
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insight into novel sulfoxide containing SABRE polarisation transfer catalysts†‡
Ben. J.
Tickner
 a,
Jennifer S.
Lewis
a,
Richard O.
John
a,
Adrian C.
Whitwood
b and
Simon B.
Duckett
*a
a,
Jennifer S.
Lewis
a,
Richard O.
John
a,
Adrian C.
Whitwood
b and
Simon B.
Duckett
*a
aCenter for Hyperpolarization in Magnetic Resonance (CHyM), University of York, Heslington, York YO10 5NY, UK. E-mail: simon.duckett@york.ac.uk
bDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK
First published on 24th September 2019
Abstract
Signal Amplification By Reversible Exchange (SABRE) is a hyperpolarisation technique that commonly uses [Ir(H)2(carbene)(substrate)3]Cl complexes to catalytically transfer magnetisation from para-hydrogen derived hydride ligands to coordinated substrates. Here, we explore the reactivity of a novel class of such catalysts based on sulfoxide containing [IrCl(H)2(carbene)(DMSO)2], which are involved in the hyperpolarisation of pyruvate using SABRE. We probe the reactivity of this species by NMR and DFT and upon reaction with sodium pyruvate establish the formation of two isomers of [Ir(H)2(η2-pyruvate)(DMSO)(IMes)]. Studies with related disodium oxalate yield [Ir2(H)4(IMes)2(DMSO)2(η2-κ2-Oxalate)] that is characterised by NMR and X-ray diffraction.
Introduction
Small molecule bond activation is an important area of inorganic chemistry that finds a central role in a range of applications in biochemistry,1 recycling,2 and organic synthesis.3 In fact, many rather inert reagents such as CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and R3CH5 can be activated by light6 or transition metal systems.7,8 The activation of small homonuclear diatomic molecules such as H2 and O2 by oxidative addition to metal centres has been known for many years.9,10 It typically occurs via a concerted mechanism wherein the newly introduced groups are located in a cis relationship.11 Activation of more polar molecules such as CH3I is, however, more often complex and this difference is revealed in the trans relationship between the two newly introduced groups.9 Orbital overlap effects during H2 addition, as revealed through density function theory (DFT), are also complex with repulsive interactions between filled orbitals on the metal and H2 yielding a barrier to the oxidative addition process that must be overcome.12
4 and R3CH5 can be activated by light6 or transition metal systems.7,8 The activation of small homonuclear diatomic molecules such as H2 and O2 by oxidative addition to metal centres has been known for many years.9,10 It typically occurs via a concerted mechanism wherein the newly introduced groups are located in a cis relationship.11 Activation of more polar molecules such as CH3I is, however, more often complex and this difference is revealed in the trans relationship between the two newly introduced groups.9 Orbital overlap effects during H2 addition, as revealed through density function theory (DFT), are also complex with repulsive interactions between filled orbitals on the metal and H2 yielding a barrier to the oxidative addition process that must be overcome.12
Many catalytic processes exploit the oxidative addition of H2 to a transition metal centre by enabling the subsequent transfer of the two hydrogen atoms into an unsaturated centre13,14 However, recent examples in main group chemistry involving frustrated Lewis pairs mean that direct addition is also possible.15 Understanding of these reactions can be improved by exploiting a technique known as para-Hydrogen Induced Polarisation (PHIP) to detect reaction intermediates.16,17 This process incorporates the two protons of a single para-hydrogen (p-H2) molecule into a substrate via a hydrogenation reaction to see the PHIP effect. p-H2 is a spin isomer of H2 and exists as a singlet state with a nuclear spin order of zero. H2 gas can be enriched (>98%) in its para state by simply cooling it in the presence of a paramagnetic catalyst such as Fe2O3 or activated charcoal.18–20 If the spin orientation of p-H2 survives the ensuing hydrogenation reaction the NMR properties of the product can be harvested to increase product detectability. This effect was exemplified in 1986 by Bowers and Weitekamp21,22 with Eisenberg and Bargon23,24 producing similar independent observations around the same time. As the net nuclear spin of p-H2 is zero it is invisible to an NMR experiment but once the symmetry of its two protons is broken the resulting spin order can be detected. In fact, the NMR signal intensity of the now NMR visible, and hyperpolarised, product is derived from the resulting large non-Boltzmann populations that lie across its nuclear spin energy levels. Since its introduction PHIP has been used to detect low concentration analytes and true intermediates in the field of catalysis.22,25–27 Excitement has resulted from the hydrogenation of some unsaturated clinical agents which has led to their in vivo MRI detection.28–30
Since 2009, the p-H2 based method Signal Amplification By Reversible Exchange (SABRE) has been used to harness signal gains from p-H2 without the need for the direct hydrogenation of a substrate (sub).31 It achieves the catalytic transfer of magnetisation into a substrate through the formation of a J-coupled network within the associated catalyst.32 The first step in SABRE typically involves the conversion of a stable 16 electron precursor such as [IrCl(COD)(IMes)] (1) (where IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene and COD = cis,cis-1,5-cyclooctadiene) into an intermediate [Ir(COD)(IMes)(sub)]Cl which then undergoes hydrogen addition to form highly reactive [Ir(H)2(COD)(IMes)(sub)]Cl.33–35 Subsequent hydrogenation of the COD ligand then leads to a SABRE catalyst such as [Ir(H)2(IMes)(sub)3]Cl which importantly reversibly binds both H2 and sub. In SABRE, H2 addition to square planar [Ir(COD)(IMes)(Sub)]Cl typically occurs over the COD-Ir-Sub axis rather than the COD-Ir-IMes axis.36 This observation has been explained in terms of both reduced steric interactions with the bulky carbene ligand and electronic effects in terms of orbital interactions.36 It has been suggested that H2 addition is favoured over axis containing ligands with π acceptor orbitals which provide additional stabilising interactions as the geometry of the complex changes during the addition step.37
The most common substrate molecules used in SABRE have proven to feature N-donor sites that readily coordinate to iridium. N-Heterocycles such as pyridine,31,38–41 nicotinamides,31,42,43 pyrazines,39,40 and pyrazoles41 reflect common examples although other N-functionalities found in nitriles44 and amines45 have been used. It has recently been reported that in the presence of a stabilising sulfoxide coligand, the reversible coordination and subsequent hyperpolarisation of oxygen ligating pyruvate can occur.46
The direct hyperpolarisation of pyruvate was made possible in this case by the formation of the novel polarization transfer catalyst [Ir(H)2(η2-pyruvate)(DMSO)(IMes)]. In addition to this active catalyst, [IrCl(H)2(DMSO)2(IMes)], is also present in solution and is expected to be critical in improving the efficiency of this important hyperpolarisation transfer process. In this work we investigate the formation, behaviour, and ligand exchange processes exhibited by this sulfoxide containing polarisation transfer catalyst. We do this in solvents that were purchased from Sigma and used without further purification as our aim is to study reactivity without taking any special precautions. This is reflective of the most likely scenario when used by the non-specialist. We extend this method to include an examination of the behaviour of oxalate with the aim of developing the range of materials that can be hyperpolarised with sulfoxide containing SABRE polarisation transfer catalysts.
