
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Balancing charge-transfer strength and triplet states for deep-blue thermally activated delayed fluorescence with an unconventional electron rich dibenzothiophene acceptor†
Rongjuan
Huang‡
a,
Nadzeya A.
Kukhta‡
 b,
Jonathan S.
Ward
b,
Andrew
Danos
a,
Andrei S.
Batsanov
b,
Martin R.
Bryce
*b and
Fernando B.
Dias
*a
b,
Jonathan S.
Ward
b,
Andrew
Danos
a,
Andrei S.
Batsanov
b,
Martin R.
Bryce
*b and
Fernando B.
Dias
*a
aDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: f.m.b.dias@durham.ac.uk
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 14th October 2019
Abstract
Manipulation of the emission properties of deep-blue emitters exhibiting thermally activated delayed fluorescence (TADF) through molecular design is challenging. We present an effective strategy to probe deeper into the role of localized (LE) and charge transfer (CT) states in the reverse intersystem crossing (RISC) mechanism. In a series of donor–acceptor–donor (D–A–D) blue emitters the dibenzothiophene functionality is used as an unconventional acceptor, while derivatives of 9,10-dihydro-9,9-dimethylacridine are used as electron-donors. tert-Butyl and methoxy substituents in the para-positions of the donor greatly enhance the donor strength, which allows exploration of different energy alignments among CT and LE triplet states. In the tert-butyl substituted compound the low energy triplet is localized on the acceptor unit, with the RISC mechanism (kRISC = 0.17 × 105 s−1) likely involving the mixture of CT and LE triplet states that are separated by less than 0.09 eV. An optimized organic light-emitting diode (OLED) based on the tBu-compound presents a maximum external quantum efficiency of 10.5% and deep-blue emission with Commission Internationale de l'Eclairage coordinates of (0.133, 0.129). However, when methoxy substituents are used, the low-energy triplet state moves away from the emissive 1CT singlet increasing the energy gap to 0.24 eV. Despite a larger ΔEST, a faster RISC rate (kRISC = 2.28 × 105 s−1) is observed due to the upper-state RISC occurring from the high-energy triplet state localized on the D (or A) units. This work shows the importance of fine-tuning the electronic interactions of the donor and acceptor units to control the TADF mechanism and achieve a deep-blue TADF OLED.
Introduction
Organic molecules with emission in the visible and near-infrared regions have attracted significant interest, owing to their application in organic light-emitting diodes (OLEDs).1–5 The potential to fabricate flexible, thin, low-weight light-emitting devices, at a fraction of the cost of existing inorganic-based LEDs is highly desirable. OLEDs are capable of generating electroluminescence with high efficiency and colour quality, with fast response. Therefore OLEDs have the potential to revolutionise the display and lighting markets, including the development of wearable technologies for sensing and medical treatment.6–12 For these applications to advance further, it is necessary to develop materials that emit light with high efficiency in the blue, green and red regions of the visible spectrum. Materials must also show enhanced stability, particularly under electrical excitation.13–16Simple fluorescent emitters were the first widely investigated materials for application in OLEDs.6,16,17 Due to spin statistics,18,19 the external quantum efficiency (EQE) in pure fluorescent OLEDs is limited to 5%, since only the 25% singlet excitons created under electrical excitation contribute to the device electroluminescence. Phosphorescent emitters using heavy-metals were introduced later and offer much improved EQE and brightness.20–22 Despite this improvement, the scarcity of noble metals such as iridium and platinum, and degradation issues (especially for emitters in the blue region) is a significant problem.23,24 Additional environmental problems that arise from heavy-metal processing following product disposal make the utilization of phosphorescent OLEDs less than ideal.25
The third generation of OLED emitters use all-organic molecules that exhibit thermally activated delayed fluorescence (TADF).26 This photophysical mechanism was originally demonstrated in Eosin in 1961,27 and was applied in efficient OLEDs by Adachi and co-workers in 2012.28 TADF materials provide a promising alternative to phosphors in achieving high device efficiency without using heavy-metals, and show great promise in terms of the cost-efficiency, brightness and external quantum efficiency (EQE).
Unlike conventional fluorescent and phosphorescent emitters, TADF compounds can utilize up to 100% of excitons via a reverse intersystem crossing (RISC) mechanism that is able to promote the low-energy triplet states (T1) up to the singlet excited state manifold (Sn).29,30 This process requires a small singlet–triplet energy splitting (ΔEST), which can be achieved in donor–acceptor molecules where the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have minimal overlap. In such TADF molecules, the transition between the ground and excited states (promoted by photon absorption or electrical excitation) invokes a significant alteration to the electronic density and dipole moment. Excited states with such characteristics are known as charge-transfer states (CT) and involve minimal electronic exchange interactions which result in very small ΔEST.
Ideal TADF emitters with high RISC rates should also have a fast 1CT radiative rate constant to promote strong luminescence and a short excited state lifetime, which is highly beneficial to device stability.31,32 Rigid chromophores that ensure effective control of the dihedral angle between the D and A units, while still allowing dynamic rocking about the D–A bridge, are preferred to obtain fast RISC rates and minimize non-radiative internal conversion.33,34 However, a trade-off exists between the luminescence and the triplet harvesting efficiencies. Maximizing the RISC rate results in TADF molecules designed with excited states that have strong CT character, which is detrimental to radiative decay rates. Nonetheless, TADF molecules can in principle be designed with sufficiently fast ISC/RISC rates and even faster singlet radiative decay. Unfortunately, no clear rules currently exist to guide molecular design of high efficiency TADF emitters with sufficiently fast radiative decay to avoid the build-up of triplet states and non-radiative channels.35
A multitude of various colour TADF emitters have been reported to date.36–42 The majority are composed of well-decoupled D and A units, which create excited states with strong CT character. This molecular design works remarkably well for the preparation of red, green and even sky-blue TADF emitters.29,37,38,43–45 However, developing design rules to effectively match D and A units in order to maximize TADF emission and simultaneously achieve fast radiative decay rates remains challenging, especially in the deep-blue region most desirable for applications.36,40,46,47 A small ΔEST requires careful consideration and matching of electron acceptor and donor strengths, but emission with strong CT character is almost inevitably shifted to longer wavelengths and shows slow radiative decay rate. In contrast, with weaker electron acceptor or donor units, the CT excited states are created with more blue emission and faster radiative rates, but a larger ΔEST is created due to a larger HOMO–LUMO overlap and a slower RISC rate is obtained. This results in longer-lived triplet excited states that are more susceptible to suffer from luminescence quenching, leading to pronounced efficiency roll-off and potential device instability.
