
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of alicyclic substituents on the extraction ability of new family of 1,10-phenanthroline-2,9-diamides†
Pavel S. Lemport *a,
Petr I. Matveeva,
Alexander V. Yatsenkoa,
Mariia V. Evsiuninaa,
Valentine S. Petrova,
Boris N. Tarasevicha,
Vitaly A. Roznyatovskya,
Pavel V. Dorovatovskiib,
Victor N. Khrustalevcd,
Sergey S. Zhokhova,
Vitaly P. Solov'eve,
Leonid A. Aslanova,
Vladimir G. Petrova,
Stepan N. Kalmykova,
Valentine G. Nenajdenkoa and
Yuri A. Ustyniuka
*a,
Petr I. Matveeva,
Alexander V. Yatsenkoa,
Mariia V. Evsiuninaa,
Valentine S. Petrova,
Boris N. Tarasevicha,
Vitaly A. Roznyatovskya,
Pavel V. Dorovatovskiib,
Victor N. Khrustalevcd,
Sergey S. Zhokhova,
Vitaly P. Solov'eve,
Leonid A. Aslanova,
Vladimir G. Petrova,
Stepan N. Kalmykova,
Valentine G. Nenajdenkoa and
Yuri A. Ustyniuka
aChemistry Department, Lomonosov Moscow State University, 119991, Moscow, Russia. E-mail: lemport.pavel@yandex.ru
bNational Research Center “Kurchatov Institute”, Russia
cDepartment of Inorganic Chemistry, Peoples' Friendship University of Russia (RUDN University), Russia
dN.D. Zelinsky Institute of Organic Chemistry of Russian Academy of Sciences, Russia
eFrumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Russia
First published on 10th July 2020
Abstract
Development of efficient extractants for the separation of actinides and lanthanides in the technologies of nuclear fuel cycle is one of the most urgent and complex tasks in modern nuclear energetics. New family of 4,7-dichloro-1,10-phenanthroline-2,9-dicarboxylic acid diamides based on cyclic amines was synthesized and shown to exhibit high selectivity in the La/Am pair separation (SF (Am/La ≈ 10)) and in the Am/Eu pair separation (SF (Am/Eu ≈ 12)). It was shown that pyrrolidine derived diamide is more efficient extractant for americium, curium and lanthanides from highly acidic HNO3 solution than its non-cyclic N,N,N′,N′-tetraalkyl analogues. The structures of synthesized compounds were studied in details by IR, NMR spectroscopy, and single crystal X-ray diffraction. According to spectroscopy data, incorporation of aromatic rings to the amide fragment of ligand leads to complex dynamic behavior in solutions what is believed to strongly affect the extraction ability of synthesized ligands.
Introduction
More than 10% of the world's electricity in 2020 will be supplied by nuclear power plants and the contribution of nuclear power into world's energy production is steadily growing as one of the sources with lowest carbon dioxide emissions.1 The most serious drawbacks of nuclear power is the need to reprocess and dispose spent nuclear fuel (SNF) due to its long-term and high radiotoxicity generated by the minor actinides (mAn = Am, Cm, Np) and plutonium. Partitioning and transmutation strategy (P&T)2–4 is an approach aimed at the separation (partitioning) of the SNF into its components, followed by transmutation of mAn into radionuclides with shorter half-times that makes them less hazardous in long term perspective. However, due to their high neutron-poisoning capacity, the lanthanides (Ln) need to be separated from SNF prior to transmutation of mAn. Due to their very similar chemical properties, the partitioning of mAn and Ln is the most challenging hydrometallurgical separation known.2–5Solvent extraction relied on the use of complexing organic ligands which is a common technique for the separation of mAn and Ln. Therefore, the molecular design of such compounds determines the efficiency and selectivity of their binding to metal in an aqueous solution.6
To date, a large number of chelators and extractants have been proposed for this purpose.3,5,7 Extraction chemistry of diglycolamides (DGA) and bis-triazinyl N-heterocycles is described in separate reviews8,9
1,10-Phenanthroline-2,9-dicarboxylic acid and its diamides is an important class of N,N′,O,O′-tetradentate ligands. According to Pearson HSAB concept they contain both “hard” (carbonyl oxygen atoms) and “soft” (heterocyclic nitrogen atoms) donor coordination centers. Having moderate Brønsted basicity but high Lewis basicity, these ligands are capable to effective binding with metal cations in strongly acidic media and form stable complexes with cations of transition and post-transition metals, which are soluble in polar organic solvents.10–14 Their high resistance in respect to oxidation and hydrolysis allows one to use them as perspective extractants for the separation of metal cations of very similar nature in “organic solvent/acidic aqueous solution” two-phase systems.11,12,15–18
Diamides of 1,10-phenanthroline-2,9-dicarboxylic acid have been found to demonstrate high values of selectivity factors for actinides and lanthanides separation.15,16 Extraction properties of these compounds can be customized by varying the substituents in phenanthroline core and/or at the amide nitrogen atoms. In spite of the fact that a number of variations are known (N,N′-dialkyl-,19,20 N,N′-diaryl-,21 N,N-dialkyl-,22 N,N,N′,N′-tetraalkyl-19,23,24 and N,N′-dialkyl-N,N′-diaryl amides of 1,10-phenanthroline-2,9-dicarboxylic acid17,18,25) it is still difficult to link the structural features of these ligands with their extraction properties.
A deep search allows us to find only few examples of amides of 1,10-phenanthroline-2,9-dicarboxylic acid with alicyclic substituents in the amide function, however without any extraction tests performed.26
In the present work we have synthesized a new family of 4,7-dichloro-1,10-phenanthroline-2,9-dicarboxamides based on commercially available cyclic secondary amines. We also performed extraction experiments in the partitioning of lanthanides, americium and curium to evaluate the effects of structure variations on the selectivity and efficiency of the separation.
The incorporation of chlorine atoms into the phenanthroline core was carried out in order to reduce the Brønsted basicity of the synthesized ligands and to increase their solubility in extraction tests. Such modification permits to have ligands 100 times less basic than nonsubstituted phenanthroline which is important for the separation of f-elements in highly acidic solutions.27 In order to satisfy the CHON principle,28 chlorine atoms can be subsequently replaced with more radiation-resistant CN-groups using cross-coupling reactions catalyzed by palladium salts,29,30 maintaining required extraction properties and solubility of resulting ligands.
Results and discussion
Synthesis of 1,10-phenanthroline-2,9-dicarboxamides
Synthesis of 4,7-dichloro-1,10-phenanthroline-2,9-dicarboxylic acid 1 has been described previously.18Diamides 3a–h were prepared by the interaction of acyl dichloride 2 with the corresponding cyclic secondary amines in CH2Cl2 media with yields up to 90% (Table 1). An excess of the corresponding secondary cyclic amine was used as an acceptor of HCl for the synthesis of 3a–e, whereas triethylamine was used for the synthesis of 3f–h. As a result, new family of ligands was prepared very efficiently to give target amides in up to 90% yield.
|
||||
|---|---|---|---|---|
| Code | Starting amine | Yield, % | Solubility in “F-3”, mol L−1 | Solubility in CHCl3, mol L−1 |
| 3a | Pyrrolidine | 82 | 0.015 | 0.33 |
| 3b | Piperidine | 81 | 0.020 | 0.40 |
| 3c | Azepane | 82 | 0.023 | 0.67 |
| 3d | Morpholine | 71 | 0.015 | 0.44 |
| 3e | N-Methyl-piperazine | 79 | 0.018 | 0.62 |
| 3f | Indoline | 76 | 0.0008 | 0.03 |
| 3g | 1,2,3,4-Tetra-hydroquinoline | 90 | 0.002 | 0.20 |
| 3h | 9H-Carbazole | 63 | 0.0002 | 0.0002 |
Compounds 3a–h are white or slightly colored high-melting solids. Diamides 3a–e are soluble in dichloromethane and acetone and sparingly soluble in hexane. The solubility of diamides 3f–h is significantly lower being compared with 3a–e.
It was a matter of practical interest to measure the solubility of diamides 3a–h in solvent “F-3” (3-nitrobenzotrifluoride), which is widely used for its high solvation ability towards actinide complexes and its ability to reveal the best extraction properties of N-heterocyclic diamides.31,32
Expectedly, the solubility increases in the rows 3a–3c and 3f–3g with increase of lipophilicity (Table 1).
It should be noted, that solubility of diamides 3a–3e within a range of 0.015–0.023 mol L−1 is sufficient for the intended use in extraction systems for separation of Ln and mAn.33 However, the solubility of diamides 3f–h is significantly lower in comparison to known N,N,N′,N′-tetraalkyl and N,N′-dialkyl-N,N′-diarylsubstituted diamides.23–25
The structures of 3a–h were confirmed by IR, 1H NMR, 13C NMR spectrometry and high-resolution mass spectrometry (HRMS). An assignment of signals in NMR spectra was made using two-dimensional 1H/13C hetero-nuclear correlation techniques (HSQC, HMBC). The structures of diamides 3a, 3b, 3f and starting acid 1 were also determined by X-ray diffraction.