Results and discussion
Formation of [Ir(COD)(IMes)(OH2)]Cl (2) from [IrCl(COD)(IMes)] (1)
When the SABRE precatalyst [IrCl(COD)(IMes)] (1) is dissolved in 0.6 mL of methanol-d4 at 298 K, 1H NMR resonances can be observed for 1 in addition to a minor product, 2 which forms at the ∼2% level. The ratio of this product increases to 13% after the addition of 50 μL H2O, as shown in Fig. 1 thereby confirming a role of the original H2O contaminant in methanol-d4. When this experiment was repeated in a solution of 0.6 mL methanol-d4 doped with 5 μL DMSO, the ratio of 2 formed after the addition of 50 μL of H2O proved to be just 6%. It is noteworthy that no evidence for [IrCl(COD)(DMSO)(IMes)] was apparent. A similar 1H NMR measurement of 1 in a solution of CDCl3 revealed the presence of 2 at the 3% level. Subsequent addition of H2O increases this to 5% but the immiscibility of CDCl3 and H2O prevents the use of high water concentrations. Hence, the equilibrium position between 1 and 2 is dependent on solvent choice and water concentration. 2 is assigned as [Ir(COD)(IMes)(OH2)]Cl which is further confirmed by 2D NMR characterisation data (see ESI‡) at 245 K which shows a NOE between the bound aqua ligand at δ 8.15 and the IMes backbone at δ 2.16. 1H NMR resonances for hexa-aqua species are often found between δ 8 and δ 11.47 The formation of 2 is not unexpected as iridium aqua complexes are known and the reactions performed throughout this work are not completed under anhydrous conditions because our aim is to achieve SABRE catalysis using an air stable precursor with minimum end-user challenge.48–50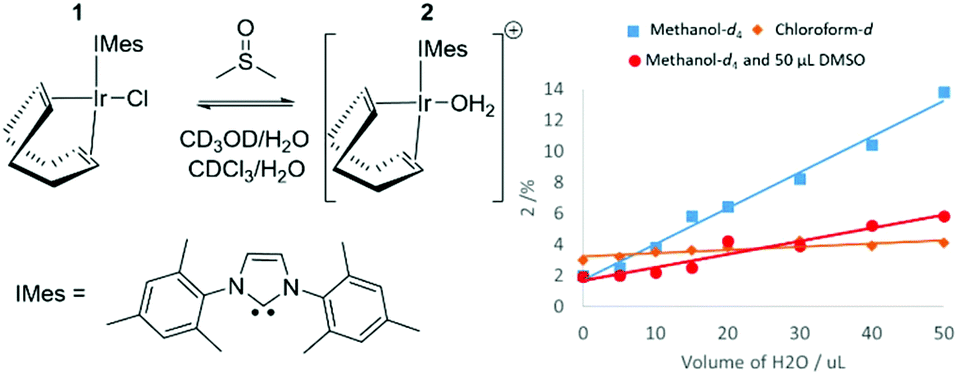 | ||
| Fig. 1 Demonstrating the proportion of 2 in solution increases with the level of added water in the solvents methanol and chloroform in addition to a methanol–DMSO mixture thereby confirming its identity as [Ir(COD)(IMes)(OH2)]Cl as shown. | ||
DFT calculations have been used to further confirm these product assignments, with their predicted relative energies detailed in Table 1 (see ESI‡ for details). These calculations used full models of the complexes, [Ir(COD)(IMes)(X)], and are relative to a zero point which includes all species (indicated complex, DMSO, H2O and CH3OH) in an equimolar amount. They do not reflect transition state barriers and excess reagents are not accounted for. These calculations confirm that 1, where X = Cl, is most stable in agreement with experiment. [Ir(COD)(IMes)(OH2)]Cl, 2 is predicted to be more stable than the corresponding methanol or sulfoxide adducts, again supporting our NMR observations.
| X | H/kJ mol−1 | G/kJ mol−1 |
|---|---|---|
| Cl | 0 | 0 |
| OH2 | 52.8 | 57.0 |
| S-DMSO | 42.3 | 68.9 |
| HOCH3 | 56.1 | 69.4 |
Formation of [IrCl(H)2(DMSO)2(IMes)] (3)
When H2 is added to an equilibrium mixture of 1 and 2 containing 4 equivalents of DMSO in methanol-d4 at 245 K the initial 1H NMR spectra reveal two hydride resonances at δ −15.49 and δ −21.51 which share a mutual J coupling of 5.5 Hz. Low temperature NMR characterisation of the complex yielding these signals confirms it to be of the form [Ir(H)2(IMes)(DMSO)2(L)], 3.46 The formation of iridium(III) sulfoxide complexes bound through sulphur for use in SABRE hyperpolarisation studies has been reported previously.51 When this hydrogenation reaction is examined at 245 K in CDCl3 or dichloromethane-d2 (DCM-d2) hydride resonances appear at δ −15.50, δ −21.12 and δ −15.67, δ −21.36, respectively. The similarity in these signals suggests the common presence of 3 in both cases. This chemistry is complicated, however, by the fact 3 is unstable in solution over long timescales, as detailed in the ESI.‡The identity of 3 was further confirmed by repeating this experiment using [IrBr(COD)(IMes)], as the corresponding reaction product [IrBr(H)2(DMSO)2(IMes)] yields hydride signals at δ −15.67 and δ −20.45 in methanol-d4 which are clearly different to those of 3. Furthermore, when [Ir(CH3CN)(COD)(IMes)]PF6 is used instead, [Ir(H)2(CH3CN)(DMSO)2(IMes)]PF6 forms which yields signals at δ −15.76 and δ −19.83 at 255 K; we note there was no evidence for the displacement of CH3CN by DMSO in the associated NMR spectra. Hence, the hydride resonance in these complexes shift according to the identity of the ligand that is trans to it thereby confirming the indicated product identities. The identity of 3 was also studied by DFT as detailed in the results shown in Table 2. [IrCl(H)2(DMSO)2(IMes)] was found to be lower in energy than the corresponding species where chloride is replaced by water, methanol or sulfoxide.
| X | H/kJ mol−1 | G/kJ mol−1 |
|---|---|---|
| Cl | 0 | 0 |
| OH2 | 24.4 | 34.3 |
| S-DMSO | 39.1 | 70.8 |
| ODCH3 | 29.3 | 45.4 |
When the reaction of H2 with an equilibrium mixture of 1 and 2 in methanol-d4 containing DMSO was monitored at 245 K (Fig. 2) no evidence for the formation of any H2 addition products except 3 is observed. This suggests that H2 addition is slow, and subsequent COD hydrogenation in intermediate 1-H2 of Fig. 2 is fast. Interestingly, the proportion of 2 remains roughly constant as this conversion proceeds thereby suggesting any equilibrations involving it are also slow. The route to 3 is therefore most likely to involve direct H2 addition to 1 rather than 2. DFT confirms that H2 addition to 1 proceeds over its COD-Ir–Cl axis rather than the COD-Ir-IMes axis according to the relative energies of the corresponding products (see ESI‡). This is supported by the fact that when this H2 addition reaction is monitored in CDCl3 at 245 K, resonances corresponding to 1-H2, at δ −13.39 and δ −18.42, are detected in addition to those of 3. The resonances for 1-H2 rapidly disappear upon warming this solution to 298 K where 3 then forms.