Despite the current reasonable understanding of the RISC mechanism, many aspects are still unclear. In particular, the role of localized triplet and charge transfer states is not comprehensively understood. In the present work, a new strategy to design an efficient deep-blue TADF emitter is presented. The new emitter (DAc-DBT) has the unconventional dibenzothiophene (DBT) unit as a weak electron acceptor and 9,10-dihydro-9,9-dimethylacridine (Ac) derivatives as the donor units in a D–A–D configuration. The motivation for the present work was to exploit the high energy triplet levels of both DBT and Ac. The strategy was to align both the localized triplet states of the donor and acceptor (3LED and 3LEA respectively) and the singlet 1CT emissive state of the DAc-DBT molecule, to generate deep-blue TADF with high efficiency. DBT is renowned for its rigid planar structure, narrow emission spectrum, and strong intersystem crossing properties due to the sulfur atom.48 DBT is thus expected to enhance ISC/RISC rates. Moreover, the DBT fragment was utilized previously as an electron acceptor in a series of carbazole-based host materials,49 but we are not aware of its previous use as an acceptor in TADF emitter materials. As DBT and Ac units possess similar triplet levels, both 3LED and 3LEA are expected to participate in the RISC mechanism for highly efficient triplet-harvesting. The fine-tuning of the donor strength by substitution on the Ac units allows modulation of the CT energy to probe varying energy alignments between LE and CT states. Investigations into how the different states contribute to RISC are presented.
Three DAc-DBT derivatives are discussed. The analogue of the parent DAc-DBT molecule substituted with tert-butyl groups, DtBuAc-DBT, shows ΔEST of 0.09 eV compared to 0.20 eV in DAc-DBT in zeonex. When the strength of the CT character is further enhanced, by replacing the tert-butyl with methoxy groups in DOMeAc-DBT, the 3CT state is shifted below the local triplet states. ΔEST then increases to 0.24 eV in DOMeAc-DBT. Weaker TADF is observed in DOMeAc-DBT, compared with DtBuAc-DBT, but surprisingly with a faster TADF decay (10.5 μs), compared to 136.4 μs in DtBuAc-DBT. As the TADF decays with the triplet state lifetime, and the rate of non-radiative processes (IC and ISC) is faster in DtBuAc-DBT than in DOMeAc-DBT, the observation of a faster TADF decay in DOMeAc-DBT is attributed to a faster RISC rate. However, this raises a puzzling question: How can a faster RISC rate be compatible with a larger ΔEST and observation of weaker TADF contribution? In this manuscript we unravel how upper triplet–singlet state crossing and self-quenching of the triplet population by a low energy triplet supports this scenario. To the best of our knowledge an acceleration of the RISC rate when ΔEST increases has not been reported and interpreted in detail before. Moreover, the deep-blue OLED derived from DtBuAc-DBT with a smaller ΔEST achieved a maximum EQE of 10.5% and EQE of 9.8% at the practical brightness of 100 cd m−2, with narrow emission spectrum. This device also shows Commission Internationale de l'Eclairage (CIE) coordinates of (0.133, 0.129).
Results and discussion
Molecular design and synthesis
The molecular structures of the DAc-DBT derivatives studied in this work are presented in Fig. 1. The design of the DAc-DBT derivatives is inspired by a previously synthesized molecule (DPTZ-DBT), which used DBT as the electron acceptor and phenothiazine (PTZ) as the electron donors to promote efficient room-temperature phosphorescence (RTP).48 The fast ISC rate50 and high triplet level48 of DBT aligned with the singlet 1CT state of DPTZ-DBT, efficiently populates a lower triplet state localized on the PTZ unit. The low triplet level of PTZ resulted in an inefficient RISC rate and thus TADF was not observed, instead making DPTZ-DBT an excellent RTP emitter. | ||
| Fig. 1 Chemical structures of the DAc-DBT derivatives. | ||
Using inspiration from the previous DPTZ-DBT work, it was recognised that the singlet and triplet states of the D and A constituents could be aligned together in DAc-DBT emitters. This strategy has facilitated RISC, promoted high TADF efficiency, and shifted the emission into the deep-blue region. Firstly, the low triplet level of PTZ was replaced by a high triplet of the Ac unit to achieve a small ΔEST. Secondly, the donor strength of the Ac unit was tuned by functionalisation with electron-donating tert-butyl and methoxy groups, which simultaneously protect the 2,7-positions of Ac to enhance the chemical and electrochemical stability. The 1CT state of DAc-DBT is aligned with its triplet excited states localized on the Ac and DBT units, thereby promoting fast ISC/RISC rates.
The synthetic procedures of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT utilizing palladium-catalyzed Buchwald–Hartwig chemistry51 are presented in the ESI† (Section S1). Their structures and high purity were established using 1H and 13C{1H} NMR spectroscopy, mass spectrometry, elemental analysis and single-crystal X-ray crystallography.
The X-ray molecular structures of DAc-DBT and DtBuAc-DBT are presented in Fig. 2 and Fig. S1–S3 (ESI†). Due to the possible disorder of the methoxy substituents, growth of crystals suitable for X-ray crystallography using DOMeAc-DBT was not possible. The DBT unit is planar in DAc-DBT and slightly twisted (by 5° between the arene rings) in DtBuAc-DBT. The dihydroacridine moieties are practically perpendicular to the DBT (dihedral angles ranging from 83 to 90°) and show variable folding angles (θ) along the N⋯C(9) vector. Above 170 K, the DAc-DBT·CD2Cl2 solvate has a single molecule per asymmetric unit; one dihydroacridine has θ = 30.5° and the other is disordered (57 to 43%) between two conformations with θ = 22.3° and 10.5°, respectively. Below 170 K, the crystal structure is modulated with doubled a parameter and two ordered molecules per asymmetric unit, one showing θ = 30.8° and 25.4°, the other θ = 32.7° and 8.7°. In DtBuAc-DBT, which has essentially the same crystal structure at 200 and 120 K, the dihydroacridine folding is smaller and more symmetrical, θ averaging 15.3° at 200 K and 13.7° at 120 K. Such flexibility is in agreement with the literature: the November 2018 issue of the Cambridge Structural Database52 lists 19 structures with 26 unique 9,9-dimethyldihydroacridine moieties with a C(sp2) substituent at N, the θ varying from 1.2° to 48.3°; indeed, two independent molecules in the structure of N-(4-tert-butylphenyl)-1,8-naphthalimide-9,9-dimethyl-9,10-dihydroacridine53 show θ = 4.8° and 21.9°.
