent and cannot be detected inside cells. The siRNA-carrying nanoparticles were characterized by UV-Vis-spectroscopy and high-resolution transmission electron microscopy (HRTEM). DCS turned out to be not applicable because the large organic shell reduced the effective density of the nanoparticle that no sedimentation occurred at the given rotational speed of the centrifuge within reasonable times (>12 h). Dynamic light scattering (DLS) did not show the ultrasmall nanoparticle (core too small) but again confirmed the absence of larger (plasmonic) or agglomerated gold nanoparticles. The nanoparticle gold core was analyzed by HRTEM. Fourier transformation analysis gave the d-spacings of elemental gold (fcc). Due to the low scattering contrast of the organic ligands, only the metallic core was visible in HRTEM. HRTEM showed a mostly uniform shape of the gold core without agglomerates. An average diameter of 2.2 ± 0.6 nm for Au-siRNA-eGFP nanoparticles and 2.2 ± 0.5 nm for Au-siRNA-eGFP-Cy5 nanoparticles was found (Fig. 2). We have demonstrated earlier that the click reaction does not influence the size of the gold core,46–48 a result that is confirmed here.
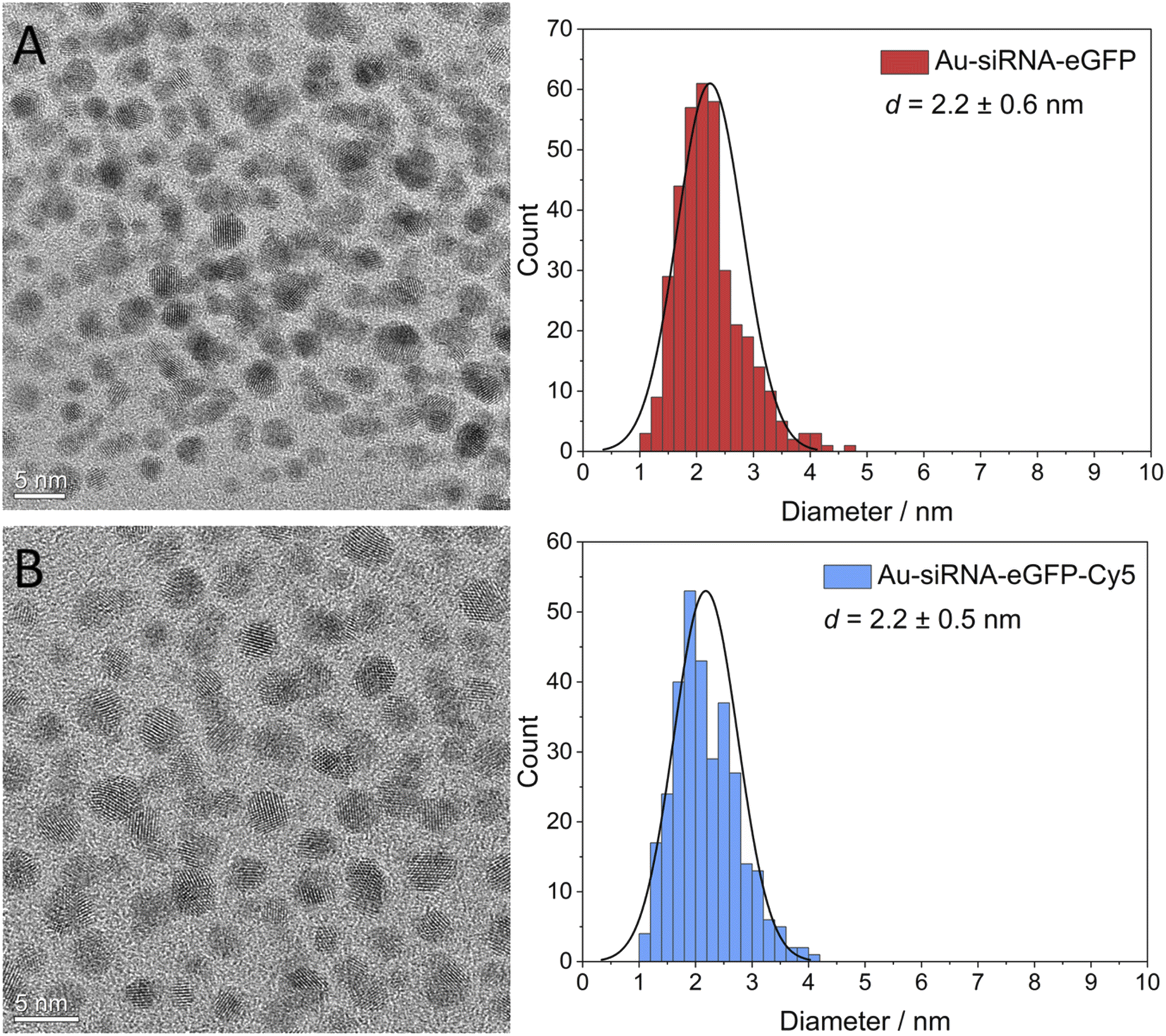 | ||
| Fig. 2 High-resolution transmission electron micrographs of Au-siRNA-eGFP (A) and Au-siRNA-eGFP-Cy5 (B) nanoparticles and corresponding particle size distribution diagrams. | ||
A UV-Vis-spectrum of Au-siRNA-eGFP and Au-siRNA-eGFP-Cy5 nanoparticles showed the absorption band of the attached siRNA molecules (absorption band from 228 to 303 nm, Fig. 3) and confirmed the covalent binding of siRNA to the azide-terminated gold nanoparticles. Again, no absorption by larger (plasmonic) gold nanoparticles was found. For each synthesis, 129 nmol of alkyne-terminated siRNA and 11.9 nmol of azide-terminated gold nanoparticles were used (molar ratio 11:1), assuming a surface loading as with molecular tweezers.48 The click efficiency of the reaction was determined by analysis of the siRNA-loaded gold nanoparticles. Between 6 and 10 siRNA molecules were attached to the surface of each nanoparticle, with good agreement of the data from the UV absorption of RNA and Cy5. Depending on the siRNA used, at least 58% of the applied siRNA-alkyne was bound to the nanoparticle. This is of considerable importance due to the high price of siRNA. The recovery of gold was between 60 and 90%, i.e. also comparatively high. The number of clicked molecules determines the molecular footprint on the nanoparticle surface. For a spherical nanoparticle with an average diameter of 2 nm, the nominal surface area is 12.6 nm2.46 Thus, the siRNA ligands had a molecular footprint between 1.35 and 1.96 nm2, in good agreement with clicked dye molecules like FAM (1.6 to 2.1 nm2),46,47 or AlexaFluor-647 (1.15 nm2).46 Lach et al. found a footprint of 0.20 nm2 for quaternary ammonium ligands on 3.4 nm gold nanoparticles.77 This is also in line with Girard et al. who showed that a ssDNA molecule (18 bases) required a surface area of about 1 nm2 on a 2 nm gold nanoparticle, i.e. geometrically, each 2 nm-nanoparticle can carry about 12 ssDNA molecules.24 All analytical data of the nanoparticles are summarized in Table 1. The nanoparticles have a composition of about Au∼250GSH∼125(siRNA)8. With molar masses of Au (197 g mol−1), GSH (306 g mol−1) and siRNA (∼14000 g mol−1), we obtain a molar mass of one nanoparticle of about 199500 g mol−1 with weight fractions of Au = 25 wt%, GSH = 19 wt%, and siRNA = 56 wt%.
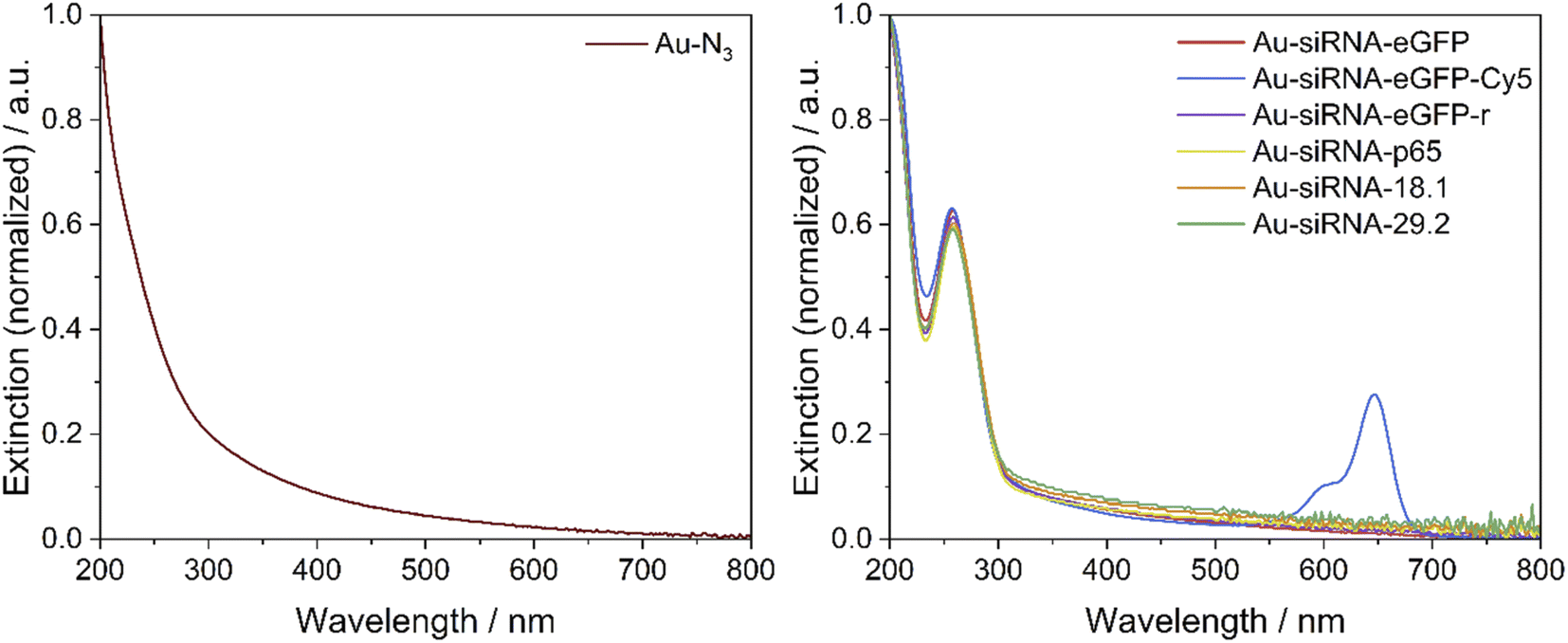 | ||
| Fig. 3 UV-Vis spectra of azide-terminated gold nanoparticles (Au-N3, left) and of siRNA-functionalized gold nanoparticles (right). The absorption of RNA is visible by the band at 258 nm. The absorption of Cy5 is visible by the band at 647 nm. | ||
| Ligands per nanoparticle (1H-NMR and UV-Vis spectroscopy) | Recovery of gold/% | Yield of siRNA attachment/% | Gold core diameter (HRTEM)/nm | Ligand footprint/nm2 | Hydrodynamic diameter (DCS)/nm | |
|---|---|---|---|---|---|---|
| Au-GSH46 | 125 (1H-NMR) | 72 | — | 1.7 ± 0.9 | 0.10 | 1.5 ± 0.2 |
| Au-N3 (ref. 46) | 118 (1H-NMR) | 86 | 94 | 2.0 ± 0.4 | 0.11 | 1.5 ± 0.2 |
| Au-siRNA-eGFP | 9.5 (UV-Vis) | 89 | 86 | 2.2 ± 0.6 | 1.35 | — |
| Au-siRNA-eGFP-Cy5 | 8.9 (UV-Vis, RNA)/8.7 (UV-Vis, Cy5) | 79 | 81 (RNA)/79 (Cy5) | 2.2 ± 0.5 | 1.41 (RNA)/1.45 (Cy5) | — |
| Au-siRNA-eGFP-r | 8.3 (UV-Vis) | 75 | 76 | — | 1.50 | — |
| Au-siRNA-p65 | 8.7 (UV-Vis) | 62 | 79 | — | 1.45 | — |
| Au-siRNA-18.1 | 6.4 (UV-Vis) | 76 | 58 | — | 1.96 | — |
| Au-siRNA-29.2 | 6.8 (UV-Vis) | 75 | 63 | — | 1.83 | — |
This is a considerably higher loading of siRNA than that reported for “standard” gold nanoparticles prepared by the Turkevich method (citrate reduction), followed by ligand exchange.45 Yamankurt et al. prepared siRNA-coated gold nanoparticles (13 nm, “spherical nucleic acids”) with a maximum of 98 siRNA duplex molecules per particle.29 This corresponds to a molecular footprint of about 5.4 nm2 per siRNA duplex. In comparison, the surface density of siRNA after clicking is about 3 to 4 times higher than after ligand exchange. In terms of the particle composition, the composition of a 13 nm gold nanoparticle is about Au70000(siRNA)98. This leads to a molar mass of one nanoparticle of about 15.1∙106 g mol−1 with weight fractions of Au = 91 wt% and siRNA = 9 wt% (assuming 14 kDa per siNA duplex as in our case). In terms of siRNA loading, ultrasmall nanoparticles carry about 2.2:0.1 = 22 times more siRNA per mass of gold applied.
siRNA for silencing the eGFP gene is a useful model because the extent of gene silencing can be easily demonstrated by analyzing the eGFP fluorescence intensity by fluorescence microscopy. Moreover, we also focused on siRNA for silencing nuclear factor-kappaB (NF-κB) dimer gene expression, since such gene products are important cellular players responsible for many regulatory processes including inflammatory and cancerous diseases.78–82
The uptake of fluorescently labelled Au-siRNA-eGFP-Cy5 nanoparticles was observed with confocal laser scanning microscopy (CLSM) HeLa-eGFP cells. All slices shown are taken from the focal plane in the middle of the whole cell volume (z-stacks). Based on the z-stacks, the nanoparticles were well taken up by all cells but did not enter the cell nucleus. They were found in the cytoplasm or decorated the nucleus (Fig. 4).
