
Collective enantioselective total synthesis of (+)-sinensilactam A, (+)-lingzhilactone B and (−)-lingzhiol: divergent reactivity of styrene†
Da-Wei
Zhang
ab,
Hui-Lan
Fan
ab,
Wenzhao
Zhang
c,
Cheng-Ji
Li
ab,
Sanzhong
Luo
 *cd and
Hong-Bo
Qin
*a
*cd and
Hong-Bo
Qin
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Yunnan Key Laboratory of Natural Medicinal Chemistry, Kunming 650201, P. R. China. E-mail: qinhongbo@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cInstitute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
dCenter of Basic Molecular Science, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: luosz@tsinghua.edu.cn
First published on 23rd July 2020
Abstract
The collective total synthesis of (+)-sinensilactam A, (+)-lingzhilactone B, (+)-lingzhilactone C and (−)-lingzhiol has been accomplished from a common epoxide intermediate 9. Chemoselective epoxy opening with either an aryl or alkene moiety of styrene led to different carbon skeletons, which can be advanced to a divergent and concise total synthesis of four meroterpenoids.
During the past 7 years, Ganoderma meroterpenoids have attracted increasing attention among the phytochemical community. With meroterpenoids being increasingly identified, this type of molecule has become the third most abundant constituent of Ganoderma. Sinensilactam A, lingzhilactone B and lingzhiol, isolated from the genus Ganoderma by Cheng,1–3 are reno-protective by inhibition of Smad3 phosphorylation in TGF-β1-induced cellular assays. In 2018, Yang's group reported the first racemic total synthesis of sinensilactam A using rhodium catalyzed [3+2] cycloaddition as the key step.4 However, the synthesis required 18 steps involving harsh conditions such as high pressure (14 MPa) and long-term heating (150 °C, 48 h). A concise synthetic route under mild conditions is still highly desirable. On the other hand, we and others have previously reported the total syntheses of lingzhiol and lingzhilactone B.5–12,27 In this communication, we have reported a distinctive strategy for the collective synthesis of four meroterpenoids, (+)-sinensilactam A, (+)-lingzhilactone B, (+)-lingzhilactone C and (−)-lingzhiol. The key step involves non-metal promoted selective nucleophilic opening of epoxide by an alkene moiety of styrene, which has not been explored in total synthesis, though a metal-catalyzed reaction of epoxide with styrene is known.13–15
Sinensilactam A can be obtained from dimethyl lingzhilactone B 15 by annulation with 5-hydroxy-2-pyrrolidinone. Lingzhilactone B has one more carbon at C8′ compared to lingzhiol; therefore, styrene is a reasonable equivalent of aryl ketone because it can be either oxidatively cleaved into a dicarbonyl compound, or oxidized into aryl ketone (Fig. 1). The retrosynthetic analysis is outlined below. Ketoaldehyde 15 can be obtained by the ozonolysis of styrene 12, which could be synthesized through an epoxy ring opening reaction with the double-bond of styrene 9. Alternatively, the epoxy ring opening by the aryl moiety would lead to lingzhiol. Styrene 9 could be obtained sequentially by the alkylation of ethyl 2-oxocyclopentane carboxylate with aryl acetyl chloride, twice Wittig reaction, regioselective allylic oxidation and the epoxidation of allylic alcohol.