 | ||
| Fig. 2 X-ray crystal structures of DAc-DBT and DtBuAc-DBT. | ||
 | ||
| Fig. 3 Optimized molecular geometries with HOMO (red) and LUMO (black) levels and energies of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT (rBMK/6-31G(d) level of theory). | ||
Theoretical calculations
DFT calculations were carried out at the rBMK level of theory.54 The BMK functional was preferred for the geometry optimization and the prediction of the optical properties of all the studied molecules as this functional was shown to be suitable for the description of the low energy absorption band of D–A CT compounds.54–56 For additional insight, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of the D and A fragments were calculated (Fig. S4a, ESI†). While all the units possess planar and rigid molecular skeletons, the wave-function distribution and frontier orbital energies show clear differences. DBT has a shallow ELUMO (−0.29 eV) and a very deep EHOMO (−6.62 eV) suggesting that it could act as a weaker acceptor when connected to a stronger Ac donor.36 While the LUMO remains almost unchanged with the introduction of substituents on the Ac derivatives, the HOMO energy is raised from −5.59 eV (Ac) to −5.09 eV in OMe-Ac. The methoxy substituents induce a relatively larger electron density delocalization throughout the molecule. Increased electron density in OMe-Ac is consistent with the red-shifted theoretical absorption (onset at 330 nm for OMe-Ac and 300 nm for Ac, tBu-Ac and DBT) (Fig. S5, ESI†). The DFT calculations establish that OMe-Ac is the strongest electron donor of the three units. The singlet and triplet energy diagram (Fig. S4b, ESI†) indicates that tert-butyl groups on the Ac donor slightly lower the S1 level while maintaining the T1 energy close to that of the DBT acceptor. In contrast, the methoxy substituents lower the S1 energy significantly and only slightly affect the T1 energy, leading to a smaller energy-gap between S1 and T1.The overall geometry of the DAc-DBT derivatives is determined by the rigid skeleton of the DBT acceptor, to which planar Ac donors are attached almost orthogonally (Fig. 2). All the Ac donors show planar geometries in the calculated structures in contrast to the X-ray results (folded Ac units). The difference in geometry between gas phase and X-ray data is likely due to the influence of crystal packing. More importantly, the HOMO/LUMO wave functions vary significantly in the molecular series despite similar geometries. The HOMO and LUMO are effectively decoupled in all cases, consistent with the CT character of the excited state, but with the HOMO expanding slightly onto the DBT unit with the addition of electron donating substituents (Fig. 3 and Table S2, ESI†). The HOMO levels increase in the D–A–D series, with DAc-DBT (−5.65 eV) < DtBuAc-DBT (−5.47 eV) < DOMeAc-DBT (−5.14 eV), while the LUMO is localized solely on the DBT unit and remains almost unchanged. Thus, the energy of the CT states is lower in DOMeAc-DBT compared to DtBuAc-DBT.
The UV-vis absorption spectra of the DBT derivatives were calculated (Fig. S6, ESI†). The absorption profiles of DAc-DBT and DtBuAc-DBT are similar: both feature two peaks. In contrast, DOMeAc-DBT shows only a single absorption band with Gaussian shape. The onset of the DOMeAc-DBT absorption band is red-shifted (353 nm vs. 331 nm for DAc-DBT and DtBuAc-DBT, respectively), confirming stronger CT character in DOMeAc-DBT. The nature of the lowest energy band was described by means of natural transition orbitals (NTOs)57 for a selected set of pertinent excited states (S0 → S1–3) (Fig. S7, ESI†). The hole and particle are well separated in all transitions (S0 → S1–3) demonstrating the CT character of the excited states.
The calculated triplet energy of the three target compounds varies significantly. The introduction of tert-Bu groups does not significantly affect the T1 value (3.37 eV and 3.36 eV for DAc-DBT and DtBuAc-DBT, respectively), while the methoxy groups on the Ac lower the triplet energy of DOMeAc-DBT significantly to 3.18 eV. Triplet NTOs (Fig. 4a) indicate that DAc-DBT and DtBuAc-DBT have the T1 localized on the DBT acceptor, demonstrating the LE character of the triplet state. However, the triplet hole and particle of DOMeAc-DBT involve both DBT and OMe-Ac units, thus indicating a CT character for the lowest triplet state. Interestingly, the higher triplet states (T2,3) are located in a close proximity to the S1 level and possess more delocalized character (Fig. S8 and S9, ESI†). It can be clearly seen that the triplet level of DtBuAc-DBT almost coincides with the local triplets of the DBT acceptor and the tBu-Ac donor (Fig. 4b), suggesting that both units might participate in triplet harvesting. Thus, the difference in triplet energies is expected to be a significant contributor to the differences in the observed TADF efficiency.
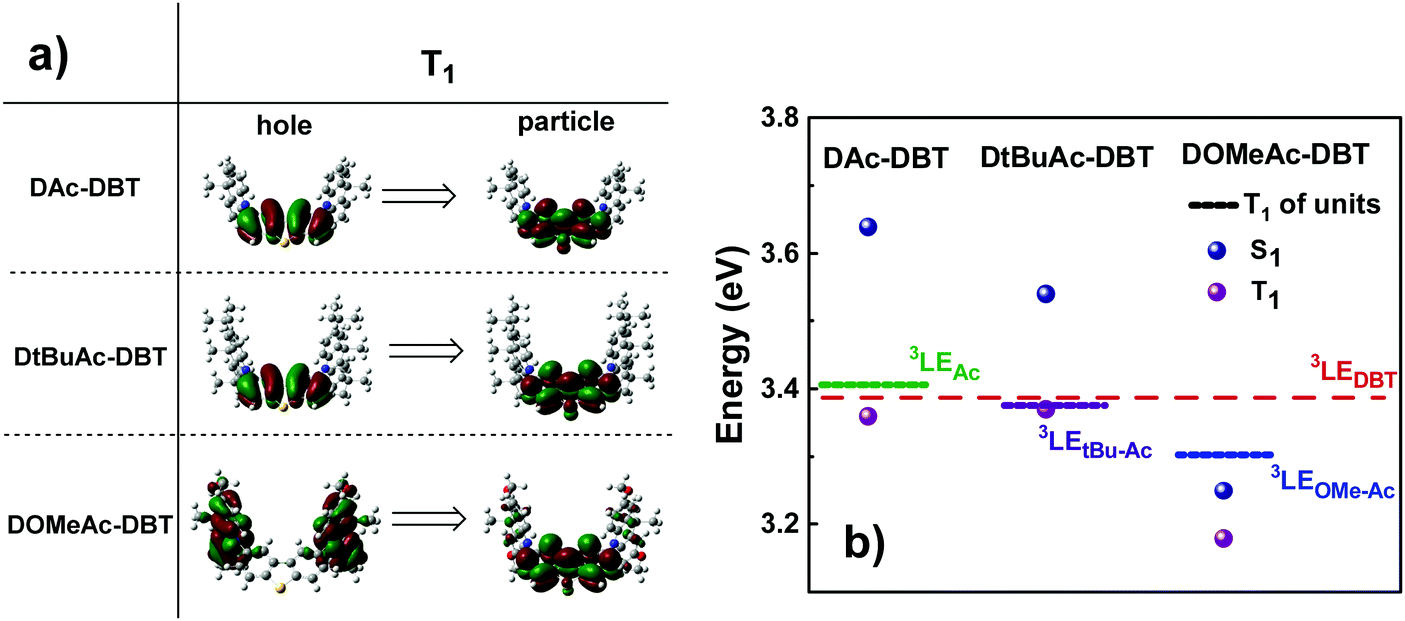 | ||
| Fig. 4 (a) Natural transition orbitals (NTO) corresponding to the first triplet transitions and (b) singlet and triplet energy diagram of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT along with the energy levels of the individual units (TD-DFT rBMK/6-31G(d)). Hole refers to the highest occupied natural transition orbital (HONTO), while particle refers to lowest unoccupied natural transition orbital (LUNTO).57 | ||
Cyclic voltammetry (CV) measurements pointed at the improvement of the electrochemical stability of the DAc-DBT derivatives with the introduction of the substituents (Section S5, ESI†). The increase of the experimentally obtained HOMO values is in line with the calculated data.