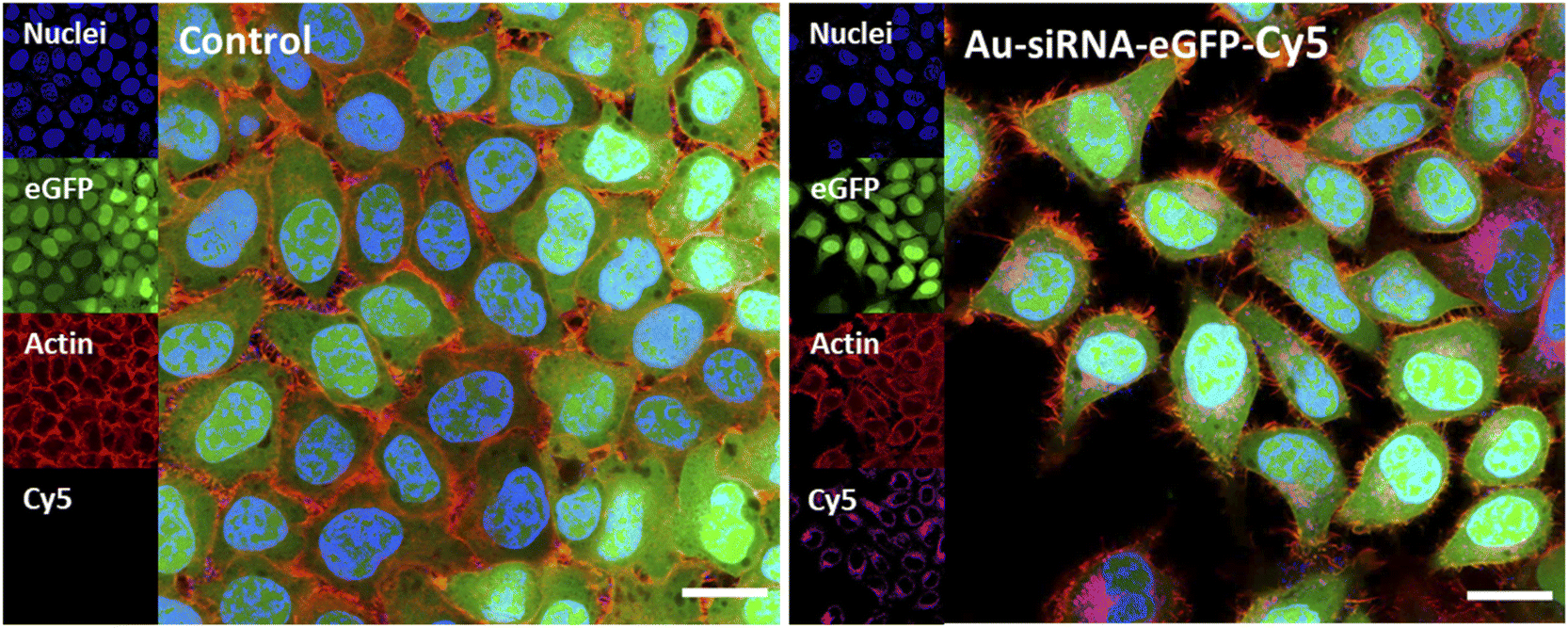 | ||
| Fig. 4 Confocal laser scanning images showing the uptake of ultrasmall Au-siRNA-eGFP-Cy5 nanoparticles by HeLa-eGFP cells. The cells were incubated with 5 μg mL−1 gold nanoparticles per well (13.7 μg siRNA mL−1) for 24 h, washed, fixed, and stained. Control: untreated HeLa-eGFP cells (no nanoparticles). Nuclei: stained with DAPI; actin: stained with AlexaFluor™ 568 Phalloidin; Cy5: Au-siRNA-eGFP-Cy5 nanoparticles. Scale bars are 20 μm. Data represent the mean of three individual experiments with the standard deviation (N = 3). | ||
The viability of the nanoparticle-treated HeLa-eGFP cells was determined with an MTT cell viability assay. The siRNA-loaded nanoparticles did not lead to a significant change in cell viability when compared to the untreated control. In contrast to the nanoparticles, Lipofectamine/siRNA-based formulations showed a significant decrease in cell viability. This is well known and due to the adverse nature of Lipofectamine (Fig. 5).83
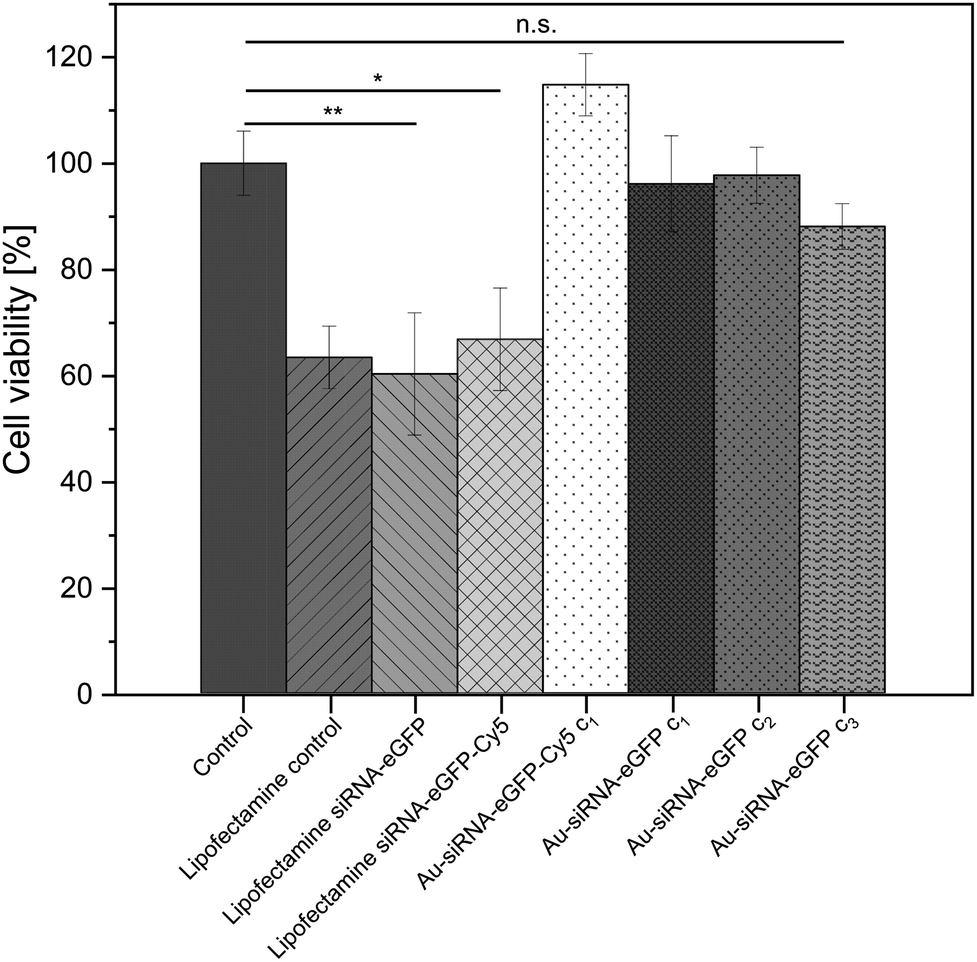 | ||
| Fig. 5 MTT viability assay with HeLa-eGFP cells, incubated with Lipofectamine/siRNA-eGFP or water-dispersed Au-siRNA-eGFP nanoparticles. Nanoparticle dispersions were tested at three concentrations, i.e. c1: 14.8 μg siRNA mL−1 (13.7 μg siRNA mL−1 for siRNA-eGFP-Cy5); c2: 74 μg siRNA mL−1; c3: 148 μg siRNA mL−1. The incubation time was 24 h, except for Lipofectamine where it was 5 h. Control: untreated HeLa-eGFP cells. One-way ANOVA was used, followed by Tukey's comparison test: n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Data represent the mean of three individual experiments with the standard deviation (N = 3). | ||
The nanoparticles were able to silence the production of eGFP in HeLa-eGFP cells as shown by fluorescence microscopy (Fig. 6). The mean fluorescence intensity (MFI) in these images was used to quantify the eGFP gene silencing efficiency. Only fully confluent parts of a well were monitored. Interestingly, Au-siRNA-eGFP-Cy5 nanoparticles remained for a much longer time inside the cells than Lipofectamine-transported siRNA-eGFP-Cy5 which was barely visible after the incubation time of 72 h. The fluorescent label Cy5 on Au-siRNA-eGFP-Cy5 nanoparticles decreased the gene silencing efficiency. This underscores the fact that labels on biomolecules may compromise their biological effect. A possible explanation could be that the functionalization of an alkyne and Cy5 at the 5′- and 3′-ends of the siRNA sequence made the siRNA inaccessible for the RISC. Thus, the separation of the sense and antisense strands could have been blocked by the design of the siRNA-eGFP-Cy5 (for sequence designs see Table 2) when coupled to the surface of the gold nanoparticles. It has been reported that the conjugation of a fluorophore to a bioactive molecules can change its subcellular localization and function.84
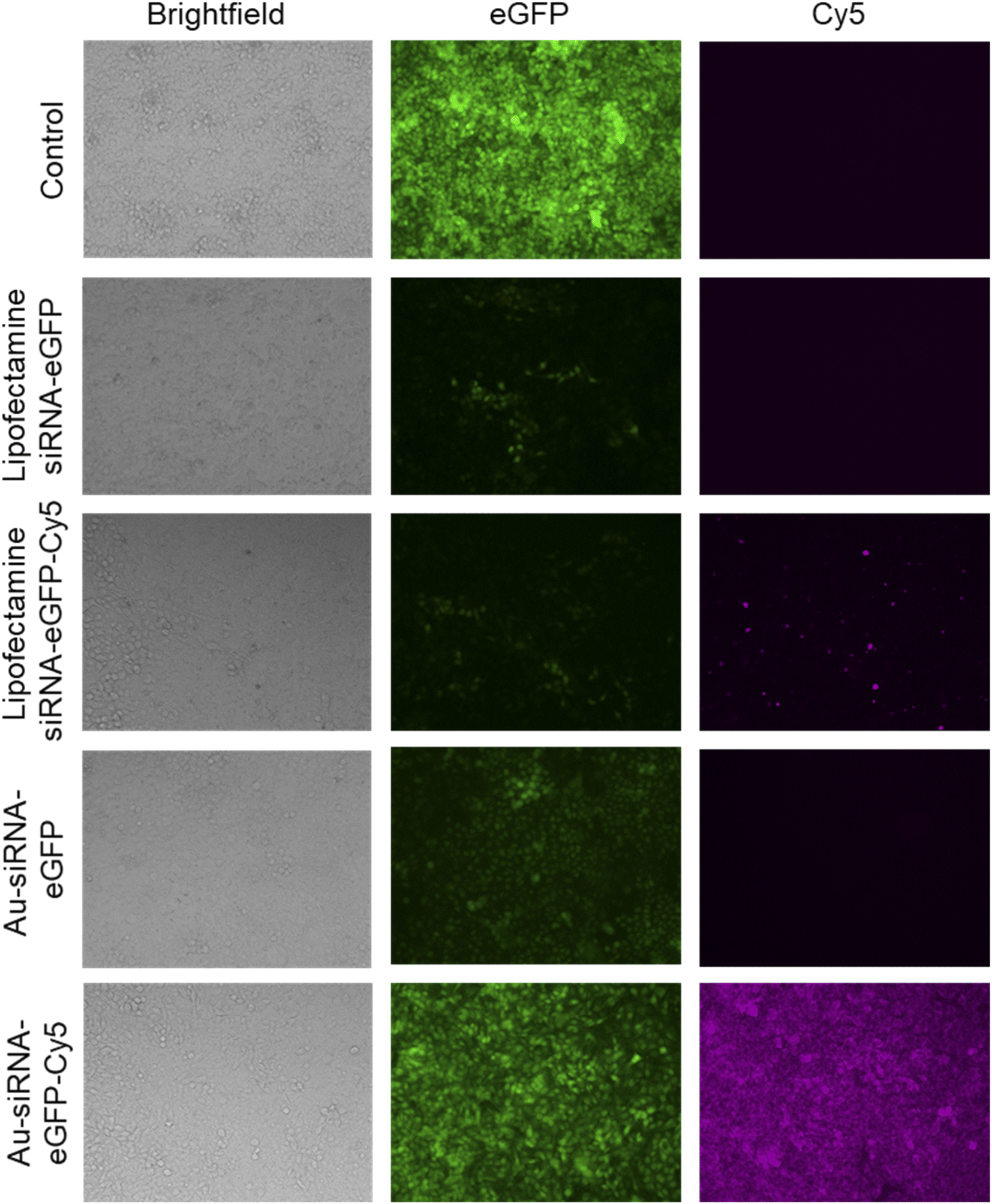 | ||
| Fig. 6 Representative fluorescence images showing the silencing of eGFP expression in HeLa-eGFP cells by Lipofectamine/siRNA-eGFP polyplex (3.2 μg mL−1 siRNA/Lipofectamine) and by Au-siRNA-eGFP (14.8 μg siRNA mL−1) or Au-siRNA-eGFP-Cy5 (13.7 μg siRNA mL−1) nanoparticle treatment. Data represent the mean of three individual experiments with the standard deviation (N = 3). | ||
| siRNA code | siRNA target | Sequence 5′-3′ of sense and antisense strand of duplex siRNA |
|---|---|---|
| siRNA-eGFP | eGFP | rGrCrArArGrCrUrGrArCrCrCrUrGrArArGrUrUrCrArU (sense) |
| (M = 14414 g mol−1) |
eGFP | 5-Octadinyl dU/rArUrGrArArCrUrUrCrArGrGrGrUrCrArGrCrUrUrGrC (antisense) |
| siRNA-eGFP-Cy5 | eGFP | 5-Cy5/rGrCrArArGrCrUrGrArCrCrCrUrGrArArGrUrUrCrArU (sense) |
| (M = 14946 g mol−1) |
eGFP | 5-Octadinyl dU/rArUrGrArArCrUrUrCrArGrGrGrUrCrArGrCrUrUrGrC (antisense) |
| siRNA-eGFP-r | eGFP | 5-Octadinyl dU/rGrCrArArGrCrUrGrArCrCrCrUrGrArArGrUrUrCrArU (sense) |
| (M = 14414 g mol−1) |
eGFP | rArUrGrArArCrUrUrCrArGrGrGrUrCrArGrCrUrUrGrC (antisense) |
| siRNA-p65 | NF-κB | 5-Octadinyl dU/rCrCrArUrGrGrArGrUrUrCrCrArGrUrArCrUrUTT (sense) |
| (M = 13694 g mol−1) |
NF-κB | rArArGrUrArCrUrGrGrArArCrUrCrCrArUrGrGTT (antisense) |
| siRNA-18.1 | HSV-UL18.1 | 5-Octadinyl dU/rGrCrArCrCrGrUrUrArArCrCrUrUrCrGrCrArATT (sense) |
| (M = 13694 g mol−1) |
HSV-UL18.1 | rUrUrGrCrGrArArGrGrUrUrArArCrGrGrUrGrCTT (antisense) |
| siRNA-29.2 | HSV-UL29.2 | 5-Octadinyl dU/rCrUrUrUrCrGrCrArArUrCrArArUrUrCrCrArA (sense) |
| (M = 12448 g mol−1) |
HSV-UL29.2 | rUrUrGrGrArArUrUrGrArUrUrGrCrGrArArArG (antisense) |
Based on the fluorescence microscopy images as shown in Fig. 6 the mean fluorescence intensity data were calculated and are presented in Fig. 7A. The nanoparticles were remarkably efficient in silencing the eGFP gene in HeLa-eGFP cells, but still less efficient than Lipofectamine (Fig. 7B). However, it is advantageous that they were considerably less cytotoxic than Lipofectamine. Higher nanoparticle concentrations did not substantially increase the gene silencing efficiency. There was also no significant difference between the nanoparticles where the antisense strand was attached to the gold (siRNA-eGFP) compared to the case when the sense strand was attached to the gold (siRNA-eGFP-r). This might have been expected because the antisense strand is considered to be the active agent, but is obviously not the case.85
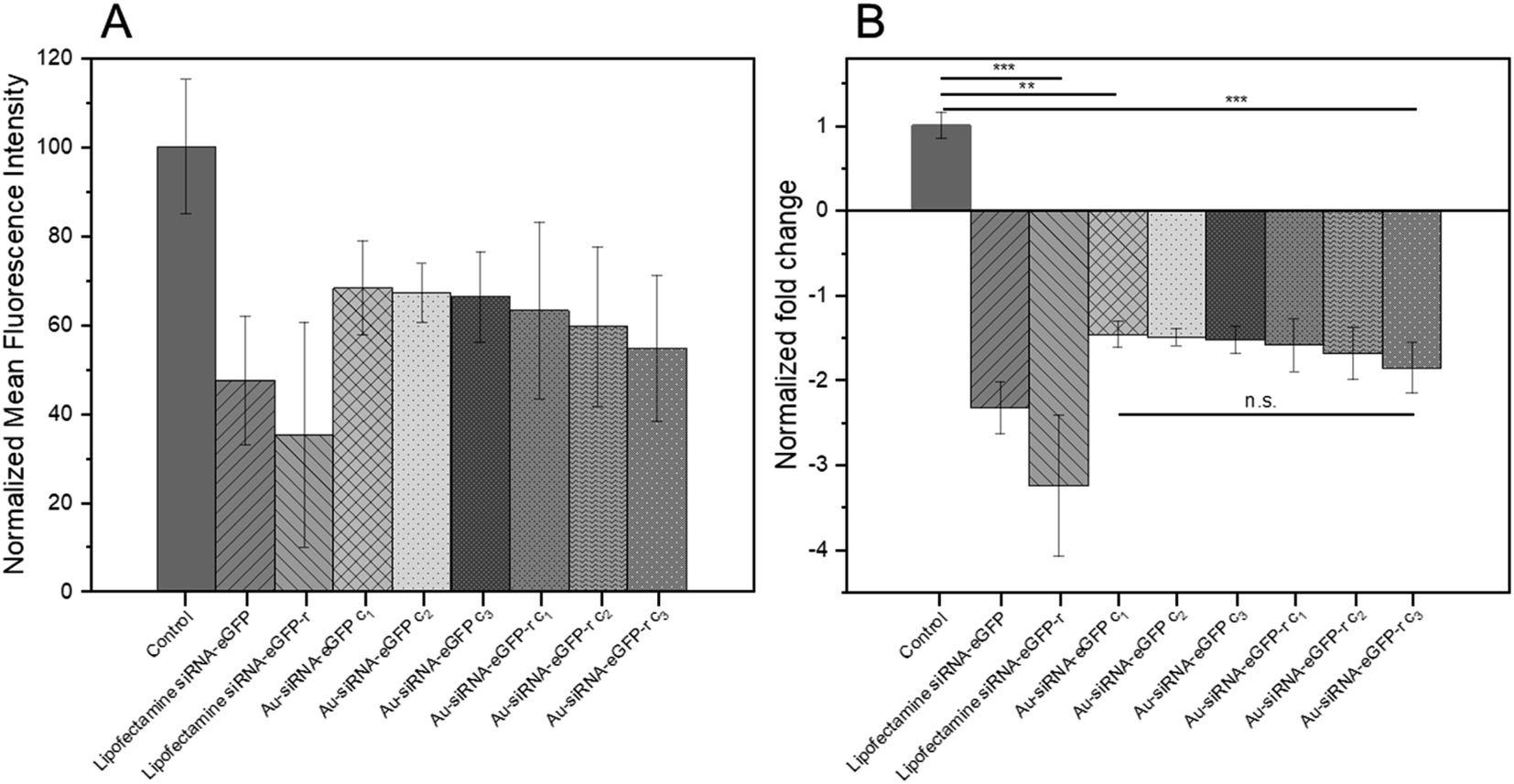 | ||
| Fig. 7 (A) Normalized mean fluorescence intensity as calculated from cell culture images as shown in Fig. 6. (B) Normalized fold decrease of eGFP gene expression in HeLa-eGFP cells. Nanoparticle dispersions were tested in three concentrations, i.e. c1: 14.8 μg siRNA mL−1; c2: 74 μg siRNA mL−1; c3: 148 μg siRNA mL−1. One-way ANOVA was used, followed by Tukey's comparison test: n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Data represent the mean of three individual experiments with the standard deviation (N = 3). | ||
Yamankurt et al. have studied the mechanism of the intracellular processing of gold-conjugated siRNA in detail. They reported that the enzyme Dicer was cleaving the duplex from the gold surface, thereby liberating the siRNA for intracellular gene silencing.29 It is likely that a similar mechanism occurs with siRNA-functionalized ultrasmall nanoparticles as well. This could also explain the higher stability of nanoparticle-conjugated siRNA towards enzymatic degradation in comparison to dissolved siRNA. They reported gene silencing efficiencies of the protein HER2 in SKOV-3 cells to about 15% on the RNA level by nanoparticle treatment. However, they also reported that the nanoparticles had a significant cytotoxicity if applied at doses above about 1 nmol L−1.29
As medically relevant protein, we chose nuclear factor (NF)-κB. This protein plays an important role in inflammation-associated diseases because numerous NF-κB target genes encode proinflammatory proteins.82 The principal means to control the NF-κB activity is by retention of inactive forms of heterogeneous transcription factor proteins in the cell's cytoplasm by inhibitors of κB (IκB) molecules.78 Upon activation by a diverse range of stimuli (immunoreceptors, cytokines, growth factors, and other stress impulses),80 the freely released NF-κB proteins immediately form homo- and hetero-conjugations, which bind to specific DNA regions. The active NF-κB protein monomers are termed p50, p52, p65 (RelA), c-Rel, and RelB. Especially the Rel proteins can form several different NF-κB dimers. Of these, the p50/p65 heterodimer is physiologically the most abundant.79 Importantly, the combination of subunits within the NF-κB protein components results in distinct DNA-sequence-specific transactivation properties.81
It has been shown that a key regulator of macrophage behavior is NF-κB signaling, via nuclear translocation and binding of p65/p50 transcription factors. In consequence, there is increasing evidence that increased NF-κB activity in macrophages polarizes them towards an M1-like phenotype.86–88 In our study after incubation of inflamed (LPS-primed) macrophages with Au-siRNA-p65 nanoparticles carrying anti-p65 NF-κB siRNA (1 μg siRNA mL−1, 72 h), a considerable number of cells was shown to be nanoparticle-positive (Fig. 8A; see also ESI, Fig. 1†). We chose to target p65 NF-κB siRNA over other NF-κB dimers on the basis of our previous experience with calcium phosphate nanoparticles for gene silencing.89–91 Moreover, the presence of nanoparticles did not reduce the viability of the target cells (Fig. 8B). Protein expression analyses revealed a statistically significant depression not only of p65 NF-κB, but also of other NF-κB dimers like p50, p100, and p105 (the latter two not significant). In contrast, the impact on p52 was inconspicuous (Fig. 7C). Since the mentioned NK-κB proteins are responsible for encoding numerous pro-inflammatory proteins, cytokines, and growth factors,92 we expect that the nanoparticle-mediated anti-p65 siRNA inhibition in macrophages will lead to a decreased pro-inflammatory activity. In particular, the protein expression of the two most common NF-κB dimer components p65 and p50 was depressed. These are known to regulate many physiological activities.79
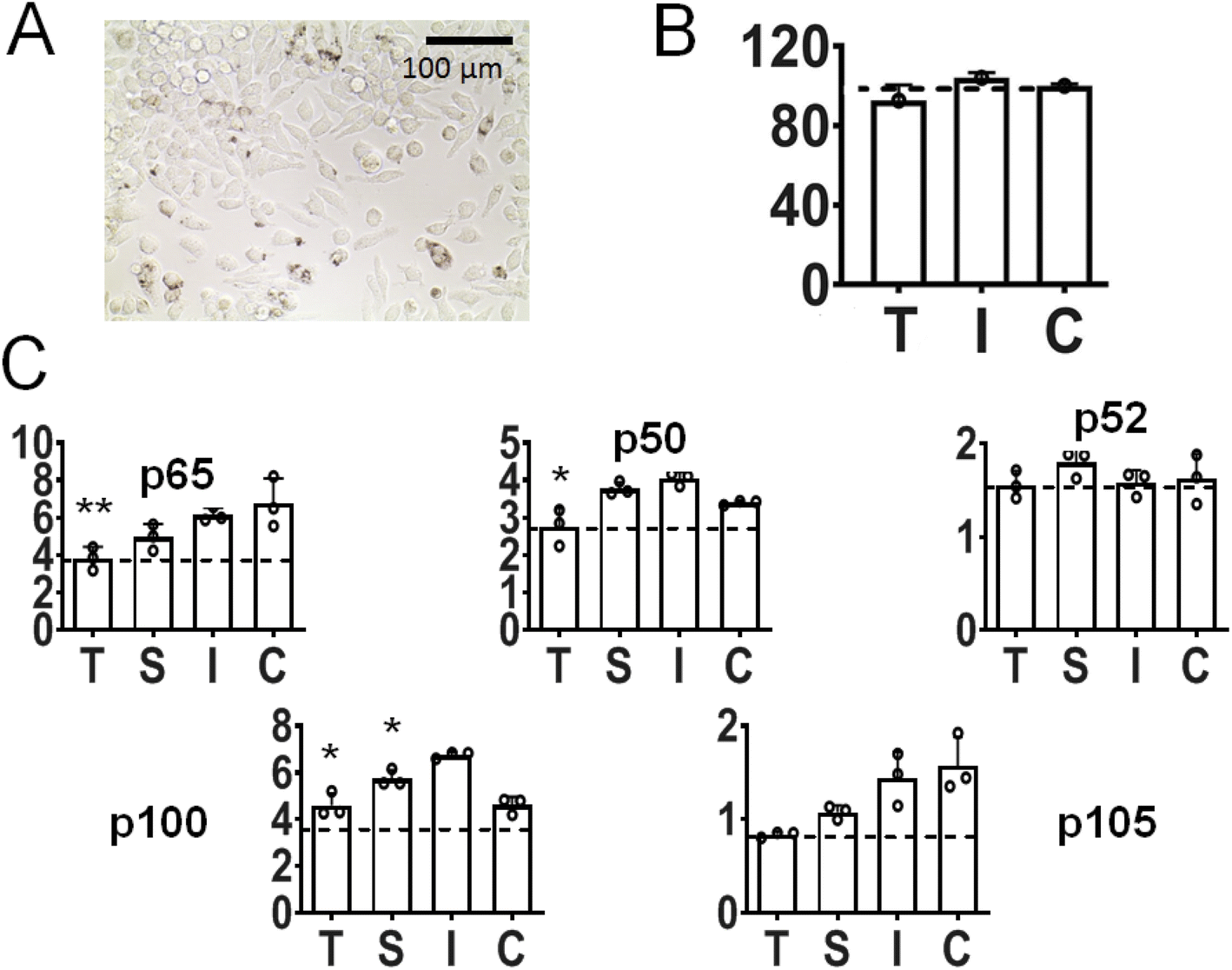 | ||
| Fig. 8 Impact of Au-siRNA-p65 nanoparticles in inflamed murine macrophages. (A) Light microscopy image of inflamed macrophages (J774.A1, LPS-primed) after nanoparticle exposure. (B) Cell viability after nanoparticle exposure. (C) Protein expression (western and immunoblot) relative to GAPDH of different NF-κB members, T: inflamed cells treated with anti-p65 NK-κB Au-siRNA-p65 nanoparticles (1 μg siRNA mL−1, 72 h), S: inflamed cells treated with Lipofectamine/siRNA-p65 (1 μg siRNA mL−1, 72 h), I: non-treated inflamed cells, C: non-treated, non-inflamed cells. *: statistically different compared to I with p < 0.05 (*) or p < 0.01 (**) (Student's t-test). Data represent the mean of three individual experiments with the standard deviation (N = 3). | ||
Finally, we demonstrate that gold nanoparticles carrying siRNA can be used to combat viral infections by gene silencing. Herpes simplex viruses type 1 (HSV-1) and type 2 (HSV-2) belong to the most common viruses affecting humans.93,94 Upon primary infection, the viruses establish a life-long latent infection in the sensory neurons.95,96 Periodic reactivations of the viruses may lead to genital, orofacial, or ocular infections that are clinically indistinguishable between HSV-1 and HSV-2.97 Most recurrent episodes are mild and self-limiting.98 However, besides severe, life-threatening reactivations in immunocompromised patients,99 HSV-infections of the eye represent a serious risk to human health.100 Frequent recurrent manifestations of the virus at the cornea may lead to the development of herpetic stromal keratitis and finally to blindness.100 The increasing incidence of acyclovir (ACV)- and multidrug-resistant strains in patients with corneal HSV-infections makes the treatment increasingly challenging.101,102 Clearly, novel strategies for the topical treatment of HSV-infections of the cornea are urgently needed. Paradowska et al. have recently reported that citrate-coated 10–15 nm gold nanoparticles can interact with HSV-1 in an unspecific way.103
To investigate whether siRNA-functionalized gold nanoparticles can be used to treat viral infections like genital HSV-infections of the eye, we examined the antiviral efficacy of Au-siRNA-18.1 or Au-siRNA-29.2 nanoparticles against HSV-2 in cell culture, respectively. siRNA-18.1 targets the capsid encoding gene UL18 104 and siRNA-29.2 targets the DNA-binding protein UL29.105 We found that Au-siRNA-18.1 or Au-siRNA-29.2 both strongly inhibited HSV-2 replication in cell culture in a dose-dependent way (Fig. 9). A complete inhibition of the viral replication was observed at a concentration of 2 pmol μL−1 Au-siRNA-18.1 (26 μg siRNA mL−1) and 4 pmol μL−1 Au-siRNA-29.2 (52 μg siRNA mL−1). In contrast, dissolved siRNA-18.1 and siRNA-29.2 had no effect on the viral replication. These results clearly emphasize that siRNA-functionalized gold nanoparticles are a promising platform for the development of novel anti-invective drugs. Conventionally, siRNAs such as anti-HSV-siRNA 18.1 or 29.2 need to be formulated with transfection reagents to achieve uptake into target cells.104,105 siRNA-functionalized gold nanoparticles clearly avoid the need for an additional transfection agent to achieve the desired gene silencing.
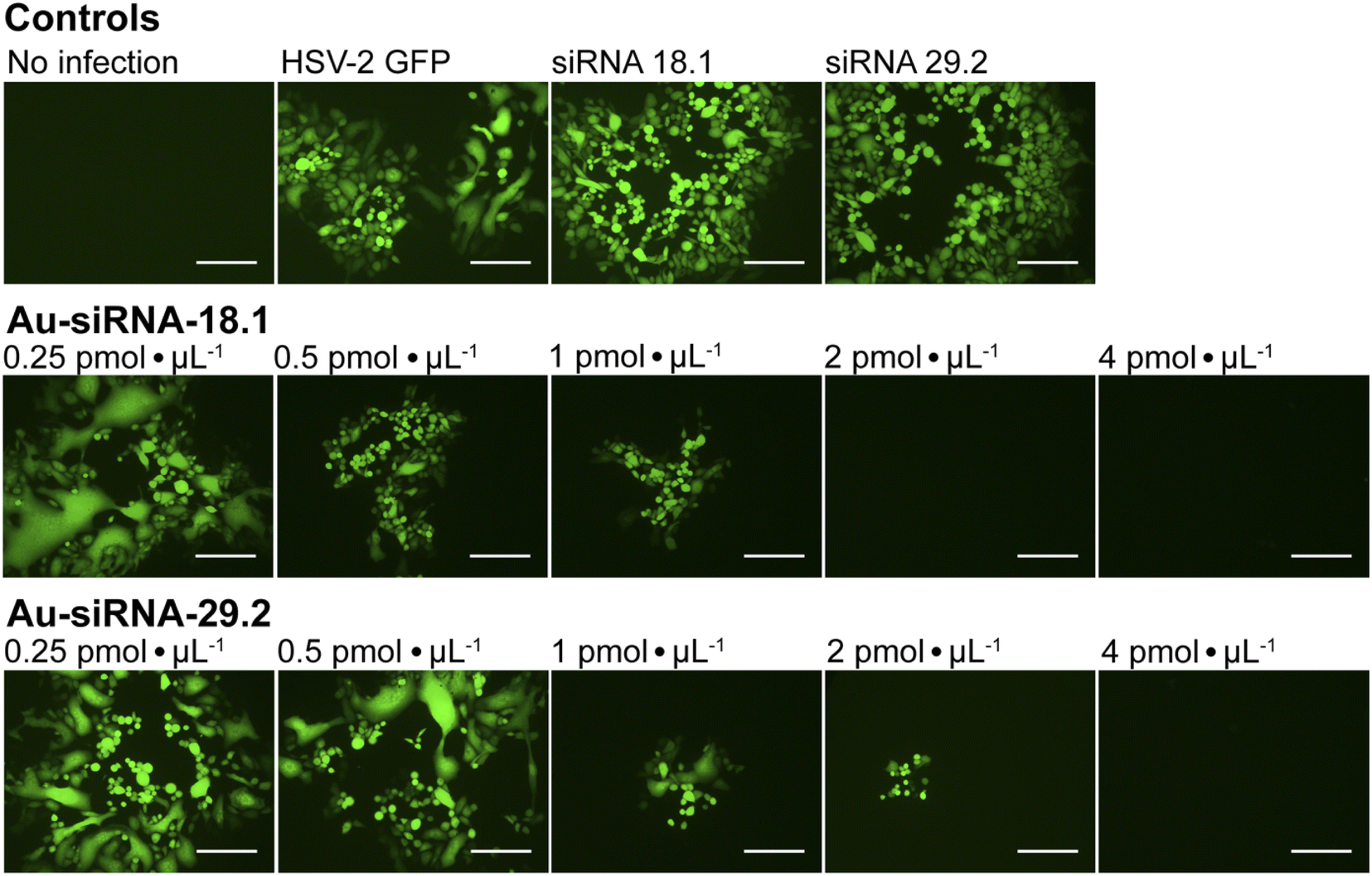 | ||
| Fig. 9 Inhibition of HSV-2 replication by Au-siRNA-18.1 and Au-siRNA-29.2. Vero cells were pre-incubated for 4 h with various concentrations of Au-siRNA-18.1 or Au-siRNA-29.2 nanoparticles (0.25 pmol siRNA μL−1 to 4 pmol siRNA μL−1) and subsequently infected with 25 TCID50 units of HSV-2 GFP reporter virus. siRNA-18.1 and siRNA-29.2 dissolved in cell culture medium (“naked”) at a concentration of 4 pmol siRNA μL−1 were used as control. After 48 h, the cytopathic effects (CPE) were assessed by fluorescence microscopy. Representative immunofluorescence images acquired at a 100-fold magnification from three independent experiments (N = 3) are shown. Scale bar 200 μM. | ||
The copper-catalyzed azide–alkyne click reaction (CuAAC) obviously does not lead to a structural damage of the siRNA as it is still able to silence the target genes, demonstrated here for a number of cases. Whereas a detailed investigation of the chemical integrity of the clicked siRNA is difficult to impossible (NMR does not work; detachment may lead to chemical damage as well), it is obvious that at least a significant fraction of siRNA has preserved its chemical nature. This is supported by literature data on click reactions of nucleic acids by CuAAC. It has been reported that CuAAC as a mild and orthogonal click reaction as defined by Sharpless106 does not damage nucleic acids and is therefore a suitable means for their modification.107–110
Conclusions
Ultrasmall gold nanoparticles (2 nm core diameter) can be functionalized with siRNA duplexes by clicking them to the particle surface with a surface density that is 3 to 4 times higher compared to ligand exchange reactions. Due to the small particle diameter, the weight ratio of siRNA to gold is about 22 times higher than with “classical” 13 nm gold nanoparticles. The particles were easily taken up by HeLa cells and led to a strong silencing of eGFP in HeLa-eGFP cells. This effect is therapeutically relevant to treat inflammations as demonstrated with LPS-stimulated macrophages. Their treatment with gold nanoparticles carrying siRNA led to a significant silencing of the two most common NF-κB dimer components p65 and p50. Furthermore, the treatment of Vero cells that were previously infected with herpes simplex virus 2 (HSV-2) with such nanoparticles fully inhibited HSV-2 replication in a dose-dependent way. The fact that the nanoparticle-attached siRNA was still active in gene silencing indicates that it was not significantly damaged by the click reaction. Thus, siRNA functionalized Au-nanoparticles Au-siRNA-18.1 and Au-siRNA-29.2 represent an efficient tool for the topical treatment of HSV-infections such as ACV-resistant genital herpes or HSV-infections of the cornea. The cytotoxicity of the nanoparticles was generally low at the applied doses. We conclude first that surface-clicking is a superior method to attach siRNA to a gold nanoparticle surface. Second, ultrasmall gold nanoparticles are especially suitable to transport high amounts of siRNA into cells for gene silencing with a minimum gold dose.In control studies, the dissolved siRNA did not lead to the desired gene silencing effect because it was not taken up by the cells. We ascribe the fact that nanoparticle-attached siRNA is much more active in gene silencing than dissolved siRNA to two reasons. First, it can enter a cell together with a nanoparticle much more easily than in dissolved form due to the nanoparticle-specific uptake mechanisms. Second, it appears to be better protected against enzymatic degradation by RNAses. The reason for this effect is still unknown, but steric effects associated with the dense packing of nucleic acids on the nanoparticle surface could play an important role.
Materials and methods
Chemicals and reagents
Elemental gold (>99%) was dissolved in aqua regia to prepare tetrachloroauric acid (HAuCl4). Hydrochloric acid (HCl, 37%), nitric acid (HNO3, 67%), and sodium hydroxide (NaOH, 1 M) were obtained from Bernd Kraft (Duisburg, Germany). Sodium borohydride (NaBH4, >96%), sodium L-ascorbate (>99%), copper(II)sulfate pentahydrate (>99%), deuterium oxide (D2O, 99%), sodium ascorbate (99%), and Spin-X® UF 30k MWCO PES spin filters (Corning®) were obtained from Sigma-Aldrich (Steinheim, Germany). Sucrose (99%) was obtained from VWR Chemicals (Langenfeld, Germany). Methanol (99.8%) was obtained from Fisher Scientific (Geel, Belgium). Aminoguanidine hydrogen carbonate (98%) was obtained from Alfa Aesar (Kandel, Germany). Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, >95%) was obtained from Jena Bioscience (Jena, Germany). Dodecane (99%) and 2-(4,5-dimethylthiazol-2-yl)-3,5-diphenyl-2H-tetrazol-3-ium bromide (MTT), and Invitrogen™ Lipofectamine™ 2000 transfection reagent were obtained from Thermo Fisher Scientific (Schwerte, Germany). Dimethyl sulfoxide (DMSO, >99.5%) was obtained from Carl Roth (Karlsruhe, Germany). The PVC nanoparticle calibration dispersion (Lot#149, 1.385 g L−1) for differential centrifugal sedimentation (DCS) was obtained from CPS Instruments Inc. (Oosterhout, The Netherlands).All siRNA duplexes were obtained from Integrated DNA Technologies (Coralville, Iowa, USA) in purified form (standard desalting or HPLC purification for dye-modified sequences). They were obtained with an alkyne group for clicking and in one case with an additional Cy5 group for fluorescent labelling. All siRNA sequences are given in Table 2. Note that siRNA against eGFP was tested in two sequence modifications. siRNA was attached to the nanoparticle at the 5′ ends of the sequences, i.e. the antisense strand (siRNA-eGFP) or the sense strand (siRNA-eGFP-r) (r stands for reverse).
For the synthesis and purifications of Au-GSH nanoparticles, ultrapure water (Purelab ultra instrument from ELGA, Celle, Germany) was used. All glassware was cleaned with boiling aqua regia and washed twice with ultrapure water before use. UltraPure™ water (distilled water, DNase/RNase free, Invitrogen) was used for the reconstitution of siRNA. For synthesis, purifications, and analyses of Au-N3 and all Au-siRNA nanoparticles, UltraPure™ water was used. To create conditions that were as RNase-free as possible after the synthesis of the Au-GSH nanoparticles, all solutions were prepared with sterile pipette tips in sterile vessels and filtered using non-pyrogenic sterile syringe filters (0.2 μm, Sarstedt). In addition, all glassware was sterilized before use at 200 °C for at least 2 h before use. All working surfaces were treated beforehand with RNase AWAY™ (Carl Roth).
Methods
Atomic absorption spectroscopy (AAS) was used to determine the gold concentration of the nanoparticle dispersion. Nanoparticle dispersions (5 μL) were dissolved in aqua regia (450 μL) and diluted with ultrapure water (3.5 mL) for each measurement. A Thermo Electron M-Series spectrometer (graphite tube furnace; operated according to DIN EN ISO/IEC 17025:2005) was used.Analytical disc centrifugation (differential centrifugal sedimentation; DCS) was carried out with a CPS Instruments DC 24000 disc centrifuge (24000 rpm, 29000 relative centrifugal force; rcf). A density gradient of sucrose solutions with different concentrations (8 and 24 wt%) was applied. For stabilization, the solution was covered with 0.5 mL dodecane. The calibration was performed before each run with 100 μL of a dispersion of poly(vinyl chloride) (PVC) latex in water with a defined hydrodynamic diameter of 483 nm. 100 μL nanoparticle dispersion was used for each run. To compute the hydrodynamic diameter, the density of elemental gold (19300 kg m−3) was used.
UV-Vis spectroscopy was performed with a Genesis 50 instrument (Thermo Scientific) in quartz glass cuvettes after background correction from 200 nm to 800 nm. Sample volumes of 600 μL were analyzed.
High-resolution transmission electron microscopy (HRTEM) was performed with an aberration-corrected FEI Titan transmission electron microscope equipped with a Cs-probe corrector (CEOS Company), operating at 300 kV.111 The nanoparticle dispersion was drop-cast on a copper grid, coated with an ultrathin amorphous carbon film.
Synthesis of gold nanoparticles
Ultrasmall gold nanoparticles (diameter 2 nm) were covalently functionalized with siRNA as described below, following the procedure for click reaction reported earlier in detail.46 First, glutathione-stabilized gold nanoparticles (Au-GSH) were synthesized by reduction of tetrachloroauric acid with sodium tetraborohydride. Next, they were then modified by converting the amino group of the ligand into an azide group.46 Azide-terminated nanoparticles (Au-N3; 118 azide groups per 2 nm gold nanoparticle) were then conjugated with alkyne-terminated double-stranded siRNA by copper-catalyzed azide–alkyne cycloaddition (CuAAC).46 This ensured the formation of a stable covalent bond between siRNA and particle surface. Au-N3 nanoparticles (1 eq., 1.4 μmol azide groups, 11.9 nmol Au-N3, 0.58 mg Au) and (as example) alkyne-terminated siRNA-eGFP (0.09 eq. to N3, 129 nmol, 1.86 mg) were dissolved in 100 μL of water. A solution of copper sulphate (CuSO4, 7.5 μmol, 1.2 mg) in 6 mL water together with tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 119 μmol, 63 mg) and aminoguanidine hydrogen carbonate (7.5 μmol, 1.0 mg) was prepared. 98 μL of this solution (CuSO4, 0.07 eq. to N3, 15.7 μg; THPTA, 2.2 eq. to N3, 1.65 mg; aminoguanidine hydrogencarbonate, 0.07 eq. to N3, 13.3 μg) were added to the dispersion to start the click reaction. Next, 28.1 μL of a 10 mM sodium ascorbate solution (0.2 eq. to N3, 55.6 μg) was added. The dispersion was stirred overnight for 16 h. Then, the nanoparticle dispersion was centrifuged five times by 30 kDa Spin-X® UF spin filters at 4000 rpm (2500 g) for 45 min to remove any unbound siRNA. The full characterization data of Au-GSH and Au-N3 nanoparticles, including the calculation of the number of GSH groups per Au-NP (125) and of N3 groups per gold nanoparticle (118), has been reported in ref. 46, including 1D and 2D NMR spectra.The following alkyne-terminated siRNAs were clicked to Au-N3 nanoparticles in this way: siRNA-eGFP-Cy5 (0.09 eq. to N3, 129 nmol, 1.93 mg), siRNA-eGFP-r (0.09 eq. to N3, 129 nmol, 1.86 mg), siRNA-p65 (0.09 eq. to N3, 129 nmol, 1.77 mg), siRNA-18.1 (0.09 eq. to N3, 129 nmol, 1.77 mg), and siRNA-29.2 (0.09 eq. to N3, 129 nmol, 1.61 mg).
For all types of nanoparticles, the gold content of the nanoparticle dispersions was determined by atomic absorption spectroscopy (AAS) and converted to the number of gold nanoparticles, using the density of gold and the average particle diameter (solid core) of 2 nm. As each gold nanoparticle has a mass of 8.08∙10−20 g, 1 g of gold nanoparticles corresponds to 1.24∙1019 particles.46 The amount of siRNA was measured by UV-Vis spectroscopy with calibration curves of siRNA-alkyne solutions. The quantification of siRNA was performed by the absorbance of ribonucleic acids (integral from 228 to 303 nm), and additionally with Cy5 for siRNA-eGFP-Cy5 (integral from 500 to 700 nm). To determine the concentration of the gold nanoparticles, atomic absorption spectroscopy (AAS) was used as described above. The ratio of siRNA molecules to gold nanoparticles (diameter 2 nm) in a given dispersion gave the number of siRNA molecules per nanoparticle. For a typical sample, the siRNA concentration per nanoparticle was calculated as follows:
Concentration of gold nanoparticles (2 nm) from the molar gold concentration by AAS:
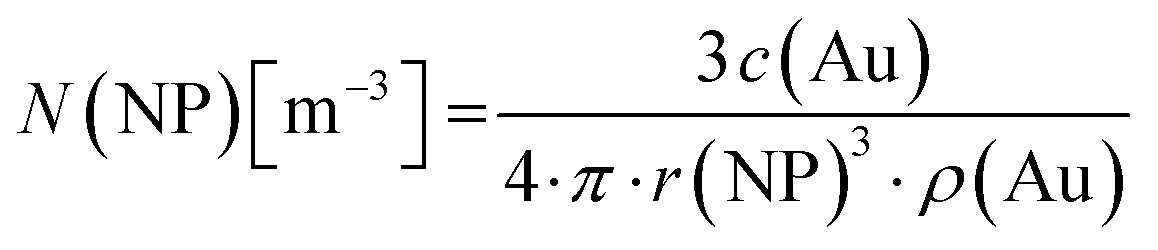 | (1) |
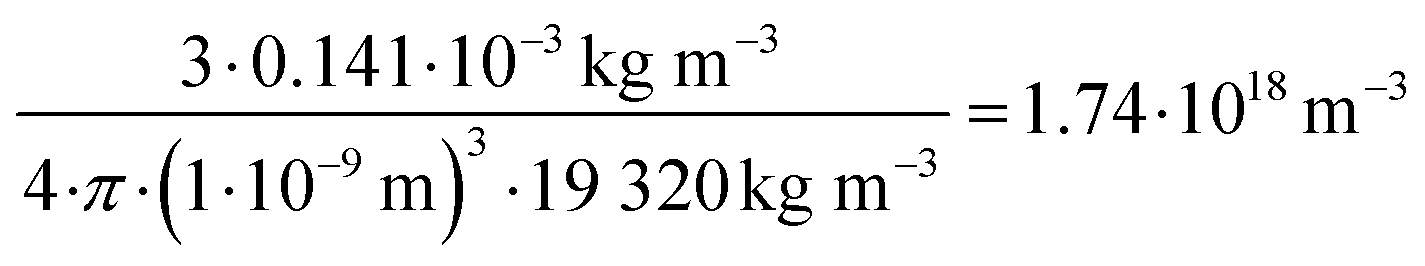 | (2) |
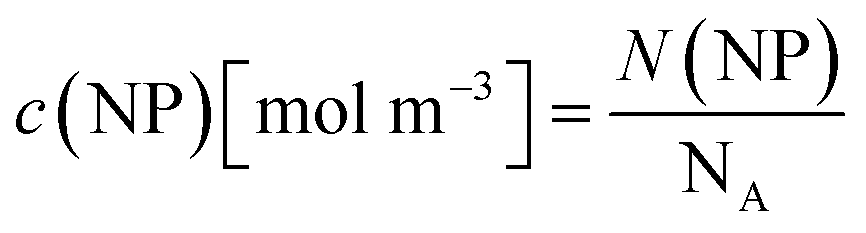 | (3) |
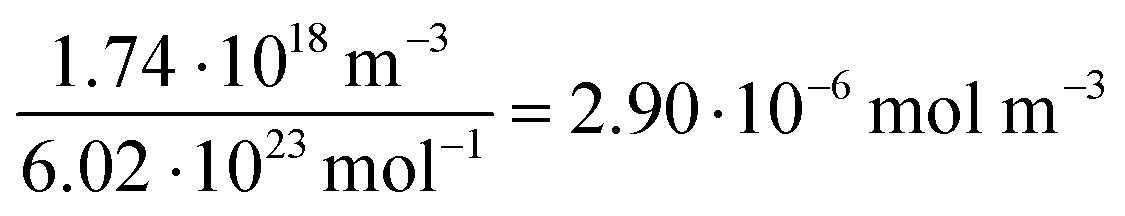 | (4) |
Concentration of Cy5 in an Au-siRNA-Cy5 dispersion (1 Cy5 per siRNA) by UV spectroscopy, using Lambert–Beer's law after recording previous calibration curves (500–700 nm):
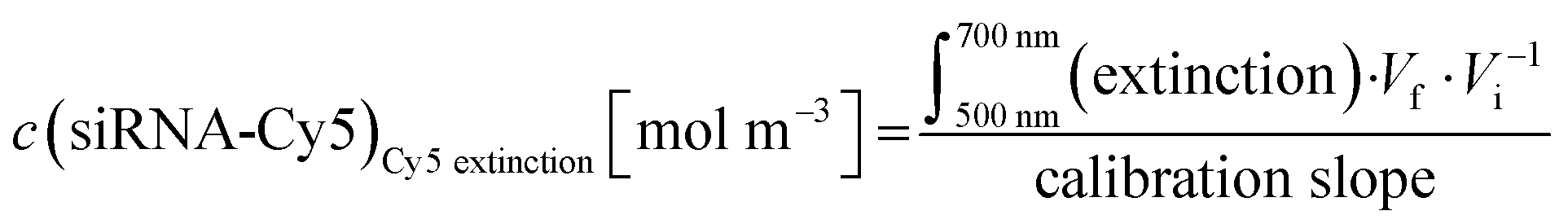 | (5) |
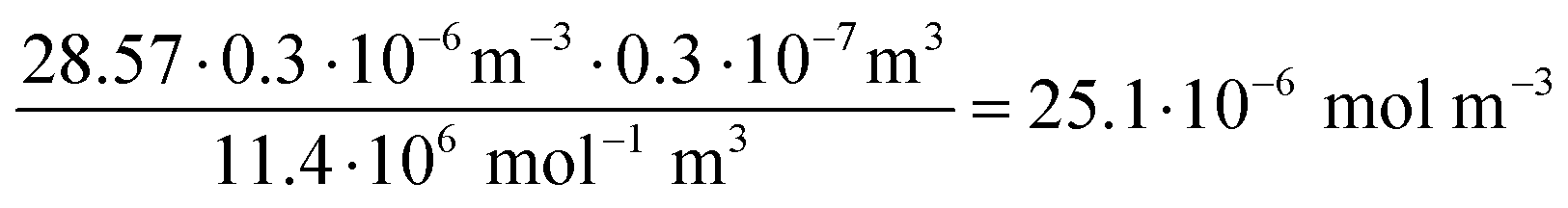 | (6) |
Concentration of siRNA in an Au-siRNA-Cy5 dispersion by UV spectroscopy, using Lambert–Beer's law after recording previous calibration curves (234–297 nm; absorption of nucleobases):
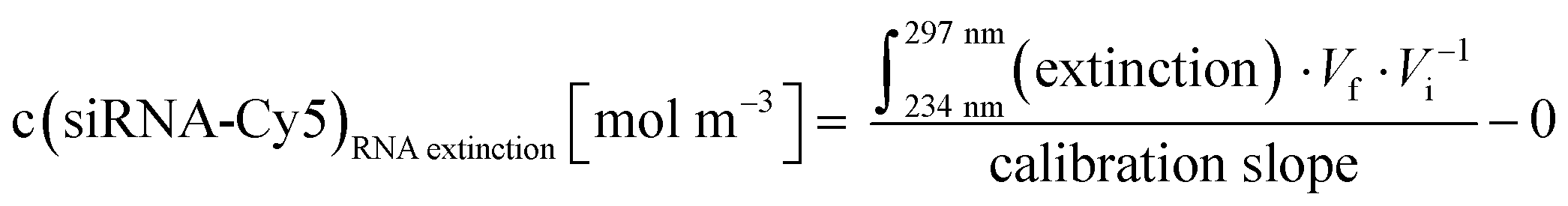 | (7) |
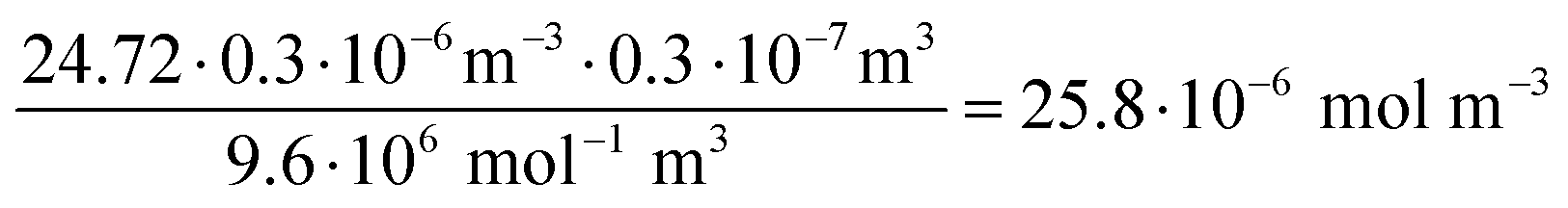 | (8) |
Number of siRNA molecules per nm from Cy5 absorption:
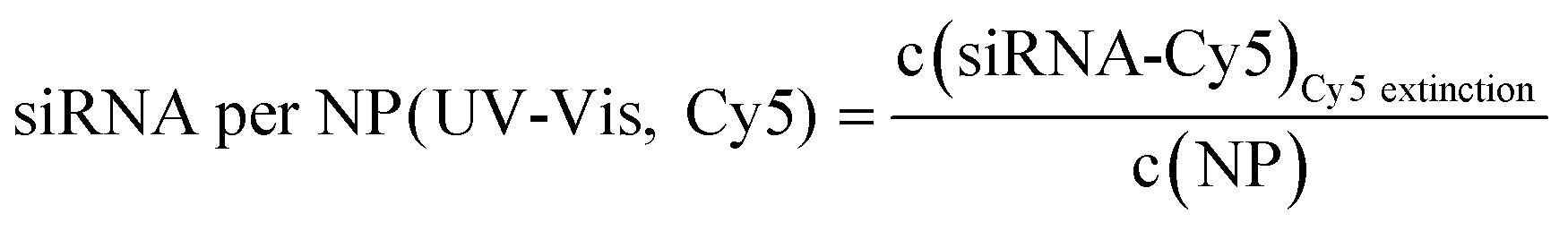 | (9) |
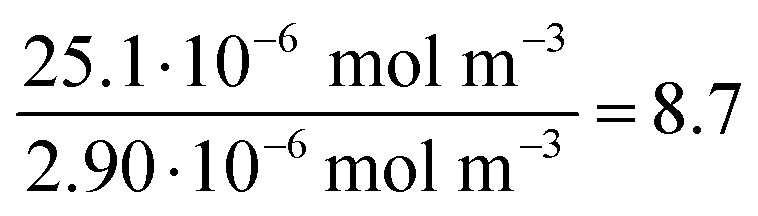 | (10) |
Number of siRNA molecules per nm from nucleobase absorption:
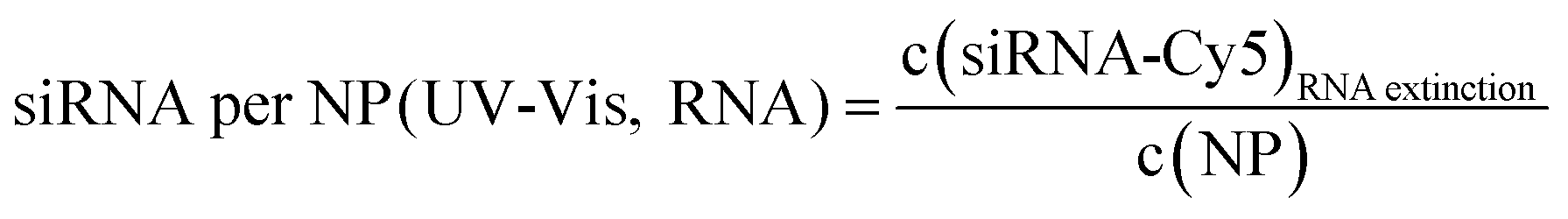 | (11) |
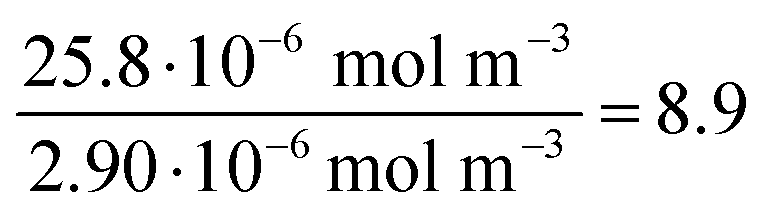 | (12) |
Cells and viruses
The eGFP-expressing human cervix carcinoma cell line HeLa-eGFP was generated in our group. The cells were cultivated at 37 °C and 5% CO2 in Gibco™ Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific), 50 μg mL−1 Gibco™ Geneticin (G418 sulfate, Thermo Fisher Scientific), 1 mM Gibco™ sodium pyruvate (Thermo Fisher Scientific), and 1 mM Gibco™ GlutaMAX (Thermo Fisher Scientific). Cells were passaged either at 70–90% confluency or every two to three days by trypsinization with 0.05% Gibco™ Trypsin-EDTA (Thermo Fisher Scientific). The cells were washed two to three times with Gibco™ Dulbecco's buffered saline (DPBS, Thermo Fisher Scientific) between the individual steps of each experiment.Vero cells (American Type Culture Collection, ATCC, CCL81, Rockville, MD, USA) were cultivated in Dulbecco's modified Eagle medium (DMEM, Life Technologies Gibco, Darmstadt, Germany), supplemented with 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin and 10% (v/v) fetal calf serum (FCS; Life Technologies Gibco), at 37 °C and 5% CO2. HSV-2-(333)-GFP (further referred to as HSV-2 GFP) was provided by B. Sodeik (Institute of Virology, Medical University of Hannover, Germany). Viral titers were determined by a standard endpoint dilution assay and calculated as 50% tissue culture infectious dose (TCID50) mL−1 as described earlier.112
The murine monocyte cell line J774A.1 (CLS Cell Lines Service GmbH, Eppelheim, Germany) was cultured in DMEM/F-12 (1:1) (+)-L-glutamine (Gibco, Darmstadt, Germany) supplemented with 10 vol% fetal calf serum (FCS; Gibco). To induce an inflammation status, the monocytes were stimulated with lipopolysaccharide (LPS, Sigma-Aldrich; 1 μg mL−1) 4 h prior to investigation (denoted as “LPS-primed” in the following).
Viability of HeLa cells
The cell viability was determined by an MTT assay. First, HeLa-eGFP cells were seeded at a density of 50000 cells per well in a 24-well plate and incubated with 0.5 mL DMEM overnight at 37 °C in 5% CO2 atmosphere. Next, the cells were incubated either with water-dispersed siRNA-clicked ultrasmall gold nanoparticles or with Lipofectamine/siRNA. Lipofectamine/siRNA was applied at a concentration of 3.2 μg siRNA (siRNA-eGFP/siRNA-eGFP-Cy5/siRNA-eGFP-r) mL−1 per well according to the manufacturer's instructions. Nanoparticle dispersions were tested at three concentrations, i.e. c1: 5 μg Au mL−1 and 14.8 μg siRNA mL−1 (13.7 μg siRNA mL−1 for Au-siRNA-eGFP-Cy5), c2: 25 μg Au mL−1 and 74 μg siRNA mL−1, c3: 50 μg Au mL−1 and 148 μg siRNA (siRNA-eGFP/-eGFP-Cy5/-eGFP-r) mL−1. The control groups for cell viability were cells treated with 500 μL of a Lipofectamine/DMEM mixture (1:1000 Lipofectamine:DMEM) and HeLa-eGFP cells cultivated in medium alone. After the incubation for 5 h (only for Lipofectamine) or 24 h (all other cases), the cells were washed three times with DPBS to remove weakly adhering lipoplexes and nanoparticles. For the staining solution, 5 mg MTT were dissolved in 1 mL PBS and then diluted with 4 mL DMEM to a final concentration of 1 mg mL−1. 0.3 mL of the staining solution was pipetted into each well, and the cells were incubated for 1 h. Next, the solution was replaced with 0.3 mL DMSO and incubated for another 30 min. The dissolved formazan was quantitatively determined in a 96-well plate with a Multiscan plate reader (Thermo Fisher Scientific GmbH) at 570 nm.
Nanoparticle uptake and gene silencing in HeLa-eGFP cells
The uptake of Au-siRNA-eGFP-Cy5 nanoparticles by HeLa-eGFP cells was analyzed by multiple focal plane (z-stacks, interval 0.5 μm) confocal laser scanning microscopy (CLSM) in an 8-well chamber polymer slide surface modified with ibiTreat for tissue culture applications (μ-Slide 8-well, ibidi). CLSM was performed with a TCS SP8X Falcon instrument (Leica Microsystems) with a 63×/1.2 water immersion objective. 20000 cells were seeded per well and incubated with 0.2 mL DMEM overnight at 37 °C in 5% CO2 atmosphere. The cells were incubated with siRNA-eGFP-Cy5-clicked ultrasmall gold nanoparticles (final concentration 25 μg Au mL−1 per well). The control group were HeLa-eGFP cells cultivated in medium alone. After incubation for 24 h, the cells were washed three times with DPBS, and fixed with 4 vol% formaldehyde solution according to standard protocols. For post-fixation, the actin skeleton of the cells was stained with AlexaFluor™ 568 Phalloidin. Nuclei were stained with DAPI according to the manufacturer's protocol. The excitation wavelengths were 405 nm for DAPI (emission: 420 to 460 nm), 488 nm for eGFP (emission: 495 to 515 nm), 568 nm for AlexaFluor™ 568 Phalloidin (emission: 590 to 610 nm), and 647 nm for Cy5 (emission: 660 to 700 nm).
Gene silencing in HeLa-eGFP cells was analyzed by fluorescence microscopy with a Biorevo BZ-9000 instrument (Keyence). Images were recorded with the filter cubes for FITC (excitation: 470/40 nm, emission: 525/50 nm) and Cy5 (excitation: 620/60 nm, emission: 700/75 nm) and 10× and 20× air objectives together with the BZ-II viewer software. Gene silencing of eGFP in HeLa-eGFP cells was quantified by calculating the mean fluorescence intensity (MFI) in the fluorescence images and the fold change in the decrease of signal intensity. 50000 cells per well were seeded in a 24-well plate (Sarstedt). The cells were incubated with siRNA-functionalized ultrasmall nanoparticles for 24 h or with Lipofectamine/siRNA (siRNA-eGFP/-eGFP-Cy5/-eGFP-r) for 5 h. The lipoplex formation was carried out according to the manufacturer's protocol with a final concentration of 3.2 μg siRNA mL−1 per well. Nanoparticle dispersions were tested in three concentrations of applied siRNA, i.e. c1: 6–8 μg Au mL−1 and 14.8 μg siRNA mL−1, c2: 30–40 μg Au mL−1 and 74 μg siRNA mL−1, c3: 60–80 μg Au mL−1 and 148 μg siRNA mL−1. After incubation, the cells were washed twice with DPBS and submerged in fresh DMEM. All cells were fixed 72 h after the initial treatment. Next, four images of cell-confluent areas were taken per well by fluorescence microscopy. The mean fluorescence intensity (MFI) of the images was analyzed with the program Image J version 1.53k.113 Then, the average eGFP MFI of the untreated control group was divided by the MFI of the siRNA-treated cells. The fold change in signal decrease (gene silencing) of eGFP and was expressed as follows:
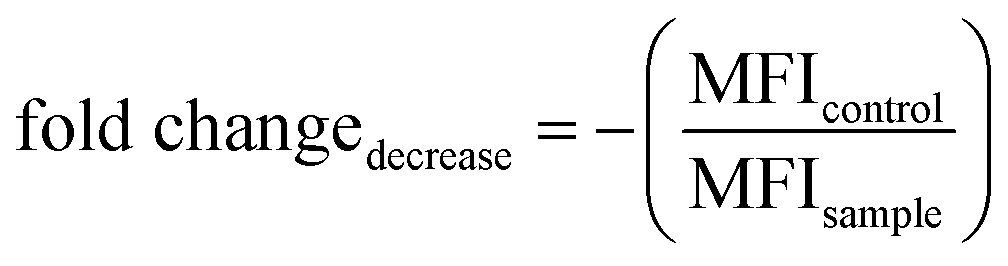 | (13) |
Only fully confluent areas with cells were analyzed to compute the mean fluorescence intensity, therefore the eGFP expression could be compared between the samples.
Cell viability determination on inflamed murine monocytes
For cell viability determination, LPS-primed monocytes were seeded in untreated 96-well cell culture plates and pre-incubated for 24 h. Au-siRNA-p65 nanoparticles were added with concentration of 1 μg siRNA-p65 mL−1 and incubated for 72 h. Non-treated LPS-primed and non-treated native cells were used as controls. Afterwards, the cells were incubated with the AlamarBlue®Cell Proliferation Reagent (Life Technologies GmbH, Darmstadt, Germany) according to the manufacturer's instruction. After 1 h of incubation, the fluorescence emission (λexc/λem: 560/590 nm) was measured. The cell viability was calculated by normalizing fluorescence data to that of non-treated native cells.Gene silencing in inflamed murine monocytes
To study the impact of gold nanoparticles carrying anti-p65 siRNA on J774A.1 monocytes, the following experimental routes were used: LPS-primed cells exposed to Au-siRNA-p65 nanoparticles (1 μg siRNA mL−1, 72 h); free siRNA-p65 (1 μg mL−1, 72 h) together with Lipofectamine® (Thermo Fisher); non-treated but LPS-primed monocytes (native inflamed monocytes); non-treated cells without LPS-priming (non-inflamed monocytes). Cells were lysed with RIPA buffer for subsequent analysis of protein expression. The total protein concentration was measured with the Bradford assay.Proteins were separated in multiple 10% (w/v) SDS-PAGE assays (10 to 20 μg total protein per lane) and transferred to an Immobilon-P membrane (Merck Millipore, Darmstadt, Germany). A multi-strip western blotting was performed.91 After blocking (PUREBlock™ Blocking Buffer, Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany; overnight, 4 °C), the membranes were probed with the corresponding primary antibodies (p65, p105/p50, and p100/p52, all mouse anti-rabbit, 1:1000 from Cell Signaling Technology, Danvers, USA; or GAPDH 1:3000 from Santa Cruz Biotechnology, Texas, USA, all overnight at 4 °C), washed, and finally incubated with the corresponding HRP-coupled secondary mouse anti-rabbit antibody (1:10000, 1 h at RT, Dianova GmbH, Hamburg, Germany). Chemiluminescence was detected by PURECL and Fusion FX7 Edge (Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany). To quantify the expression of the proteins of the different experimental groups, a densitometric analysis of western blot bands was performed with the software Bio1D (Vilber Lourmat Deutschland GmbH, Eberhardzell, Germany). Data were normalized to β-actin (loading control).
Inhibition of HSV-2 replication by gene silencing
Small interfering RNA 18.1 (siRNA-18.1) targeting the capsid encoding gene UL18 (ref. 104) and siRNA-29.2 targeting the DNA-binding protein UL29 (ref. 105) show a potent antiviral activity against HSV-2.104,105 Ultrasmall gold nanoparticles were functionalized with siRNA-18.1 siRNA-29.2 as described above. The impact of Au-siRNA-18.1 and Au-siRNA-29.2 nanoparticles on HSV-2 replication was tested in Vero cell culture. Serial dilutions of Au-siRNA-18.1 and Au-siRNA-29.2 nanoparticle stock dispersions (0.25 to 4 pmol siRNA μL−1) of Au-bound siRNA were added to confluent Vero cell monolayers grown in 96-well microtiter plates. Dissolved siRNA-18.1 and siRNA-29.2 in cell culture medium at concentrations of 4 pmol siRNA μL−1, respectively, were used as controls. After 4 h of incubation, the cells were infected with 25 TCID50 HSV-2 GFP. Non-infected Vero cells and mock-treated but HSV-2 infected cells served as controls. After 48 h, the cytopathic effects (CPE) were assessed by fluorescence microscopy. Immunofluorescence images were acquired with a Zeiss Observer Z1 fluorescence microscope (Carl Zeiss, Oberkochen, Germany) at 100-fold magnification. The concentrations of Au-siRNA-18.1 and Au-siRNA-29.2 required for reducing virus-induced CPE by 100% were defined as the completely neutralizing titer. With a molar mass of siRNA of about 13000 g mol−1 (Table 2), a concentration of 1 pmol siRNA μL−1 = 1 nmol siRNA mL−1 corresponds to about 13 μg siRNA mL−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Robin Meya and Beate Römer for elemental analysis, Julia Göring for Western and immunoblotting, Susann Burgold for excellent technical assistance, Dr Torsten Schaller and Dr Felix Niemeyer for assistance with NMR spectroscopy. We also thank the Imaging Center Campus Essen (ICCE) for access to confocal laser scanning microscopy on the instrument Leica TCS SP8X FALCON (Leica Instruments), which was funded by the DFG (Major Research Instrumentation Program as per Art. 91b GG, INST 20876/294-1 FUGG). This study was also supported by the Rudolf Ackermann Foundation (awarded to O. Witzke).References
- M. M. Xie, Y. L. Xu, J. Huang, Y. C. Li, L. Y. Wang, L. L. Yang and H. Mao, WIREs Nanomed. Nanobiotechnol., 2020, 12, e1644 CrossRef.
- M. Fan, Y. Han, S. T. Gao, H. Y. Yan, L. Z. Cao, Z. H. Li, X. J. Liang and J. C. Zhang, Theranostics, 2020, 10, 4944–4957 CrossRef CAS PubMed.
- K. M. Rice, G. K. Ginjupalli, N. Manne, C. B. Jones and E. R. Blough, Nanotechnology, 2019, 30, 372001 CrossRef CAS PubMed.
- K. A. Willets, A. J. Wilson, V. Sundaresan and P. B. Joshi, Chem. Rev., 2017, 117, 7538–7582 CrossRef CAS PubMed.
- L. A. Dykman and N. G. Khlebtsov, Biomaterials, 2016, 108, 13–34 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- Y. Ding, Z. W. Jiang, K. Saha, C. S. Kim, S. T. Kim, R. F. Landis and V. M. Rotello, Mol. Ther., 2014, 22, 1075–1083 CrossRef CAS PubMed.
- L. C. Cheng, X. M. Jiang, J. Wang, C. Y. Chen and R. S. Liu, Nanoscale, 2013, 5, 3547–3569 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- W. J. Stark, Angew. Chem., Int. Ed., 2011, 50, 1242–1258 CrossRef CAS PubMed.
- C. M. Cobley, J. Y. Chen, E. C. Cho, L. V. Wang and Y. N. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC.
- M. Homberger and U. Simon, Philos. Trans. R. Soc., A, 2010, 368, 1405–1453 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- Y. G. Srinivasulu, Q. F. Yao, N. Goswami and J. P. Xie, Mater. Horiz., 2020, 7, 2596–2618 RSC.
- C. J. Zeng, Pure Appl. Chem., 2018, 90, 1409–1427 CrossRef CAS.
- J. Langer, S. M. Novikov and L. M. Liz-Marzan, Nanotechnology, 2015, 26, 322001 CrossRef.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion and A. P. Alivisatos, Nano Lett., 2001, 1, 32–35 CrossRef CAS.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion, S. C. Williams and A. P. Alivisatos, J. Phys. Chem. B, 2002, 106, 11758–11763 CrossRef CAS.
- J. R. McMillan, J. D. Brodin, J. A. Millan, B. Lee, M. O. de la Cruz and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 1754–1757 CrossRef CAS PubMed.
- M. Girard, S. Z. Wang, J. S. Du, A. Das, Z. Y. Huang, V. P. Dravid, B. Lee, C. A. Mirkin and M. O. de la Cruz, Science, 2019, 364, 1174 CrossRef CAS PubMed.
- S. D. Huo, N. Q. Gong, Y. Jiang, F. Chen, H. B. Guo, Y. L. Gan, Z. S. Wang, A. Herrmann and X. J. Liang, Sci. Adv., 2019, 5, eaaw6264 CrossRef CAS PubMed.
- S. Z. Wang, J. S. S. Du, N. J. Diercks, W. J. Zhou, E. W. Roth, V. P. Dravid and C. A. Mirkin, J. Am. Chem. Soc., 2019, 141, 20443–20450 CrossRef CAS PubMed.
- C. R. Laramy, M. N. O'Brien and C. A. Mirkin, Nat. Rev. Mater., 2019, 4, 201–224 CrossRef CAS.
- C. A. Mirkin, Inorg. Chem., 2000, 39, 2258–2272 CrossRef CAS PubMed.
- G. Yamankurt, R. J. Stawicki, D. M. Posadas, J. Q. Nguyen, R. W. Carthew and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1312 CrossRef CAS.
- K. L. Young, A. W. Scott, L. L. Hao, S. E. Mirkin, G. L. Liu and C. A. Mirkin, Nano Lett., 2012, 12, 3867–3871 CrossRef CAS PubMed.
- W. L. Zhang, B. Meckes and C. A. Mirkin, ACS Cent. Sci., 2019, 5, 1983–1990 CrossRef CAS PubMed.
- S. S. Zhu, H. Xing, P. Gordiichuk, J. Park and C. A. Mirkin, Adv. Mater., 2018, 30, 1707113 CrossRef PubMed.
- A. M. Gorabi, N. Kiaie, S. Aslani, T. Jamialahmadi, T. P. Johnston and A. Sahebkar, J. Autoimmun., 2020, 114, 102529 CrossRef CAS PubMed.
- B. Kim, J. H. Park and M. J. Sailor, Adv. Mater., 2019, 31, 1903637 CrossRef CAS PubMed.
- B. Hu, L. Zhong, Y. Weng, L. Peng, Y. Huang, Y. Zhao and X. J. Liang, Signal Transduction Targeted Ther., 2020, 5, 101 CrossRef CAS PubMed.
- A. A. Sousa, P. Schuck and S. A. Hassan, Nanoscale Adv., 2021, 3, 2995–3027 RSC.
- Z. H. Liu, Z. N. Wu, Q. F. Yao, Y. T. Cao, O. J. H. Chai and J. P. Xie, Nano Today, 2021, 36, 101053 CrossRef CAS.
- Y. X. Du, H. T. Sheng, D. Astruc and M. Z. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- R. A. Revia, Z. R. Stephen and M. Zhang, Acc. Chem. Res., 2019, 52, 1496–1506 CrossRef CAS PubMed.
- M. Kopp, S. Kollenda and M. Epple, Acc. Chem. Res., 2017, 50, 1383–1390 CrossRef CAS PubMed.
- K. Zarschler, L. Rocks, N. Licciardello, L. Boselli, E. Polo, K. P. Garcia, L. De Cola, H. Stephan and K. A. Dawson, Nanomedicine, 2016, 12, 1663–1701 CrossRef CAS PubMed.
- N. H. Kim, M. J. Hackett, J. Park and T. Hyeon, Chem. Mater., 2014, 26, 59–71 CrossRef.
- L. Shang, S. J. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- R. Yu, L. M. Liz-Marzan and F. J. Garcia de Abajo, Chem. Soc. Rev., 2017, 46, 6710–6724 RSC.
- J. Turkevich, P. C. Stevenson and J. Hilliery, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- K. Klein, K. Loza, M. Heggen and M. Epple, ChemNanoMat, 2021, 7, 1330–1339 CrossRef CAS.
- S. B. van der Meer, K. Loza, K. Wey, M. Heggen, C. Beuck, P. Bayer and M. Epple, Langmuir, 2019, 35, 7191–7204 CrossRef CAS PubMed.
- S. B. van der Meer, I. Hadrovic, A. Meiners, K. Loza, M. Heggen, S. K. Knauer, P. Bayer, T. Schrader, C. Beuck and M. Epple, J. Phys. Chem. B, 2021, 125, 115–127 CrossRef CAS PubMed.
- K. Klein, M. Hayduk, S. Kollenda, M. Schmiedtchen, J. Voskuhl and M. Epple, Molecules, 2022, 27, 1788 CrossRef CAS PubMed.
- N. Bialas, V. Sokolova, S. B. van der Meer, T. Knuschke, T. Ruks, K. Klein, A. M. Westendorf and M. Epple, Nano Sel., 2022 DOI:10.1002/nano.202200049.
- L. Yang, L. Shang and G. U. Nienhaus, Nanoscale, 2013, 5, 1537–1543 RSC.
- C. Carrillo-Carrion, A. I. Bocanegra, B. Arnaiz, N. Feliu, D. C. Zhu and W. J. Parak, ACS Nano, 2019, 13, 4631–4639 CrossRef CAS PubMed.
- V. Sokolova, G. Mekky, S. B. van der Meer, M. C. Seeds, A. J. Atala and M. Epple, Sci. Rep., 2020, 10, 18033 CrossRef CAS.
- V. Sokolova, G. Nzou, S. B. van der Meer, T. Ruks, M. Heggen, K. Loza, N. Hagemann, F. Murke, B. Giebel, D. M. Hermann, A. J. Atala and M. Epple, Acta Biomater., 2020, 111, 349–362 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef.
- D. Mahl, C. Greulich, W. Meyer-Zaika, M. Köller and M. Epple, J. Mater. Chem., 2010, 20, 6176–6181 RSC.
- G. Schmid, W. G. Kreyling and U. Simon, Arch. Toxicol., 2017, 91, 3011–3037 CrossRef CAS.
- A. Leifert, Y. Pan-Bartnek, U. Simon and W. Jahnen-Dechent, Nanoscale, 2013, 5, 6224–6242 RSC.
- J. Broda, G. Schmid and U. Simon, Struct. Bonding, 2014, 161, 189–242 CrossRef.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS PubMed.
- L. Dong, M. Li, S. Zhang, J. Li, G. Shen, Y. Tu, J. Zhu and J. Tao, Small, 2015, 11, 2571–2581 CrossRef CAS PubMed.
- S. K. Boda, J. Broda, F. Schiefer, J. Weber-Heynemann, M. Hoss, U. Simon, B. Basu and W. Jahnen-Dechent, Small, 2015, 11, 3183–3193 CrossRef CAS PubMed.
- Y. Pan, A. Leifert, M. Graf, F. Schiefer, S. Thoröe-Boveleth, J. Broda, M. C. Halloran, H. Hollert, D. Laaf, U. Simon and W. Jahnen-Dechent, Small, 2013, 9, 863–869 CrossRef CAS PubMed.
- S. Son, V. G. Deepagan, S. Shin, H. Ko, J. Min, W. Um, J. Jeon, S. Kwon, E. S. Lee, M. Suh, D. S. Lee and J. H. Park, Acta Biomater., 2018, 79, 294–305 CrossRef CAS PubMed.
- A. A. Sousa, S. A. Hassan, L. L. Knittel, A. Balbo, M. A. Aronova, P. H. Brown, P. Schuck and R. D. Leapman, Nanoscale, 2016, 8, 6577–6588 RSC.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang, S. Jin, Y. Gan, P. C. Wang, S. He, X. Zhang and X. J. Liang, ACS Nano, 2012, 6, 4483–4493 CrossRef CAS PubMed.
- J. Bourquin, D. Septiadi, D. Vanhecke, S. Balog, L. Steinmetz, M. Spuch-Calvar, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, ACS Nano, 2019, 13, 7759–7770 CrossRef CAS.
- L. M. Liz-Marzan, Chem. Commun., 2013, 49, 16–18 RSC.
- H. Fissan, S. Ristig, H. Kaminski, C. Asbach and M. Epple, Anal. Methods, 2014, 6, 7324–7334 RSC.
- T. Ruks, C. Beuck, T. Schaller, F. Niemeyer, M. Zähres, K. Loza, M. Heggen, U. Hagemann, C. Mayer, P. Bayer and M. Epple, Langmuir, 2019, 35, 767–778 CrossRef CAS PubMed.
- G. Salassa and T. Burgi, Nanoscale Horiz., 2018, 3, 457–463 RSC.
- C. Guo and J. L. Yarger, Magn. Reson. Chem., 2018, 56, 1074–1082 CrossRef CAS PubMed.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- O. Wetzel, O. Prymak, K. Loza, N. Gumbiowski, M. Heggen, P. Bayer, C. Beuck, C. Weidenthaler and M. Epple, Inorg. Chem., 2022, 61, 5133–5147 CrossRef CAS.
- O. Wetzel, S. Hosseini, K. Loza, M. Heggen, O. Prymak, P. Bayer, C. Beuck, T. Schaller, F. Niemeyer, C. Weidenthaler and M. Epple, J. Phys. Chem. B, 2021, 125, 5645–5659 CrossRef CAS PubMed.
- M. Lach, C. Strelow, A. Meyer, A. Mews and T. Beck, ACS Appl. Mater. Interfaces, 2022, 14, 10656–10668 CrossRef CAS PubMed.
- P. A. Baeuerle and D. Baltimore, Science, 1988, 242, 540–546 CrossRef CAS PubMed.
- T. Huxford and G. Ghosh, Cold Spring Harbor Perspect. Biol., 2009, 1, a000075 Search PubMed.
- A. Israel, Cold Spring Harbor Perspect. Biol., 2010, 2, a000158 Search PubMed.
- C. Kunsch, S. M. Ruben and C. A. Rosen, Mol. Cell. Biol., 1992, 12, 4412–4421 CAS.
- T. Liu, L. Zhang, D. Joo and S. C. Sun, Signal Transduction Targeted Ther., 2017, 2, 17023 CrossRef PubMed.
- S. Chernousova and M. Epple, Gene Ther., 2017, 24, 282–289 CrossRef CAS PubMed.
- H. H. Szeto, P. W. Schiller, K. Zhao and G. Luo, FASEB J., 2005, 19, 118–120 CrossRef CAS PubMed.
- J. Kurreck, Angew. Chem., Int. Ed., 2009, 48, 1378–1398 CrossRef CAS PubMed.
- M. G. Dorrington and I. D. C. Fraser, Front. Immunol., 2019, 10, 705 CrossRef CAS.
- E. B. Glass, S. Masjedi, S. O. Dudzinski, A. J. Wilson, C. L. Duvall, F. E. Yull and T. D. Giorgio, ACS Omega, 2019, 4, 16756–16767 CrossRef CAS PubMed.
- R. A. Ortega, W. Barham, K. Sharman, O. Tikhomirov, T. D. Giorgio and F. E. Yull, Int. J. Nanomed., 2016, 11, 2163–2177 CrossRef CAS PubMed.
- N. Bialas, E. K. Müller, M. Epple and I. Hilger, Biomaterials, 2021, 276, 121013 CrossRef CAS PubMed.
- E. K. Müller, N. Bialas, M. Epple and I. Hilger, Biomedicines, 2022, 10, 1571 CrossRef PubMed.
- E. K. Müller, N. Bialas, M. Epple and I. Hilger, Pharmaceutics, 2022, 14, 419 CrossRef.
- M. Mussbacher, M. Salzmann, C. Brostjan, B. Hoesel, C. Schoergenhofer, H. Datler, P. Hohensinner, J. Basilio, P. Petzelbauer, A. Assinger and J. A. Schmid, Front. Immunol., 2019, 10, 85 CrossRef CAS PubMed.
- D. I. Bernstein, A. R. Bellamy, E. W. Hook III, M. J. Levin, A. Wald, M. G. Ewell, P. A. Wolff, C. D. Deal, T. C. Heineman, G. Dubin and R. B. Belshe, Clin. Infect. Dis., 2013, 56, 344–351 CrossRef CAS PubMed.
- K. J. Looker, G. P. Garnett and G. P. Schmid, Bull. W. H. O., 2008, 86A, 805–812 CrossRef PubMed.
- J. T. Vrabec and R. L. Alford, J. NeuroVirol., 2004, 10, 216–222 CrossRef CAS PubMed.
- R. Whitley, D. W. Kimberlin and C. G. Prober, in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, ed. A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley and K. Yamanishi, Cambridge, 2007 Search PubMed.
- D. Bauer, M. Alt, M. Dirks, M. Kasper, A. Buch, U. Dittmer, A. Goergens, B. Sodeik, A. Heiligenhaus, R. Michael and A. Krawczyk, Invest. Ophthalmol. Visual Sci., 2016, 57, 3300 Search PubMed.
- P. Sen and S. E. Barton, BMJ, 2007, 334, 1048–1052 CrossRef CAS PubMed.
- U. Meyding-Lamade and C. Strank, Ther. Adv. Neurol. Disord., 2012, 5, 279–296 CrossRef PubMed.
- S. Burrel, D. Boutolleau, G. Azar, S. Doan, C. Deback, I. Cochereau, H. Agut and E. E. Gabison, J. Clin. Virol., 2013, 58, 321–324 CrossRef.
- A. V. Farooq and D. Shukla, Surv. Ophthalmol., 2012, 57, 448–462 CrossRef PubMed.
- R. Duan, R. D. de Vries, J. M. van Dun, F. B. van Loenen, A. D. M. E. Osterhaus, L. Remeijer and G. M. G. M. Verjans, J. Infect. Dis., 2009, 200, 1402–1414 CrossRef CAS PubMed.
- E. Paradowska, M. Studzinska, A. Jablonska, V. Lozovski, N. Rusinchuk, I. Mukha, N. Vitiuk and Z. J. Lesnikowski, Molecules, 2021, 26, 5960 CrossRef CAS PubMed.
- F. Jin, S. Li, K. Zheng, C. Zhuo, K. Ma, M. Chen, Q. Wang, P. Zhang, J. Fan, Z. Ren and Y. Wang, PLoS One, 2014, 9, e96623 CrossRef PubMed.
- D. Palliser, D. Chowdhury, Q. Y. Wang, S. J. Lee, R. T. Bronson, D. M. Knipe and J. Lieberman, Nature, 2006, 439, 89–94 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- T. Efthymiou, W. Gong and J. P. Desaulniers, Molecules, 2012, 17, 12665–12703 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258–1267 CrossRef CAS PubMed.
- S. Kath-Schorr, Top. Curr. Chem., 2016, 374, 4 CrossRef PubMed.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef.
- A. Thust, J. Barthel and K. Tillmann, J. Large-scale Res. Fac., 2016, 2, A41 CrossRef.
- J. L. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI, Fig. 1 shows images of protein bands from inflamed murine macrophages after exposure with Au-siRNA-p65-NPs. See https://doi.org/10.1039/d2na00250g |
| ‡ Both authors share first authorship. |
| This journal is © The Royal Society of Chemistry 2022 |