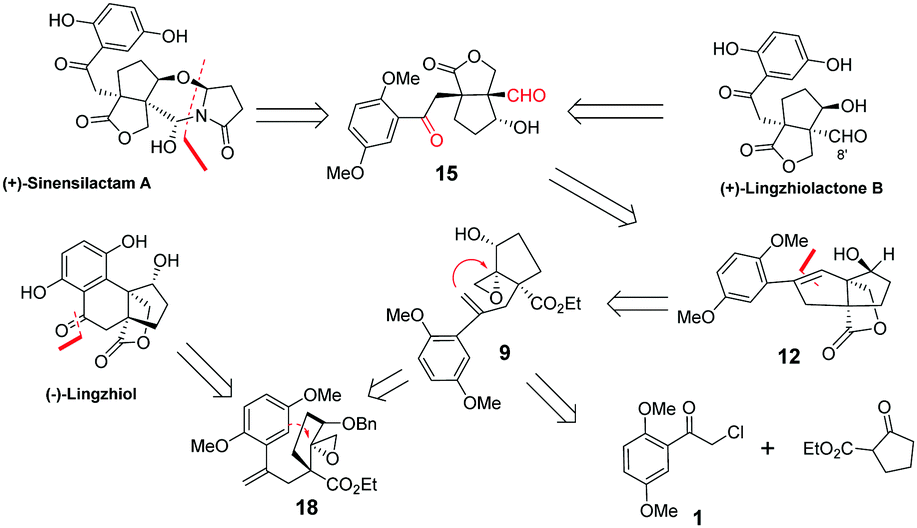 | ||
| Fig. 1 Structures of several meroterpenoids and retrosynthetic analysis. | ||
We started our total synthesis with the assembly of the chiral intermediate 9 (Scheme 1). The alkylation of 2,5-dimethoxyphenyl-acetyl chloride 1 with ethyl 2-oxocyclopentanecarboxylate proceeded smoothly to give diketone 2 in 94% yield. A resolution by baker's yeast only recovered the starting material. Therefore, we adopted a detour of a kinetic resolution of the known alkyne ketoester 4. The absolute configuration of the recovered ketone was determined to be R by its transformation into the known (−)-lingzhiol.1 The addition of 2,5-dimethoxyphenylboronic acid to chiral alkyne (R)-4 gave styrene 6 in 80% yield.16 The Wittig reaction of ketone 6 with the ylide generated from t-BuOK and triphenyl methyl bromide in THF afforded diene 7 in 81% yield as an optically pure enantiomer.17 Regioselective allylic oxidation with selenium dioxide and t-butyl hydroperoxide converted 7 into allylic alcohol 8 in 69% yield. The regioselectivity can be attributed to the steric hindrance of quaternary carbon and the cis-stereoselectivity possibly arose from the chelation of selenium with the ester group. Next, Sharpless epoxidation18 produced cis-epoxy secondary alcohol 9 regioselectively in 82% yield, which is an advanced intermediate with versatile reactivity.
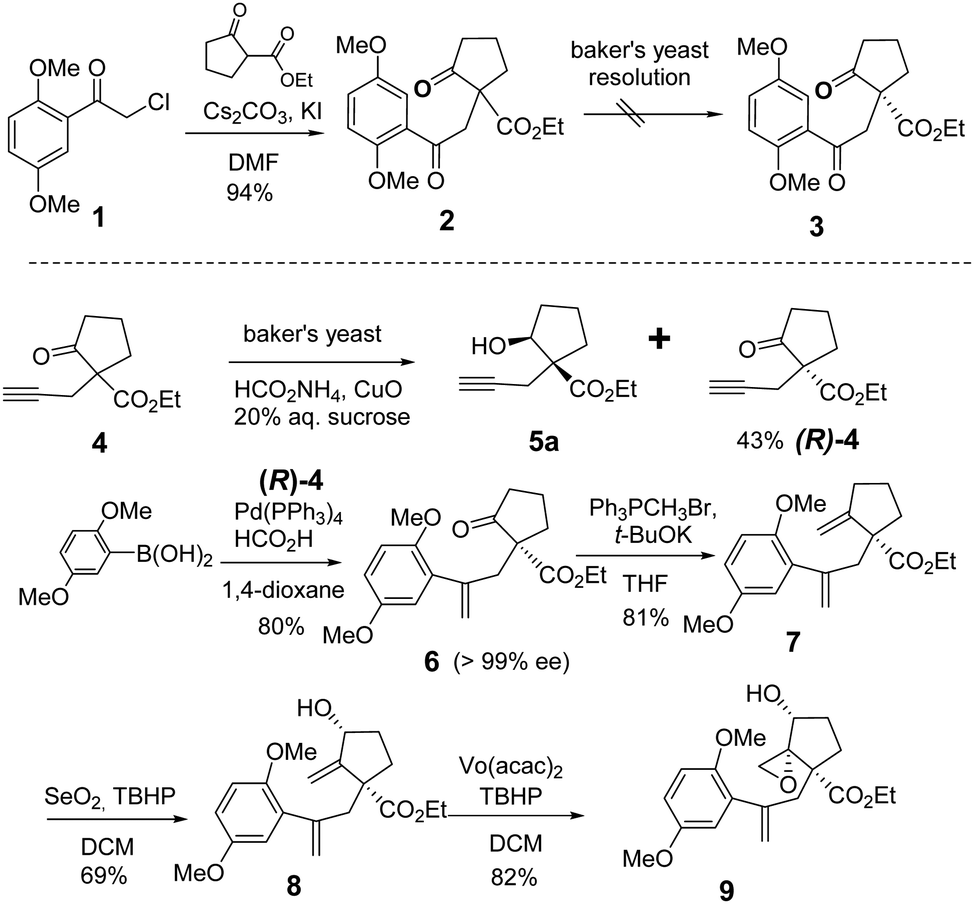 | ||
| Scheme 1 Construction of a chiral common intermediate 9. | ||