The compounds under study displayed outstanding thermal stability which was probed by thermal gravimetric analysis (TGA) (Section S6, ESI†). The decomposition temperature was found to increase in the sequence DAc-DBT < DOMe-DBT < DtBuAc-DBT.
Optical and photophysical characterization
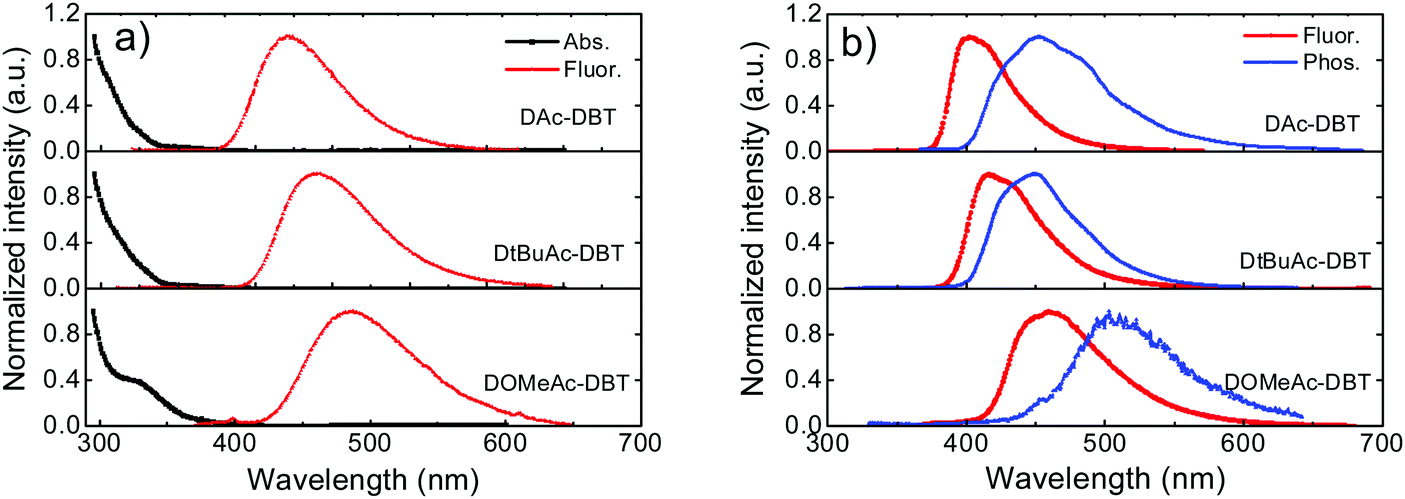 | ||
| Fig. 5 (a) UV-vis absorption and fluorescence spectra of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT in toluene solutions (1.0 × 10−4 M) measured at room temperature. (b) Steady-state fluorescence collected at room temperature and time-resolved phosphorescence spectra collected with delay time of 56 ms at 80 K for the three molecules in zeonex films (1 wt%). Excitation at 355 nm. | ||
As shown in Fig. 5b, the fluorescence spectra of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT observed in zeonex films show very similar spectral shapes as in non-polar methylcyclohexane (Fig. S14, ESI†). This indicates that excimers or aggregates are unlikely to be formed in the solid state at low concentrations in steady-state conditions. With the increasing donor strength, DAc-DBT, DtBuAc-DBT and DOMeAc-DBT films show red-shifted emission, with the peak emission wavelength remaining in the blue region (398 nm, 414 nm and 460 nm, respectively). The 1CT energies calculated from the high-energy onsets of the emission spectra are 3.28 eV, 3.17 eV and 3.00 eV, for DAc-DBT, DtBuAc-DBT and DOMeAc-DBT, respectively.
The phosphorescence spectra of the three molecules in zeonex films at 80 K are shown in Fig. 5b. DAc-DBT and DtBuAc-DBT show well-resolved phosphorescence spectra with clear vibronic structure, indicating the localized character of their triplet excited states (3LE). The triplet energies of DAc-DBT and DtBuAc-DBT in zeonex were determined from the onsets of their phosphorescence spectra to be 3.08 eV, both isoenergetic with the triplet of the DBT unit. The phosphorescence spectrum of DtBuAc-DBT shows contributions from both the donor tBu-Ac and acceptor DBT units (Fig. S15, ESI†). Thus, the tert-butyl-substituted molecule DtBuAc-DBT represents an ideal case with 3D and 3A at essentially identical energies.
In contrast, the introduction of the methoxy substituents significantly enhances the donor strength and efficiently increases the HOMO energy in DOMeAc-DBT. A rather broad and unstructured phosphorescence spectrum is observed, indicating that the lowest energy T1 state can be identified as a 3CT state. The CT character is further confirmed by the solvatochromism of the DOMeAc-DBT phosphorescence spectra in hosts of different polarity (Fig. S17, ESI†). This is also in agreement with the analysis of the triplet NTOs, which show donor-localized hole and acceptor-localized particle for the S0 → T1 excitation. Consequently, the low energy singlet and triplet states in DOMeAc-DBT are both of CT character. The phosphorescence of DOMeAc-DBT appears at a lower energy (2.76 eV) compared to DAc-DBT and DtBuAc-DBT and is no longer isoenergetic with either D or A unit (Fig. S16, ESI†). Therefore, it is very interesting to evaluate the effect of the triplet character on the efficiency of the RISC mechanism. A schematic energy diagram of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT based on all the reported data is presented in Fig. 6.