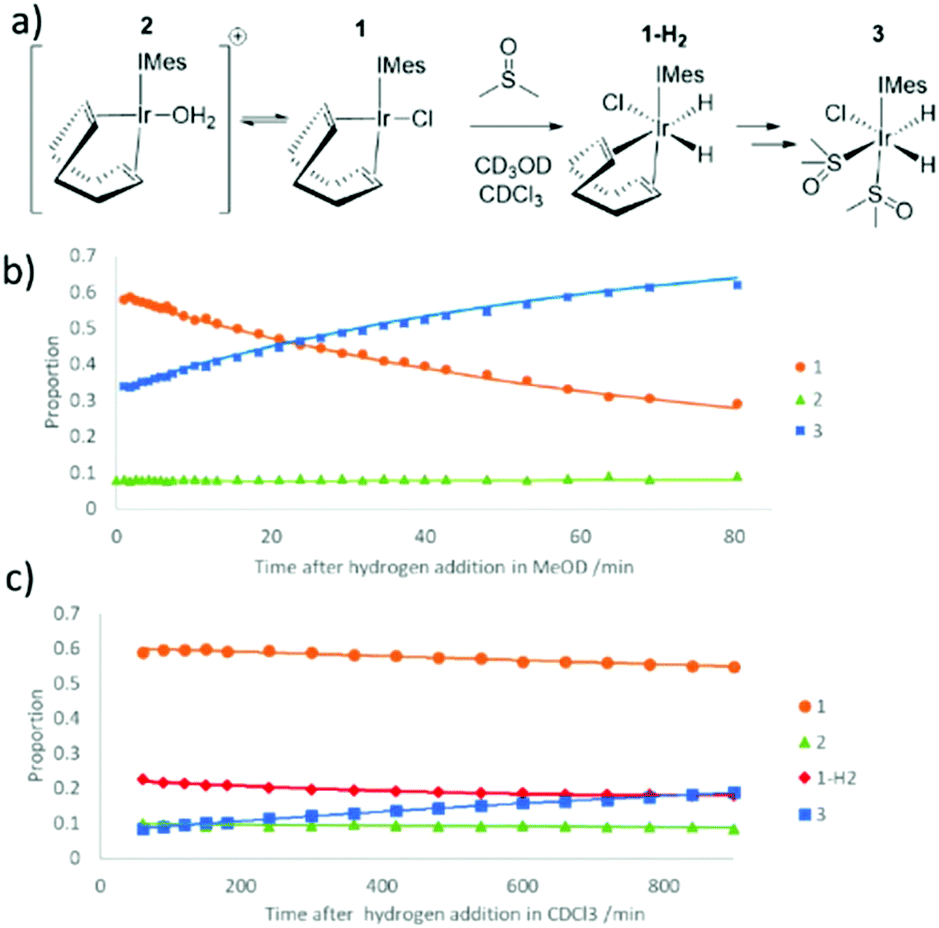 | ||
| Fig. 2 (a) Reaction steps involved in the conversion of 1 to 3 in the presence of DMSO and H2, (b) time course for the reaction, as determined in methanol-d4 solution when the starting concentrations are 5 mM [IrCl(COD)(IMes)], 1 μL DMSO and 3 bar hydrogen pressure and (c) analogous reaction time course data in CDCl3 at 245 K. | ||
Ligand exchange processes of [IrCl(H)2(DMSO)2(IMes)] (3)
When 3 is formed from a methanol-d4 solution containing 2 mg of 1, 1 μL of DMSO and 3-bar H2, its hydride resonances appear very broad at 298 K (line width of ∼120 Hz) which is consistent with rapid hydrogen loss. This deduction is confirmed upon shaking methanol-d4 solutions of 3 with 3-bar p-H2 for 10 seconds at 65 G at 298 K. Its hydride resonances exhibit PHIP signal enhancement alongside a free DMSO signal that yields a weak SABRE 1H NMR signal enhancement of 7-fold (see ESI‡). This is consistent with the reversible pairwise addition of H2 to 3 and loss of DMSO. Product 3 actually yields four distinct 1H NMR signals for the methyl groups of its two DMSO ligands. The DMSO ligand cis to carbene yields a pair of inequivalent CH3 signals at δ 2.83 and 3.12 while DMSO trans to carbene resonates at δ 3.19 and 3.27. Upon selective excitation of bound DMSO resonances cis to carbene in 3, evidence for chemical exchange into free DMSO is observed. In contrast, we observe no exchange of the DMSO ligand that lies trans to carbene on this timescale although we do observe interchange of the inequivalent CH3 groups. This suggests that loss of the DMSO ligand cis to IMes accounts for this scrambling of the inequivalent CH3 resonances of the DMSO trans to IMes.Measuring the change in these resonance's signal intensities as a function of time after the initial r.f. excitation step allowed the associated ligand loss rates to be calculated. The rate of reversible hydrogen loss, kH2, was found to be 3.31 ± 0.25 s−1 at 263 K and is faster than that seen in many related systems.52 Loss, and hence exchange of the DMSO ligand cis to IMes, was found to occur on a similar timescale with kDMSO 3.35 ± 0.01 s−1 at 263 K. Repeating these measurements at different temperatures allowed the transition state barriers for these two processes to be determined as ΔH±(H2) 78 ± 6 kJ mol−1, ΔS±(H2) 60 ± 25 J K−1 mol−1 and ΔH±(DMSO) 79 ± 6 kJ mol−1, ΔS±(DMSO) 66 ± 8 J K−1 mol−1 respectively. This enthalpic barrier to hydrogen loss is comparable to those for similar complexes, overall the entropy change suggests that a dissociative pathway is adopted.53
The effects of H2 pressure and DMSO concentration on these exchange processes were also studied in methanol-d4 at 243 K for differing reagent concentrations. kH2 proved to increase at higher H2 concentrations before reaching a plateau as detailed in the ESI.‡ The rate of DMSO exchange is unaffected by increasing DMSO or H2 concentration. We therefore propose that DMSO and H2 loss proceed via formation of the common 16-electron intermediate [IrCl(H)2(DMSO)(IMes)] in what is a dissociative first step. H2 exchange then proceeds via the formation of [IrCl(H)2(H2)(DMSO)(IMes)]; such indirect H2 exchange, rather than direct H2 loss, has been reported for several similar systems.51,54 When similar EXSY data were collected in CD2Cl2, the rate of hydrogen and DMSO loss proved to be slower than those in methanol-d4, as summarised in Table 3.
| Solvent | Process | k (263 K)/s−1 | ΔH≠/kJ mol−1 | ΔS≠/J K−1 mol−1 |
|---|---|---|---|---|
| Methanol-d4 | Hydrogen exchange (kH2) | 3.31 ± 0.25 | 78 ± 6 | 60 ± 25 |
| DMSO exchange (kDMSO) | 3.35 ± 0.04 | 79 ± 6 | 66 ± 8 | |
| Dichloromethane-d2 | Hydrogen exchange (kH2) | 1.16 ± 0.04 | 83 ± 8 | 73 ± 29 |
| DMSO exchange (kDMSO) | 1.56 ± 0.01 | 84 ± 2 | 78 ± 8 |
These deductions were confirmed through further DFT calculations that revealed the products of direct H2 loss to form a 16 electron product as being highly energetically unfavourable, as shown in Table 4. The five coordinate product formed by loss of DMSO trans to carbene proved highly unstable, undergoing spontaneous rearrangement to form an intermediate with a vacancy in the equatorial plane; this is consistent with the EXSY data. In contrast, the predicted ligand dissociation pathway involves the formation of five-coordinate [IrCl(H)2(DMSO)(IMes)] via loss of the DMSO ligand that lies cis to carbene.
| Loss of | H/kJ mol−1 | G/kJ mol−1 |
|---|---|---|
| H2 | 89.0 | 58.5 |
| Cl | 39.6 | 3.9 |
| DMSO cis carbene | 32.7 | −10.1 |
Even at low temperature (245–265 K), H/D exchange leading to [IrCl(D)(H)(DMSO)2(IMes)] (3-d) and [IrCl(D)2(DMSO)2(IMes)] was evident. Binding of solvent methanol to [IrCl(H)2(DMSO)(IMes)] is an obvious route to deuterium exchange and the formation of these species alongside HD and D2 gas. While we do not directly observe methanol bound adducts, they are suggested to form in similar systems and indeed proposed to account for the hydrogen isotope exchange reaction that is often observed to run in parallel to SABRE.50,55 Experimentally, at low temperature it proved possible to reliably and selectively excite the hydride resonance of 3 or 3-d. The selective excitation of the hydride resonances for these two species revealed their selective chemical exchange into H2 and HD, from 3 and 3-d respectively. Therefore, kH2 and kHD can be determined as previously described. These values proved to be the same within error and hence, there is little or no kinetic isotope effect which is consistent with other reports.56,57 We note that exchange between 3 and 3-d is not observed in the associated EXSY data which provides confirmation that the underlying deuterium exchange processes involving methanol-d4 are slow.
Formation of [Ir(H)2(η2-pyruvate)(DMSO)(IMes)], (4)
When sodium pyruvate (5 equivalents relative to iridium) is added to a solution of preformed 3 in methanol-d4 at 245 K, two new hydride ligand containing products form, which correspond to isomers 4a and 4b of [Ir(H)2(η2-pyruvate)(DMSO)(IMes)] as detailed in Fig. 3a. Logically, a further isomer of 4a is possible, 4c, in which the orientation of the pyruvate is rotated but this is not observed in solution. These products are differentiated from each other by the geometry of the coordinated pyruvate ligand and their proportion is both reaction time and temperature dependent (Fig. 3b). DFT (Table 5) is used to confirm that 4a exhibits the ligand geometry shown in Fig. 3a and that 4b is the most stable species. Despite observation of a 10 Hz coupling between the pyruvate COOH group and the hydride trans to oxygen, according to DFT the pyruvate CH3 and DMSO groups of 4a are arranged in a cis fashion as shown in Fig. 3a. This deduction was further confirmed by the observation of an NOE peak between the pyruvate CH3 group and the phenyl protons on the mesityl group of the carbene ligand.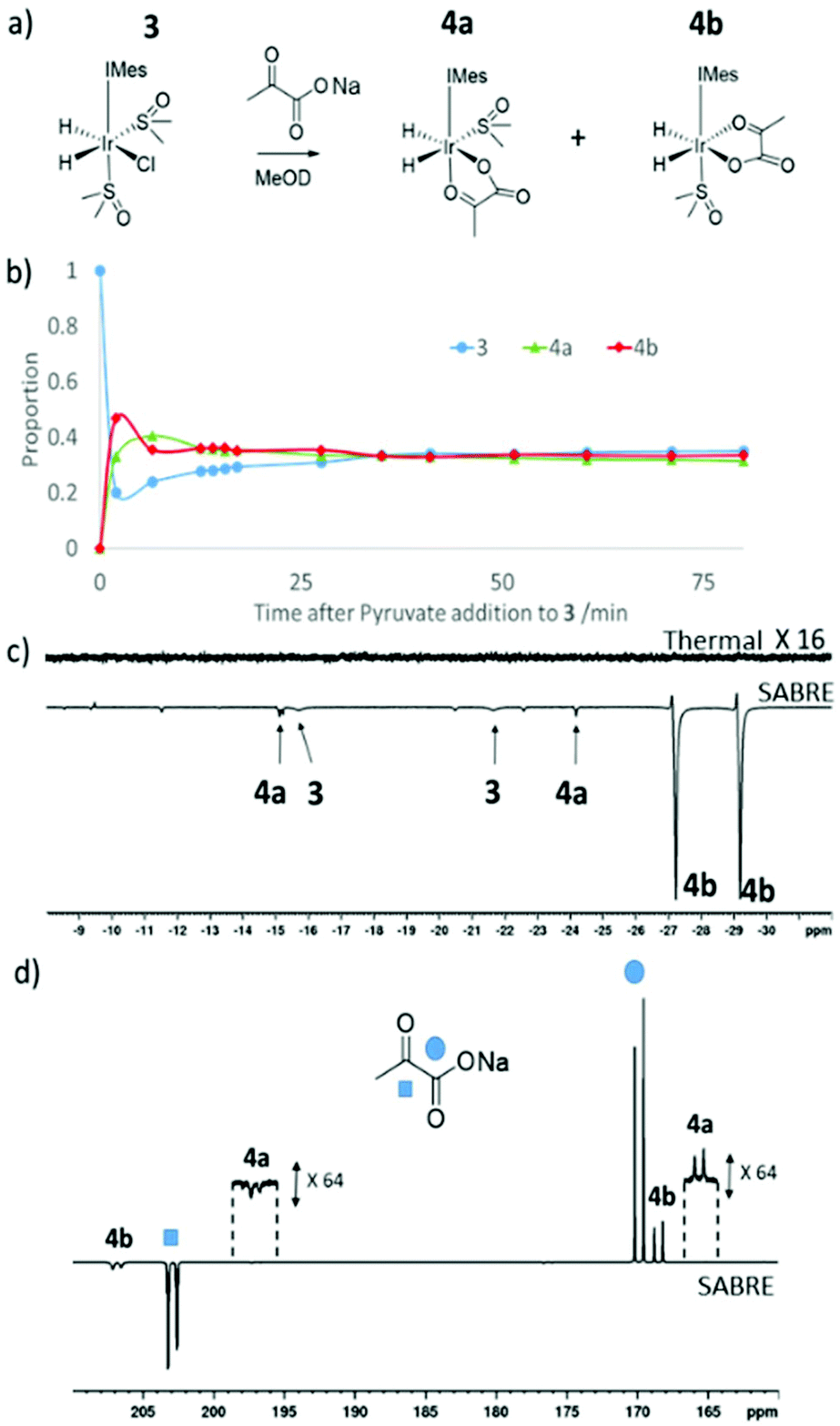 | ||
| Fig. 3 (a) Reaction of 3 and pyruvate to form 4 (b) monitoring pyruvate addition to 3 using 1H NMR spectroscopy. Pyruvate addition was made at room temperature to a solution of preformed 3 at 245 K before it was introduced into the spectrometer whose probe was at 245 K. There is therefore a rapid temperature change at the start of this data series. Upon shaking a solution of 3 and 4 with 3-bar p-H2 for 10 seconds at (c) 65 G or (d) in a mu metal shield strongly hyperpolarised 1H (c) or 13C (d) resonances of 4b are observed compared to the thermal trace. | ||
| Complex | H/kJ mol−1 | G/kJ mol−1 |
|---|---|---|
| 4a | 8.5 | 14.8 |
| 4b | 0 | 0 |
| 4c | 22.6 | 28.1 |
When this solution was examined with a 32 scan 1H NMR measurement at 298 K, unlike the 245 K data of Fig. 3b, the main hydride containing complex observed is 4b and resonances for 3 and 4a cannot be discerned. 4a and 4b have previously been implicated in pyruvate polarisation using SABRE and their NMR characterisation data has been reported.464 is also detected immediately after H2 addition to a solution of 1 containing pyruvate (5 equivalents relative to iridium) and DMSO (10 equivalents) at 298 K. When an equilibrium mixture of 3 and 4 is shaken with 3 bar p-H2 at 65 G enhanced hydride resonances are observed in the 1H NMR spectrum as shown in Fig. 3c, which are strongest for 4b.
If sodium-1,2-pyruvate-[13C2] is used as the substrate and the p-H2 shaking process is performed under SABRE-SHEATH conditions in a mu metal shield (see Experimental) an enhanced 13C response for the free material at δ 169 and δ 203 can also be readily detected.46 Two enhanced signals can be also seen for the bound pyruvate ligand in 4b at δ 168 and δ 207 in these NMR spectra. It is also possible to observe extremely weak hyperpolarised signals for the bound pyruvate ligand in 4a at δ 161 and δ 198, as shown in Fig. 4d. Interestingly, when EXSY measurements are used to probe these signals, the selective excitation of the hydride resonances of 4b reveals no exchange into H2 on the NMR timescale. Furthermore, when 13C-EXSY is used to probe the bound signals of 4b, no exchange is seen into free pyruvate on this timescale. Hence 4b appears to be relatively stable in agreement with the DFT study, but this is not consistent with the strong hydride polarisation that is evident for 4b in Fig. 3. Consequently, a role for 3 which is present and undergoes rapid H2 exchange as discussed earlier in its formation is indicated. It is by this route that the delivery of fresh p-H2 into 4 and the resulting enhancement of hydride and pyruvate ligand signals is achieved. As the NMR relaxation times of the 13C resonances in pyruvate are much longer than those of 1H, slow exchange is still commensurate with the build-up of pyruvate polarisation in solution. Related relayed p-H2 enhancement effects have been described recently.58,59
 | ||
| Fig. 4 (a) 128 scan 13C spectrum of a solution of 1, DMSO and sodium-1,2-oxalate-[13C2] (1:5:5) in methanol-d4 (b) 13C NMR signals seen after shaking an NMR tube with 3-bar p-H2 in a mu metal shield for 30 seconds. | ||
An equilibrium mixture containing 4 can also be formed if sodium-1,2-pyruvate-[13C2] is added to a preformed methanol-d4 solution of [IrBr(H)2(DMSO)2(IMes)] (3-Br) or [Ir(H)2(CH3CN)(DMSO)2(IMes)]PF6 (3-Acn). When these samples are shaken with p-H2 for 10 seconds at 65 G strongly hyperpolarised hydride resonances can again be observed for 4b in the corresponding high field NMR measurement. These resonances appear with significantly lower intensity than those achieved with 3 derived systems (35% and 31% of this signal intensity respectively). Furthermore, when the p-H2 shaking step is repeated in a mu metal shield, hyperpolarised resonances for 13C-pyruvate are again visible. The corresponding 13C-pyruvate signal enhancements are typically 580-fold and 120-fold for the 3-Br and 3-Acn derived systems respectively. These are lower than those achieved for the analogous 3 system (1070-fold) which is consistent with the reduced amount of 4b which is present at equilibrium when these precursors are used (22 and 23% respectively when compared to 92% with 3).
We note that the linewidths of the hydride resonances of 4b at 298 K are similar in each of these samples (43–45 Hz). Similarly, the linewidths of 3, 3-Br and 3-Acn are comparable at 245 K (3–5 Hz). Therefore, we do not expect that differences in hydride polarisation levels seen for 4b in these solutions are due to different hydrogen exchange rates in 3, but rather the different binding strengths of the coligands that must be displaced by pyruvate to form 4 from 3. Hence, whilst there appears to be a link between pyruvate polarisation level and 4b concentration, the identity of L in [Ir(H)2(DMSO)2(IMes)L] must play a large effect on the level of pyruvate signal enhancement. These data further confirm that 3 is important in mediating efficient H2 exchange within the 3/4 hyperpolarisation mixture.
Using [IrCl(H)2(DMSO)2(IMes)] (3) to hyperpolarise sodium-1,2-oxalate-[13C2]
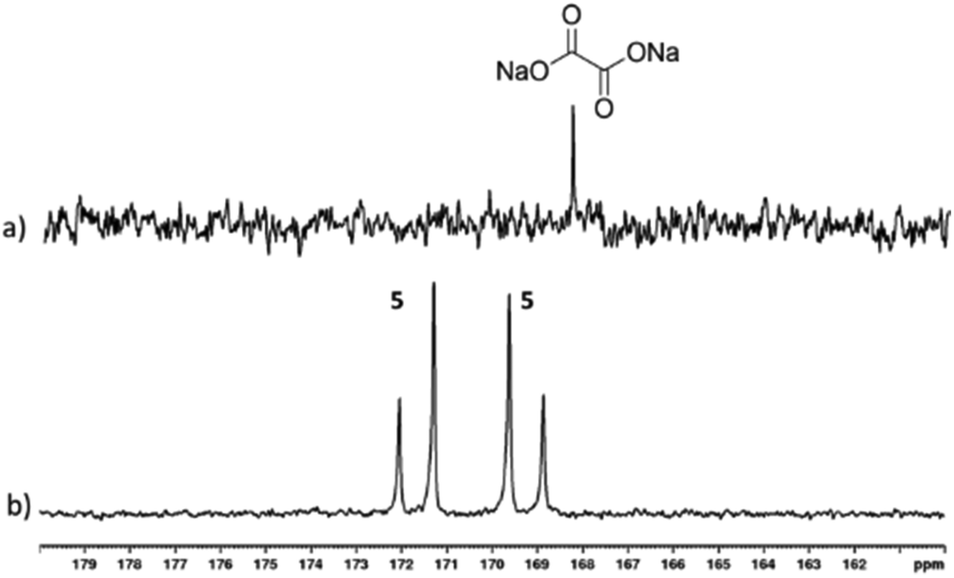
We have shown how 3 can be formed in situ and subsequently used as a precursor to form [Ir(H)2(η2-pyruvate)(DMSO)(IMes)], (4) which delivers SABRE enhancement to pyruvate. This α-keto acid motif also features in oxalate, a metabolic product that binds mineral ions in the body and is found in many foods.60 Indeed, Levitt and coworkers have reported the creation of a long lived hyperpolarised 13C2 singlet spin pair for a deuterated ester derivative of oxalate using DNP.61 Consequently, we now describe tests on sodium-1,2-oxalate-[13C2]. This involved shaking a mixture of 1, DMSO, and sodium-1,2-oxalate-[13C2] (1:5:5) with 3-bar p-H2 in 0.6 mL methanol-d4 for 30 seconds in a mu metal shield. Hyperpolarised 13C resonances were observed at δ 169.20 and δ 171.66 that share a JCC coupling of 76 Hz, as shown in Fig. 4. These signals cannot be due to free sodium-1,2-oxalate-[13C2] as a single resonance is expected.
While the complexation dynamics of oxalate are complex,62–64 based on the pyruvate observations described earlier it should be possible to form a species such as [Ir(H)2(η2-oxalate)(DMSO)(IMes)], 5 which would account for this observation. In fact, the 13C2 NMR signal profile shown in Fig. 4 is indicative of Zeeman magnetisation in such a product rather than singlet spin order. This indicates that while 13C polarisation transfer from a set of p-H2 derived hydride ligands initially results this must be associated with a [AA′BB′] spin system, which converts to the [AMSX] type with inequivalent hydride ligands in high field. A product with ligand arrangement analogous to 4a would match with this hypothesis.46 Unfortunately, examination of the hydride region of the resulting 1H NMR spectra reveal signals for 3 at δ−15.45 and δ −21.53 alongside over seven minor hydride containing complexes. None of the hydride resonances of these species correlated to these two 13C signals in an overnight 2D measurement at 243 K. Studying this reaction again at 315 K, proved to be of no benefit as conversion of 3 into the same range of hydride containing species results.
In contrast, when this experiment was repeated in DCM-d2 the hydride resonances of 3 are again observed at δ −15.71 and δ −21.27 but no 13C oxalate derived polarisation is seen. There is very low solubility of sodium oxalate in this medium. As a consequence, samples of 3 in CD2Cl2 or CH3OD were prepared and reacted with 50 μL D2O solutions of sodium-1,2-oxalate-[13C2]. Now a new product forms cleanly, that yields a hydride signal at δ −27.1 alongside diagnostic resonances for the IMes at δ 2.10, δ 2.34, δ 6.99, δ 7.15 and DMSO at δ 2.92 whose relative signal intensities suggest the presence of a [Ir(H)2(IMes)(DMSO)(L)n]x species, 6. Upon shaking with p-H2 the hydride resonances for 6 do not enhance, however, the addition of 3 mL of degassed hexane results in the growth of single crystals. Subsequent X-ray diffraction studies confirmed that the product 6 corresponds to the dimer [Ir2(H)4(IMes)2(DMSO)2(η2-κ2-Oxalate)] with structure shown in Fig. 5 (full X-ray and NMR characterisation data are given in the in the ESI‡). The ability of oxalate to form such dimers restricts its dissociation and thereby limits its use in these hyperpolarisation studies. Nonetheless, the structure of 6 reflects a common binding mode of oxalate with transition metals.63,65,66
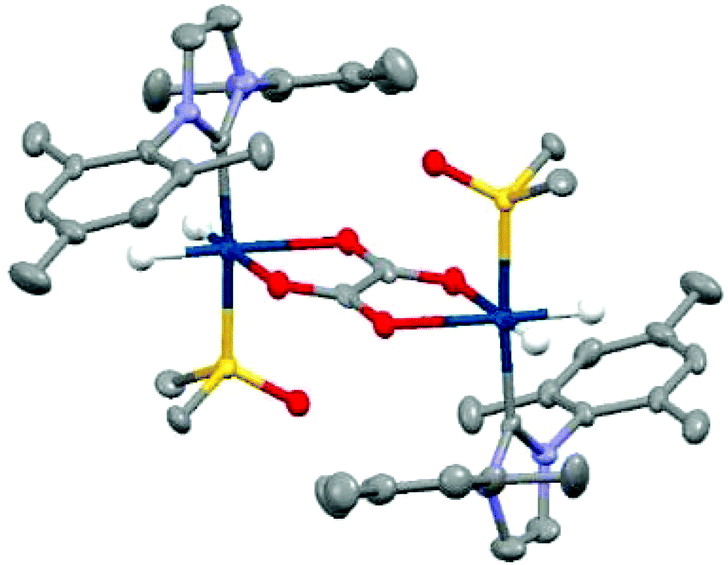 | ||
| Fig. 5 Structure of [Ir2(H)4(IMes)2(DMSO)2(η2-κ2-Oxalate)], 6, determined by X-ray crystallography. All non-hydride hydrogen atoms and solvent of crystallisation have been omitted for clarity. | ||
Experimental
All NMR measurements were carried out on a 400 MHz Bruker Avance III spectrometer using solutions at room temperature (298 K) unless otherwise stated. Para-hydrogen (p-H2) was produced by passing hydrogen gas over a spin-exchange catalyst (Fe2O3) and used for all hyperpolarisation experiments. This method produces constant p-H2 with ca. 93% purity. 1H (400 MHz) and 13C (100.6 MHz) NMR spectra were recorded with an internal deuterium lock. Chemical shifts are quoted as parts per million and referenced to the solvent. 13C NMR spectra were recorded with broadband proton decoupling. Coupling constants (J) are quoted in Hertz.Samples were prepared containing 2 mg [IrCl(COD)(IMes)] precatalyst (where IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazole-2-ylidene and COD = cis,cis-1,5-cyclooctadiene) in 0.6 mL of deuterated methanol-d4 unless otherwise stated in a 5 mm NMR tube that was fitted with a J. Young's tap. All commercial compounds were purchased from Sigma-Aldrich, Fluorochem, or Alfa-Aesar and used as supplied. [IrCl(COD)(IMes)] was synthesized according to a literature procedure.67 The resulting solutions were degassed by two freeze–pump–thaw cycles before the addition of 3-bar H2.
The shake and drop method was employed for recording hyperpolarised NMR spectra. This involves filling NMR tubes with p-H2 at 3 bar pressure and shaking them vigorously for 10 seconds in a 65 G magnetic field if 1H NMR spectra are to be recorded at 9.4 T. A mu metal shield is used if 13C NMR spectra are to be recorded. Unless otherwise stated multiple shake and drop measurements are undertaken and average signal enhancement values quoted. Signal enhancements and exchanges rates were calculated according to literature procedures.68
Conclusions
In conclusion, we have confirmed that the complex [IrCl(H)2(DMSO)2(IMes)] can be used as a polarisation transfer catalyst for hyperpolarising pyruvate. Complexes of this type undergo rapid hydrogen exchange which through a mechanistic investigation involving EXSY and DFT is expected to proceed dissociatively via five coordinate [IrCl(H)2(DMSO)(IMes)]. When this process is completed in the presence of an α-ketoacid, such as pyruvate, reaction to form [Ir(H)2(η2-pyruvate)(DMSO)(IMes)], which exists in two coordination isomers, occurs readily. Isomer 4b, in which the hydride ligands lie trans to pyruvate, is implicated in strong SABRE hyperpolarisation despite it undergoing slow ligand exchange. Consequently, a role for [IrCl(H)2(DMSO)2(IMes)], which undergoes rapid H2 and DMSO loss, is indicated in this process. When pyruvate is replaced by oxalate, stronger and more complex ligand binding is evident. The trapping of oxalate within the dimer 6 is expected to explain its much less efficient hyperpolarisation. Nonetheless, we predict that variations of the sulfoxide and NHC will enable future hyperpolarisation of oxalate and those of a wider range of other α-keto acids.Conflicts of interest
B. J. T. and S. B. D. (and others) are inventors on a patent application filed by the University of York related to this work (patent no. GB1818171.9, filed 7 November 2018).Acknowledgements
We thank Dr Peter Rayner, Hannah Kettle, and Dr Victoria Annis for synthesis of the [IrCl(COD)(IMes)], [IrBr(COD)(IMes)] and [Ir(CH3CN)(COD)(IMes)]PF6 precatalysts. Dr Wissam Iali is thanked for early stage discussions. Financial support from the Wellcome Trust (Grants 092506 and 098335), the MRC (MR/M008991/1) and the EPSRC (B. J. T. studentship and Impact Accelerator Award G0025101) is gratefully acknowledged.References
- T. S. Leyh, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 515–542 CrossRef CAS PubMed.
- X. Liu, L. He, Y.-M. Liu and Y. Cao, Acc. Chem. Res., 2013, 47, 793–804 CrossRef PubMed.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed.
- D. M. Ermert and L. J. Murray, Dalton Trans., 2016, 45, 14499–14507 RSC.
- R. Pretorius, M. R. Fructos, H. Müller-Bunz, R. A. Gossage, P. J. Pérez and M. Albrecht, Dalton Trans., 2016, 45, 14591–14602 RSC.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed.
- L.-S. Wang, R. McDonald and M. Cowie, Inorg. Chem., 1994, 33, 3735–3744 CrossRef CAS.
- M. Álvarez, E. Álvarez, M. R. Fructos, J. Urbano and P. J. Pérez, Dalton Trans., 2016, 45, 14628–14633 RSC.
- P. B. Chock and J. Halpern, J. Am. Chem. Soc., 1966, 88, 3511–3514 CrossRef CAS.
- W. H. Thompson and C. T. Sears, Inorg. Chem., 1977, 16, 769–774 CrossRef CAS.
- K. Searles, M. Pink, K. G. Caulton and D. J. Mindiola, Dalton Trans., 2012, 41, 9619–9622 RSC.
- C. E. Johnson and R. Eisenberg, J. Am. Chem. Soc., 1985, 107, 3148–3160 CrossRef CAS.
- N. Wang, M. Wang, Y. Wang, D. Zheng, H. Han, M. r. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2013, 135, 13688–13691 CrossRef CAS PubMed.
- C. R. Landis and T. W. Brauch, Inorg. Chim. Acta, 1998, 270, 285–297 CrossRef CAS.
- S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS.
- K. Sorochkina, V. V. Zhivonitko, K. Chernichenko, V.-V. Telkki, T. Repo and I. V. Koptyug, J. Phys. Chem. Lett., 2018, 9, 903–907 CrossRef CAS PubMed.
- V. V. Zhivonitko, K. Sorochkina, K. Chernichenko, B. Kótai, T. Földes, I. Pápai, V.-V. Telkki, T. Repo and I. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 27784–27795 RSC.
- S. Wagner, Magn. Reson. Mater. Phys., Biol. Med., 2014, 27, 195–199 CrossRef CAS PubMed.
- D. Canet, C. Aroulanda, P. Mutzenhardt, S. Aime, R. Gobetto and F. Reineri, Concepts Magn. Reson., Part A, 2006, 28, 321–330 CrossRef.
- P. M. Richardson, R. O. John, A. J. Parrott, P. J. Rayner, W. Iali, A. Nordon, M. E. Halse and S. B. Duckett, Phys. Chem. Chem. Phys., 2018, 20, 26362–26371 RSC.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- R. Eisenberg, Acc. Chem. Res., 1991, 24, 110–116 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548–10556 CrossRef CAS.
- K. V. Kovtunov, I. E. Beck, V. I. Bukhtiyarov and I. V. Koptyug, Angew. Chem., Int. Ed., 2008, 47, 1492–1495 CrossRef CAS PubMed.
- P. Bhattacharya, E. Y. Chekmenev, W. H. Perman, K. C. Harris, A. P. Lin, V. A. Norton, C. T. Tan, B. D. Ross and D. P. Weitekamp, J. Magn. Reson., 2007, 186, 150–155 CrossRef CAS PubMed.
- N. M. Zacharias, H. R. Chan, N. Sailasuta, B. D. Ross and P. Bhattacharya, J. Am. Chem. Soc., 2011, 134, 934–943 CrossRef PubMed.