We embarked on the first asymmetric synthesis of sinensilactam A and lingzhilactone B because their enantiomers have different bioactivities.3 To accomplish this, the double bond instead of the aryl ring should preferentially engage in nucleophilic attack (Scheme 2). A similar aryl-epoxide opening has previously been reported, and it was found that the aryl attack occurs from the opposite face of epoxide.6,10 In our case, the space-demanding cis-geometry of the neighboring hydroxyl group may reinforce this aryl preference. To overturn this preference, we sought a stereoelectronic approach by oxidizing the cyclopentanol to cyclopentanone. By doing so, the reactivity of the epoxide moiety is enhanced due to the electron-withdrawing effect, facilitating the less-nucleophilic alkene-attack. In addition, the flattened cyclopentanone ring would disfavor the aryl attack as a result of steric repulsion between the approaching aryl methoxy group and the ketone moiety.
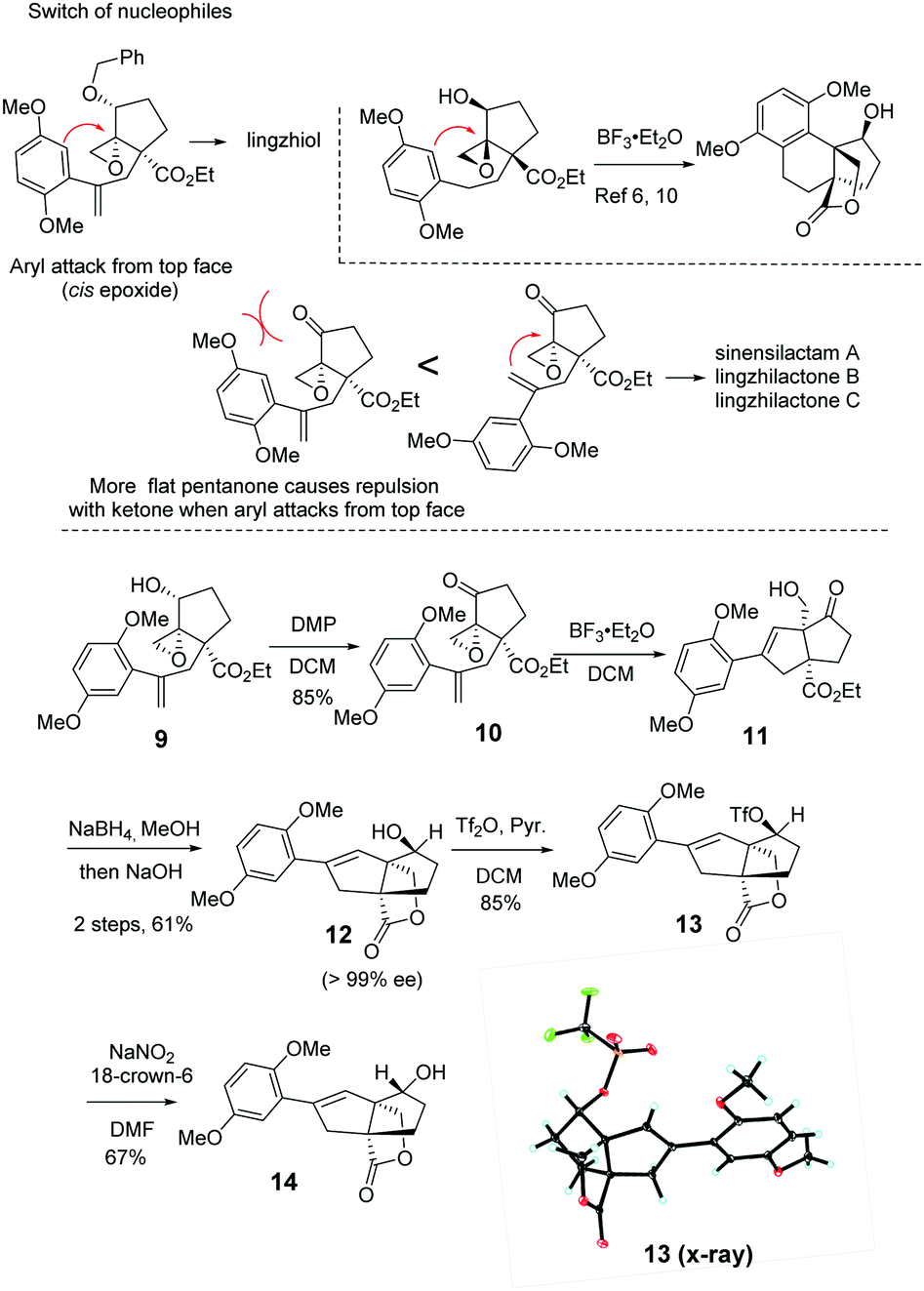 | ||
| Scheme 2 Construction of bicyclic styrene 12. | ||
To our delight, after oxidation into ketone with DMP, the desired styrene-addition adduct 11 was obtained when treated with BF3·Et2O. Lactone 12 was obtained through the reduction of ketone with NaBH4, followed by lactonization upon treatment with NaOH in MeOH. The hydroxyl group was trans to the lactone due to the directing effect of the adjacent hydroxyl group (Scheme 2, 11) during reduction. Considering the relative configurations of sinensilactam A and lingzhilactone B, the inversion of the configuration of the hydroxyl group is needed. Our extensive examination of the