 | ||
| Fig. 6 A schematic energy diagram of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT. All energies are given in eV and were calculated from the onsets of the corresponding spectra in zeonex films, compared with their individual D and A moieties. No indication of 3CT states is obtained in DAc-DBT and DtBuAc-DBT from calculations, neither are they observed in our experiments, therefore, their 3CT states are not represented in this diagram. | ||
As oxygen is a well-known triplet quencher, delayed fluorescence (DF) appearing as a result of triplet harvesting is not able to contribute to the overall emission when oxygen is present.60 The effect of oxygen-quenching on the PL spectra of DAc-DBT, DtBuAc-DBT and DOMeAc-DBT was thus analysed in toluene solution and zeonex films to quantitatively determine the contribution of DF to the total emission (Fig. S18, ESI†). A larger DF contribution is observed in DtBuAc-DBT and can be easily explained. The introduction of tert-butyl groups increases the donor strength and shifts the 1CT state to a lower energy, narrowing the energy gap (ΔEST) to 0.09 eV in DtBuAc-DBT in comparison to DAc-DBT (0.20 eV). Therefore, the alignment of singlet and triplet excited states in DtBuAc-DBT gives rise to an efficient RISC rate for a higher DF contribution. The high triplet contribution and deep-blue emission in DtBuAc-DBT makes it a very promising deep-blue TADF emitter.
The methoxy groups on DOMeAc-DBT were introduced to red-shift and tune the 1CT state closer to the two localized triplet states. However, the donating ability of the two methoxy groups is too strong and the 3CT drops even lower in energy, which decreases the DF contribution. This leads to a relatively small increase of the PL quantum yields (PLQYs) in DOMeAc-DBT zeonex film from 35% in air to 48% in nitrogen, compared to DtBuAc-DBT with well-aligned energy states (20% in air and 54% in nitrogen). Calculations also show that a further increase of donor strength induced by the presence of the methoxy groups leads to an expanded HOMO distribution, resulting in a relatively large overlap between the frontier orbitals. This is consistent with the experimental results, which show a large ΔEST of 0.24 eV. This in turn explains the observation of a higher PLQY in air (35% in DOMeAc-DBTvs. 20% in DtBuAc-DBT), the reduced formation of triplet states, and less DF contribution to the overall emission. However, the PLQYs of DAc-DBT are very low (1% in air), as it is unstable as shown by time-resolved measurements.
Time-dependent spectroscopy
To further investigate the photophysical properties, time-resolved emission decays were collected. DAc-DBT was also measured but found to undergo irreversible laser induced breakdown over the timescale of the measurements, which is confirmed by the redshifted prompt emission appearing at a very early delay time (Fig. S19, ESI†). The instability in DAc-DBT is also evidenced by the extra CV feature of a new oxidation peak (Fig. S10, ESI†) and the lowest TGA degradation temperature (Fig. S11, ESI†) compared with the substituted molecules. As the emission decays very rapidly (in the nanosecond region), we tentatively suggest that this may be due to the dimerization by intermolecular π–π stacking, which occurs even in zeonex at low concentrations but is too weak to be detected in steady-state emission (Fig. S20a, ESI†). Therefore, the DF spectra and decay lifetimes of DAc-DBT are unreliable and are not included. Such shifts were not observed for the substituted compounds of the series (Fig. S21 (ESI†) and Fig. 8a), suggesting that blocking the 2,7-positions of acridine can help to prevent possible aggregation.The time-resolved emission decays of DtBuAc-DBT and DOMeAc-DBT zeonex films are shown in Fig. 7. The nanosecond timescale prompt fluorescence (PF) decay component shows no significant change with temperature, as expected. In contrast, the microsecond DF and millisecond phosphorescence (PH) components show clear temperature dependence. The DF intensity increases with increasing temperature, indicating a TADF mechanism in both DtBuAc-DBT and DOMeAc-DBT. The strictly linear power dependence on the excitation dose also confirms this scenario (Fig. S22, ESI†). The PH component shows the opposite dependence on temperature, also as expected since non-radiative processes dominate at higher temperatures in this time region. The PF component shows a shorter fluorescence lifetime in DtBuAc-DBT (5.9 ns), compared with that of DOMeAc-DBT (9.6 ns). The radiative rate (kf) and non-radiative rate (knr) including both internal conversion (kIC) and intersystem crossing (kISC), are determined from the PLQY measurements in the presence of oxygen and from the PF lifetime61 (see Table 1). For DtBuAc-DBT, kf and knr are 3.4 × 107 s−1 and 14 × 107 s−1, respectively, indicating that in DtBuAc-DBT the triplet formation rate effectively competes with the radiative rate, thus favouring ISC and RISC processes. Time-resolved area normalized spectra of DtBuAc-DBT in zeonex matrix at 290 K and 80 K are shown in Fig. S21 (ESI†). In both cases, the DF spectra perfectly match the PF emission (delay time at 1.2 ns), indicating an efficient triplet up-conversion to the lowest singlet state. At 80 K, no delayed fluorescence is observed, with only PH occurring from the 3LEA state.