The literature search provides the structures of a large number of complexes of 1,10-phenanthroline-2,9-dicarboxylic acid and its diamides with cations of transition and post-transition metals.11,13,14,34–38 However, information about the structures of the acid itself, its diamides and/or derivatives with substituents in the phenanthroline core, is very limited. The search in the Cambridge Crystallographic Database (CSD release 2020.0) revealed several hits, namely: DMSO solvate of 1,10-phenanthroline-2,9-dicarboxylic acid39 N,N′-bis(2-pyridyl)-1,10-phenanthroline-2,9-dicarboxamide,21 N,N′-bis(2-amino-6-pyridyl)-1,10-phenanthroline-2,9-dicarbox-amide,21 methanol solvate of N,N′-diethyl-N,N′-ditolyl-1,10-phenanthroline-2,9-dicarboxamide,17 hemihydrate of 4,7-bis(2-methylpropoxy)-N,N′-diphenyl-1,10-phenanthroline-2,9-dicarbox-amide and 4,7-bis(2-methylpropoxy)-9-[(1-phenylethyl)-carbamoyl]-2-(phenylcarbamoyl)-1,10-phenanthrolin-1-ium trifluoromethane-sulfonate.20
Molecular and crystal structures of acid 1 and its diamides
In the present work molecular and crystal structures of starting dicarboxylic acid 1 and its diamides 3a, 3b, 3f were examined by single-crystal X-ray diffraction.The X-ray data and full description of molecular and crystal structures of 1·H2O·DMF are given in ESI (see Fig. S4, Tables S2–S8†).
Molecular structures and packing diagrams of three diamide ligands are presented in Fig. 1–3, and full data on bond lengths, flat and dihedral angles and intermolecular contacts in the studied structures are given in ESI (Tables S10–S15, S17–S22 and S24–S30†). Compound 3b crystallizes as the 3b·H2O hydrate, with the water molecules being rotationally disordered as mentioned below in Experimental section.
 | ||
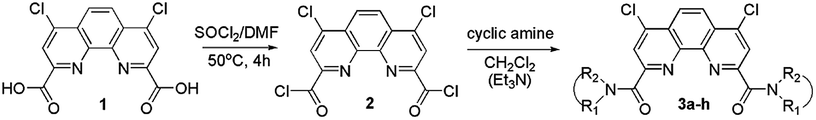
| Fig. 1 Molecular structure of 3a (a) and packing diagram showing the dimeric arrangement (b). Thermal ellipsoids are drawn at the 50% probability level. In the packing diagram, all H atoms are omitted. | ||
 | ||
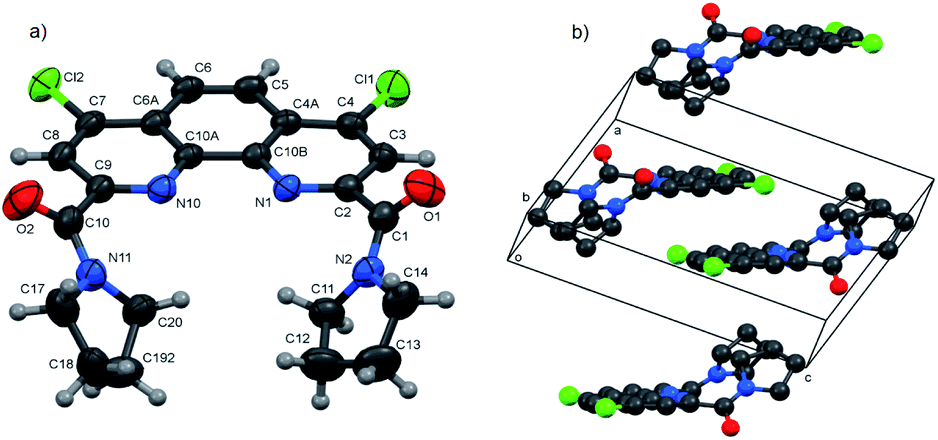
| Fig. 2 Asymmetric part of 3b·H2O (a) and packing diagram drawn along the b axis direction showing stacks and layers (b). Thermal ellipsoids are drawn at the 50% probability level. In the packing diagram, all H atoms are omitted. | ||
 | ||
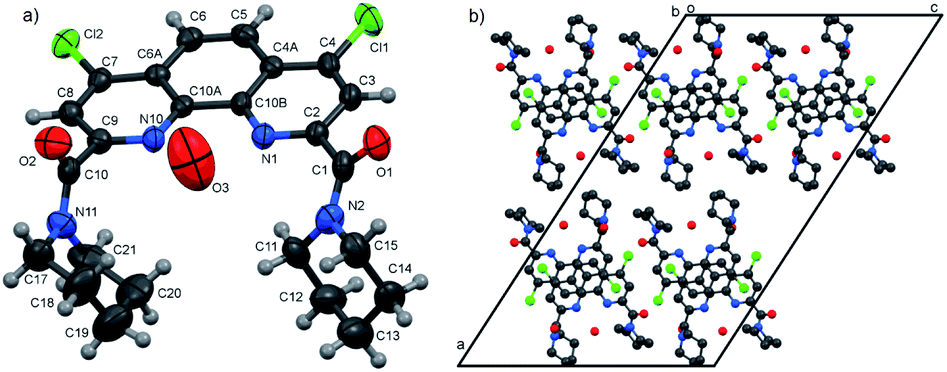
| Fig. 3 Molecular structure (a) and packing diagram (b) of 3f. Thermal ellipsoids are drawn at the 70% probability level. The contacts C–H⋯O are presented by violet lines, other H atoms are omitted for clarity. | ||
In all three structures, the phenanthroline cores are essentially planar, with the mean deviations of atoms from the planes not exceeding 0.05 Å. All compounds crystallize in the twisted asymmetric conformations, in which the oxygen atoms of the amide groups are positioned far away from each other. These conformations significantly differ from those adopted by the ligands in the complexes with metal ions, where the carbonyl oxygen atoms are brought together and deviate only slightly from phenanthroline tricycle plane in syn–syn conformation. The amide groups are nearly planar, with π-conjugation in the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N fragments evident from the C–N and CO bond lengths. Their least-squares planes are inclined to the phenanthroline fragments at 51.07(9)° and 41.1(1)° (3a), 48.16(9)° and 81.26(9)° (3b), and 64.1(4)° and 74.0(4)° (3f). In 3a and 3b, the amide oxygen atoms lie in periplanar position with respect to the phenanthroline fragments, whereas in 3f they are oriented anti-periplanar.
C–N fragments evident from the C–N and CO bond lengths. Their least-squares planes are inclined to the phenanthroline fragments at 51.07(9)° and 41.1(1)° (3a), 48.16(9)° and 81.26(9)° (3b), and 64.1(4)° and 74.0(4)° (3f). In 3a and 3b, the amide oxygen atoms lie in periplanar position with respect to the phenanthroline fragments, whereas in 3f they are oriented anti-periplanar.
The five-membered amide cycles in 3a and 3f adopt the “envelope” conformations, whereas the six-membered cycles in 3b have the “chair” conformations. The angles C–N–C in the amide five- and six-membered cycles increase in the row 3f < 3a < 3b (see ESI, Table S30†). Because of their large size, the piperidine groups in 3b hinder free rotation about the C–C and C–N bonds. In the molecule of 3f, the five-membered cycles are compact, but rotation about the C–C bonds by 360° is hindered by the bulk and rigid aromatic groups. Consequently, molecule of 3a seems to be the most flexible.
It is obvious that in the absence of crystal packing effects, the rotation around the C2,9–C(O) bonds between the phenanthroline fragments and the amide C atoms gives rise to a number of conformers, as confirmed by the spectral data for solutions presented below.
Another typical feature of the crystal structures of diamides under study is the formation of dimers and stacks due to the π–π interactions between the phenanthroline fragments. In crystals, the molecules of 3a are packed into the centrosymmetric dimers (Fig. 1b), with the distance between the planes of phenanthroline nuclei of 3.414(1) Å. In the structure of 3b·H2O, the molecules related by the screw axis 21 form stacks with the shortest intercentroid separation of 3.641(1) Å between the central aromatic ring and the terminal ring containing the N10 atom. In the crystals of 3f, the molecules are connected by the inversion centers into the stacks along the a axis direction, the distances between the phenanthroline planes being 3.316(2) and 3.368(2) Å. Thus, the formation of π–π associates in aqueous solutions due to such interactions is also possible.
Thus, we unambiguously confirmed the structure of synthesized diamides and found out some features of their crystal packaging.
Extraction of lanthanides, americium and curium by diamides 3‡
Separation of a pair of Am3+ and Eu3+ cations is usually acts as a model test when evaluating the extraction properties organic ligands. Saturated solutions of diamides 3 in “F-3” were used in the extraction of these cations from 3 mol L−1 solution of nitric acid. The values of the D between the organic and aqueous phases are shown in Fig. 4.§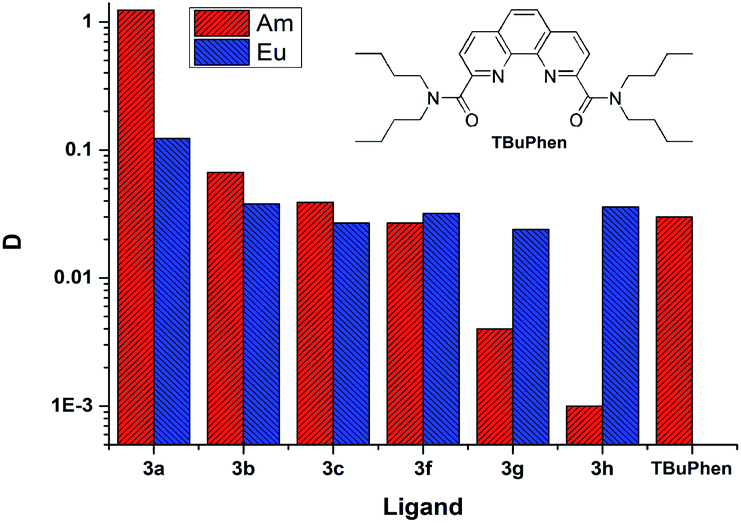 | ||
| Fig. 4 The distribution ratios D (Am) and D (Eu) for extraction from 3 mol L−1 nitric acid. The organic phase is saturated solutions of ligands in “F-3”. | ||
D is the distribution ratio of the metal cation in a biphasic system which is calculated as follows: D = [M]org/[M]aq, where [M]org and [M]aq is metal cation concentration in organic and aqueous phases, respectively.