commonly used Mitsunobu conditions was futile,19–23 and no inversion occurred. Eventually, when compound 12 was treated with Tf2O and pyridine, triflate 13 was converted into cis isomer 14 in the presence of NaNO2 and 18-crown-6.24 The structure of 13 was unambiguously determined by single-crystal X-ray diffraction analysis.
From styrene lactone 14, the total synthesis was achieved following Yang's procedure (Scheme 3). Dihydroxylation and the subsequent Pb(OAc)4-mediated cleavage of the resulting diol proceeded smoothly to afford the key intermediate 15, which was then deprotected with BBr3 to complete the total synthesis of (+)-lingzhilactone B. The further protection of aldehyde into acetal gave (+)-lingzhilactone C. From ketoaldehyde 15, we tried to utilize 5-ethoxy-2-pyrrolidinone, instead of 5-hydroxy-2-pyrrolidinone, in order to improve the yield. As is well-known, the in situ generation of an N-acyliminium intermediate from 5-ethoxy-2-pyrrolidinone proceeded much faster at lower temperature (1 h vs. 5 h, 0 °C vs. 40 °C). Unfortunately, we encountered serious issues during purification as its Rf value was the same as that of the product. Finally, the total synthesis of (+)-sinensilactam A was achieved by the condensation of 15 with 5-hydroxy-2-pyrrolidinone (66% yield) and subsequent demethylation with BBr3 (52% yield). The synthetic samples have rotations of +50.3 (c 0.07, MeOH) [literature value: +35.7 (c 0.11, MeOH)]3 for (+)-sinensilactam A, +154.8 (c 0.08, acetone) for (+)-lingzhilactone B and +114.3 (c 0.06, DMSO) for (+)-lingzhilactone C.