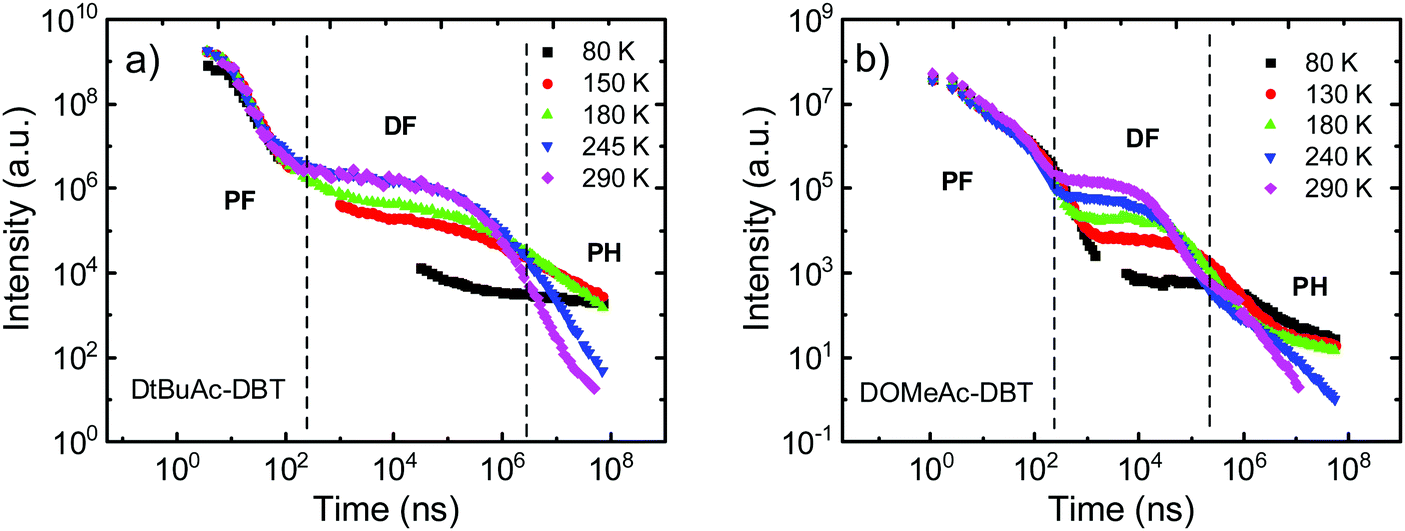 | ||
| Fig. 7 Temperature dependent time-resolved decays of (a) DtBuAc-DBT and (b) DOMeAc-DBT in zeonex films (1 wt%) collected at various temperatures from 290 K to 80 K. | ||
| Compound | λ em (nm) | S1/T1b (eV) | ΔESTc (eV) | Φ f/ΦPLd (±5%) | τ f (ns) | τ DF (μs) | k f (×107 s−1) | k nr (×107 s−1) | k RISC (×105 s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a The peak of photoluminescence at room temperature. b S1 and T1 energies calculated from the onsets of the fluorescence (290 K) and phosphorescence spectra (500 ms at 80 K), respectively. c Experimentally determined singlet–triplet energy splitting (ΔEST). d PLQY measured in air (Φf) and in nitrogen (ΦPL) atmosphere at room temperature. e Lifetimes of PF and DF components fitting from the time-resolved decays at room temperature. f Calculated rate constants of fluorescence (kf), non-radiative decay (knr) including intersystem crossing (kISC) and internal conversion (kIC), and reverse intersystem crossing (kRISC). | |||||||||
| DAc-DBT | 398 | 3.28/3.08 | 0.20 | 1/6 | — | — | — | — | — |
| DtBuAc-DBT | 414 | 3.17/3.08 | 0.09 | 20/54 | 5.9 | 136.4 | 3.4 | 14 | 0.2 |
| DOMeAc-DBT | 460 | 3.00/2.76 | 0.24 | 35/48 | 9.6 | 10.5 | 3.6 | 6.8 | 2.3 |
For DOMeAc-DBT, using the data obtained kf = 3.6 × 107 s−1, similar to the rate determined for DtBuAc-DBT. However, knr for DOMeAc-DBT is slower at 6.8 × 107 s−1. The shorter PF lifetime of DtBuAc-DBT is thus consistent with the faster ISC or IC pathways in this compound. The ISC rate (kISC) in DtBuAc-DBT can be estimated using the DF/PF ratio obtained from the comparison between PLQY measurements in nitrogen atmosphere and under air.61 Using the PLQY values given in Table 1, the ISC rate in DtBuAc-DBT is determined as kISC = 10 × 107 s−1, which is faster than the total rate of non-radiative decay for DOMeAc-DBT, further explaining the shorter fluorescence lifetime of DtBuAc-DBT and consistent with the smaller ΔEST (0.09 eV). Moreover, a faster RISC rate in DtBuAc-DBT is also expected compared to DOMeAc-DBT, as the S1–T1 gap is much larger in DOMeAc-DBT (0.24 eV).
Surprisingly, DOMeAc-DBT shows a shorter TADF lifetime τDF = 10.5 μs, than DtBuAc-DBT, τDF = 136.4 μs. As the non-radiative processes are slower in DOMeAc-DBT (knr = 6.8 × 107 s−1) than in DtBuAc-DBT (knr = 14 × 107 s−1), note also that the phosphorescence decays in the same ms time range in both compounds (Fig. 7), the faster TADF decay in DOMeAc-DBT suggests a faster RISC rate in this compound. However, the TADF contribution to the overall emission is smaller. This apparent discrepancy is puzzling and therefore warranted further study of the TADF mechanism in DOMeAc-DBT.
The 1CT and the 3LED states in DOMeAc-DBT are very well aligned, showing an energy gap of <0.01 eV (<0.08 between 1CT and 3LEA as seen in Fig. 6), while the 3CT state is at a much lower energy (ΔE1CT–3CT = 0.24 eV). RISC originating directly from the low-lying 3CT state is thus inefficient. However, any triplets occupying the 3LEA (or 3LED) states would be able to undergo down-hill RISC to 1CT proceeding very rapidly as the ΔE1CT–3LE < 0.01 eV. This ordering of excited states therefore explains why the DF lifetime is faster in DOMeAc-DBT, but the overall TADF contribution is smaller. We propose that rapid upper-state crossing RISC between 3LE and 1CT in DOMeAc-DBT competes with 3LE triplets relaxing to the low energy 3CT triplet. This IC relaxation to a non-emissive triplet state quenches TADF contribution, resulting in a smaller contribution to the total emission despite a faster RISC rate.
Crucially, the time-resolved spectra shown in Fig. 8b and c, obtained for time delays longer than 5 ms, where no TADF is observed, even at RT, give strong evidence to support the role of the high-energy triplet state in the emission process. The isoemissive point observed in Fig. 8b shows unequivocally that two different triplet states are involved in the phosphorescence emission of DOMeAc-DBT, one that perfectly matches the phosphorescence of the OMe-Ac single unit, at higher energy, and another at lower energy which we assign to the 3CT phosphorescence, in agreement with the frontier orbital distributions shown in the DFT calculations (see Fig. S8 and S9 in ESI†). At 290 K, the emission spectrum shows practically no shift over the entire time range from 1.1 ns to 11.2 ms, showing that TADF mostly dominates the long-lived emission at RT. However, at a low temperature an emission intensity increase is observed in the millisecond time region (Fig. 7b), showing that phosphorescence instead of TADF is dominant. The emission of DOMeAc-DBT is therefore composed of PF at early times (<100 ns), TADF in the microsecond range, and PH at later times (ms). Strikingly, the fluorescence and phosphorescence spectra at RT are very similar to each other, which is only possible if the PH originates from the triplet state localized on the donor unit (3LED) which is isoenergetic with the 1CT state. Consistently, the emission spectrum of DOMeAc-DBT at very-long delay times, obtained at room temperature, matches the PH of the OMe-Ac single fragment (Fig. S16, ESI†). The PH from the 3CT state is not observed at RT as this state has a much slower radiative rate, thus longer lifetime, and can be easily quenched. However, importantly, PH from both 3LE and 3CT states are observed at low temperature, as shown in Fig. 8b. At delay time of 5 ms, the PH of DOMeAc-DBT shows features from both the 3LED state localized on the OMe-Ac donor unit and the 3CT state (Fig. 8c). The 3LE phosphorescence appears at a higher energy and relaxes over time, whereas the 3CT phosphorescence appears at a lower energy and shows longer lifetime, as expected. The time-resolved spectra therefore give full support to the role of the 3LE state in mediating RISC, and assist in revealing the upper-state RISC mechanism in DOMeAc-DBT.