- F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. Green, J. Chem. Phys., 2009, 131, 194505 CrossRef PubMed.
- A. J. Ruddlesden, R. E. Mewis, G. G. Green, A. C. Whitwood and S. B. Duckett, Organometallics, 2015, 34, 2997–3006 CrossRef CAS PubMed.
- A. J. Ruddlesden and S. B. Duckett, Chem. Commun., 2016, 52, 8467–8470 RSC.
- L. S. Lloyd, A. Asghar, M. J. Burns, A. Charlton, S. Coombes, M. J. Cowley, G. J. Dear, S. B. Duckett, G. R. Genov and G. G. R. Green, Catal. Sci. Technol., 2014, 4, 3544–3554 RSC.
- K. M. Appleby, R. E. Mewis, A. M. Olaru, G. G. R. Green, I. J. S. Fairlamb and S. B. Duckett, Chem. Sci., 2015, 6, 3981–3993 RSC.
- A. L. Sargent and M. B. Hall, Inorg. Chem., 1992, 31, 317–321 CrossRef CAS.
- M. Fekete, O. Bayfield, S. B. Duckett, S. Hart, R. E. Mewis, N. Pridmore, P. J. Rayner and A. Whitwood, Inorg. Chem., 2013, 52, 13453–13461 CrossRef CAS.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. Cockett, S. B. Duckett, G. G. Green, J. A. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- H. Zeng, J. Xu, J. Gillen, M. T. McMahon, D. Artemov, J.-M. Tyburn, J. A. Lohman, R. E. Mewis, K. D. Atkinson and G. G. Green, J. Magn. Reson., 2013, 237, 73–78 CrossRef CAS PubMed.
- E. B. Dücker, L. T. Kuhn, K. Münnemann and C. Griesinger, J. Magn. Reson., 2012, 214, 159–165 CrossRef PubMed.
- M. L. Truong, T. Theis, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Phys. Chem. C, 2015, 119, 8786–8797 CrossRef CAS PubMed.
- P. Spannring, I. Reile, M. Emondts, P. P. Schleker, N. K. Hermkens, N. G. van der Zwaluw, B. J. van Weerdenburg, P. Tinnemans, M. Tessari and B. Blümich, Chem. – Eur. J., 2016, 22, 9277–9282 CrossRef CAS PubMed.
- R. E. Mewis, R. A. Green, M. C. Cockett, M. J. Cowley, S. B. Duckett, G. G. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- W. Iali, P. J. Rayner, A. Alshehri, A. J. Holmes, A. J. Ruddlesden and S. B. Duckett, Chem. Sci., 2018, 9, 3677–3684 RSC.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., 2019, 131, 10377–10381 CrossRef.
- A. F. Oliveri, L. A. Wills, C. R. Hazlett, M. E. Carnes, I.-Y. Chang, P. H.-Y. Cheong and D. W. Johnson, Chem. Sci., 2015, 6, 4071–4085 RSC.
- L. Dadci, H. Elias, U. Frey, A. Hoernig, U. Koelle, A. E. Merbach, H. Paulus and J. S. Schneider, Inorg. Chem., 1995, 34, 306–315 CrossRef CAS.
- S. Ogo, N. Makihara, Y. Kaneko and Y. Watanabe, Organometallics, 2001, 20, 4903–4910 CrossRef CAS.
- S. Knecht, S. Hadjiali, D. A. Barskiy, A. Pines, G. Sauer, A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya and G. Buntkowsky, J. Phys. Chem. C, 2019, 123, 16288–16293 CrossRef CAS.
- B. J. Tickner, R. O. John, S. S. Roy, S. J. Hart, A. C. Whitwood and S. B. Duckett, Chem. Sci., 2019, 10, 5235–5245 RSC.
- A. M. Olaru, A. Burt, P. J. Rayner, S. J. Hart, A. C. Whitwood, G. G. Green and S. B. Duckett, Chem. Commun., 2016, 52, 14482–14485 RSC.
- B. J. Tickner, W. Iali, S. S. Roy, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2019, 20, 241–245 CrossRef CAS PubMed.
- B. E. Hauger, D. Gusev and K. G. Caulton, J. Am. Chem. Soc., 1994, 116, 208–214 CrossRef CAS.
- O. Semenova, P. M. Richardson, A. J. Parrott, A. Nordon, M. E. Halse and S. B. Duckett, Anal. Chem., 2019, 91, 6695–6701 CAS.
- P. Zhou, A. A. Vitale, J. San Filippo Jr. and W. H. Saunders Jr., J. Am. Chem. Soc., 1985, 107, 8049–8054 CrossRef CAS.
- F. Abu-Hasanayn, A. S. Goldman and K. Krogh-Jespersen, J. Phys. Chem., 1993, 97, 5890–5896 CrossRef CAS.
- B. J. Tickner, R. O. John, S. S. Roy, S. Hart, A. C. Whitwood and S. B. Duckett, Chem. Sci., 2019, 10, 5235–5245 RSC.
- S. S. Roy, K. M. Appleby, E. J. Fear and S. B. Duckett, J. Phys. Chem. Lett., 2018, 9, 1112–1117 CrossRef CAS PubMed.
- H. Sidhu, R. Gupta, S. K. Thind and R. Nath, Ann. Nutr. Metab., 1987, 31, 354–361 CrossRef CAS PubMed.
- C. Laustsen, G. Pileio, M. C. D. Tayler, L. J. Brown, R. C. D. Brown, M. H. Levitt and J. H. Ardenkjaer-Larsen, Magn. Reson. Med., 2012, 68, 1262–1265 CrossRef CAS PubMed.
- N. Hao, E. Shen, Y. G. Li, E. B. Wang, C. W. Hu and L. Xu, Eur. J. Inorg. Chem., 2004, 2004, 4102–4107 CrossRef.
- B. Modec, J. V. Brenčič and J. Koller, Eur. J. Inorg. Chem., 2004, 2004, 1611–1620 CrossRef.
- M. Gruselle, C. Train, K. Boubekeur, P. Gredin and N. Ovanesyan, Coord. Chem. Rev., 2006, 250, 2491–2500 CrossRef CAS.
- B. Modec, J. V. Brenčič, D. Dolenc and J. Zubieta, Dalton Trans., 2002, 4582–4586 RSC.
- M. E. Robinson, J. E. Mizzi, R. J. Staples and R. L. LaDuca, Cryst. Growth Des., 2015, 15, 2260–2271 CrossRef CAS.
- L. D. Vazquez-Serrano, B. T. Owens and J. M. Buriak, Inorg. Chim. Acta, 2006, 359, 2786–2797 CrossRef CAS.
- B. J. Tickner, W. Iali, S. S. Roy, A. C. Whitwood and S. B. Duckett, ChemPhysChem, 2019, 20, 241–245 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Robin N. Perutz on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1941589. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02951f |
| This journal is © The Royal Society of Chemistry 2019 |