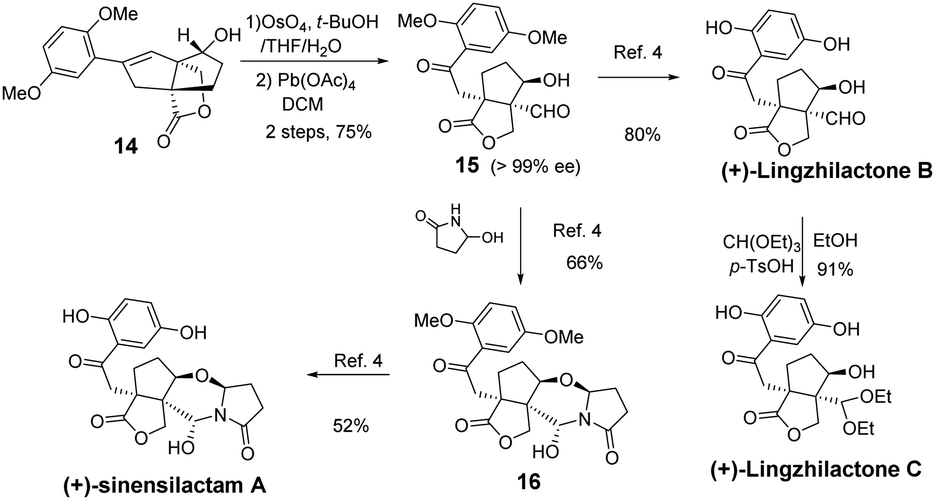 | ||
| Scheme 3 Completion of the synthesis of (+)-sinensilactam A. | ||
Next, the total synthesis of lingzhiol was investigated from epoxy styrene 9 (Scheme 4). The hydroxyl group of 9 was protected as benzyl ether 17 in 75% yield, with the purpose of preventing the undesired semipinacol rearrangement under Lewis acid catalysis.25 Benzyl ether 17 underwent an aryl-epoxide opening reaction to give the expected tetracyclic styrene 18 by TMSOTf catalysis.6 An initial experiment with BF3·Et2O gave the desired product in 40% isolated yield. After further screening the Lewis acid and temperature, the optimal 51% isolated yield was obtained with 2.5 equiv. TMSOTf in DCM at 0 °C. Under the same conditions, BF3·Et2O afforded the desired product in 45% yield. No alkene-attack product was observed in these cases. Routine OsO4-catalyzed dihydroxylation and subsequent oxidative cleavage of diol with NaIO4 afforded ketoester 19 in 74% yield. In this way, we took a detour of benzylic oxidation, which was usually not effective.8 Finally, the global deprotection of dimethoxy and benzyloxy groups furnished (−)-lingzhiol in 52% yield. Our synthesis took 10 steps and the overall yield was 2.3%.
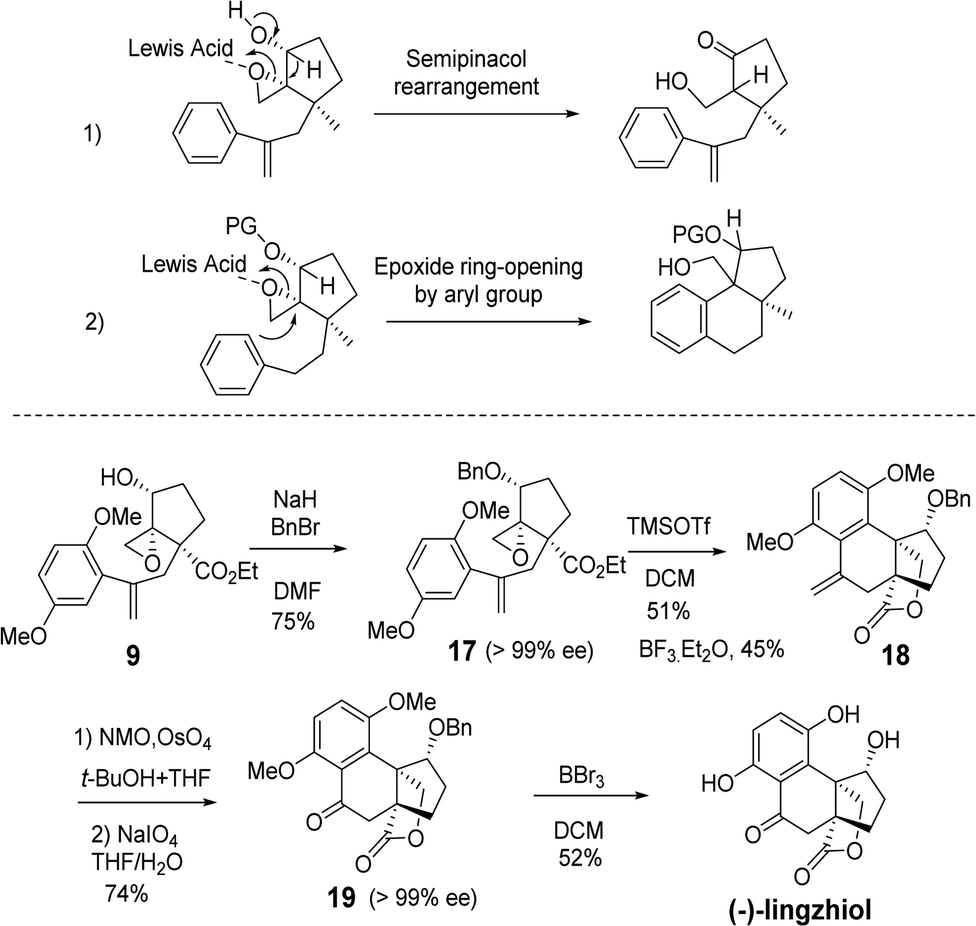 | ||
| Scheme 4 Total synthesis of (−)-lingzhiol. | ||
The synthetic strategy is straightforward and the conditions are mild. The synthetic sample has a rotation of −60.7 (c 0.12, MeOH) [literature value: −94.2 (c 0.36, MeOH)].1 Our NMR data of the synthetic samples are all in agreement with those of the literature.
To demonstrate the utility of our synthetic strategy, the (+)-sinensilactam A isomer and (+)-epi-lingzhilactone B, which are C6′ epimers of natural products, were also prepared following similar operations (Scheme 5). From styrene 12, a similar transformation of double bonds afforded 20 in 78% yield. It was then demethylated to give (+)-epi-lingzhilactone B in good yield (81%). When similar operations of pyrrolidinone installation and demethylation were performed, the (+)-sinensilactam A isomer was isolated in a comparable yield. The structure of 21 was confirmed by single-crystal X-ray diffraction analysis.
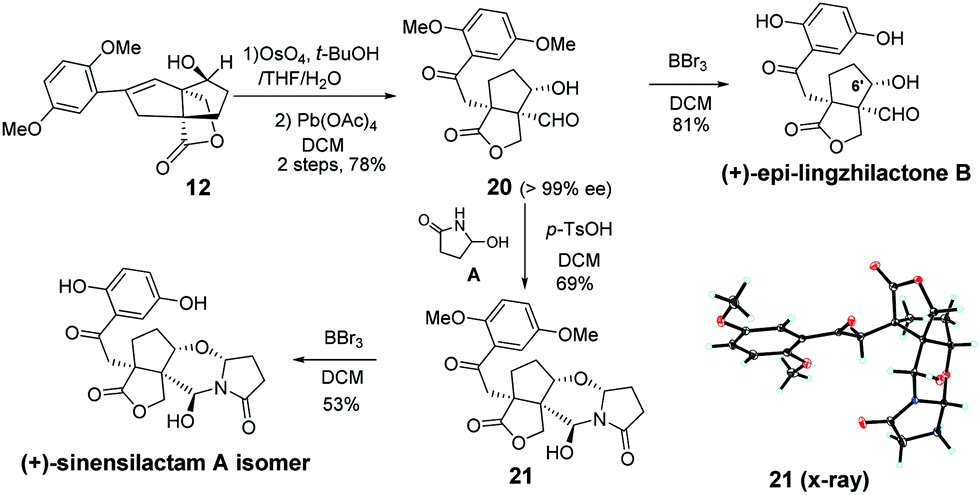 | ||
| Scheme 5 Synthesis of the two isomers. | ||
Finally, we sought an alternative asymmetric synthesis of the key chiral intermediate 3 using the state-of-the-art enantioselective photocatalytic process. Based on our previously developed chiral primary amine catalyzed photolytic alkylation reaction,23 the catalytic asymmetric synthesis of the chiral quaternary diketoester 3, which can be readily transformed into 7, was investigated (Scheme 6). Under the optimal conditions,26 up to 99% ee was realized with 71% isolated yield at 100 mg scale. It can be transformed into key diene intermediate 7 under similar Wittig reaction conditions, possibly shortening the whole synthesis by two steps.
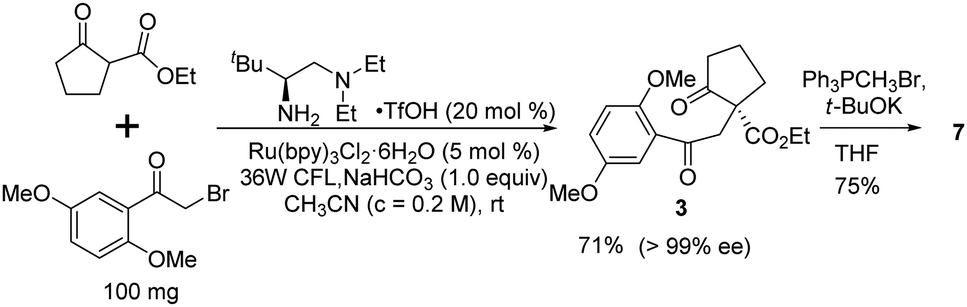 | ||
| Scheme 6 Catalytic asymmetric construction of the quaternary stereocenter. | ||
In summary, we have developed a collective strategy for the concise synthesis of four meroterpenoids based on the divergent reactivity of styrene, which is significantly different from the widely used semipinacol rearrangement strategy in the syntheses of similar meroterpenoids. A regioselective epoxy ring-opening reaction with the double bond of styrene provided the key lactone styrene 12, leading to the total synthesis of (+)-sinensilactam A for the first time in 14 steps and 1.1% overall yield. Starting with the same intermediate, (+)-lingzhilactone B was obtained in 13 steps and 2.7% overall yield. On the other hand, the key reaction to (−)-lingzhiol features a one-step construction of a 5/5/6/6 ring system by simultaneous epoxy ring-opening and lactonization promoted by TMSOTf in 10 steps with an overall yield of 2.3%. The current strategy provides a detour from the problematic benzylic oxidation in the total synthesis of this series of natural products. Finally, a catalytic asymmetric synthesis of the key chiral intermediate 3 was also achieved by using the established chiral primary aminocatalytic protocol. It should be noted that the construction of all-carbon quaternary centers is quite efficient under rather mild conditions, and may have further applications in total synthesis.
This work was financially supported by the High End Talent Program of Yunnan Province to Dr H.-B. Q. (2015HA028), the NSFC (21372229, 21861132003 and 21672217), the State Key Laboratory of Phytochemistry and Plant Resources in West China (Y83802A), and the Kunming Institute of Botany, CAS.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y.-M. Yan, J. Ai, L. L. Zhou, A. C. K. Chung, R. Li, J. Nie, P. Fang, X.-L. Wang, J. Luo, Q. Hu, F.-F. Hou and Y.-X. Cheng, Org. Lett., 2013, 15, 5488–5491 CrossRef CAS PubMed.
- Y.-M. Yan, X.-L. Wang, L.-L. Zhou, F.-J. Zhou, R. Li, Y. Tian, Z.-L. Zuo, P. Fang, A. C. K. Chung and F.-F. Hou, J. Ethnopharmacol., 2015, 176, 385–393 CrossRef CAS PubMed.
- Q. Luo, L. Tian, L. Di, Y.-M. Yan, X.-Y. Wei, X.-F. Wang and Y.-X. Cheng, Org. Lett., 2015, 17, 1565–1568 CrossRef CAS PubMed.
- W. Shao, J. Huang, K. Guo, J. Gong and Z. Yang, Org. Lett., 2018, 20, 1857–1860 CrossRef CAS PubMed.
- R. Long, J. Huang, W. Shao, S. Liu, Y. Lan, J. Gong and Z. Yang, Nat. Commun., 2014, 5, 5707 CrossRef CAS PubMed.
- D. Chen, H.-M. Liu, M.-M. Li, Y.-M. Yan, W.-D. Xu, X.-N. Li, Y.-X. Cheng and H.-B. Qin, Chem. Commun., 2015, 51, 14594–14596 RSC.
- D. Chen, W. D. Xu, H. M. Liu, M. M. Li, Y. M. Yan, X. N. Li, Y. Li, Y. X. Cheng and H. B. Qin, Chem. Commun., 2016, 52, 8561–8564 RSC.
- K. Sharmah Gautam and V. B. Birman, Org. Lett., 2016, 18, 1499–1501 CrossRef CAS PubMed.
- L.-M. Mehl and M. E. Maier, J. Org. Chem., 2017, 82, 9844–9850 CrossRef CAS PubMed.
- R. Rengarasu and M. E. Maier, Asian J. Org. Chem., 2017, 6, 108–117 CrossRef CAS.
- X. Li, X. Liu, X. Jiao, H. Yang, Y. Yao and P. Xie, Org. Lett., 2016, 18, 1944–1946 CrossRef CAS PubMed.
- D. Chen, X.-M. Li, H.-M. Liu, M.-M. Li, Y.-X. Cheng and H.-B. Qin, Tetrahedron Lett., 2016, 57, 2877–2879 CrossRef CAS.
- Y. Ikeda, H. Yorimitsu, H. Shinokubo and K. Oshima, Adv. Synth. Catal., 2004, 346, 1631–1634 CrossRef CAS.
- G. P. Cerai and B. Morandi, Chem. Commun., 2016, 52, 9769–9772 RSC.
- S. H. Teng, M. E. Tessensohn, R. D. Webster and J. S. Zhou, ACS Catal., 2018, 8, 7439–7444 CrossRef CAS.
- C. H. Oh, H. H. Jung, K. S. Kim and N. Kim, Angew. Chem., Int. Ed., 2003, 42, 805–808 CrossRef CAS PubMed.
- R. Rengarasu and M. E. Maier, Asian J. Org. Chem., 2017, 6, 108–117 CrossRef CAS.
- K. B. Sharpless and R. C. Michaelson, J. Am. Chem. Soc., 1973, 95, 6136–6137 CrossRef CAS.
- P. O'Brien, C. M. Rosser and D. Caine, Tetrahedron Lett., 2003, 44, 6613–6615 CrossRef.
- F. Mazraati Tajabadi, R. H. Pouwer, M. Liu, Y. Dashti, M. R. Campitelli, M. Murtaza, G. D. Mellick, S. A. Wood, I. D. Jenkins and R. J. Quinn, J. Med. Chem., 2018, 61, 6609–6628 CrossRef CAS PubMed.
- J. A. Davy, J. W. Mason, B. Moreau and J. E. Wulff, J. Org. Chem., 2012, 77, 6332–6339 CrossRef CAS PubMed.
- S.-Z. Jiang, T. Lei, K. Wei and Y.-R. Yang, Org. Lett., 2014, 16, 5612–5615 CrossRef CAS PubMed.
- P. Yates, R. S. Grewal, P. C. Hayes and J. F. Sawyer, Can. J. Chem., 1988, 66, 2805–2815 CrossRef CAS.
- A. Nakazato, K. Sakagami, A. Yasuhara, H. Ohta, R. Yoshikawa, M. Itoh, M. Nakamura and S. Chaki, J. Med. Chem., 2004, 47, 4570–4587 CrossRef CAS PubMed.
- Z. L. Song, C. A. Fan and Y. Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed.
- Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed.
- P. S. Riehl, A. D. Richardson, T. Sakamoto and C. S. Schindler, Org. Lett., 2020, 22, 290–294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1888466 and 1888467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04064a |
| This journal is © The Royal Society of Chemistry 2020 |