 | ||
| Fig. 8 DOMeAc-DBT in zeonex film: time-resolved area normalized emission spectra at (a) 290 K and (b) 80 K. (c) Phosphorescence spectra recorded at delay times of 5 ms and 500 ms, compared with the that of the donor OMe-Ac unit measured at 80 K. | ||
Devices
To evaluate the electroluminescent (EL) characteristics of DtBuAc-DBT, OLEDs were fabricated by vacuum thermal evaporation using the following device structure (Fig. 9a): ITO/NPB (40 nm)/TSBPA (10 nm)/DPEPO:x%DtBuAc-DBT (30 nm)/DPEPO (10 nm)/TPBi (40 nm)/LiF/Al, which was recently employed successfully for other similar blue TADF materials.62 Here N,N′-bis-(naphthalene-1-yl)-N,N′-bis(phenyl)benzidine (NPB), 4,4-(diphenylsilanediyl)bis(N,N-diphenylaniline) (TSBPA), and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) were employed as hole-injecting, hole-transporting and electron-transporting layers, respectively. Bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO) was chosen as a suitable host for the blue material based on its high energy triplet state. An additional 10 nm DPEPO layer was used as a hole-blocking layer. The doping concentration of DtBuAc-DBT in DPEPO was optimized at 22% after screening concentrations of 10–35%. As DPEPO has a very poor hole conductivity,63 the hole current in the emissive layer mainly depends on the hole transport of the emitter. Thus, a higher doping concentration is required for a better charge balance and efficiency. | ||
| Fig. 9 (a) EQE vs. luminance characteristics. Inset shows the device structure and energy levels. (b) The comparison of PL spectrum of DPEPO:10%DtBuAc-DBT film and EL spectra of 10% and 22% doping concentration in blue devices collected at 9 V. The inset shows the CIE coordinates of the devices. | ||
The device shows a deep-blue emission peaking at 455 nm with a full width at half maximum of 84 nm, which is almost identical with the PL spectrum of the DPEPO:10%DtBuAc-DBT film (Fig. 9b), and results in high colour purity. The spectra are also independent of the driving voltages (Fig. S23a, ESI†), revealing that the charge recombination is well confined within the emissive layer in the EL process. The current density–voltage–luminance curves are shown in Fig. S23b (ESI†). The device shows a turn-on voltage of 4.0 V, the current and power efficiencies are 13.9 cd A−1 and 9.7 lm W−1, respectively. A maximum external quantum efficiency (EQE) of 10.5% is obtained as shown in Fig. 9a, which is in agreement with the measured PLQYs assuming there is no outcoupling enhancement. Remarkably, the EQE only drops slightly (9.8%) at the brightness of 100 cd m−2. Noteworthy, the device shows CIE chromaticity coordinates of (0.133, 0.129) as shown in the inset of Fig. 9b, which is deeper blue than most of the recent devices of TADF emitters.62,64–68
Conclusions
In summary, the synthesis and in-depth photophysical characterization of a series of blue TADF emitters with a D–A–D architecture utilizing the unconventional electron-rich DBT unit as the acceptor and Ac derivatives as donor units are reported. The introduction of tert-butyl or methoxy groups to the D unit enhances the donor strength of the Ac and helps to tune the singlet and triplet energy levels. Excellent energy alignment between the 3LED and 3LEA triplet states and the 1CT singlet state is obtained in the tert-butyl substituted compound DtBuAc-DBT. This leads to a decrease in the energy gap (ΔE1CT–3LE) from 0.20 eV in DAc-DBT to 0.09 eV in DtBuAc-DBT, which greatly facilitates the RISC process and results in triplet harvesting with great efficiency. Surprisingly, despite the excellent singlet–triplet energy alignment between the low energy 3LE triplet and the 1CT state in DtBuAc-DBT, the RISC rate is slower than in other highly efficient TADF emitters where RISC proceeds with rates of approximately 106 s−1. However, the prototype proof-of-concept OLED based on DtBuAc-DBT emitter exhibits a deep-blue EL spectrum peaking at 455 nm with the CIE coordinates of (0.133, 0.129) and a maximum EQE of 10.5%.Interestingly, when the donor strength is further increased by using methoxy groups in DOMeAc-DBT, a small energy gap of <0.01 eV between 1CT and the high-energy triplet 3LED is obtained. However, a lower energy triplet state of CT character is also observed, showing an energy gap of 0.24 eV to the 1CT state. DOMeAc-DBT shows a smaller TADF contribution in comparison to DtBuAc-DBT, but notably with a much faster RISC rate, (kRISC = 2.3 × 105 s−1), despite a larger ΔEST. The faster RISC is ascribed to an upper-state crossing from 3LED to 1CT promoted by the small ΔE1CT–3LE = 0.01 eV. The small TADF contribution in DOMeAc-DBT is thus attributed to the competition between the RISC rate (from 3LED to 1CT) and the rate of triplet 3LED relaxation to the low 3CT state by rapid IC. We have shown here that even when a material is designed in ways that the singlet 1CT and triplet 3LE states are specifically energy aligned to maximize the RISC rate, the low-energy 3CT may act as a quenching state in certain conditions, competing with RISC and having a detrimental effect on the triplet harvesting outcome. This work expands the range of acceptor units that can be used in TADF emitters and shows the importance of precisely tuning the local and triplet states by carefully selecting complementary donor and acceptor units to achieve efficient TADF.
Author contributions
R. H. and N. A. K. contributed equally to this manuscript. N. A. K. and J. S. W. designed, synthesized and characterized the studied compounds under the supervision of M. R. B. R. H. carried out the photophysical characterization of all compounds under the supervision of F. B. D. N. A. K. performed and analysed all the DFT calculations. R. H. and N. A. K. lead the writing and coordination of the manuscript with additional contribution from all authors. F. B. D. lead the interpretation of the experimental photophysical data with assistance from R. H. A. D. performed and interpreted PLQY and OLED measurements with R. H. A. S. B. collected X-ray diffraction data and performed data analysis. The manuscript was read, corrected and approved by all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
R. H. acknowledges a PhD grant funded by Durham University. N. A. K., J. S. W., A. D. and M. R. B. thank EU Horizon 2020 Grant Agreement No. 732103 (HyperOLED) for funding. F. B. D. acknowledges Samsung-SAIT for funding this work using their Global Research Outreach (GRO) Program, and F. B. D. and M. R. B. acknowledge the EPSRC for funding under grant numbers EP/L02621X/1 and EP/N028511/1.References
- K. Wang, Y. Z. Shi, C. J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S. L. Tao, C. S. Lee, X. M. Ou and X. H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31515–31525 CrossRef CAS PubMed.
