
Green synthesis of reduced Ti3C2Tx MXene nanosheets with enhanced conductivity, oxidation stability, and SERS activity†
Tej B.
Limbu
 a,
Basant
Chitara
a,
Jason D.
Orlando
a,
Martha Y.
Garcia Cervantes
a,
Shalini
Kumari
b,
Qi
Li
b,
Yongan
Tang
c and
Fei
Yan
*a
a,
Basant
Chitara
a,
Jason D.
Orlando
a,
Martha Y.
Garcia Cervantes
a,
Shalini
Kumari
b,
Qi
Li
b,
Yongan
Tang
c and
Fei
Yan
*a
aDepartment of Chemistry and Biochemistry, North Carolina Central University, Durham, NC 27707, USA. E-mail: fyan@nccu.edu; tejblimbu@gmail.com
bDepartment of Physics, The Pennsylvania State University, University Park, PA 16802, USA
cDepartment of Mathematics and Physics, North Carolina Central University, Durham, NC 27707, USA
First published on 4th March 2020
Abstract
Transition metal carbides (MXenes) are an emerging family of highly conductive two-dimensional materials with additional functional properties introduced by surface terminations. Further modification of the surface terminations makes MXenes even more appealing for practical applications. Herein, we report a facile and environmentally benign synthesis of reduced Ti3C2Tx MXene (r-Ti3C2Tx) via a simple treatment with L-ascorbic acid at room temperature. r-Ti3C2Tx shows a six-fold increase in electrical conductivity, from 471 ± 49 for regular Ti3C2Tx to 2819 ± 306 S m−1 for the reduced version. Additionally, we show an enhanced oxidation stability of r-Ti3C2Tx as compared to regular Ti3C2Tx. An examination of the surface-enhanced Raman scattering (SERS) activity reveals that the SERS enhancement factor of r-Ti3C2Tx is an order of magnitude higher than that of regular Ti3C2Tx. The improved SERS activity of r-Ti3C2Tx is attributed to the charge transfer interaction between the MXene surface and probe molecules, re-enforced by an increased electronic density of states (DOS) at the Fermi level of r-Ti3C2Tx. The findings of this study suggest that reduced MXene could be a superior choice over regular MXene, especially for the applications that employ high electronic conductivity, such as electrode materials for batteries and supercapacitors, photodetectors, and SERS-based sensors.
1. Introduction
Since their discovery in 2011,1 titanium carbides, also called MXenes, a two-dimensional (2D) layered material, have attracted increasing attention from the scientific community, due to their exciting properties such as high metallic-like conductivity, good environmental and chemical stability, and controllable surface hydrophilicity.2,3 Subsequently, several other species of the MXene family4–8 have been discovered and synthesized in laboratories by top-down approaches starting with a three-dimensional parent material known as MAX. MAX phases are transition metal carbides, nitrides, or carbonitrides with the general formula Mn+1AXn, where n = 1, 2, or 3, M is a transition metal, A is an A group element (mainly 13 and 14 group), and X is C and/or N. After removal of the A layers in MAX phases by chemical etching, the obtained 2D materials are regarded as MXenes, which are usually represented by the general formula Mn+1XnTx, where Tx is the surface termination acquired during the chemical etching process. Depending upon the type of M and X elements, and surface terminations, various members of the MXene family are known to exhibit quite different physical properties and chemical behavior; from metallic to semiconducting, or topological insulators, and even magnetic.6,7,9 Due to their diverse and exciting properties, MXenes have been explored for a myriad of applications, including energy storage devices,10–12 catalysis,5,13,14 water desalination,15 transparent conductors,16,17 light absorbers,3 electromagnetic interference shielding,18 photodetection,19 and surface-enhanced Raman scattering (SERS) detection,4,20,21 among others.Among a wide variety of MXene species, Ti3C2Tx MXene is reported to have shown the highest electrical conductivity22 and has been studied most extensively. It possesses impressive metallic-like conductivity comparable to those of reduced graphene oxide (GO) and carbon nanotubes,23,24 and for most applications, such as electrode materials for batteries, supercapacitors, transparent conductors, plasmonic devices, photodetectors, and SERS substrates, high electrical conductivity is desired. Interestingly, the conductivity of MXenes can be further increased by de-intercalation and surface de-functionalization.6 Hart et al.6 demonstrated by in situ vacuum annealing, electrical biasing, spectroscopic analysis, and transmission electron microscopy (TEM) that the loss of –OH and –F terminations in Ti3C2Tx MXene improves electronic conductivity. This result is consistent with a literature report25 that by density functional theory simulation showed that surface functionalization with –OH, –H, –F, and –O on Ti3C2 MXene reduces the density of states (DOS) at the Fermi level, thereby reducing the conductivity. This implies that there exists plenty of room for further enhancement of the electrical conductivity of MXene by surface modification. Reduction of MXenes could be a new approach to tailor the properties and conductivity enhancement, in analogy with the reduction of insulating GO to highly conducting reduced-graphene oxide (r-GO).26,27 Yoon et al.2 showed that Li-ethylenediamine (Li-EDA) treatment on Ti3C2Tx removes most of the F and OH terminations and Ti4+ chemical states are changed to Ti3+, and the reduction of Ti3C2Tx MXene produces a Pauli paramagnetic character. Although this work showed an effective method of Ti3C2Tx MXene reduction, the use of Li-EDA as a reducing agent required a glove-box environment for safety purposes. This indicates a need for a safer, reproducible, and easily accessible method for the MXene reduction process.
Among several potential applications, MXenes have shown promise for making cheap and efficient SERS substrates.4,20,21,28 SERS is a non-destructive and highly sensitive technique for molecular detection at trace amounts. Although noble metal nanostructure-based SERS substrates have shown an ultrahigh enhancement factor (EF) of 1014,29 the high cost of production limits their wide use for practical purposes. MXenes, which are produced by simple solution processing methods with relatively cheap precursors, can be an alternative material for commercial SERS substrates. Sarycheva et al.20 showed that Ti3C2Tx MXene-based SERS substrates enhanced the Raman signal of common organic dyes by as much as 106. Soundiraraju et al.4 reported even more interesting SERS results with titanium nitride (Ti2NTx) MXene. They found an EF of 1012 using rhodamine 6G as a probe compound on a SERS substrate made by coating paper with Ti2NTx. Such a high EF is good enough for single molecule detection, indicating a potential use of MXenes for replacing expensive SERS substrates made from noble metal nanostructures.
Despite their exciting physical and electrochemical properties, it is widely known that MXenes have a weak chemical and environmental stability,30–35 which can limit their use for practical applications. Because of the spontaneous and rapid oxidation issue in water and air, better ways of storing MXenes have also been explored and suggested.30,32,35 However, it would be even more interesting if the cause of oxidation could be identified and the MXene crystals could be engineered to make them shelf-stable, or if one could discover a state-of-the-art route to synthesize chemically and environmentally stable MXenes.
In this work, we demonstrate a green protocol for the room temperature synthesis of reduced Ti3C2Tx MXene (r-Ti3C2Tx) via a simple L-ascorbic acid treatment, which is facile and highly reproducible. We show a significant modification of the properties of r-Ti3C2Tx with enhanced electrical conductivity and oxidation stability as compared to that of the Ti3C2Tx MXene (Ti3C2Tx) prepared by conventional methods,1,20,36 showing reduced MXene as a primary choice for use in several applications that employ high electronic conductivity. We also report an application of r-Ti3C2Tx as a SERS substrate and demonstrate an improvement in the SERS EF by an order of magnitude compared to that of Ti3C2Tx.
2. Experimental section
2.1 Synthesis of Ti3C2Tx
To synthesize r-Ti3C2Tx, we started with etching of Al layers in Ti3AlC2 MAX powder purchased from Carbon-Ukraine (Y-Carbon, Ltd). The reduction process is preceded by etching and MXene delamination. The etching of Al layers and delamination of MXene were carried out following the methods described in a published report36 with a slight modification. Briefly, 0.5 g of Ti3AlC2 MAX powder was slowly added to 10 ml of 30% hydrofluoric acid (HF) (ACROS Organics, 48–51% solution in water) under a gentle and constant stirring condition with the help of a Teflon magnetic bar, and the stirring was continued for 7 hours. The solution was then washed one time with deionized (DI) water by centrifugation at a speed of 3500 rpm for 30 minutes and the water-like supernatant was removed. The sediment was then further washed with at least 5 liters of DI water via vacuum-assisted filtration using a polyvinyl difluoride (PVDF) filter membrane with 0.22 μm pore size (Durapore, Millipore) until the pH of the filtrate was lowered to ∼6.5. The cleaned sediment was then dried in the same filter cup by placing it in a vacuum desiccator for 24 hours. For the delamination process, 100 mg of Ti3C2Tx powder was put into 20 ml of 1% tetramethylammonium hydroxide (TMAOH) (Alfa Aesar, Electronic Grade, 99.9999% (metal basis) liquid) solution and was shaken for 24 hours. After 24 hours of shaking, the solution was centrifuged at 5000 rpm for 30 minutes to separate out the unwanted brown solution containing TMAOH. The centrifugation process was repeated several times until the pH of the supernatant solution was decreased to ∼7. The MXene solution was then centrifuged at 3500 rpm for 5 minutes to separate out the bigger and unetched particles, and the stable MXene colloidal solution was collected for further processing.2.2 Green synthesis of r-Ti3C2Tx
For the reduction process, first, DI water was added to the obtained MXene colloidal solution to make a total volume of 40 ml. The solution was shaken and divided into two equal volumes, 20 ml each for Ti3C2Tx and r-Ti3C2Tx. One part was further centrifuged to extract a slurry and freeze-dried to obtain a powder material for characterization. The obtained Ti3C2Tx powder was weighed to be ∼25 mg. The other part, which was expected to contain the same amount of Ti3C2Tx (∼25 mg) in 20 ml of water was mixed with 1 g of L-ascorbic acid (Fisher Scientific, Reagent Grade) and shaken for 5 hours. After 5 hours of treatment with L-ascorbic acid, the solution was centrifuged several times at 5000 rpm for 30 minutes to remove the acidic solution. Finally, a slurry of r-Ti3C2Tx nanosheets was extracted and freeze-dried to obtain the r-Ti3C2Tx powder for characterization. In order to obtain Ti3C2Tx and r-Ti3C2Tx colloidal solutions for film preparation on SiO2/Si and paper, the synthesis process was repeated, and instead of freeze-drying, the slurries of MXene nanosheets were dissolved in a suitable amount of methanol.2.3 Fabrication of Ti3C2Tx/r-Ti3C2Tx films
To fabricate the films of Ti3C2Tx and r-Ti3C2Tx on SiO2/Si substrates, the slurries of Ti3C2Tx and r-Ti3C2Tx nanosheets were dissolved in a suitable amount of methanol separately in two different glass vials to obtain dense colloidal solutions. The dense colloidal solutions of Ti3C2Tx and r-Ti3C2Tx nanosheets were drop casted on oxygen plasma-cleaned SiO2/Si substrates followed by subsequent heating at 60 °C by placing the samples on a hot plate. Ti3C2Tx and r-Ti3C2Tx films on paper substrates were fabricated on PVDF filter paper with a pore size of 0.22 μm (Durapore, Millipore) employing vacuum-assisted filtration.2.4 Characterization of Ti3C2Tx/r-Ti3C2Tx
Characterization of Ti3C2Tx/r-Ti3C2Tx samples was performed by using Raman spectroscopy (Horiba LabRAM Evolution RAMAN microscope), atomic force microscopy (AFM) (Horiba Smart SPM Atomic Force Microscope), UV-visible spectroscopy (VWR, UV-3100PC Spectrophotometer), scanning electron microscopy (SEM) (FEI Verios 460L), TEM (FEI Talos F200X, 200 KV accelerating voltage), X-ray diffractometry (XRD) (Rigaku SmartLab X-ray Diffractometer), and X-ray photoelectron spectroscopy (XPS) (SPECS FlexMod XPS, Mg Kα excitation (1254 eV)). Ti(20 nm)/Au(300 nm) contact pads were deposited on the fabricated MXene films by using electron beam evaporation. Electrical properties were studied by employing transmission line measurements with two probes in a probe station (EVERBEING) equipped with a Keithley analyzer (4200A-SCS). For the test of oxidation stability, the dried and stored Ti3C2Tx and r-Ti3C2Tx powders were re-dispersed in water at a concentration of 15 mg/10 ml, sonicated for 10 minutes and left open in an ambient atmosphere for several days. The stability test was repeated with another concentration, 5 mg/10 ml of water for confirmation. The SERS measurements were performed using a Raman spectrometer (Horiba LabRAM Evolution RAMAN microscope) with 532 nm laser excitation and a 100× objective lens with an integration time of 20 s. Raman enhancement of Ti3C2Tx and r-Ti3C2Tx was examined with three dye molecules, i.e., crystal violet (CV) (ACROS Organics), methylene blue (MB) (ACROS Organics), and rhodamine 6G (R6G) (ACROS Organics).3. Results and discussion
3.1 Synthesis and characterization
Fig. 1 schematically summarizes the synthesis route of r-Ti3C2Tx. Etching of Al layers in the Ti3AlC2 MAX phase (Fig. 1a) with HF produces 2D crystals of titanium carbide with –O, –OH, and –F terminations (Fig. 1b). The Ti3C2Tx layers are still bound to each other by a weak hydrogen or van der Waals force between Tx (OH, F, O) terminations of two adjacent MXene layers.1,36 Upon intercalation with tetramethylammonium ions during delamination using TMAOH, the Ti3C2Tx layers are separated into free standing particles (Fig. 1c). Treatment of colloidal Ti3C2Tx solution with L-ascorbic acid changes the surface chemistry of MXene nanosheets to give rise to reduced Ti3C2Tx (Fig. 1d). We note that we have followed the model presented by Yoon et al.2 to describe the surface chemistry changes in the 2D Ti3C2Tx due to HF etching and reduction processes as depicted in Fig. 1b–d, which will be discussed later in detail.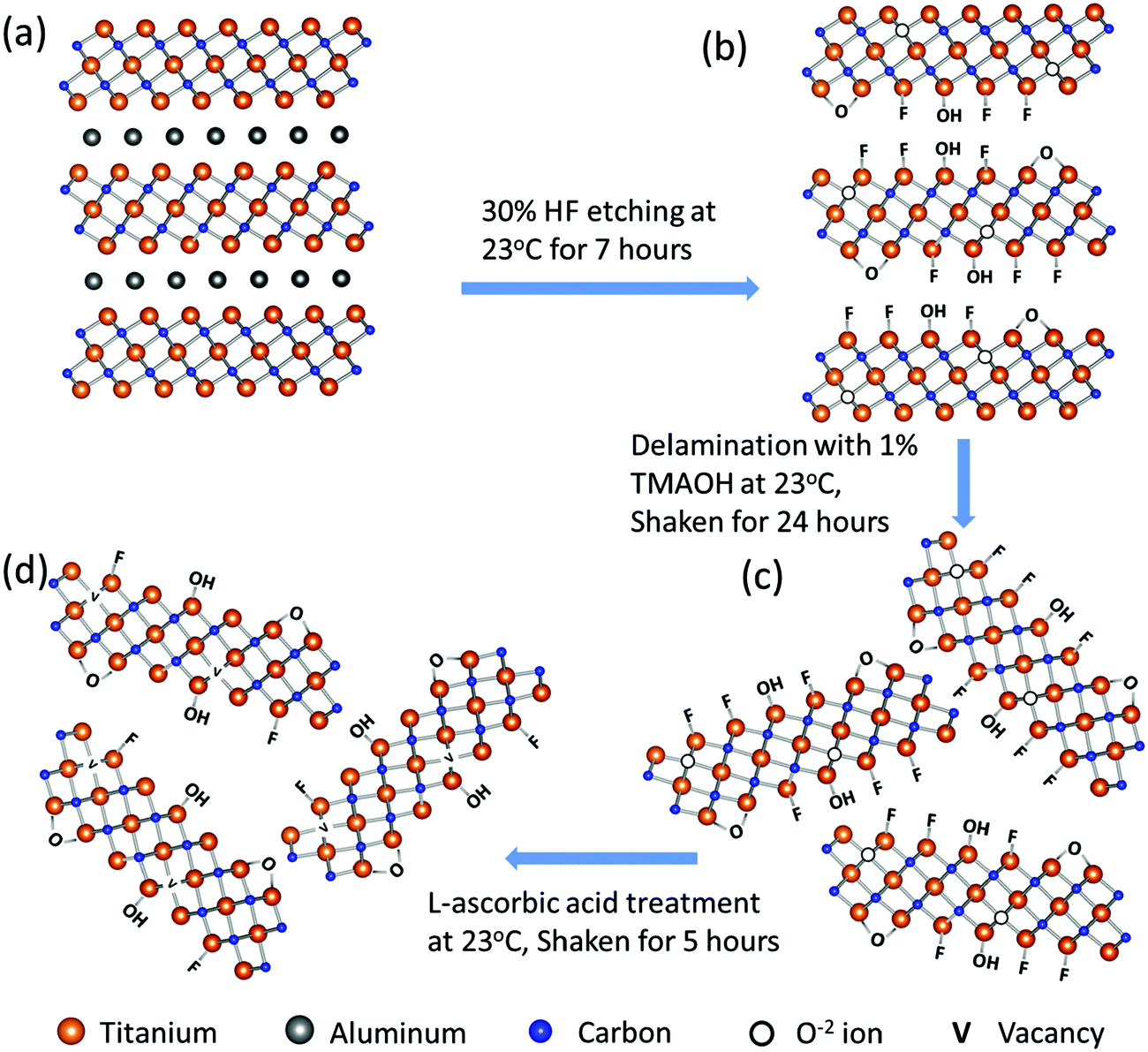 | ||
| Fig. 1 Schematic diagram of r-Ti3C2Tx synthesis. | ||
 | ||
| Fig. 2 (a and b) SEM micrographs of Ti3C2Tx after HF-etching of Ti3AlC2 MAX phase, (c and d) SEM micrographs of Ti3C2Tx and r-Ti3C2Tx powders, respectively, (e) Tapping mode AFM image of Ti3C2Tx nanosheets on SiO2/Si, and (f) high magnification TEM image of a Ti3C2Tx nanosheet suspended over a holey carbon grid. The inset shows the selected area electron diffraction (SAED) pattern. | ||
SEM images in Fig. 2a and b show that 3D Ti3AlC2 MAX particles are changed to accordion-like multilayer Ti3C2Tx structures indicating that Al layers in the Ti3AlC2 MAX phase are etched out by HF. Fig. 2c and d show fabric-like SEM micrographs of Ti3C2Tx powder before and after L-ascorbic acid treatment, respectively. The tapping mode AFM image (Fig. 2e) of Ti3C2Tx nanosheets deposited onto SiO2/Si substrates shows that the majority of the sheets are large with a lateral dimension of about 1 μm and 2 to 6 nm height corresponding to 1 to 3 layers. Fig. 1f shows a high magnification TEM image of a 2D Ti3C2Tx nanosheet suspended over a holey carbon grid, and its inset shows the selected area electron diffraction pattern with a six-fold reflection spot corresponding to the hexagonal crystal symmetry of Ti3C2Tx.
XPS analysis of the samples shows a clear difference among the Ti3AlC2 MAX phase, Ti3C2Tx, and r-Ti3C2Tx. The XPS survey spectrum (Fig. 3a) of the Ti3AlC2 MAX phase (black) shows a peak at 77.0 eV corresponding to Al, which, however, is below the detection limit in the spectra of Ti3C2Tx (red), and r-Ti3C2Tx (blue). This indicates that HF treatment on Ti3AlC2 MAX powder removes most of the Al layers. The survey spectra of Ti3C2Tx show a F peak at 685.0 eV with 8.1 atomic%, due to the addition of fluorine during HF etching.36 After treatment with L-ascorbic acid, the fluorine amount significantly decreases to 3.8%. This shows that L-ascorbic acid treatment significantly changes the surface chemistry of Ti3C2Tx, similar to the results in a published report,2 where Li-EDA treatment is shown to remove a significant amount of F termination from Ti3C2Tx. Fig. 3b depicts a high-resolution peak of Ti2p ranging from 454.8 to 464.8 eV for Ti3AlC2 MAX phase (black), Ti3C2Tx (red), and r-Ti3C2Tx (blue). Deconvolution (Fig. S1a and b, ESI†) shows that Ti2p3/2 peaks appear between 454.8 and 459.0 eV and the peaks beyond 459.0 eV are their corresponding doublets (Ti2p1/2). These peaks have been assigned to Ti, Ti2+, Ti3+, and Ti4+ chemical states as in the literature.2,37,38 The peaks appearing at 454.8 eV (460.3 eV) and 459.0 eV (464.9 eV) for Ti3C2Tx are assigned to Ti bonded to C of Ti3C2 crystal and TiO2 resulting from surface oxidation, respectively.37,38 For Ti3C2Tx, the component peak at 459.0 eV (464.9 eV) is also largely contributed by Ti4+ chemical states of Ti atoms surrounded by the O2− ions in the lattice, which occurs due to the replacement of some carbons by oxygen in the crystal during the etching process.2 This fact is supported by the increased intensity ratio of Ti4+ to Ti–C peaks for Ti3C2Tx compared to the Ti3AlC2 MAX phase. The peaks at 456.4 eV (461.5 eV) and 458.2 eV (263.5 eV) are assigned to Ti2+ and Ti3+ chemical states, respectively.2,38,39 One of the main purposes of XPS analysis is to assess the difference between Ti3C2Tx and r-Ti3C2Tx. Interestingly, we observed clear changes in the intensities of Ti3+ and Ti4+ components. In contrast to Ti3C2Tx (red), r-Ti3C2Tx (blue) shows a high intensity of Ti3+ peak at 458.2 eV (463.7 eV) with a diminished Ti4+ peak at 459.0 eV (464.7 eV), indicating a reduction of Ti4+ chemical states to Ti3+ under L-ascorbic acid treatment.
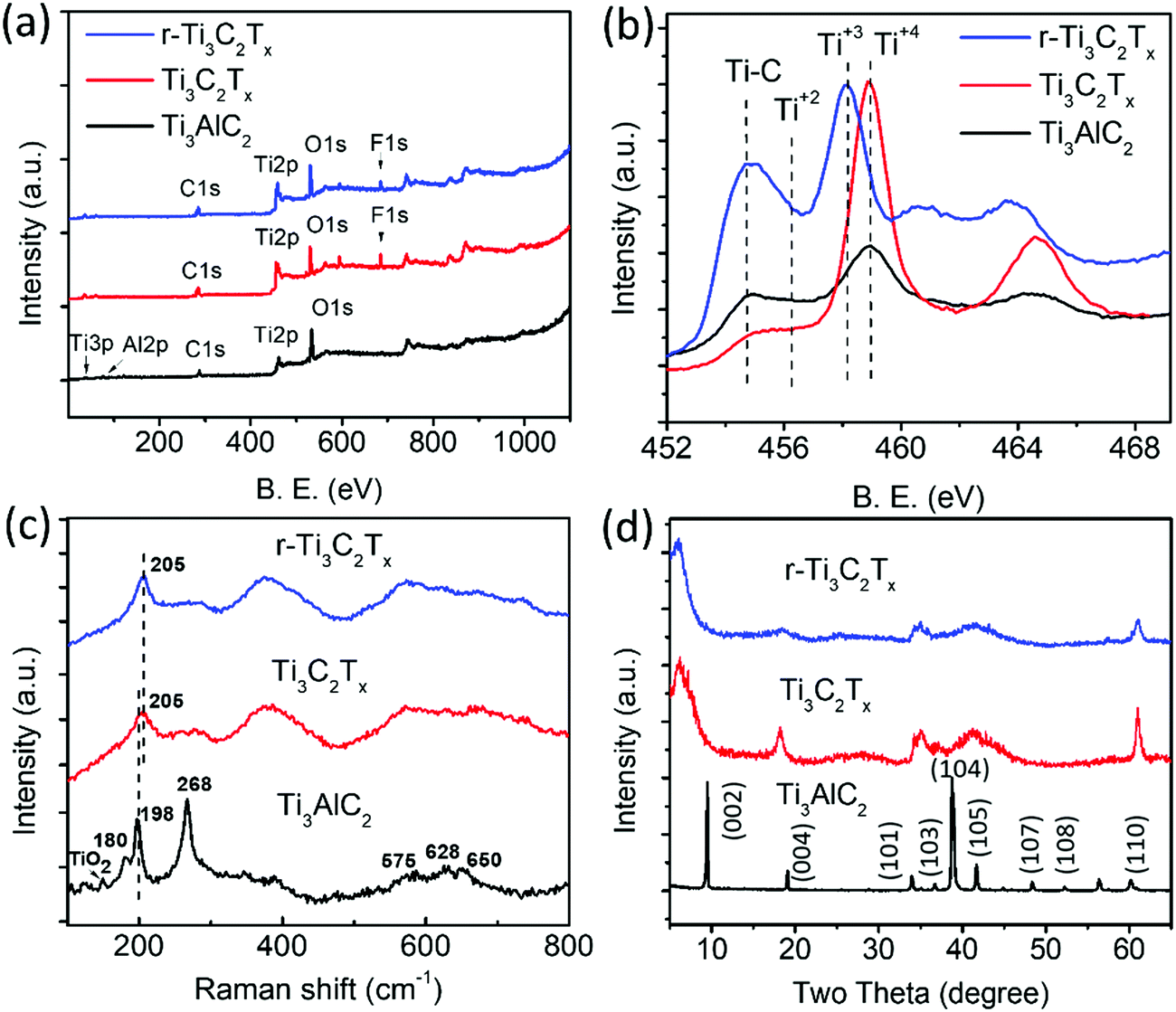 | ||
| Fig. 3 (a) XPS survey spectra, (b) high resolution XPS spectra of Ti2p region, (c) Raman spectra, and (d) XRD patterns, respectively, of Ti3AlC2 MAX phase (black), Ti3C2Tx (red), and r-Ti3C2Tx (blue). | ||
The O1s fine spectra (Fig. S1c and d, ESI†) of both Ti3C2Tx and r-Ti3C2Tx have been fitted with four different components peaking at 530.4, 531.6, 532.6, and 533.7 eV and are assigned to TiO2, Ti–O, Ti–OH, and adsorbed H2O molecules, respectively. The deconvolution clearly shows that the TiO2 component decreases upon reduction of Ti3C2Tx with L-ascorbic acid, and the center of gravity of the O1s fine spectrum of r-Ti3C2Tx is slightly shifted to a higher energy with respect to Ti3C2Tx, consistent with a published report.2 Some changes can also be seen between C1s XPS spectra of Ti3C2Tx and r-Ti3C2Tx (see Fig. S1e and f, ESI†). The C1s fine spectrum of Ti3C2Tx has been deconvoluted into five components peaking at 282.4, 284.5, 285.6, 286.8, and 288.9 eV and they are assigned to Ti–C bonds in Ti3C2Tx crystal, C–C, CHx, C–O, and COOH, respectively. The spectrum of r-Ti3C2Tx has been deconvoluted with the same components, but the peak intensity ratio of C bonds in the components C–C, CHx, C–O, and COOH altogether to that of Ti–C alone increases compared to Ti3C2Tx, similar to the results in a published report.2
Fig. 3c shows the normalized Raman spectra of Ti3AlC2 MAX phase (black), Ti3C2Tx (red), and r-Ti3C2Tx (blue). Ti3AlC2 MAX phase shows six distinct Raman bands, among which three intense bands occurring at 180, 198, and 268 cm−1 are associated with the vibrations of Ti and Al bonds and three shallow bands occurring at 575, 628, and 650 cm−1 are associated with Ti–C bonds.1,40 A small peak at 149 cm−1 is the Eg vibrational mode of anatase phase TiO2 particles41 formed due to spontaneous oxidation of surface titanium atoms. Note that this peak is very intense relative to the MXene band intensity when there is a sufficient amount of TiO2,41 and hence even with a small amount present, it is easily detectable. Ti3C2Tx and r-Ti3C2Tx show a broadened peak at 205 cm−1, which is assigned to the Ti–Al vibrational mode occurring at 198 cm−1 in MAX phase and is slightly blue-shifted, due to a phonon stiffening effect as the crystals are thinned down to the nanoscale. Apparently, our observation suggests that this Raman peak is visible even at a small amount of Al in MXene crystals, which is below the detection limit of XPS. The three peaks occurring at 575, 628, and 650 cm−1 in MAX phase are merged and red-shifted in Ti3C2Tx and r-Ti3C2Tx, as observed previously.1,40
The XRD patterns (Fig. 3d) show that several peaks are not observed for Ti3C2Tx (red), including the most intense one appearing at 39.0° due to the disappearance of the non-basal crystal planes upon removal of Al layers by HF etching. Consistent with the reports,1,28 we observed a broadening, loss of intensity, and shift of the peaks to lower angles for (00l) peaks such as (002) and (004). The peaks appearing in the range of 33–45° broaden so significantly that they look merged together. The most prominent change is a huge shift of the (002) peak from 9.5° in Ti3AlC2 MAX to 6.5° in Ti3C2Tx, owing to the removal of Al layers and the introduction of surface terminations (–F, –O, –OH).36 r-Ti3C2Tx shows similar XRD peaks to Ti3C2Tx, except that the (002) peak is further shifted to a slightly lower angle, 6.0° in r-Ti3C2Tx, which could be attributed to a further increase of the c-lattice spacing upon removal of F terminations.
3.2 Enhanced electrical conductivity of r-Ti3C2Tx
Fig. 4a shows a SEM micrograph of r-Ti3C2Tx with Ti/Au contact pads. More detailed images of Ti3C2Tx and r-Ti3C2Tx are presented in Fig. S2, ESI.† High magnification SEM images of the Ti3C2Tx and r-Ti3C2Tx films are presented in the insets of Fig. 4b and c, respectively. Fig. 4b and c present the plots of the measured total resistance vs. contact pad spacing for Ti3C2Tx and r-Ti3C2Tx, respectively. Neglecting the small value of contact pad to probe resistance (∼12 Ω) measured in our case, we have used the following relation for total measured resistance: RT = (Rs/W)L + 2Rc, where Rs is the sheet resistance of the films, L and W are spacing and width of the metal pads, respectively, and Rc is the metal pad to MXene film contact resistance. Hence, from slopes of the fitted lines in Fig. 4b and c, we obtained sheet resistance values of 1415 ± 125 and 288 ± 27 Ω sq−1 for Ti3C2Tx and r-Ti3C2Tx, respectively. With the measured thicknesses of 1.5 and 1.2 μm for Ti3C2Tx and r-Ti3C2Tx films (see Fig. S3, ESI†), the corresponding conductivity values are 471 ± 49 and 2819 ± 306 S m−1, respectively. The contact resistance of the Ti/Au pad with MXene films obtained from the y-intercept values in Fig. 4b and c are 4.2 ± 0.4 and 0.5 ± 0.05 KΩ for Ti3C2Tx and r-Ti3C2Tx films, respectively, and the corresponding contact resistivities are (2.6 ± 0.3) × 10−4 and (3.3 ± 0.4) × 10−5 Ω m2. These values are high compared to the values for continuous graphene–metal contacts,42,43 but reasonable considering the nature of the films made by drop casting of the MXene sheets.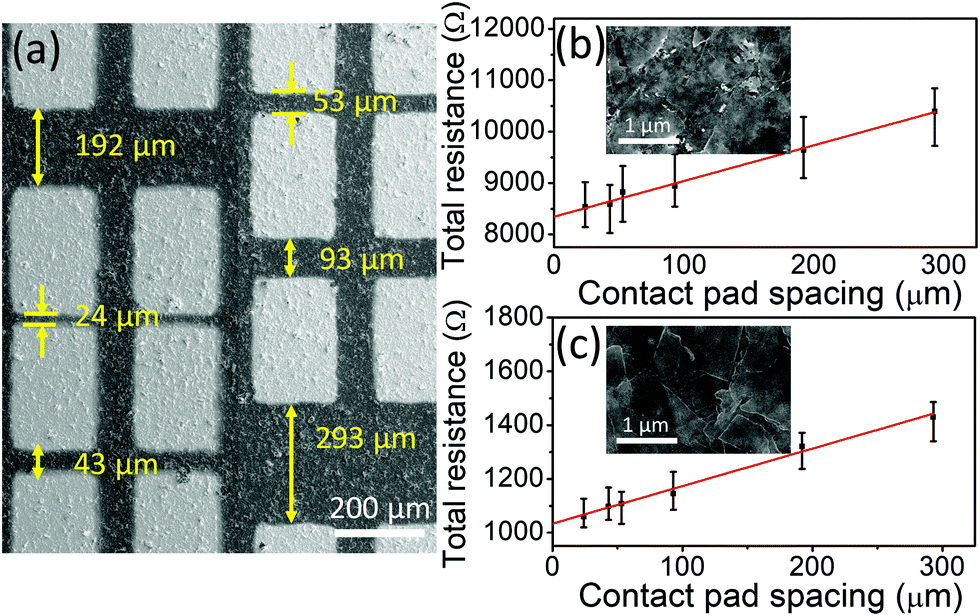 | ||
| Fig. 4 SEM image of Au/Ti contact pads on r-Ti3C2Tx films on SiO2/Si (a), plots of total resistance vs. contact pad spacing of (b) Ti3C2Tx and (c) r-Ti3C2Tx films on SiO2/Si. Insets in figures b and c are the high magnification SEM images of the corresponding films. | ||
Although our conductivity values for Ti3C2Tx and r-Ti3C2Tx films fall within widely scattered values of 2D titanium carbide MXene films reported previously,16,24,33,37,44,45 these values are smaller than most of the previously reported values for Ti3C2Tx. Since the conductivity of MXene films is synthesis and processing dependent, the relatively small electrical conductivity of our Ti3C2Tx can mainly be attributed to the use of HF as an etchant instead of a mixture of HCl and alkali metal fluoride salts such as LiF, as suggested in a published report.36 Moreover, the choice of intercalant for delamination, flake size and thickness, and film processing method could also alter the conductivity. Most importantly, it is exciting to find that the electrical conductivity of Ti3C2Tx is enhanced six-fold upon treatment with L-ascorbic acid, suggesting a great value of the reduction process in MXene processing. Our result suggests that the reduction of Ti3C2TxviaL-ascorbic treatment significantly enhances the electrical conductivity regardless of the initial conductivity value. An eight-fold reduction in the contact resistivity of r-Ti3C2Tx with respect to Ti3C2Tx also suggests that r-Ti3C2Tx is a more suitable material than Ti3C2Tx for fabricating devices that require metal–MXene contacts, such as electronic and optoelectronic devices. The reason for the enhanced conductivity in r-Ti3C2Tx can be explained as a consequence of the changes in chemistry of the MXene crystals during HF and L-ascorbic acid treatments. First, the carbon atoms that are replaced by O−2 ions during the HF etching process2 are subsequently removed by L-ascorbic acid treatment creating vacancies in the crystal lattice and this provides a localized electron in the vacant site. Next, the XPS result shows that a significant loss of fluorine terminations occurs during the reduction process. These modifications in the crystal chemistry cause redistribution of the missing hybridized Ti3d–C2p and Ti3d–F2p states to Ti–Ti metallic bond states near the Fermi energy, thereby increasing the DOS at the Fermi level. With this argument, a significantly large electrical conductivity of each r-Ti3C2Tx nanosheet is expected compared to an untreated Ti3C2Tx sheet.
3.3 Oxidation stability
Fig. 5(a–f) show photographs of the aqueous solutions of Ti3C2Tx (left) and r-Ti3C2Tx (right) collected on different days. We reiterate that the aqueous solutions (15 mg/10 ml) of Ti3C2Tx and r-Ti3C2Tx were prepared by re-dispersing the dried powders of the corresponding materials in water to study oxidation stability. Both Ti3C2Tx and r-Ti3C2Tx aqueous solutions look black initially, but upon exposure to water for several days, Ti3C2Tx aqueous solution turns whitish faster than r-Ti3C2Tx aqueous solution, indicating a faster degradation of Ti3C2Tx crystals. The repeated experiment with a lower concentration (5 mg/10 ml) of Ti3C2Tx and r-Ti3C2Tx in water results in the same observation (see Fig. S4, ESI†). As shown in Fig. 5b, the MXene nanosheets allowed to oxidize for 3 days do not look distinctively different to the eye, but the difference can easily be detected by Raman spectroscopic analysis of the corresponding dried powders (Fig. 5g and h). The features in the Raman spectra for each of Ti3C2Tx and r-Ti3C2Tx exposed to water for a different length of time are different. With the increased number of days, the degraded MXene powders show a gradual increase of the Eg vibrational band intensity of anatase TiO2 occurring at ∼149 cm−1 (indicated with stars) and a decrease of the MXene peak occurring at ∼205 cm−1, as observed by Zhang et al.31 It is evident from Fig. 5g and h that the ratio of anatase TiO2 to MXene band intensities increases faster for Ti3C2Tx than r-Ti3C2Tx, which indicates that r-Ti3C2Tx nanoflakes are more stable in oxygen-rich environments such as water. SEM micrographs (Fig. S5, ESI†) further confirm that nucleation and growth of TiO2 nanoparticles take place faster on Ti3C2Tx surfaces than on r-Ti3C2Tx surfaces.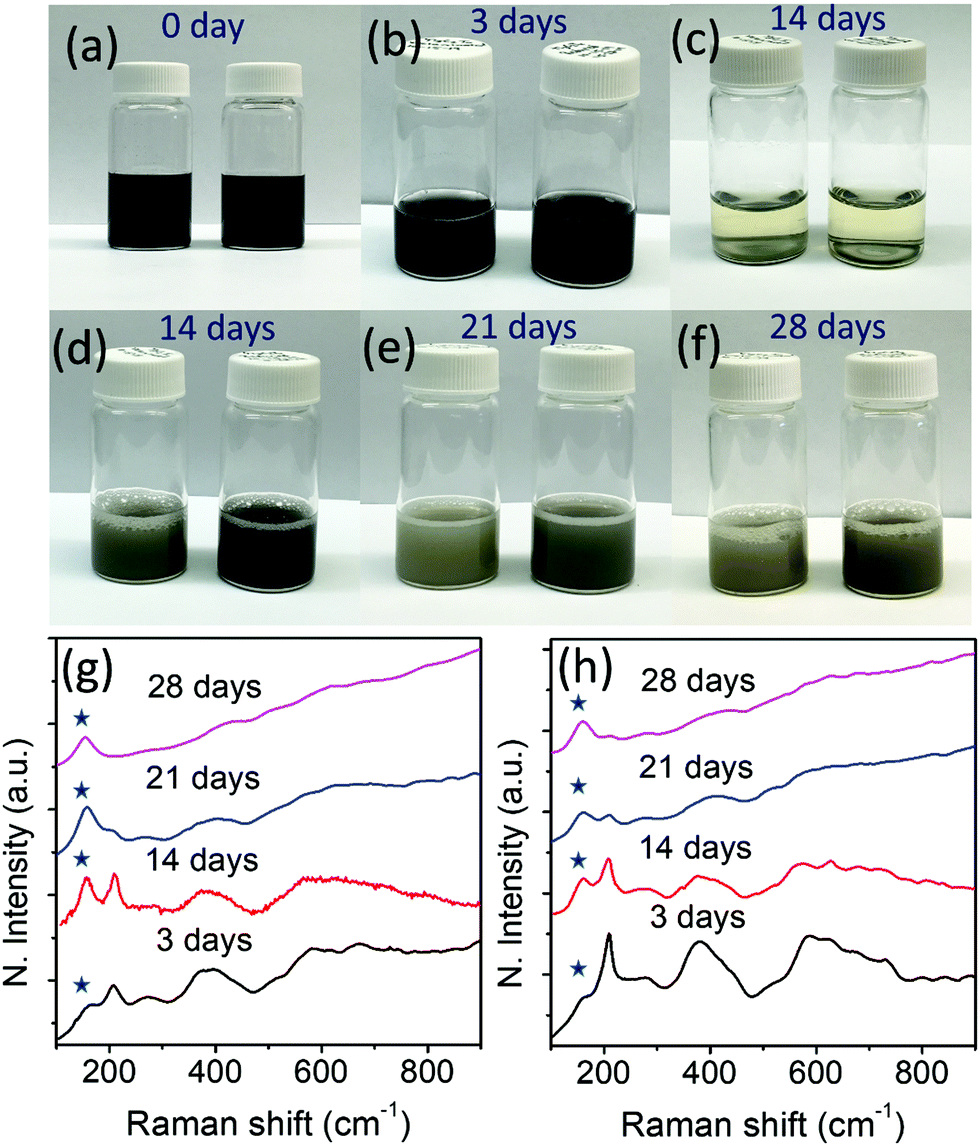 | ||
| Fig. 5 Photographs of the aqueous solutions of Ti3C2Tx (left) and r-Ti3C2Tx (right) collected after (a) 0 day, (b) 3 days, (c) 14 days before shaking, (d) 14 days, (e) 21 days, and (f) 28 days. The concentrations of Ti3C2Tx and r-Ti3C2Tx solutions were 15 mg in 10 ml of water. Normalized Raman spectra collected on dried powder from the aqueous solutions of (g) Ti3C2Tx, and (h) r-Ti3C2Tx kept for different days. | ||
It is evident that after treatment with L-ascorbic acid, Ti3C2Tx nanosheets become more stable in the oxygen rich environment. This result is consistent with a recent work30 that uncovered that Ti3C2Tx nanosheets were protected from severe oxidation up to six months when stored in a sodium ascorbate aqueous solution. The average lateral size of the Ti3C2Tx and r-Ti3C2Tx nanosheets in the SEM images in Fig. S2c, d and S5 presented in the ESI† are ∼1.6 and ∼1.5 μm, respectively. Hence, a faster degradation of Ti3C2Tx nanosheets compared to r-Ti3C2Tx in water is not because of the smaller flake size as reported previously.31 The enhanced oxidation stability of r-Ti3C2Tx can be explained based on the reduction of the Ti3C2Tx nanosheets caused by L-ascorbic acid molecules. As described above, O2− ions replace a significantly large number of carbons from the Ti3C2Tx lattice during the Al layer etching process, as shown in Fig. 1b. These oxygen sites in the Ti3C2Tx crystal act as nucleation centers for TiO2 nanoparticle growth. Upon exposure of the Ti3C2Tx nanosheets to water, reactive oxygen species such as hydroxyl radical or oxygen molecules react with the nanosheets breaking the Ti–C bonds and forming Ti–O bonds.30 This process accelerates the TiO2 nanoparticle growth. The carbons released during this process are bonded to each other forming disordered carbon material (see Fig. S6, ESI†). Hence, the MXene nanosheets continue to degrade while TiO2 nanoparticles continue to increase in size. This argument is supported by the SEM micrographs (see Fig. S5, ESI†) of the MXene sheets, where TiO2 nanoparticles are observed to increase in size with continued exposure to water. The TiO2 nanoparticle growth on the MXene surface also confirms that MXene degradation not only starts at the Ti3C2Tx sheet edges as claimed in the literature,30,31 but it also occurs simultaneously at the basal plane. However, L-ascorbic acid treatment on Ti3C2Tx removes the O2− ions sitting in the carbon sites, and the possible nucleation sites for TiO2 particle growth are minimized. This argument is supported by a much smaller density and size of the TiO2 nanoparticles grown on the basal plane of r-Ti3C2Tx compared to Ti3C2Tx (see Fig. S5, ESI†). Such an enhanced oxidation stability makes r-Ti3C2Tx a promising material for use in applications.
3.4 SERS activity
Fig. 6a–c show SERS spectra of the probe molecules, crystal violet (CV) at 2 × 10−6 M, methylene blue (MB) at 1 × 10−6 M, and rhodamine 6G (R6G) at 1 × 10−7 M, respectively, collected on Ti3C2Tx/SiO2/Si (black) and r-Ti3C2Tx/SiO2/Si (red) substrates. It is apparent that the r-Ti3C2Tx/SiO2/Si substrate shows about an order of magnitude higher Raman signal intensity compared to Ti3C2Tx/SiO2/Si for all three probe molecules. Raman enhancement on SERS substrates prepared with Ti3C2Tx and r-Ti3C2Tx on PVDF filter membrane (see photographs Fig. S7, ESI†) showed similar results. | ||
| Fig. 6 SERS spectra of (a and d) CV, (b and e) MB, and (c and f) R6G collected on Ti3C2Tx/SiO2/Si (black) and r-Ti3C2Tx/SiO2/Si (red), and bare SiO2/Si (blue) substrates. Concentration of dyes on SiO2/Si substrate is 1 M in water. | ||
The SERS EF was calculated using the following equation: EF = (ISERS/CSERS)/(Iref/Cref), where ISERS is the Raman intensity of a selected vibrational mode of a dye with a concentration of CSERS, and Iref is the intensity of the same vibrational mode of the dye on a SERS inactive substrate (SiO2/Si) with a concentration of Cref. The calculated SERS EFs for different dyes are presented in Table 1.
| Probe molecules | SERS substrates | Raman shift (cm−1) | Peak assignment | Dye concentration (M) | EF |
|---|---|---|---|---|---|
| CV | Ti3C2Tx/SiO2/Si | 1613 | In-plane stretching of C–C ring46 | 2.0 × 10−6 | 2.1 × 105 |
| r-Ti3C2Tx/SiO2/Si | 1.6 × 106 | ||||
| MB | Ti3C2Tx/SiO2/Si | 1623 | In-plane stretching of C–C ring47 | 1.0 × 10−6 | 1.0 × 105 |
| r-Ti3C2Tx/SiO2/Si | 7.8 × 105 | ||||
| R6G | Ti3C2Tx/SiO2/Si | 1361 | Carboxylate stretches48 | 1.0 × 10−7 | 1.3 × 106 |
| r-Ti3C2Tx/SiO2/Si | 1.0 × 107 |
It is evident that the SERS EF is an order of magnitude higher on r-Ti3C2Tx/SiO2/Si than on Ti3C2Tx/SiO2/Si for all three dyes used. The calculated SERS EFs for Ti3C2Tx/SiO2/Si are similar to that of the SERS substrates fabricated with Ti3C2Tx on glass and silicon in the literature20 and are much larger than those of Ti2NTx-based SERS substrates fabricated on glass and silicon reported by Soundiraraju et al.4
As shown in Fig. 6d–f, some of the vibrational peak positions in the SERS spectra of CV, MB, and R6G are red-shifted with respect to their original positions, i.e., Raman peak positions measured on the non-SERS substrate, SiO2/Si. For example, the in-plane stretching mode of the C–C ring of CV and MB appears at 1619 and 1629 cm−1 on SiO2/Si, respectively, which shift to 1613 and 1623 cm−1 on MXene substrates. For R6G, the peak associated with the in-plane stretching mode of C–C ring appears at 1196 cm−1 on bare SiO2/Si, which is shifted to 1187 cm−1 on MXene substrates. We also noticed that there was no shift in some peaks, such as those appearing at 1173 cm−1 and 1032 cm−1 corresponding to the in-plane bending mode of C–H in CV46 and MB,47 respectively, and 1361 cm−1 corresponding to the carboxylate stretch mode48 in R6G. The shift in the peak positions of dye molecules on Ti3C2Tx and r-Ti3C2Tx SERS substrates is attributed to charge transfer interaction49,50 of the MXenes with the probe molecules. To understand our results, it is reasonable to expect the transfer of electrons from electron rich MXenes to C–C rings of the dye molecules, which slightly weakens C–C bonds of the rings, causing a red-shift of the associated vibrational peaks. However, no charge transfer from MXenes to the C–H bond of CV and MB, and to the carboxylate group of R6G takes place. Hence, a peak shift associated with these bonds was not observed. Band shift due to charge transfer interaction is common in several other spectroscopies, such as XPS,51 nuclear magnetic resonance spectroscopy,52 and infrared absorption spectroscopy (IR).52,53 In Raman spectroscopy, the degree of charge transfer is reflected by the magnitude of wavenumber shift53,54 and has a positive correlation with Raman signal enhancement.55 Hence, based on the magnitude of the band shift, the degree of the charge transfer is found to be slightly greater in R6G than in CV and MB. This is supported by the higher Raman EF of R6G compared to those of CV and MB. Moreover, the laser excitation wavelength, 532 nm, is much closer to the light absorption peak position of R6G (527 nm) (see Fig. S8 for UV visible spectra, ESI†) than the absorption peak positions of CV (591 nm) and MB (663 nm). Hence, the matching of the excitation beam energy with a molecular transition is another factor that accounts for the higher EF of R6G than those of CV and MB.
Now, we discuss briefly why the r-Ti3C2Tx/SiO2/Si SERS substrate enhances the Raman signal of dye molecules more than the Ti3C2Tx/SiO2/Si substrate. As mentioned above, L-ascorbic acid treatment removes more than 50% of the F terminations on Ti3C2Tx, exposing a much larger number of surface-Ti atoms to the adsorbing dye molecules. This allows a larger population of adsorbing dye molecules to interact and form charge transfer complexes with r-Ti3C2Tx. Consequently, a higher EF factor is achieved. Additionally, as described above, the increased electronic DOS at the Fermi level in r-Ti3C2Tx facilitates electronic charge transfer to dye molecules, causing an enhanced Raman EF. Despite the increased metallic property or DOS at the Fermi level in r-Ti3C2Tx, no plasmonic absorption was observed in the UV visible spectrum (see Fig. S9, ESI†). Hence, the observed SERS enhancement of the dye molecules cannot be attributed directly to the electromagnetic contribution.
4. Conclusions
We have developed a facile and green protocol for the room temperature synthesis of reduced Ti3C2Tx MXene nanosheets via a simple treatment with L-ascorbic acid. The results show that r-Ti3C2Tx has a 6-fold higher electrical conductivity and has a better oxidation stability than Ti3C2Tx, making r-Ti3C2Tx a more promising material for most applications. Furthermore, r-Ti3C2Tx offers a much smaller contact resistance with Ti/Au contact pads, which encourages the use of r-Ti3C2Tx in fabricating devices that require metal-MXene contacts, such as electronic and optoelectronic devices.The study demonstrates that r-Ti3C2Tx enhances the Raman signal of dye molecules by an order of magnitude compared to Ti3C2Tx. The SERS enhancement factor obtained for R6G on r-Ti3C2Tx is as high as 107 using the laser excitation wavelength of 532 nm, indicating a potential use of r-Ti3C2Tx for making practical SERS substrates. The outstanding SERS activity of r-Ti3C2Tx has been attributed to a larger number of Ti atoms exposed due to the loss of F terminations allowing a larger population of dye molecules to interact with r-Ti3C2Tx. The higher SERS activity of r-Ti3C2Tx is further re-enforced by the increased electronic DOS at the Fermi level. The findings of this study suggest that reduced MXene could be a superior choice over MXene prepared by regular methods, especially for applications that employ high electronic conductivity, such as electrode materials for batteries and supercapacitors, photodetectors, and SERS-based sensors.
Abbreviations
| 2D | Two-dimensional |
| TMAOH | Tetramethylammonium hydroxide |
| Li-EDA | Li-ethylenediamine |
| r-Ti3C2Tx | Reduced Ti3C2Tx MXene |
| CV | Crystal violet |
| MB | Methylene blue |
| R6G | Rhodamine 6G |
| SERS | Surface-enhanced Raman scattering |
| AFM | Atomic force microscopy |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| SAED | Selected area electron diffraction |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffractometry |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for the financial support of this project by the U.S. National Science Foundation (Awards # 1831133 and #1523617). Q. L. and S. K. would like to thank NSF Award #1905833 and DOE FG02-08ER46531. This work was performed in part at the Analytical Instrumentation Facility (AIF) and Nanofabrication facility (NNF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-1542015). The AIF and NNF are members of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI). The authors thank Dr Spyridon Pavlidis and Mr Rohan Sengupta for assisting in electrical measurements.References
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248 CrossRef CAS PubMed.
- Y. Yoon, T. A. Le, A. P. Tiwari, I. Kim, M. W. Barsoum and H. Lee, Nanoscale, 2018, 10, 22429 RSC.
- K. Chaudhuri, M. Alhabeb, Z. Wang, V. M. Shalaev, Y. Gogotsi and A. Boltasseva, ACS Photonics, 2018, 5, 1115 CrossRef CAS.
- B. Soundiraraju and B. K. George, ACS Nano, 2017, 11, 8892 CrossRef CAS PubMed.
- M. H. Tran, T. Schäfer, A. Shahraei, M. Dürrschnabel, L. Molina-Luna, U. I. Kramm and C. S. Birkel, ACS Appl. Energy Mater., 2018, 1, 3908 CrossRef CAS.
- J. L. Hart, K. Hantanasirisakul, A. C. Lang, B. Anasori, D. Pinto, Y. Pivak, J. T. van Omme, S. J. May, Y. Gogotsi and M. L. Taheri, Nat. Commun., 2019, 10, 522 CrossRef CAS PubMed.
- L. Verger, V. Natu, M. Carey and M. W. Barsoum, Trends Chem., 2019, 1, 656 CrossRef.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322 CrossRef CAS PubMed.
- M. Khazaei, M. Arai, T. Sasaki, C.-Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall’Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78 CrossRef CAS PubMed.
- Q. Pan, C. Duan, H. Liu, M. Li, Z. Zhao, D. Zhao, Y. Duan, Y. Chen and Y. Wang, ACS Appl. Energy Mater., 2019, 2, 6834 CrossRef CAS.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494 CrossRef CAS.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589 CrossRef CAS.
- C. E. Ren, K. B. Hatzell, M. Alhabeb, Z. Ling, K. A. Mahmoud and Y. Gogotsi, J. Phys. Chem. Lett., 2015, 6, 4026 CrossRef CAS PubMed.
- C. (John) Zhang, B. Anasori, A. Seral-Ascaso, S.-H. Park, N. McEvoy, A. Shmeliov, G. S. Duesberg, J. N. Coleman, Y. Gogotsi and V. Nicolosi, Adv. Mater., 2017, 29, 1702678 CrossRef PubMed.
- G. Ying, A. D. Dillon, A. T. Fafarman and M. W. Barsoum, Mater. Res. Lett., 2017, 5, 391 CrossRef CAS.
- J. Liu, H.-B. Zhang, R. Sun, Y. Liu, Z. Liu, A. Zhou and Z.-Z. Yu, Adv. Mater., 2017, 29, 1702367 CrossRef PubMed.
- D. B. Velusamy, J. K. El-Demellawi, A. M. El-Zohry, A. Giugni, S. Lopatin, M. N. Hedhili, A. E. Mansour, E. D. Fabrizio, O. F. Mohammed and H. N. Alshareef, Adv. Mater., 2019, 31, 1807658 CrossRef PubMed.
- A. Sarycheva, T. Makaryan, K. Maleski, E. Satheeshkumar, A. Melikyan, H. Minassian, M. Yoshimura and Y. Gogotsi, J. Phys. Chem. C, 2017, 121, 19983 CrossRef CAS.
- E. Satheeshkumar, T. Makaryan, A. Melikyan, H. Minassian, Y. Gogotsi and M. Yoshimura, Sci. Rep., 2016, 6, 32049 CrossRef CAS PubMed.
- K. Hantanasirisakul, M. Alhabeb, A. Lipatov, K. Maleski, B. Anasori, P. Salles, C. Ieosakulrat, P. Pakawatpanurut, A. Sinitskii, S. J. May and Y. Gogotsi, Chem. Mater., 2019, 31, 2941 CrossRef CAS.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752 CrossRef CAS PubMed.
- Z. Ling, C. E. Ren, M.-Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676 CrossRef CAS PubMed.
- Y. Xie and P. R. C. Kent, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235441 CrossRef.
- M. Savchak, N. Borodinov, R. Burtovyy, M. Anayee, K. Hu, R. Ma, A. Grant, H. Li, D. B. Cutshall, Y. Wen, G. Koley, W. R. Harrell, G. Chumanov, V. Tsukruk and I. Luzinov, ACS Appl. Mater. Interfaces, 2018, 10, 3975 CrossRef CAS PubMed.
- Y. Chen, K. Fu, S. Zhu, W. Luo, Y. Wang, Y. Li, E. Hitz, Y. Yao, J. Dai, J. Wan, V. A. Danner, T. Li and L. Hu, Nano Lett., 2016, 16, 3616 CrossRef CAS PubMed.
- X. Xie, Y. Zhu, F. Li, X. Zhou and T. Xue, Sci. China: Technol. Sci., 2019, 62, 1202 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667 CrossRef CAS.
- X. Zhao, A. Vashisth, E. Prehn, W. Sun, S. A. Shah, T. Habib, Y. Chen, Z. Tan, J. L. Lutkenhaus, M. Radovic and M. J. Green, Matter, 2019, 1, 513 CrossRef.
- C. J. Zhang, S. Pinilla, N. McEvoy, C. P. Cullen, B. Anasori, E. Long, S.-H. Park, A. Seral-Ascaso, A. Shmeliov, D. Krishnan, C. Morant, X. Liu, G. S. Duesberg, Y. Gogotsi and V. Nicolosi, Chem. Mater., 2017, 29, 4848 CrossRef CAS.
- T. Habib, X. Zhao, S. A. Shah, Y. Chen, W. Sun, H. An, J. L. Lutkenhaus, M. Radovic and M. J. Green, npj 2D Mater. Appl., 2019, 3, 8 CrossRef.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- H. Ghassemi, W. Harlow, O. Mashtalir, M. Beidaghi, M. R. Lukatskaya, Y. Gogotsi and M. L. Taheri, J. Mater. Chem. A, 2014, 2, 14339 RSC.
- V. Natu, J. L. Hart, M. Sokol, H. Chiang, M. L. Taheri and M. W. Barsoum, Angew. Chem., Int. Ed., 2019, 58, 12655 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633 CrossRef CAS.
- A. Pazniak, P. Bazhin, N. Shplis, E. Kolesnikov, I. Shchetinin, A. Komissarov, J. Polcak, A. Stolin and D. Kuznetsov, Mater. Des., 2019, 183, 108143 CrossRef.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, Y. Rosen and J. Barsoum, Appl. Surf. Sci., 2016, 30, 406–417 CrossRef.
- Y. Zhang, Z. Xing, X. Liu, Z. Li, X. Wu, J. Jiang, M. Li, Q. Zhu and W. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 26851 CrossRef CAS PubMed.
- V. Presser, M. Naguib, L. Chaput, A. Togo, G. Hug and M. W. Barsoum, J. Raman Spectrosc., 2012, 43, 168 CrossRef CAS.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567 RSC.
- S. Min Song, T. Yong Kim, O. Jae Sul, W. Cheol Shin and B. Jin Cho, Appl. Phys. Lett., 2014, 104, 183506 CrossRef.
- V. Passi, A. Gahoi, E. G. Marin, T. Cusati, A. Fortunelli, G. Iannaccone, G. Fiori and M. C. Lemme, Adv. Mater. Interfaces, 2019, 6, 1801285 CrossRef.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L.-Å. Näslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374 CrossRef CAS PubMed.
- H. Kitadai, X. Wang, N. Mao, S. Huang and X. Ling, J. Phys. Chem. Lett., 2019, 10, 3043 CrossRef CAS PubMed.
- L. Pei, Y. Huang, C. Li, Y. Zhang, B. A. Rasco and K. Lai, J. Nanomater., 2014, 730915 Search PubMed.
- K.-D. Shim and E.-S. Jang, Bull. Korean Chem. Soc., 2018, 39, 936 CrossRef CAS.
- M. Liu, M. Liu, Y. Shi, Y. Shi, G. Zhang, G. Zhang, Y. Zhang, M. Wu, J. Ren and B. Man, Appl. Spectrosc., 2018, 72, 1613 CrossRef CAS PubMed.
- Y. Wang, W. Ji, H. Sui, Y. Kitahama, W. Ruan, Y. Ozaki and B. Zhao, J. Phys. Chem. C, 2014, 118, 10191 CrossRef CAS.
- S. Liu, Y. Li, X. Zhao, X. Liu and M. Chen, Spectrochim. Acta, Part A, 2011, 82, 205 CrossRef CAS PubMed.
- C. Lenser, Q. Lu, E. Crumlin, H. Bluhm and B. Yildiz, J. Phys. Chem. C, 2018, 122, 4841 CrossRef CAS.
- M. S. Refat, H. A. Saad, A. M. A. Adam, M. A. Al-Omar and A. M. Naglah, Acta Pharm., 2016, 66, 533 CAS.
- T. B. Limbu, F. Mendoza, D. Barrionuevo, J. Carpena, B. Maruyama, R. S. Katiyar, B. R. Weiner and G. Morell, AIP Adv., 2016, 6, 035319 CrossRef.
- A. C. Crowther, A. Ghassaei, N. Jung and L. E. Brus, ACS Nano, 2012, 6, 1865 CrossRef CAS PubMed.
- H. Kitadai, X. Wang, N. Mao, S. Huang and X. Ling, J. Phys. Chem. Lett., 2019, 10, 3043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Deconvolution of XPS fine peaks for Ti2p, O1s, and C1s; details of electrical measurements; calculation of electrical conductivity and contact resistivity; thickness measurement for Ti3C2Tx and r-Ti3C2Tx films; MXene oxidation stability test; SEM analysis of degraded Ti3C2Tx and r-Ti3C2Tx; Raman spectra of disordered carbon material formed on MXene; photographs of the SERS substrates on PVDF filter membranes; UV-vis absorption spectra of CV, MB, and R6B; examination of plasmonic absorption in Ti3C2Tx and r-Ti3C2Tx. See DOI: 10.1039/c9tc06984d |
| This journal is © The Royal Society of Chemistry 2020 |