As it seen from Fig. 4, ligand 3a extracts Am(III) and Eu(III) with the highest distribution ratios. An increase in the cycle size at the amide nitrogen atom per one methylene group (ligand 3b) leads to a drastic decrease in the extraction capacity in this solvent and to about five times decrease in selectivity towards Am/Eu pair. The same patterns are observed when extracting from nitric acid solutions of 1 mol L−1 to 5 mol L−1. The distribution ratios D and separation factors SF (SF = D1/D2, where D1 and D2 are distribution ratios for “metal 1” and “metal 2”, D1 > D2, SF > 1) for both ligands grow monotonously with an increase in the nitric acid content in this interval, which is typical for all diamides of 4,7-dichloro-1,10-phenanthroline-2,9-dicarboxylic acid16,18 (Fig. 4, 5, Table 2). Extraction data for ligand 3d can be found in ESI (Fig. S1†). Comparison data for N,N,N′,N′-tetrabutyl-1,10-phenanthroline-2,9-dicarboxamide (TBuPhen) was taken from ref. 40.
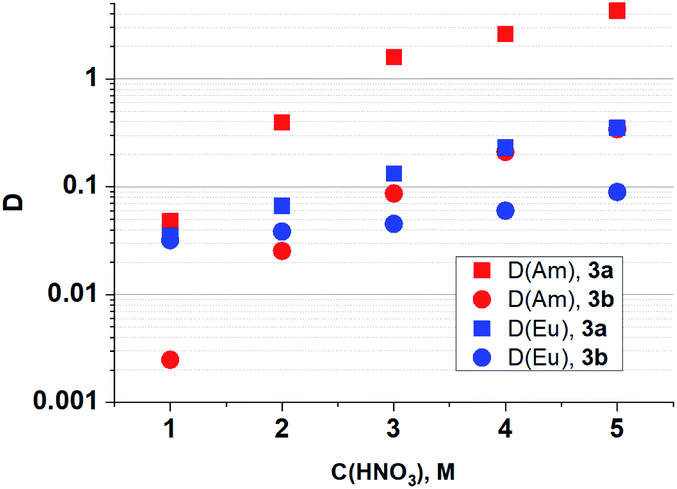 | ||
| Fig. 5 Dependences of D (Am) and D (Eu) on the concentration of HNO3 in the equilibrium water phase for extraction by 3a and 3b solutions in “F-3”. | ||
| Ligand | C(HNO3), mol L−1 | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|
| 3a | SF (Am/Eu) | 1.4 | 5.9 | 12 | 12 | 12 |
| 3b | SF (Am/Eu) | 0.1 | 0.7 | 2 | 4 | 4 |
When one and then two benzene rings are incorporated into the structure of amide fragment of the 3a and 3b ligands, their extraction ability with respect to Am(III) decreases sharply, while the extraction ability with respect to Eu(III) remains almost constant. As a result, the separation factor SFAm/Eu inverts when coming from the ligands 3a–3c to the ligands 3f–3h.
It should be noted, that for the several types of extractants among of which are diamides of pyridine-2,6-dicarboxilyc and 1,10-phenanthroline-2,9-dicarboxylic acids,40–45 the “effect of abnormal aromatic strengthening” is known. Attachment of aryl-substituent to the amide nitrogen significantly increases their extraction efficiency. An origin of the phenomenon considered in.46 Regretfully, the solubility of 3g, 3h and 3f in “F-3” is too low for correct comparison of their extraction efficiency with the same of N,N′-dialkyl-N,N′-diaryl-1,10-phenanthroline-2,9-dicarboxmides.
The stoichiometry of complexes formed during extraction in “F-3” was determined by the slope analysis of “lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D − lgC(L)” dependence.
D − lgC(L)” dependence.
The distribution ratios D (Am) and D (Eu) depending on the concentration of 3a during extraction from the 5 mol L−1 HNO3 aqueous phase are presented in ESI (Fig. S2 and Table S1†). The obtained solvate numbers, which are 1.32 for Am(III) and 1.04 for Eu(III), show that Am(III) is extracted into the organic phase as complexes of 1:1 and 1:2 composition, and Eu(III) as a complex 1:1 only.
Using the obtained lgD(Am) − lgC(L) relationship, the extraction efficiency can be extrapolated to the concentration of 0.5 mol L−1 and calculated D(Am) is about 600. In similar conditions TBuPhen40 shows D(Am) about 10. Also as it presented in Fig. 4, 3a possess much higher extraction ability towards Am when comparing it with TBuPhen in similar conditions. This clearly shows a sharp increase in extraction efficiency due to the volume reduction and hardening of substituents at the amide nitrogen atom.
The extraction of the entire set of lanthanides (except promethium) from solutions containing 2–5 mol L−1 nitric acid by 3a and 3b solutions in “F-3” (Fig. 6) demonstrates monotonous decrease in distribution ratios with increasing atomic number of Ln that has already been observed previously for diamides of 1,10-phenanthroline-2,9-dicarboxylic acid.18
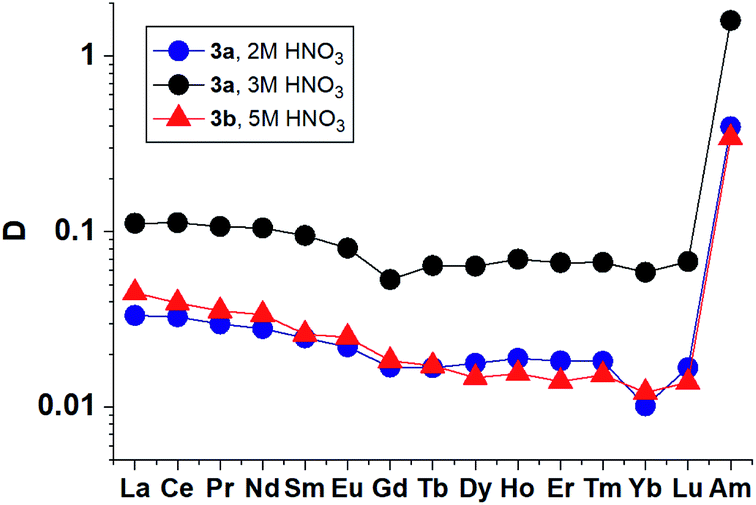 | ||
| Fig. 6 Distribution ratios for lanthanides(III) and Am(III) for extraction by 3a and 3b solution in “F-3”. | ||
The results obtained show that 3a and 3b in their selectivity to the Am/La pair (SF (Am/La) ≈ 10) are close to TBuPhen23 and derivatives of diamides of 2,2′-bipyridyl-6,6′-dicarboxylic acid.47,48
Also, we should mention that in some cases of the Cm and Ln distribution ratios are very low (≈0.01). Since the content in the organic phase was calculated as the difference in the initial water phase and the equilibrium water phase, these values should be considered as semi-quantitative (very low). However, for D(Eu), it is clear that the values obtained by two methods (ICP-OES and gamma-spectrometry) are consistent.
Extractive separation of neighboring radionuclides Am/Cm is a complicated and urgent task in modern nuclear technologies. We studied the extraction of Cm(III) from nitric acid solutions by diamides 3a and 3b in “F-3”. The values of obtained D and SF (Am/Cm) are presented in Table 3.
| Ligand | C(HNO3), mol L−1 | D (Am) | D (Cm) | SF (Am/Cm) |
|---|---|---|---|---|
| 3a | 1 | 0.125 | 0.033 | 3.8 |
| 3a | 2 | 0.3 | 0.1 | 3.0 |
| 3a | 3 | 1.24 | 0.46 | 2.7 |
| 3b | 5 | 0.37 | 0.15 | 2.6 |
Thus, diamides 3a and 3b in their selectivity are inferior to N,N′-diethyl-N,N′-di(p-hexylphenyl)-4,7-dichloro-1,10-phenanthroline-2,9-dicarboxamide16 which demonstrates SF (Am/Cm) values as high as 7.
To determine the cause of the observed unexpected “structure–property” patterns we conducted a thorough study of structural behavior of synthesized ligands 3a–3h by IR (both in solid and in solution state) and NMR spectroscopy.
Pecularities of structure and dynamics of diamides 3
Rather important general factor determining structure – extraction efficiency relationship for diamides under study is “the ligand preorganisation energy” which is a difference in energies between conformation of free ligand and its conformation in complex with metal cation. In this regard, the noticeable decrease in the distribution coefficients D found by comparing diamides 3a and 3b with diamide 3f is quite expected, since the amide fragments in 3a and 3b in the crystals are in the periplanar position, while in 3f they are anti-periplanar. However, in the solution, the diamides 3 can take on conformations different from those in the solid. In this case, the extraction properties of these ligands will be substantially determined as well by activation barriers that separate such conformations. These obvious considerations prompted us to study the structures of diamides 3 in solutions using IR and NMR spectroscopy.Fig. 7 shows the overview IR spectra of compound 3a in the solid state and in CCl4 solution. At first glance, the solid state and solution IR spectra of diamides 3a–h are very similar. A thorough study of stretch bands of CO groups in IR-spectra of diamides 3 in solid state revealed a complicated structure, as shown in Fig. 8.
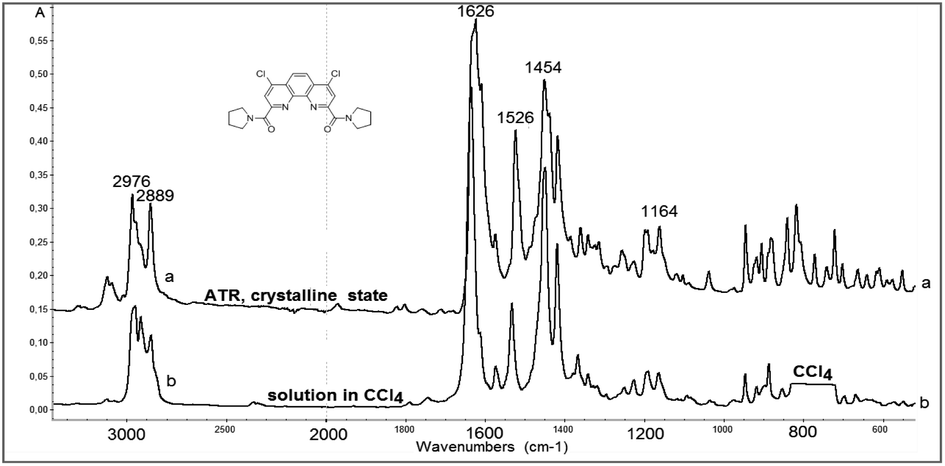 | ||
| Fig. 7 View of IR-spectra of 3a in crystalline state (above) and in CCl4 solution (below). Solvent absorption in the area of 800 cm−1 is excluded. | ||
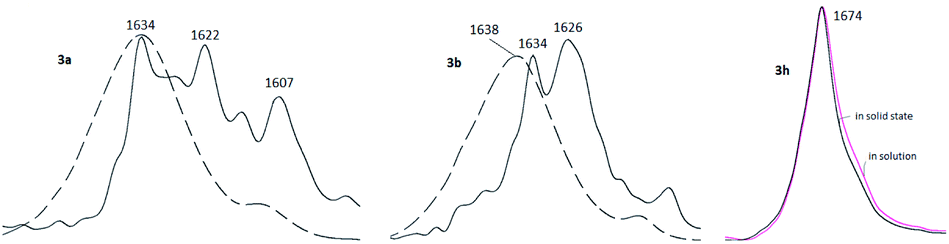 | ||
| Fig. 8 Stretch vibrations of CO groups of 3a, 3b and 3h in crystalline state (black solid lines) and in CCl4 solutions (dashed or colored lines). | ||
Complicated contours of these bands clearly indicate that carbonyl groups in crystals exist in different spatial environment. All diamides 3 contain C–C(O)–N fragment, which can be expected to provide conformational mobility with respect to the C–C(O) and N–C(O) bonds. Most likely, the complication of the spectra is associated with the formation of different rotamers with respect to these bonds during crystallization leading to the formation of polymorphic forms of crystals. The spectra can be further complicated by the Fermi resonances with overtones or combinations bands of non-planar deformation oscillations of C–H aromatic rings, which are manifested in the region of 820–870 cm−1.
In solutions, due to the conformational equilibrium, relatively wide symmetrical bands are observed, the maxima of which are slightly shifted to the short-wave region (see ESI, Table S31†). This behavior of carbonyl absorption suggests that several conformational forms coexist in solutions with close absorption maxima. Noteworthy, in case of 3h contours of bands of CO groups in solid state and in solution are very similar what may be explained by prevalence of one stable form in both media. More detailed data were obtained using dynamical NMR (DNMR).
The fact of hindered rotation around the OC–NRR′ bond in amides of carboxylic acids is well documented.49 The energy barrier of this process lies within a wide range from 13 to 30 kcal mol−1 depending on the structure of particular substance. Typically, the rotation around partially double R'C(O)–NR2 bond proceeding slowly in NMR timescale. As a result, the atoms 1H and 13C of the substituent R closest to the nitrogen atom have different chemical shifts and appear as separate signals when recording NMR spectra for symmetric amides at room temperature.
The dynamic behavior of amides and anilides of arylcarboxylic acids in solutions is more complicated. Rotation barriers for Ar–C(O) and R2N–C(O) bonds are close enough and these dynamic processes occur in concerted mode. Due to that conformation maps of such compounds can be rather complicated. Molecules of that type exist in solutions as mixtures of conformers, interconversions of which are strongly temperature dependent. DNMR is the most informative technique for their study.49–52 Diamides 3 belong to class of arylamides, but each molecule contains two amide-groups what correspondingly increases the number of possible conformers.
1H NMR spectra of diamides 3a–c indicate that at 25 °C in CDCl3 solution of these compounds the rotation around “Phen”–C(O) bonds is fast in NMR timescale whereas the rotation around “Phen”C(O)–NR2 bonds is slow. As an example 1H NMR spectrum for 3a is depicted in Fig. 9.
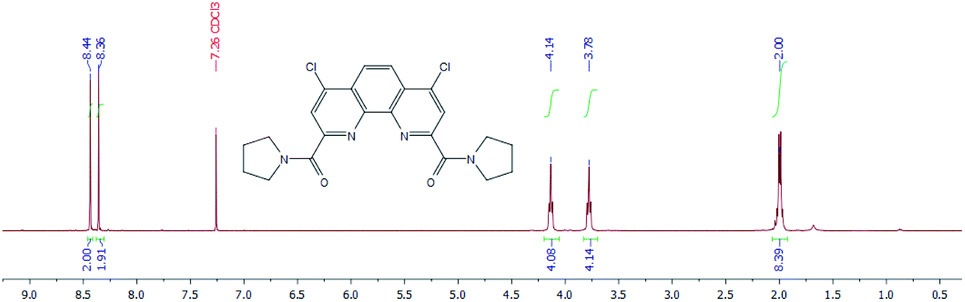 | ||
| Fig. 9 1H NMR spectrum of 3a in CDCl3 at 25 °C. | ||
The difference in proton chemical shifts of CH2-groups in α-positions to the amide nitrogen atom in diamides 3a–3c is due to the contribution of magnetic anisotropies of the CO group and aromatic ring surrounding of phenanthroline moiety.49 The value of this difference depends on the planarity of the amide substituents and their coplanarity to the phenanthroline core, as well as on the polarity of the solvent used.
Thus, in CDCl3 the biggest difference is observed for compound 3a (0.36 ppm), for 3b this value decreases to 0.07 ppm and for 3c chemical shifts of these protons are equal. Similar phenomenon was noticed previously in 1H NMR spectra of N-benzoylpirrolydine and N-benzoylpiperidine,53 but for the 3a/3b pair the effect is more strongly pronounced. Obviously, activation barrier of rotation around the amide bond decreases with increasing of volume of substituents at the amide nitrogen.49,54–57 Our single-crystal X-ray diffraction study of 3a and 3b showed (see above) that amide fragments in these compounds are close to planar. Thus, it is most probably, that the changes in chemical shifts difference of α-CH2-groups protons induced by solvents in the series 3a–c due to the changes in the dihedral angles around “Phen”–C(O)–bonds.
1H NMR spectrum of diamide 3f is more complex and temperature dependent. In CDCl3 solution at −30 °C (Fig. 10a) it contains the signals of two conformers in molar ratio about 2:1. The major conformer has a symmetrical structure in which both indoline fragments are in the same spatial environment. The signals of this conformer are marked with red arrows (Fig. 10a).
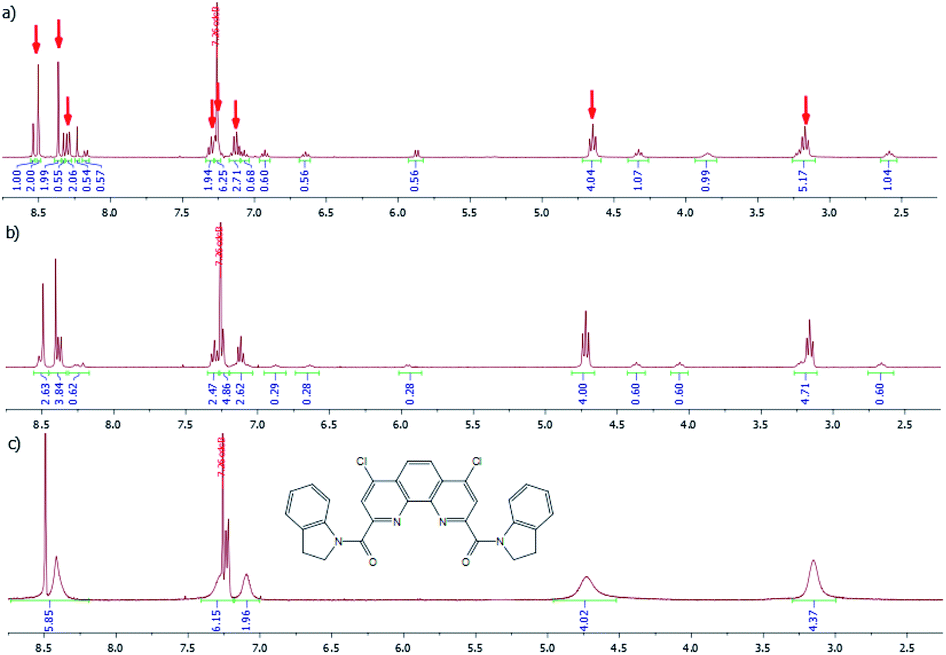 | ||
| Fig. 10 1H NMR spectra of 3f in CDCl3 at −30 °C (a), 25 °C (b) and 55 °C (c). | ||
In minor conformer spatial environment of two indoline fragments is different.
A gradual increase in temperature first leads to a progressive broadening of the signals of both conformers in the spectrum, which, as one would expect, is more pronounced for the signal of the minor conformer. Then, at 55 °C a collapse of signals of the same type of protons is observed (Fig. 10c).
These changes in the spectrum clearly indicate the acceleration of the inhibited rotation around the “Phen”–C(O) bonds, leading to the interconversion of conformers. To confirm this assumption, we used ROESY NMR technique (Fig. 11).
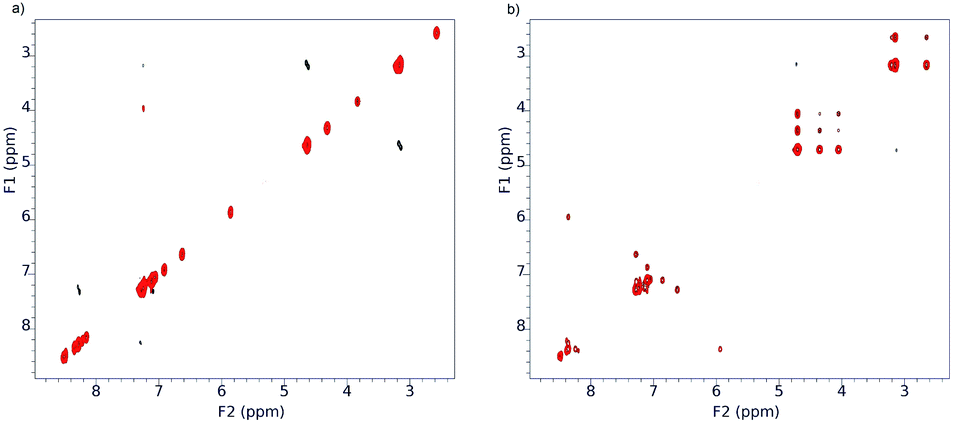 | ||
| Fig. 11 ROESY 1H NMR spectra of 3f in CDCl3 at −30 °C (a) and 25 °C (b). | ||
As it seen from Fig. 11, at −30 °C the exchange process is slow and ROESY spectrum at this temperature contains only diagonal peaks (Fig. 11a). Analysis of cross-peaks in the region 3.6–4.8 ppm in the ROESY spectrum at 25 °C (Fig. 11b) clearly shows that positional exchange between two indoline fragments in minor conformer proceeds through symmetrical conformer.
Similar behavior was observed for 1H NMR spectra of 3f in DMSO-d6, but the signals of main and minor forms are more broaden in comparison with those in CDCl3. A fragmental view of aromatic region with the assignment of signals is given in Fig. 12.
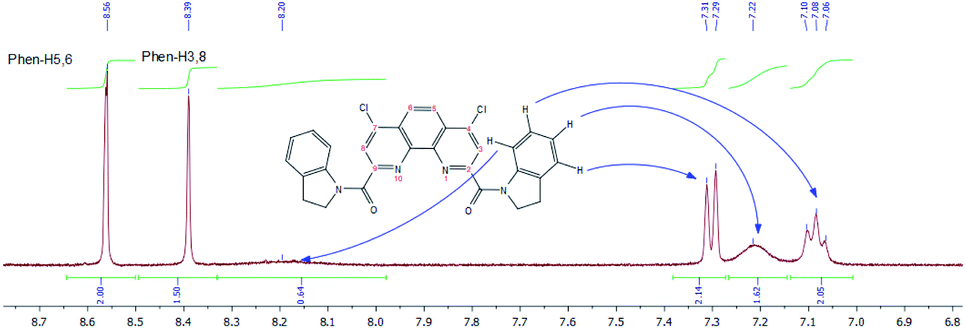 | ||
| Fig. 12 1H NMR spectra of 3f in DMSO-d6 at 55 °C (fragmental view). | ||
Such spectral changes were also found for 3g (see ESI†).
In contrast to other diamides 3, 1H NMR spectrum of bis-carbazolyl diamide 3h is quite simple and predictable (Fig. 13). The introduction of two spatially very bulky carbazolyl residues into the molecule leads to the fact that only one conformer of a symmetrical structure is stable.
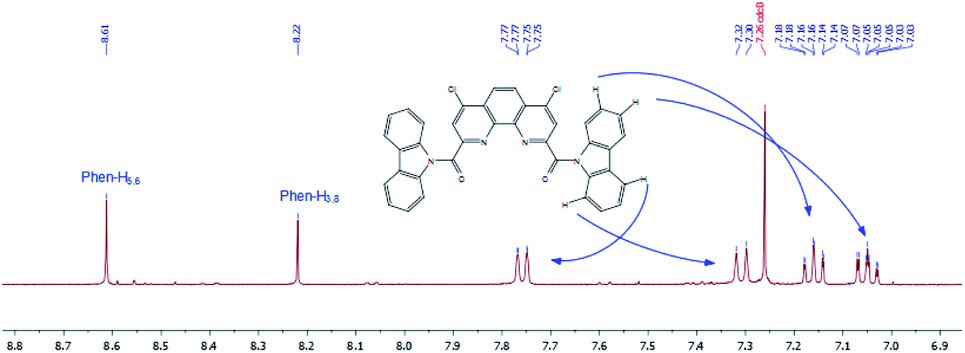 | ||
| Fig. 13 1H NMR spectrum of 3h in CDCl3 at 55 °C (region from 6.9 to 8.7 ppm). | ||
Thus, according to our study, diamides 3f and 3g exist in solutions as equilibrium mixtures of conformers that arise as a result of slow rotation around “Phen”–C(O) bonds and around R2N–C(O) amide bonds, which leads to the observed temperature dependences in 1H NMR spectra. These observations may explain the fact of a sharp decrease in selectivity towards Am/Eu pair for diamides 3f and 3g.
Another fact that needs to be explained is the sharp decrease in selectivity towards Am/Eu pair when switching from amide 3a to amide 3b. It is well known58–62 that the ability to bind metal cations for a wide range of extractants is associated with the ability to realize so called “pre-organized” conformation in solution. In the case of this class of compounds, the flexible parts of the extractant are the amide groups where oxygen atoms are involved in binding with the metal cation.
Thus, according to our study, diamides 3f and 3g exist in solutions as equilibrium mixtures of conformers that arise as a result of hindered rotation around “Phen”–C(O) bonds and around R2N–C(O) amide bonds, which leads to the observed temperature dependences in 1H NMR spectra. Sufficiently high rotation barriers around Phen–CO bonds in 3f and 3g compared with those in diamides 3a–3c may be an additional reason for the decrease in the extraction ability of the first two diamides.
Experimental section
Materials
The list of the studied diamides and their labels (3a–3h) are presented in Table 1.All syntheses were performed in argon inert atmosphere. Dichloromethane was purified by distillation over calcium hydride prior to use. Triethylamine was purified by simple distillation, previously held for 12 hours over sodium hydroxide. All the cyclic amines which are liquid under normal conditions were purified by simple or vacuum distillation. Carbazole was previously purified by recrystallization from toluene.
3-Nitrobenzotrifluoride (“F-3”) analytical grade was purchased from Rhodia (France) and was used as a solvent in the extraction experiments without further purification.
Methods
NMR spectra were recorded using standard 5 mm sample tubes on Agilent 400-MR spectrometer with operating frequencies of 400.1 MHz (1H) and 100.6 MHz (13C) and Bruker Avance-600 spectrometer with operating frequencies of 600.1 MHz (1H) and 150.6 MHz (13C). Deuterated solvents CDCl3, THF-d8, benzene-d6, acetonitrile-d3, and DMSO-d6 for NMR spectra were purchased from commercial sources and used without further purification.The ROESY spectrum was recorded on the Agilent 400-MR spectrometer using standard ROESYAD pulse sequence with a mixing time 200 ms and an acquisition time 150 ms.
IR spectra were recorded on FTIR spectrometer Nicolet iS5 (Thermo Scientific) using an internal reflectance attachment with diamond optical element – attenuated total reflection (ATR) with 45° angle of incidence. Resolution 4 cm−1, the number of scans is 32.
IR spectra in CCl4 solutions were recorded in a 0.06 cm cuvette with KBr windows at the concentrations of compounds 0.01 mol L−1 at room temperature. Ultrasonic bath was used to facilitate dissolution of diamides 3f–3h.
HRMS ESI – mass spectra were recorded on the MicroTof Bruker Daltonics and Orbitrap Elite instruments.
Single crystals of 1·DMF·H2O and 3f were obtained upon slow isothermal (25 °C) recrystallization of corresponding substances from DMF-ethyl acetate (50/50) mixture, single crystals of 3a were obtained upon slow adding of n-hexane to a solution of 3a in ethyl acetate-chloroform (50/50) mixture, and 3b·H2O – by recrystallization of 3b from acetonitrile.
X-ray diffraction experiments for 1·DMF·H2O and 3f were performed on the “Belok” beamline of the National Research Center “Kurchatov Institute” (Moscow, Russian Federation) using a Rayonix SX165 CCD detector and X-ray radiation with λ = 0.78790 Å and 0.79272 Å for 1·DMF·H2O and 3f, respectively. For 3a and 3b·H2O, the STOE STADIVARI diffractometer equipped with the fine-focus Cu GeniX 3D radiation source (λ = 1.54186 Å) and a Pilatus 100 K detector was used. For 1·DMF·H2O and 3f, data reduction procedure was carried out with the iMOSFLM program from the CCP4 program set,63 absorption correction was performed with the SCALA program,64 whereas for 3a and 3b·H2O, the STOE X-AREA program was applied. All structures were determined by direct methods and refined by full-matrix least-squares on F2 with all non-H atoms given anisotropically. Hydrogen atoms of hydroxy groups and water molecule in 1·DMF·H2O were located from difference Fourier maps and refined isotropically with Uiso(H) = 1.5Ueq(O). For 3b·H2O, we failed to determine the positions of water molecule H atoms, which seem to be disordered. All other H atoms were positioned geometrically and refined as riding at parent C atoms with Uiso(H) = 1.5Ueq(C) for methyl groups and Uiso(H) = 1.2Ueq(C) for all other H atoms. For 1·DMF·H2O and 3f, all calculations were performed with SHELXTL,65a whereas for 3a and 3b·H2O SHELXT65b was used for the structure solution and SHELXL65a – for the refinement. Full crystallographic data are deposited in Cambridge Structural Database, deposition numbers 1938798 (1·DMF·H2O), 1988714 (3a), 1983770 (3b·H2O), 1938799 (3f).† Selected crystal data for the studied compounds are presented below:
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.158(1), b = 8.352(2), c = 15.356(3) Å, α = 81.10(3), β = 82.92(3), γ = 78.62(3)°, V = 885.1(3) Å3, Z = 2, dc = 1.607 g cm−3, μ = 0.538 mm−1, F(000) = 440, T = 100(2) K, Tmin/Tmax = 0.890/0.980, 2θmax = 61.52°, 15682 reflections, 4025 unique (Rint = 0.028), R1 = 0.031 for 3711 reflections with I > 2σ(I), wR2 = 0.089 for all reflections and 268 refined parameters, GoF on F2 = 1.073., a = 7.7488(3), b = 10.1002(4), c = 13.5780(5) Å, α = 79.974(3), β = 76.276(3), γ = 83.821(3)°, V = 1014.12(7) Å3, Z = 2, dc = 1.452 g cm−3, μ = 3.11 mm−1, F(000) = 460, T = 293(2) K, Tmin/Tmax = 0.672/0.953, 2θmax = 135.95°, 8085 reflections, 3593 unique (Rint = 0.023), R1 = 0.043 for 2581 reflections with I > 2σ(I), wR2 = 0.129 for all reflections and 281 refined parameters, GoF on F2 = 1.025.181 reflections, 4179 unique (Rint = 0.113), R1 = 0.036 for 2196 reflections with I > 2σ(I), wR2 = 0.072 for all reflections and 298 refined parameters, GoF on F2 = 0.846., a = 7.2410(14), b = 11.680(2), c = 14.506(3) Å, α = 103.259(14), β = 95.030(2), γ = 97.662(18)°, V = 1174.6(4) Å3, Z = 2, dc = 1.525 g cm−3, μ = 0.420 mm−1, F(000) = 556, T = 100(2) K, Tmin/Tmax = 0.93/0.97, 2θmax = 61.92°, 18379 reflections, 5233 unique (Rint = 0.078), R1 = 0.059 for 3967 reflections with I > 2σ(I), wR2 = 0.153 for all reflections and 344 refined parameters, GoF on F2 = 1.021.
, a = 7.158(1), b = 8.352(2), c = 15.356(3) Å, α = 81.10(3), β = 82.92(3), γ = 78.62(3)°, V = 885.1(3) Å3, Z = 2, dc = 1.607 g cm−3, μ = 0.538 mm−1, F(000) = 440, T = 100(2) K, Tmin/Tmax = 0.890/0.980, 2θmax = 61.52°, 15682 reflections, 4025 unique (Rint = 0.028), R1 = 0.031 for 3711 reflections with I > 2σ(I), wR2 = 0.089 for all reflections and 268 refined parameters, GoF on F2 = 1.073., a = 7.7488(3), b = 10.1002(4), c = 13.5780(5) Å, α = 79.974(3), β = 76.276(3), γ = 83.821(3)°, V = 1014.12(7) Å3, Z = 2, dc = 1.452 g cm−3, μ = 3.11 mm−1, F(000) = 460, T = 293(2) K, Tmin/Tmax = 0.672/0.953, 2θmax = 135.95°, 8085 reflections, 3593 unique (Rint = 0.023), R1 = 0.043 for 2581 reflections with I > 2σ(I), wR2 = 0.129 for all reflections and 281 refined parameters, GoF on F2 = 1.025.181 reflections, 4179 unique (Rint = 0.113), R1 = 0.036 for 2196 reflections with I > 2σ(I), wR2 = 0.072 for all reflections and 298 refined parameters, GoF on F2 = 0.846., a = 7.2410(14), b = 11.680(2), c = 14.506(3) Å, α = 103.259(14), β = 95.030(2), γ = 97.662(18)°, V = 1174.6(4) Å3, Z = 2, dc = 1.525 g cm−3, μ = 0.420 mm−1, F(000) = 556, T = 100(2) K, Tmin/Tmax = 0.93/0.97, 2θmax = 61.92°, 18379 reflections, 5233 unique (Rint = 0.078), R1 = 0.059 for 3967 reflections with I > 2σ(I), wR2 = 0.153 for all reflections and 344 refined parameters, GoF on F2 = 1.021.Detailed crystallographic data provided in ESI (see from p. S41†).
Synthesis and analytical data
General method for synthesis of diamides 3a–e
A solution of the corresponding cyclic amine (50 mmol) in 5 mL of methylene chloride was added at −20 °C under vigorous stirring to a suspension of 10 mmol (3.74 g) of chloride 2 in 25 mL of methylene chloride. Then the reaction mixture was allowed to reach room temperature and stirred overnight. Next, the reaction mixture was diluted with 50 mL of methylene chloride, washed with water (2 × 50 mL), dried over sodium sulfate, and the solvent was distilled off. The residue was treated with hexane, recrystallized from a mixture of chloroform/hexane, obtaining the desired product in the form of a white or slightly colored solid.O), 1573, 1522, 1449, 1437, 1416 (CC, CN); 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 2H), 8.31 (s, 2H), 4.09 (t, J = 6.5 Hz, 4H), 3.74 (t, J = 6.5 Hz, 4H), 2.01–1.91 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 164.9, 154.3, 145.2, 143.8, 127.5, 124.4, 124.2, 49.2, 47.3, 26.9, 24.2; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C22H21Cl2N4O2 443.1037, found 443.1034.:CH2Cl2 1:4); IR (cm−1) 3084, 3043, 2938, 2914, 2843 (C–H stretching vibrations), 1634, 1626 (CO), 1571, 1532, 1456, 1446, 1425 (CC, CN); 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 2H), 8.10 (s, 2H), 3.79 (t, J = 5.3 Hz, 4H), 3.72 (t, J = 5.3 Hz, 4H), 1.82–1.59 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 166.0, 154.5, 145.3, 143.9, 127.3, 124.3, 124.0, 48.7, 43.9, 26.7, 25.7, 24.7; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C24H25Cl2N4O2 471.1350, found 471.1335.:CH2Cl2 1:5); IR (cm−1) 3037, 2926, 2853 (C–H stretching vibrations), 1634, 1631, 1626 (CO), 1590, 1530, 1456, 1427 (CC, CN); 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 2H), 8.11 (s, 2H), 3.82–3.72 (m, 8H), 1.90 (p, J = 5.9 Hz, 4H), 1.78 (p, J = 5.9 Hz, 4H), 1.71–1.57 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 167.6, 154.9, 145.4, 143.8, 127.3, 124.1, 124.0, 49.9, 47.4, 29.6, 27.6, 27.3, 27.0; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C26H29Cl2N4O2 499.1663, found 499.1662.:CH2Cl2 1:1); IR (cm−1) 3093, 3026, 2966, 2912, 2852 (C–H stretching vibrations), 1644, 1640, 1634, 1634, 1628, (CO), 1570, 1528, 1475, 1462, 1444, 1427 (CC, CN); 1H NMR (400 MHz, CDCl3), δ 8.41 (s, 2H), 8.21 (s, 2H), 3.98 (t, J = 4.8 Hz, 4H), 3.95–3.83 (m, 8H), 3.75 (t, J = 4.8 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 165.8, 153.5, 145.1, 144.3, 127.6, 124.9, 124.3, 67.3, 67.0, 48.2, 43.3; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C22H21Cl2N4O4 475.0935, found 475.0928.O), 1531, 1460, 1446, 1425 (CC, CN);1H NMR (400 MHz, CDCl3) δ 8.39 (s, 2H), 8.17 (s, 2H), 3.98–3.87 (m, 8H), 2.55 (t, J = 5.0 Hz, 4H), 2.42 (t, J = 5.0 Hz, 4H), 2.32 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 165.8, 153.9, 145.1, 144.1, 127.5, 124.7, 124.2, 55.7, 54.8, 47.5, 46.2, 42.8; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C24H27Cl2N6O2 501.1568, found 501.1562.General method for synthesis of diamides 3f–h
Triethylamine (30 mmol, 3.04 g, 4.20 mL) was added to a suspension of 10 mmol (3.74 g) of chloride 2 in 25 mL of methylene chloride at −20 °C under vigorous stirring and then the solution of the corresponding secondary amine (22.5 mmol) in 10 mL of methylene chloride was added dropwise. After that, the reaction mixture was stirred at −20 °C for 1 hour, allowed to reach the room temperature and was additionally stirred for 24 hours. Then it was washed with water (2 × 50 mL), dried over sodium sulfate and the solvent was distilled off. The residue was washed with chloroform, giving the desired product in the form of slightly colored solid.:CH2Cl2 1:10); IR (cm−1) 3109, 3070, 3053, 2971, 2936, 2862 (C–H stretching vibrations), 1634 (CO), 1597, 1569, 1533, 1481, 1463, 1437, 1403 (CC, CN); 1H NMR (400 MHz, DMSO-d6, recorded at 343.15 K) δ 8.56 (s, 2H), 8.39 (s, 2H), 8.20 (bs, 2H), 7.30 (d, J = 7.8 Hz, 2H), 7.22 (bs, 2H), 7.08 (t, J = 7.8 Hz, 2H), 4.52 (bs, 4H), 3.17–3.05 (m, 4H); HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C30H20Cl2N4O2 539.1045, found 539.1045.:CH2Cl2 1:2); IR (cm−1) 3061, 3042, 2946, 2885, 2843 (C–H stretching vibrations), 1651, 1644, 1640, 1637 (CO), 1602, 1581, 1571, 1530, 1491, 1450, 1439, 1400 (CC, CN); 1H NMR (400 MHz, C6D6) δ 7.96 (s, 2H), 7.84 (s, 2H), 7.62 (bs, 2H), 6.93–6.79 (m, 6H), 3.86–3.78 (m, 4H), 2.49 (t, J = 6.8 Hz, 4H), 1.70–1.62 (m, 4H); 13C NMR (101 MHz, C6D6) δ 166.9, 155.5, 145.9, 143.3, 139.7, 131.4, 128.9, 127.2, 126.1, 125.3, 124.9, 124.2, 123.8, 46.2, 27.2, 24.1; HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C32H25Cl2N4O2 567.1350, found 567.1349.O), 1527, 1491, 1480, 1444, 1381 (CC, CN); 1H NMR (CDCl3), δ, ppm: 1H NMR (400 MHz, Chloroform-d) δ 8.61 (s, 2H), 8.22 (s, 2H), 7.80–7.73 (m, 4H), 7.31 (d, J = 8.5 Hz, 4H), 7.16 (td, J = 7.4, 1.1 Hz, 4H), 7.05 (ddd, J = 8.5, 7.4, 1.1 Hz, 4H); HRMS (ESI-TOF) (m/z) [M + H]+ calculated for C38H21Cl2N4O2 635.1035, found 635.1029.Solvent extraction experiments
Solvent extraction experiments were carried out as follows. 0.5 mL of organic solution of the extractant in 3-nitrobenzotrifluoride (“F-3”) and 0.5 mL of aqueous phase containing nitric acid and radionuclides were mixed in 1.5 mL polypropylene vial. Three sets of aqueous solutions were prepared for extraction experiments. The first one contained trace concentrations of 241Am (≈1500 Bq mL−1) and 152Eu (≈2500 Bq mL−1), the second set contained 241Am (≈1500 Bq mL−1) and 244Cm (≈2000 Bq mL−1). The third set contained lanthanum and all lanthanides (except promethium) in total concentration 10−4 mol L−1. The phases were stirred for 15 minutes on a vortex shaker at 25 ± 1 °C in an air thermostat. Then samples were centrifuged (5 minutes, 6000 rpm) and aliquots of each phase were taken for determination of radionuclide or lanthanide concentration.Content of 241Am (Eγ = 59.5 keV) and 152Eu (Eγ = 121.8 keV) was determined by gamma-spectrometry using a high-pure germanium detector GR 3818 (Canberra Ind.) in the first set of experiments. Second set of samples was analyzed by alpha-spectrometry (Eα(241Am) = 5637 keV, Eα(244Cm) = 5901 keV) using an alpha spectrometer Model 7401 with Si detectors (Canberra Ind.). Initial aqueous phase and equilibrium aqueous phases of the third set of samples were analyzed by ICP-OES (Agilent ICP-OES 720).
The distribution ratios (D) of metals were calculated for the first set of experiments as the ratio of the counting rate in the organic and aqueous phase. For the second and the third sets of samples activity/concentration in organic phase was calculated as difference between concentrations in initial and equilibrium aqueous phases. The separation factors (SF) were calculated as the ratio of the distribution ratios. All extraction experiments were repeated three times. Uncertainties for 0.01 < D < 100 are within ±10%.
Conclusions
A family of new diamides of 4,7-dichloro-1,10- phenanthroline-2,9-dicarboxylic acid was synthesized starting from the corresponding dichloride 2 and cyclic amines. The structures of three diamides and starting acid itself were determined by single-crystal X-ray diffraction. Diamides 3a, 3b and 3f crystallize in the twisted asymmetric conformations. In 3a and 3b, these fragments are in periplanar positions to each other, while the more bulky indoline fragments in 3f occupy anti-periplanar positions. All structures are stabilized by a network of intramolecular hydrogen bonds. In structures 3b and 3f, the π-stacking interaction between aromatic nuclei is clearly manifested. The addition of aromatic rings in the ligand structure leads to complex dynamic behavior in solutions.In solutions of all diamides 3, according to NMR data, the rotation along the R2N–C(O) amide bonds is inhibited. In diamides 3a–3c, containing small volume monocyclic groups in amide fragments, rotation along the “Phen”–CO bonds at room temperature is free. Insertion of the first aromatic ring into amide moiety complicates the dynamic behavior of the ligands 3f and 3g in solutions whereas insertion of bulky carbazolyl groups simplifies the spectra of 3h. The data of IR spectroscopy showed that in the polycrystalline state and in solutions, diamides 3a–3g exist as mixtures of conformers differing probably in the dihedral angles around the amide bonds R2N–C(O) and around CO–Phen bonds.
Liquid–liquid extraction experiments revealed exceptional properties for some of diamides 3 under study. It was shown that diamide 3a is more efficient extractant for lanthanides, americium and curium from highly acidic HNO3 solution than their non-cyclic N,N,N′,N′-tetraalkyl analogues. Also 3a shows high selectivity in the La/Am pair separation (SF (Am/La ≈ 10)) and in the Am/Eu pair separation (SF (Am/Eu ≈ 12)). Its selectivity in the Am/Cm pair separation is moderate (SF (Am/Cm ≈ 3.8). Both selectivity and extraction ability of the ligands under study decrease from 3a to 3c with the expansion of alicycle in the amide function despite increase in solubility. In the same time, there is a sharp inversion in selectivity in relation to the Am(III)/Eu(III) pair in the row 3a, 3f, 3h. These unusual properties can be explained by molecular dynamics of such compounds.
In order to unambiguously confirm our assumptions of the sharp decrease in selectivity towards Am/Eu pair when switching from amide 3a to amide 3b and to get more information about relationships of dynamic behavior of diamides 3 with their extraction abilities we initiated the study of complexation of lanthanides with diamides 3a and 3b using the direct 1H NMR and UV-vis titration and DFT calculations of structures of these ligands and structures of their metal complexes. These results will be presented in a separate paper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray structure determination was supported by the RUDN University Program “5-100”. The authors acknowledge support from M. V. Lomonosov Moscow State University Program of Development. Solvent extraction study was funded by Russian Foundation for Basic Research according to the research project No 18-33-0616/mol_a.Notes and references
- Key world energy statistics 2019, International Energy Agency, website: iea.org Search PubMed.
- Reprocessing and recycling of spent nuclear fuel, ed. R. Taylor, Woodhead publishing series in Energy number 79, Elsevier, 2015, Hardcover, ISBN: 9781782422129, eBook ISBN: 9781782422174 Search PubMed.
- M. Y. Alyapyshev, V. A. Babain and Y. A. Ustynyuk, Russ. Chem. Rev., 2016, 85, 943–961, DOI:10.1070/rcr4589.
- C. J. Maher, Current headend technologies and future developments in the reprocessing of spent nuclear fuels, Elsevier Ltd., 2015, pp. 93–124, DOI:10.1016/b978-1-78242-212-9.00005-8.
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273, 10.1039/c7cs00574a.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914, DOI:10.1021/cr00098a011.
- J. Veliscek-Carolan, J. Hazard. Mater., 2016, 318, 266–281, DOI:10.1016/j.jhazmat.2016.07.027.
- S. A. Ansari, P. Pathak, P. K. Mohapatra and V. K. Manchanda, Chem. Rev., 2012, 112, 1751–1772, DOI:10.1021/cr200002f.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113(2), 1199–1236, DOI:10.1021/cr3003399.
- D. Merrill and R. D. Hancock, Radiochim. Acta, 2011, 99, 161–166, DOI:10.1524/ract.2011.1805.
- N. J. Williams, N. E. Dean, D. G. VanDerveer, R. C. Luckay and R. D. Hancock, Inorg. Chem., 2009, 48, 7853–7863, DOI:10.1021/ic900737q.
- D. Merrill, J. M. Harrington, H.-S. Lee and R. D. Hancock, Inorg. Chem., 2011, 50, 8348–8355, DOI:10.1021/ic200905f.
- M. A. Lashley, A. S. Ivanov, V. S. Bryantsev, S. Dai and R. D. Hancock, Inorg. Chem., 2016, 55, 10818–10829, DOI:10.1021/acs.inorgchem.6b02234.
- N. E. Dean, R. D. Hancock, C. L. Cahill and M. Frisch, Inorg. Chem., 2008, 47, 2000–2010, DOI:10.1021/ic701574j.
- J. Dehaudt, N. J. Williams, I. A. Shkrob, H. Luo and S. Dai, Dalton Trans., 2016, 45, 11624–11627, 10.1039/c6dT01800a.
- M. Alyapyshev, V. Babain, L. Tkachenko, E. Kenf, I. Voronaev, D. Dar'in, P. Matveev, V. Petrov, S. Kalmykov and Y. Ustynyuk, J. Radioanal. Nucl. Chem., 2018, 316, 419–428, DOI:10.1007/s10967-018-5775-7.
- C.-L. Xiao, C.-Z. Wang, L.-Y. Yuan, B. Li, H. He, S. Wang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, Inorg. Chem., 2014, 53, 1712–1720, DOI:10.1021/ic402784c.
- Yu. A. Ustynyuk, N. E. Borisova, V. A. Babain, I. P. Gloriozov, A. Y. Manuilov, S. N. Kalmykov, M. Yu. Alyapyshev, L. I. Tkachenko, E. V. Kenf and N. A. Ustynyuk, Chem. Commun., 2015, 51, 7466–7469, 10.1039/c5cc01620g.
- D. Manna, S. Mula, A. Bhattacharyya, S. Chattopadhyay and T. K. Ghanty, Dalton Trans., 2015, 44, 1332–1340, 10.1039/c4dt02402h.
- H.-Y. Hu, J.-F. Xiang, Y. Yang and C.-F. Chen, Org. Lett., 2008, 10, 1275–1278, DOI:10.1021/ol8001688.
- D. Rais, I. R. Gould, R. Vilar, A. J. P. White and D. J. Williams, Eur. J. Inorg. Chem., 2004, 1865–1872, DOI:10.1002/ejic.200300809.
- S. Cao, J. Wang, C. Tan, X. Zhang, S. Li, W. Tian and Z. Qin, New J. Chem., 2016, 40, 10560–10568, 10.1039/c6nj02696f.
- E. Makrlík, P. Vanura, P. Selucký, V. Babain, D. Dar'in and M. Alyapyshev, Acta Chim. Slov., 2017, 64(3), 582–589, DOI:10.17344/acsi.2017.3295.
- X. Zhang, X. Kong, L. Yuan, Z. Chai and W. Shi, Inorg. Chem., 2019, 58, 10239–10247, DOI:10.1021/acs.inorgchem.9b01400.
- X. Zhang, L. Yuan, Z. Chai and W. Shi, Sep. Purif. Technol., 2016, 168, 232–237, DOI:10.1016/j.seppur.2016.05.056.
- A. P. Krapcho and A. Ali, J. Heterocycl. Chem., 2004, 41, 795–798, DOI:10.1002/jhet.5570410524.
- C. J. Hawkins, H. Duewell and W. F. Pickering, Anal. Chim. Acta, 1961, 25, 257–261, DOI:10.1016/0003-2670(61)80155-4.
- The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, 4th edn, 2010, vol. 1–6, ISBN: 978-94-007-0210-3 (HB), ISBN: 978-94-007-0211-0 (e-book) Search PubMed.
- M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 19, 3513–3526, DOI:10.1002/ejic.200300162.
- P. I. Matveev, A. A. Mitrofanov, V. G. Petrov, S. S. Zhokhov, A. A. Smirnova, Yu. A. Ustynyuk and S. N. Kalmykov, RSC Adv., 2017, 7, 55441–55449, 10.1039/c7ra11622e.
- P. Distler, M. Mindová, J. John, V. A. Babain, M. Yu. Alyapyshev, L. I. Tkachenko, E. V. Kenf, L. M. Harwood and A. Afsar, Solvent Extr. Ion Exch., 2020, 38(2), 180–193, DOI:10.1080/07366299.2019.1708004.
- M. Alyapyshev, V. Babain, I. Eliseev, E. Kenf and L. Tkachenko, J. Radioanal. Nucl. Chem., 2016, 310, 785–792, DOI:10.1007/s10967-016-4907-1.
- B. G. Modolo, A. Wilden, A. Geist, D. Magnusson and R. Malmbeck, Radiochim. Acta, 2012, 100, 715–725, DOI:10.1524/ract.2012.1962.
- D. L. Melton, D. G. VanDerveer and R. D. Hancock, Inorg. Chem., 2006, 45, 9306–9314, DOI:10.1021/ic061010p.
- D. N. Zarubin, N. S. Bushkov, H. V. Lavrov, F. M. Dolgushin, N. A. Ustynyuk and Yu. A. Ustynyuk, INEOS OPEN, 2019, 2, 130–133, DOI:10.32931/io1918a.
- H. V. Lavrov, N. A. Ustynyuk, P. I. Matveev, I. P. Gloriozov, S. S. Zhokhov, M. Yu. Alyapyshev, L. I. Tkachenko, L. G. Voronaev, V. A. Babain, S. N. Kalmykov and Yu. A. Ustynyuk, Dalton Trans., 2017, 46, 10926–10934, 10.1039/c7dt01009e.
- N. E. Borisova, A. A. Kostin, M. D. Reshetova, K. A. Lyssenko, E. V. Belova and B. F. Myasoedov, Inorg. Chim. Acta, 2018, 478, 148–154, DOI:10.1016/j.ica.2018.03.042.
- A. Moghimi, R. Alizadeh, A. Shokrollahi, H. Aghabozorg, M. Shamsipur and A. Shockravi, Inorg. Chem., 2003, 42, 1616–1624, DOI:10.1021/ic025725d.
- U. Florke, K. Stuhrenberg and M. Bauer, Private communication to CSD, deposition number 1861531, refcode SIFMEC, 2018 Search PubMed.
- M. Alyapyshev, J. Ashina, D. Dar'in, E. Kenf, D. Kirsanov, L. Tkachenko, A. Legin, G. Starova and V. Babain, RSC Adv., 2016, 6, 68642–68652, 10.1039/c6ra08946a.
- M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Eliseev, A. V. Didenko and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619–636, DOI:10.1080/07366299.2011.581049.
- N. A. Rozen, A. M. Nikolotova and Z. I. Kartasheva, Radiochemistry, 1990, 32, 70–78 Search PubMed.
- N. A. Rozen, A. M. Nikolotova and Z. I. Kartasheva, Radiochemistry, 1986, 28, 407–423 Search PubMed.
- A. N. Turanov, V. K. Karandashev and N. A. Bondarenko, Radiochemistry, 2005, 47, 74–79, DOI:10.1007/s11137-005-0051-4.
- B. F. Myasoedov, M. K. Chmutova, N. E. Kochetkova, O. E. Koiro, G. A. Pribylova and N. P. Neeterova, Solvent Extr. Ion Exch., 1986, 4, 61–81, DOI:10.1080/07366298608917853.
- Yu. A. Ustynyuk, I. P. Gloriozov, S. N. Kalmykov, A. A. Mitrofanov, V. A. Babain, M. Yu. Alyapyshev and N. A. Ustynyuk, Solvent Extr. Ion Exch., 2014, 32, 508–528, DOI:10.1080/07366299.2014.915666.
- M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, A. Paulenova, A. A. Popova and N. E. Borisova, Solvent Extr. Ion Exch., 2014, 32, 138–152, DOI:10.1080/07366299.2013.833783.
- N. E. Borisova, A. V. Ivanov, P. I. Matveev, A. A. Smirnova, E. V. Belova, S. N. Kalmykov and B. F. Myasoedov, ChemistrySelect, 2018, 3, 1983–1989, DOI:10.1002/slct.201702741.
- W. E. Stewart and T. H. Siddall, Chem. Rev., 1970, 70, 517–551, DOI:10.1021/cr60267a001.
- R. A. Bragg, J. Clayden, G. A. Morris and J. H. Pink, Chem.–Eur. J., 2002, 8, 1279–1289, DOI:10.1002/1521-3765(20020315)8:6<1279::aid-chem1279>3.0.co;2-7.
- E. A. Skorupska, R. B. Nazarski, M. Ciechaska, A. Jozwiak and A. Kłys, Tetrahedron, 2013, 69, 8147–8154, DOI:10.1016/j.tet.2013.07.046.
- S. Kim, J. Kim, J. Kim, D. Won, S.-K. Chang, W. Cha, K. Jeong, S. Ahn and K. Kwak, Molecules, 2018, 23(9), 2294, DOI:10.3390/molecules23092294.
- M. Tafazzoli, A. Ziyaei-Halimjani, M. Ghiasi, M. Fattahi and M. R. Saidi, J. Mol. Struct., 2008, 886, 24–31, DOI:10.1016/j.molstruc.2007.01.019.
- G. A. Gamov, V. V. Aleksandriiskii and V. A. Sharnin, J. Mol. Liq., 2017, 231, 238–241, DOI:10.1016/j.molliq.2017.01.078.
- T. Drakenberg, K.-I. Dahlqvist and S. Forsen, J. Phys. Chem., 1972, 76, 2178–2183, DOI:10.1021/j100659a020.
- J. V. Hatton and R. E. Richards, Mol. Phys., 1960, 3, 253–263, DOI:10.1080/00268976000100291.
- J. V. Hatton and R. E. Richards, Mol. Phys., 1962, 5, 139–152, DOI:10.1080/00268976200100141.
- N. E. Borisova and M. D. Reshetova, Russ. Chem. Bull., 2015, 64, 1882–1890, DOI:10.1007/s11172-015-1088-y.
- F. W. Lewis, L. M. Harwood, M. J. Hudson and M. G. B. Drew, Inorg. Chem., 2013, 52, 4993–5005, DOI:10.1021/ic3026842.
- L. Xu, N. Pu, Y. Li, P. Wei, T. Sun, C. Xiao, J. Chen and C. Xu, Inorg. Chem., 2019, 58, 4420–4430, DOI:10.1021/acs.inorgchem.8b03592.
- S. Jansone-Popova, A. S. Ivanov, V. S. Bryantsev, F. V. Sloop, R. Custelcean, I. Popovs, M. M. Dekarske and B. A. Moyer, Inorg. Chem., 2017, 56, 5911–5917, DOI:10.1021/acs.inorgchem.7b00555.
- G. J. Lumetta, B. M. Rapko, P. A. Garza, B. P. Hay, R. D. Gilbertson, T. J. R. Weakley and J. E. Hutchison, J. Am. Chem. Soc., 2002, 124, 5644–5645, DOI:10.1021/ja025854t.
- T. G. Battye, L. Kontogiannis, O. Johnson, H. R. Powell and A. G. W. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 271–281, DOI:10.1107/S0907444910048675.
- P. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 72–82, DOI:10.1107/S0907444905036693.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary data, synthesis and characterization of compounds, extraction and X-ray analysis data. CCDC 1938798, 1988714, 1983770 and 1938799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05182a |
| ‡ In experiments with 3e a precipitate appeared at the phase interface. The most probable explanation of this fact is fast protonation of this ligand in nitric acid media and subsequent collapse of this strongly basic compound. |
| § Note that for all the graphs below the uncertainties in a range of 0.01 < D < 10 are within ±10%. |
| This journal is © The Royal Society of Chemistry 2020 |