- G. Grybauskaite-Kaminskiene, K. Ivaniuk, G. Bagdziunas, P. Turyk, P. Stakhira, G. Baryshnikov, D. Volyniuk, V. Cherpak, B. Minaev, Z. Hotra, H. Ågren and J. V. Grazulevicius, J. Mater. Chem. C, 2018, 6, 1543–1550 RSC.
- H. F. Higginbotham, C. L. Yi, A. P. Monkman and K. T. Wong, J. Phys. Chem. C, 2018, 122, 7627–7634 CrossRef CAS.
- X. Yang, H. Guo, B. Liu, J. Zhao, G. Zhou, Z. Wu and W.-Y. Wong, Adv. Sci., 2018, 5, 1800950 CrossRef PubMed.
- J. W. Sun, J. Y. Baek, K.-H. Kim, J.-S. Huh, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, J. Mater. Chem. C, 2017, 5, 1027–1032 RSC.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- P. E. Burrows, G. Gu, V. Bulovi, Z. Shen, S. R. Forrest and M. E. Thompson, IEEE Trans. Electron Devices, 1997, 44, 1188–1203 CrossRef CAS.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- M. Gmelch, H. Thomas, F. Fries and S. Reineke, Sci. Adv., 2019, 5, 7310 CrossRef PubMed.
- N. K. Patel, S. Cina and J. H. Burroughes, IEEE J. Sel. Top. Quantum Electron., 2002, 8, 346–361 CrossRef CAS.
- X. Cai and S. J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- S. Reineke, Nat. Photonics, 2014, 8, 269–270 CrossRef CAS.
- S. Scholz, D. Kondakov, B. Lüssem and K. Leo, Chem. Rev., 2015, 115, 8449–8503 CrossRef CAS PubMed.
- R. Meerheim, B. Lüssem and K. Leo, Proc. IEEE, 2009, 97, 1606–1626 CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- H. Kuma and C. Hosokawa, Sci. Technol. Adv. Mater., 2014, 15, 034201 CrossRef PubMed.
- S. J. Su, E. Gonmori, H. Sasabe and J. Kido, Adv. Mater., 2008, 20, 4189–4194 CAS.
- A. P. Monkman, ISRN Mater. Sci., 2013, 2013, 670130 Search PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- J. Lee, C. Jeong, T. Batagoda, C. Coburn, M. E. Thompson and S. R. Forrest, Nat. Commun., 2017, 8, 15566 CrossRef CAS PubMed.
- H. Fukagawa, T. Shimizu, T. Kamada, S. Yui, M. Hasegawa, K. Morii and T. Yamamoto, Sci. Rep., 2015, 5, 9855 CrossRef CAS PubMed.
- I. R. De Moraes, S. Scholz, B. Lüssem and K. Leo, Org. Electron., 2011, 12, 341–347 CrossRef.
- R. Seifert, I. Rabelo De Moraes, S. Scholz, M. C. Gather, B. Lüssem and K. Leo, Org. Electron., 2013, 14, 115–123 CrossRef CAS.
- H. Fukagawa, T. Shimizu, Y. Iwasaki and T. Yamamoto, Sci. Rep., 2017, 7, 1735 CrossRef PubMed.
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- P. L. Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. DIas, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- P. L. dos Santos, J. S. Ward, D. G. Congrave, A. S. Batsanov, J. Eng, J. E. Stacey, T. J. Penfold, A. P. Monkman and M. R. Bryce, Adv. Sci., 2018, 5, 1700989 CrossRef PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- J. Choi, D.-S. Ahn, K. Y. Oang, D. W. Cho and H. Ihee, J. Phys. Chem. C, 2017, 121, 24317–24323 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- T. T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Beilstein J. Org. Chem., 2018, 14, 282–308 CrossRef CAS PubMed.
- I. Lee and J. Y. Lee, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- F.-M. Xie, H.-Z. Li, G.-L. Dai, Y.-Q. Li, T. Cheng, M. Xie, J.-X. Tang and X. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 26144–16151 CrossRef CAS PubMed.
- H. Tachibana, N. Aizawa, Y. Hidaka and T. Yasuda, ACS Photonics, 2017, 4, 223–227 CrossRef CAS.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- P. L. Dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS PubMed.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- R. Huang, J. S. Ward, N. A. Kukhta, J. Avó, J. Gibson, T. Penfold, J. C. Lima, A. S. Batsanov, M. N. Berberan-Santos, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2018, 6, 9238–9247 RSC.
- J. S. Kang, T. R. Hong, H. J. Kim, Y. H. Son, R. Lampande, B. Y. Kang, C. Lee, J.-K. Bin, B. S. Lee, J. H. Yang, J. Kim, S. Park, M. J. Cho, J. H. Kwon and D. H. Choi, J. Mater. Chem. C, 2016, 4, 4512–4520 RSC.
- R. Huang, J. Avó, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. C. Lima, M. R. Bryce and F. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- A. D. Boese and J. M. L. Martin, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed.
- N. A. Kukhta, D. A. Da Silva Filho, D. Volyniuk, J. V. Grazulevicius and G. Sini, Chem. Mater., 2017, 29, 1695–1707 CrossRef CAS.
- J.-L. Brédas, Chem. Mater., 2017, 29, 477–478 CrossRef.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- V. F. Pais, H. S. El-Sheshtawy, R. Fernández, J. M. Lassaletta, A. Ros and U. Pischel, Chem. – Eur. J., 2013, 19, 6650–6661 CrossRef CAS PubMed.
- C. Y. Chan, L. S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Adv. Funct. Mater., 2018, 28, 1706023 CrossRef.
- O. L. J. Gijzeman and F. Kaufman, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 721–726 RSC.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- G. W. Kim, H. W. Bae, R. Lampande, I. J. Ko, J. H. Park, C. Y. Lee and J. H. Kwon, Sci. Rep., 2018, 8, 16263 CrossRef PubMed.
- S. J. Woo, Y. Kim, S. K. Kwon, Y. H. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 7199–7207 CrossRef CAS PubMed.
- X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Adv. Mater., 2018, 30, 1705005 CrossRef PubMed.
- W. Liu, S. Ying, R. Guo, X. Qiao, P. Leng, Q. Zhang, Y. Wang, D. Ma and L. Wang, J. Mater. Chem. C, 2019, 7, 1014–1021 RSC.
- Y. Xu, X. Liang, Y. Liang, X. Guo, M. Hanif, J. Zhou, X. Zhou, C. Wang, J. Yao, R. Zhao, D. Hu, X. Qiao, D. Ma and Y. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 31139–31146 CrossRef CAS PubMed.
- D. Liu, K. Sun, G. Zhao, J. Wei, J. Duan, M. Xia, W. Jiang and Y. Sun, J. Mater. Chem. C, 2019, 7, 11005–11013 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1901568–1901571. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc02175b |
| ‡ R. H. and N. A. K. contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |