
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural characterization of arsinoamides – early transition metal (Zr and Hf) and main group metal (Al, In, Sn, and Pb) complexes†
Xiao
Chen
,
Michael T.
Gamer
 and
Peter W.
Roesky
*
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 2nd October 2019
Abstract
By reaction of MCl4 (M = Zr, Hf) with 2 equiv. of [(Mes2AsNPh){Li(OEt2)2}], the first group 4 metal arsinoamide complexes [(Mes2AsNPh)2MCl2(THF)] were synthesized. They feature two weak M–As interactions. After formally replacing the chloride atoms by the amido ligands [NMe2]−, a more diverse M–As interaction arises in [(Mes2AsNPh)2M(NMe2)2]: only one M–As contact can be observed with a substantially shorter distance. This type of interaction may originate from the steric effect of the substituents on the metal center. In addition, the coordination behavior of arsinoamide in main group metal chemistry was investigated. Thus, arsinoamide complexes of Al(III), In(III), Sn(II) and Pb(II) are reported. In contrast to the group 4 complexes, no M–As interaction can be detected in these four complexes.
Introduction
Phosphinoamides (R2P-N(H)R′) and their anionic derivatives [R2P-NR′]− have been widely employed to synthesize mono- and multimetallic complexes.1–6 In addition, due to the close proximity of a hard (N) and a soft donor atom (P), phosphinoamides may act as powerful ligands to investigate metal–metal interaction in the early/late heterobimetallic complexes.7–11 Compared to the rich results reported for phosphinoamide-supported complexes, the coordination chemistry of their heavier arsenic analogues, arsinoamides ([(R′)2As-NR]−), is nearly unknown. Recently, we have established phenylamino(dimesityl)arsane Mes2AsN(H)Ph (Mes = 2,4,6-Me3C6H2) and its alkali metal derivatives, [(Mes2AsNPh){Li(OEt2)2}], [(Mes2AsNPh){Na(OEt2)}]2 and [(Mes2AsNPh){K(THF)}]2.12 Further reaction of lithium arsinoamide with low-valent group 14 compounds ([{PhC(tBuN)2}ECl] (E = Si, Ge), GeCl2·dioxane) led either to substitution or to As–N bond insertion products.13Motivated by the well-established metal complexes of phosphinoamide, we disclose here the synthesis of bi-substituted zirconium and hafnium complexes of arsinoamide. In addition, further investigation towards the substituent effect on the M–As interaction in early transition metal chemistry is reported here. Herein, the Al(III), In(III), Sn(II) and Pb(II) complexes of arsinoamide are described.
Results and discussion
Group 4 compounds [(Mes2AsNPh)2MCl2(THF)] (1: M = Zr and 2: M = Hf) were synthesized by the reaction of MCl4 (M = Zr, Hf) with 2 equiv. of [(Mes2AsNPh){Li(OEt2)2}] via a salt metathesis reaction (Scheme 1). Compounds 1 and 2 were fully characterized by standard analytical and spectroscopic techniques and the molecular structures were established by single crystal X-ray diffraction analysis.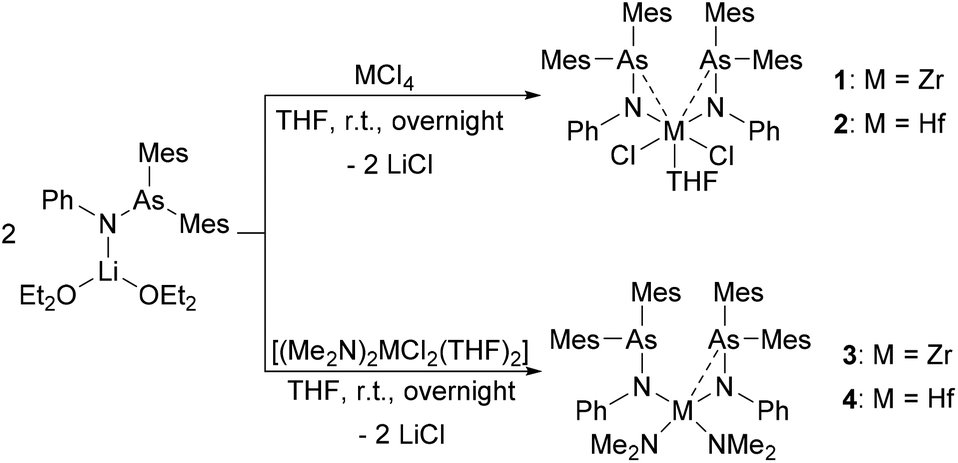 | ||
| Scheme 1 Synthesis of compounds 1–4. | ||
In general, the 1H and 13C{1H} NMR spectra of 1 and 2 show similar patterns to those observed for [(Mes2AsNPh){Li(OEt2)2}]. In the 1H NMR spectrum, compared to [(Mes2AsNPh){Li(OEt2)2}], the resonance of the ortho-methyl groups is up-field shifted (2.30 ppm (1) and 2.27 ppm (2) vs. 2.66 ppm ([(Mes2AsNPh){Li(OEt2)2}])). In the 13C{1H} NMR spectra of 1 and 2, two signals for the methyl groups and eight aromatic signals are observed, indicating highly symmetric species of 1 and 2 in solution with equivalent aryl groups on the NMR timescale.
Single crystals suitable for X-ray diffraction analysis of 1 and 2 were obtained by slowly evaporating a dichloromethane (DCM) solution of the complexes. The crystal structures of 1 and 2 (Fig. 1) reveal isostructural complexes, which may be related to the similar atomic radii of Zr and Hf. Selected bond distances and angles are summarized in Table 1. According to the molecular structures of 1 and 2, the M–As interaction can be observed in both compounds (Zr–As: 3.2101(6) and 3.2216(6) Å and Hf–As: 3.2423(4) and 3.2342(4) Å). Considering the longest reported bond length values (Zr–As: 2.9974 (7) Å in [(Cp′′2Zr)As5(ZrCp′′)] (Cp′′ = (tBu)2C5H2) and Hf–As: 2.8966(7) Å in [HfI4{o-C6H4(AsMe2)2}2]),14,15 the weakness of the arsenic metal contact in 1 and 2 is evident from the longer M–As distances. We anticipate that packing effects play a significant role in these interactions. The nitrogen atoms of the arsinoamides are bound to the metal center with average distances of 2.0375 Å (Zr–N) and 2.019 Å (Hf–N), which agree with the published values for an M–N single bond (e.g. Zr–N: 2.060(3) Å and Hf–N: 2.111(4) Å).16,17 The nitrogen atoms adopt a nearly perfect planar geometry, similar to the reported electron-poor d0 metal phosphinoamide and related complexes.18–20 As is apparent, the arsinoamide ligands of 1 and 2 are bound asymmetrically. In contrast to the trans conformation in [(Mes2AsNPh){Li(OEt2)2}],12 the arsinoamides in 1 and 2 exhibit a cis conformation, as defined by the relative orientation of the substituents on the N and As atoms. This indicates the presence of trans → cis interconversion of arsinoamide during the reaction of [(Mes2AsNPh){Li(OEt2)2}] with MCl4.
 | ||
| Fig. 1 Molecular structures of compounds 1 (left) and 2 (right). Hydrogen atoms and non-coordinating solvent molecules are omitted for clarity. | ||
| 1 | Zr–As1 | 3.2101(6) | Zr–As2 | 3.2216(6) |
| Zr–N1 | 2.039(4) | Zr–N2 | 2.036(4) | |
| N1–As1 | 1.867(3) | N2–As2 | 1.891(3) | |
| Zr–C1 | 2.827(5) | N1–Zr–N2 | 118.42(14) | |
| 2 | Hf–As1 | 3.2423(4) | Hf–As2 | 3.2342(4) |
| Hf–N1 | 2.020(3) | Hf–N2 | 2.018(3) | |
| N1–As1 | 1.870(3) | N2–As2 | 1.877(3) | |
| Hf–C1 | 2.794(3) | N1–Hf–N2 | 115.30(11) | |
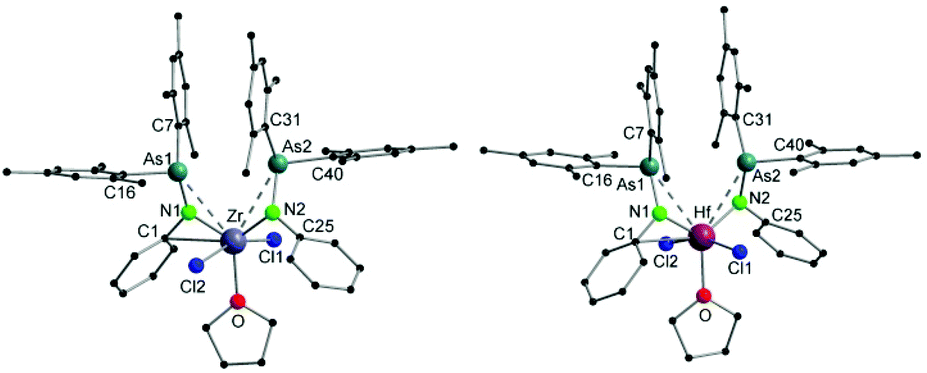
| 3 | Zr–As2 | 3.0798(4) | Zr–N1 | 2.081(2) |
| Zr–N2 | 2.096(2) | Zr–N3 | 2.040(2) | |
| Zr–N4 | 2.035(2) | N1–As1 | 1.878(2) | |
| N2–As2 | 1.863(2) | N1–Zr–N2 | 118.94(7) | |
| N3–Zr–N4 | 103.33(9) | |||
| 4 | Hf–As2 | 3.0892(3) | Hf–N1 | 2.069(2) |
| Hf–N2 | 2.078(2) | Hf–N3 | 2.028(2) | |
| Hf–N4 | 2.037(3) | N1–As1 | 1.885(2) | |
| N2–As2 | 1.870(2) | N1–Hf–N2 | 118.94(9) | |
| N3–Hf–N4 | 103.67(12) |
To further investigate the substituent effect on the M–As contact, compounds [(Mes2AsNPh)2M(NMe2)2] (3: M = Zr and 4: M = Hf) were synthesized by the reaction of [(Me2N)2MCl2 (THF)2] with 2 equiv. of [(Mes2AsNPh){Li(OEt2)2}] in a mediocre yield (Scheme 1). The 1H and 13C{1H} NMR spectra of 3 and 4 show very similar patterns. In contrast to [(Mes2AsNPh){Li(OEt2)2}], the resonance of the ortho-methyl groups of the mesityl ring is up-field shifted in the 1H NMR spectrum (2.32 ppm (3) and 2.31 ppm (4) vs. 2.66 ppm in [(Mes2AsNPh){Li(OEt2)2}]). A consistent trend can also be observed for the resonance of the amido ligands [NMe2]− (2.95 ppm (3) vs. 3.31 ppm in [(Me2N)2ZrCl2 (THF)2] and 2.97 ppm (4) vs. 3.39 ppm in [(Me2N)2HfCl2 (THF)2]). Despite the fact that the M–As coordination makes the mesityl groups on As inequivalent in the solid state (vide infra), only one aromatic signal of the mesityl ring is detected (3: 6.62 ppm and 4: 6.63 ppm). In the 13C{1H} NMR spectrum, both complexes reveal the signals of the characteristic groups (3: 44.0 (NMe2), 22.9 (o-Me), and 21.0 (p-Me) and 4: 43.5 (NMe2), 22.9(o-Me), and 20.9 (p-Me)): the observation of eight aromatic signals indicates highly symmetric species of 3 and 4 in solution.
Single crystals suitable for X-ray diffraction analysis of 3 and 4 were obtained upon cooling a diethyl ether/n-pentane (1/1) solution of the compounds at −30 °C overnight. The molecular structures of 3 and 4 reveal isostructural arrangements (Fig. 2) with the metal core lying in the center of a distorted tetrahedron with an additional M–As interaction. The metal atoms are coordinated by two nitrogen atoms of arsinoamide and two nitrogen atoms of two amido ligands. The most striking aspect of the structure is the M–As type of interaction. Only one arsenic atom is bound to the metal center (Zr–As: 3.0798(4) Å and Hf–As: 3.0892(3) Å) resulting in an η2 coordination mode of the corresponding arsinoamide ligand, while the other arsenic metal distances are too long to indicate a significant interaction. Despite the fact that only one M–As interaction can be detected in 3 and 4, their distances are substantially shorter (ca. 0.14 Å) than those in 1 and 2 (Table 1). However, packing effects may be the reason for this observation.
 | ||
| Fig. 2 Molecular structures of compounds 3 (left) and 4 (right). Hydrogen atoms are omitted for clarity. | ||
It should be noted that only two structurally characterized hafnium arsenic bonds were reported before: ([Cp′2HfCl{AsSiMe3}2] (Cp′ = MeC5H3) and [HfI4{o-C6H4(AsMe2)2}2]).15,21 The lone pairs located on the As atoms make compounds 1–4 potentially useful as ligands for the synthesis of heterobimetallic complexes, similar to the well-known Zr(Hf) complexes of phosphinoamides.7,18,22–28
To investigate the coordination behavior of arsinoamide in main group chemistry, aluminum, indium, tin, and lead were selected as metals.
Reaction of AlCl3 with 1 equiv. of [(Mes2AsNPh){Li(OEt2)2}] resulted, via a salt metathesis reaction, in the mono-substituted complex [(Mes2AsNPh)AlCl2(THF)] (5) (Scheme 2) in 57% crystalline yield. The 1H and 13C{1H} NMR spectra of compound 5 are very similar to those of [(Mes2AsNPh){Li(OEt2)2}]. For example, in the 1H NMR spectrum, the characteristic resonances of the ortho- and para-methyl groups of the mesityl ring are seen in a similar range to the starting material (2.62 ppm and 2.11 ppm in 5vs. 2.66 ppm and 2.12 ppm in [(Mes2AsNPh){Li(OEt2)2}]).
 | ||
| Scheme 2 Synthesis of compounds 5 and 6. | ||
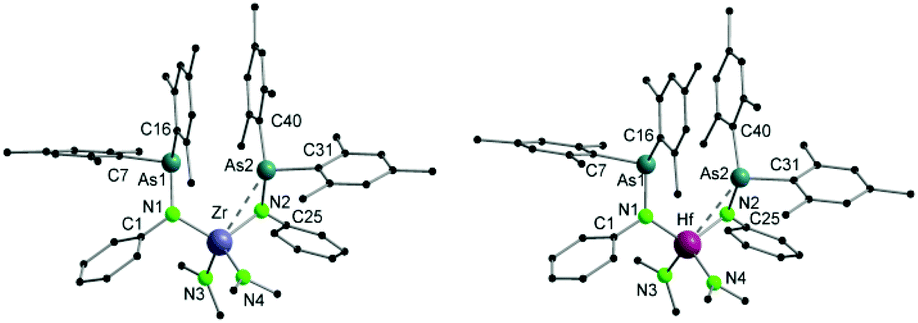
Single crystals suitable for X-ray diffraction analysis of 5 were obtained by slowly evaporating a DCM solution of the complex (Fig. 3). Selected bond distances and angles are listed in Table 2. The aluminum atom coordinates to one nitrogen atom of arsinoamide, two chloride atoms, and one oxygen atom of THF, resulting in a distorted tetrahedral geometry. The Al–N (1.819(3) Å) and Al–O (1.869(3) Å) bond lengths are in line with the reported values (e.g. Al–N: 1.792(2) Å and Al–O: 1.9246(10) Å).29–31 The three-coordinated nitrogen atom adopts an almost planar arrangement considering the sum of the bond angles around the nitrogen atom (357.4°). The As–N–Al bond angle (125.9(2)°) is consistent with As–N–Li (122.8(5)°) in the starting material. As seen in [(Mes2AsNPh){Li(OEt2)2}], arsinoamide in 5 exhibits a trans conformation.12 No Al–As interaction can be observed in the molecular structure of 5.
 | ||
| Fig. 3 Molecular structures of compounds 5 (left) and 6 (right). Hydrogen atoms are omitted for clarity. | ||
| 5 | Al–N | 1.819(3) | N–As | 1.884(3) |
| Al–Cl1 | 2.119(2) | Al–Cl2 | 2.1411(14) | |
| Al–O | 1.869(3) | As–N–Al | 125.9(2) | |
| 6 | In–N | 2.078(2) | N–As | 1.861(2) |
| In–Cl1 | 2.3884(7) | In–Cl2 | 2.3540(7) | |
| In–Cl3 | 2.3906(7) | As–N–In | 126.33(12) |
After the reaction of InCl3 with 1 equiv. of [(Mes2AsNPh){Li(OEt2)2}] in THF, [(Mes2AsNPh)InCl3][Li(THF)4] (6) was isolated in low crystalline yield, which may be explained by the instability of the product. In the 1H NMR spectrum, compared to [(Mes2AsNPh){Li(OEt2)2}], the resonance of the ortho-methyl groups is up-field shifted (2.36 ppm (6) vs. 2.66 ppm ([(Mes2AsNPh){Li(OEt2)2}])). In general, the 1H and 13C{1H} NMR spectra of 6 show the expected pattern, which is similar to the starting material [(Mes2AsNPh){Li(OEt2)2}]. Even under an inert atmosphere, slow decomposition of 6 was observed at room temperature, resulting in the formation of aminoarsane (Mes2AsN(H)Ph). In the 1H NMR spectrum, the characteristic resonances of the ortho-, para-methyl (2.35 and 2.06 ppm, respectively) and NH groups (3.99 ppm), with an integral ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:1, can be detected, which are assigned to [Mes2AsN(H)Ph] (Fig. S19†). In the IR spectrum, a single N–H stretching absorption band at 3376 cm−1 further proves the formation of [Mes2AsN(H)Ph] (3377 cm−1) (Fig. S28†). A similar decomposition has been reported during the reaction of phosphinoamide lanthanide complexes [(Ph2PNPh)4Ln][Li(THF)4] (Ln = Y, Lu) with [Pd2(C3H5)2Cl2], where the neutral aminophosphine [Ph2PN(H)Ph] was formed.10
:6:1, can be detected, which are assigned to [Mes2AsN(H)Ph] (Fig. S19†). In the IR spectrum, a single N–H stretching absorption band at 3376 cm−1 further proves the formation of [Mes2AsN(H)Ph] (3377 cm−1) (Fig. S28†). A similar decomposition has been reported during the reaction of phosphinoamide lanthanide complexes [(Ph2PNPh)4Ln][Li(THF)4] (Ln = Y, Lu) with [Pd2(C3H5)2Cl2], where the neutral aminophosphine [Ph2PN(H)Ph] was formed.10
Single crystals suitable for X-ray diffraction analysis of 6 were obtained by slowly evaporating a THF solution of the complex (Fig. 3). Single crystal X-ray diffraction analyses reveal that 6 is composed of a [Li(THF)4]+ cation and a [(Mes2AsNPh)InCl3]− anion. The indium atom is four-coordinated by three chloride and one nitrogen atoms of arsinoamide, resulting in a distorted tetrahedral geometry. The three-coordinated nitrogen atom adopts a nearly perfect planar trigonal geometry (the sum of bonding angles: 359.3°). The N–As bond distance (1.861(2) Å) does not deviate significantly from [(Mes2AsNPh){Li(OEt2)2}] (1.816(6) Å). However, the As–N–M bond angle (As–N–In: 126.33(12)°) is larger than that in [(Mes2AsNPh){Li(OEt2)2}] (As–N–Li: 112.8(5)°). No In–As interaction can be observed in the molecular structure of 6. To the best of our knowledge no comparable structurally characterized indium phosphinoamide complex is known so far.
Encouraged by the attractive reactivity of silylene and germylene with arsinoamide,13 the heavier homoleptic bi-substituted complexes of stannylene and plumbylene, [(Mes2AsNPh)2E] (E = Sn (7) and Pb (8)), were isolated by the reaction of ECl2 with 2 equiv. of [(Mes2AsNPh){Li(OEt2)2}] (Scheme 3).
 | ||
| Scheme 3 Synthesis of compounds 7 and 8. | ||
Compound 8 is extremely sensitive towards moisture and oxygen. In the solid state, compounds 7 and 8 are stable at room temperature under an inert atmosphere. Nevertheless, slow decomposition can be detected in C6D6 at room temperature. Compounds 7 and 8 show very similar patterns in the 1H and 13C{1H} NMR spectra as observed for [(Mes2AsNPh){Li(OEt2)2}]. In the 119Sn{1H} NMR spectrum, the resonance of 7 is detected at 319 ppm, which is in line with that at 291 ppm for N-heterocyclic stannylene [{CH2(CH2NDipp)2}Sn].32 The 207Pb{1H} NMR signal of 8 (3244 ppm) is in the range of reported diamido plumbylene complexes (e.g. 3504 ppm in [{DippN(CH2)3NDipp}Pb]).33
Single crystals of both 7 and 8 were grown by diffusion of a mixture of n-pentane and diethyl ether (1/1) into a solution of the complexes in toluene (Fig. 4). Single crystal X-ray diffraction analyses reveal that both compounds are isostructural. They crystallize in the tetragonal space group I41/a. Selected bond lengths and angles are listed in Table 3.
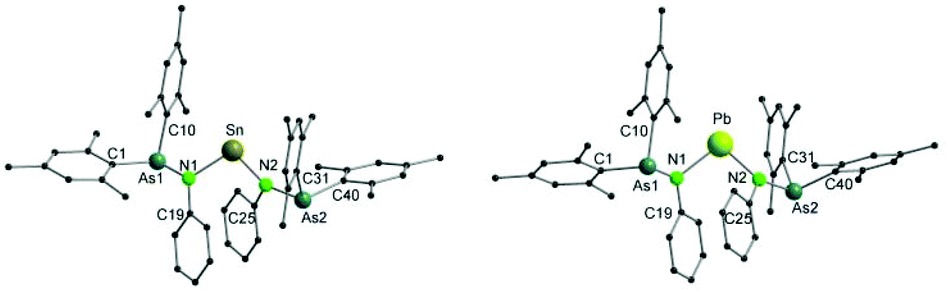 | ||
| Fig. 4 Molecular structures of 7 (left) and 8 (right). Hydrogen atoms are omitted for clarity. | ||
| 7 | Sn–N1 | 2.120(4) | Sn–N2 | 2.115(4) |
| N1–As1 | 1.880(4) | N2–As2 | 1.880(4) | |
| N1–Sn–N2 | 97.8(2) | |||
| 8 | Pb–N1 | 2.236(6) | Pb–N2 | 2.211(6) |
| N1–As1 | 1.872(6) | N2–As2 | 1.873(6) | |
| N1–Pb–N2 | 97.4(2) |
In both 7 and 8, the central metal atom is coordinated by two nitrogen atoms of the arsinoamide ligands. The E–N bond distances (Sn–N: 2.115(4) and 2.120(4) Å and Pb–N: 2.211(6) and 2.236(6) Å) fit well to the published values for the E–N single bond (e.g. 2.089(2) Å and 2.115(2) Å (Sn–N) and 2.087(5) Å and 2.298(4) Å (Pb–N)).33–36 The N–E–N bond angles (N1–Sn–N2: 97.8(2)° and N1–Pb–N2: 97.4(2)°) are smaller than the comparable N–E–N bond angles (e.g. 106.13(8)° and 104.7(2)° (N–Sn–N) and 100.60(8)° and 105.75(14)° (N–Pb–N)),37–39 which may originate from the steric effect of the ligand. The sum of bond angles around the nitrogen atoms varies from 348.9° to 356.4°, indicating a slightly disordered pyramidal geometry of the nitrogen atoms. None of the arsenic atoms shows any interaction with the metal center in 7 and 8. To the best of our knowledge, there is no comparable structurally characterized stannylene or plumbylene complexes of phosphinoamides.
Conclusions
In summary, the reaction of arsinoamide with early transition metals (Zr(IV) and Hf(IV)) and main group metals (Al(III), In(III), Sn(II) and Pb(II)) was investigated. In the case of Zr and Hf, di(arsinoamide) complexes 1–4 were established, in which the first η2 coordination mode of arsinoamide can be observed. In addition, it seems that the packing effects can significantly influence the M–As interaction. Due to the presence of the As lone pair, compounds 1–4 can act as potentially useful ligands for further synthesis of early/late heterobimetallic complexes. In the arsinoamide main group metal complexes of Al(III), In(III), Sn(II), and Pb(II) only the nitrogen atom binds to the metal center. The lone pair on As remains uncoordinated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. C. thanks the China Scholarship Council (No. 201506250062) for generous support. Financial support provided by the DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)”, projects B3 is acknowledged.Notes and references
- Z. Fei and P. J. Dyson, Coord. Chem. Rev., 2005, 249, 2056–2074 CrossRef CAS.
- P. W. Roesky, Heteroat. Chem., 2002, 13, 514–520 CrossRef CAS.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed.
- A. Stasch, Dalton Trans., 2014, 43, 7078–7086 RSC.
- A. J. Ayres, M. Zegke, J. P. Ostrowski, F. Tuna, E. J. McInnes, A. J. Wooles and S. T. Liddle, Chem. Commun., 2018, 54, 13515–13518 RSC.
- H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498–3507 CrossRef CAS.
- F. Völcker and P. W. Roesky, Dalton Trans., 2016, 45, 9429–9435 RSC.
- M. J. Sgro and D. W. Stephan, Chem. Commun., 2013, 49, 2610–2612 RSC.
- F. Völcker, F. M. Mück, K. D. Vogiatzis, K. Fink and P. W. Roesky, Chem. Commun., 2015, 51, 11761–11764 RSC.
- C. M. Thomas, J. W. Napoline, G. T. Rowe and B. M. Foxman, Chem. Commun., 2010, 46, 5790–5792 RSC.
- X. Chen, M. T. Gamer and P. W. Roesky, Dalton Trans., 2018, 47, 12521–12525 RSC.
- X. Chen, T. Simler, R. Yadav, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 9315–9318 RSC.
- M. Schmidt, A. E. Seitz, M. Eckhardt, G. B. Balázs, E. V. Peresypkina, A. V. Virovets, F. Riedlberger, M. Bodensteiner, E. M. Zolnhofer, K. Meyer and M. Scheer, J. Am. Chem. Soc., 2017, 139, 13981–13984 CrossRef CAS PubMed.
- W. Levason, M. L. Matthews, B. Patel, G. Reid and M. Webster, Dalton Trans., 2004, 3305–3312 RSC.
- L. N. Grant, M. E. Miehlich, K. Meyer and D. J. Mindiola, Chem. Commun., 2018, 54, 2052–2055 RSC.
- L. Grocholl, L. Stahl and R. J. Staples, Chem. Commun., 1997, 1465–1466 RSC.
- B. G. Cooper, C. M. Fafard, B. M. Foxman and C. M. Thomas, Organometallics, 2010, 29, 5179–5186 CrossRef CAS.
- H. Nagashima, T. Sue, T. Oda, A. Kanemitsu, T. Matsumoto, Y. Motoyama and Y. Sunada, Organometallics, 2006, 25, 1987–1994 CrossRef CAS.
- M. A. Rankin and C. C. Cummins, Dalton Trans., 2012, 41, 9615–9618 RSC.
- F. Lindenberg, J. Sieler, E. Hey-Hawkins, U. Müller and A. Pilz, Z. Anorg. Allg. Chem., 1996, 622, 683–688 CrossRef CAS.
- T. Sue, Y. Sunada and H. Nagashima, Eur. J. Inorg. Chem., 2007, 2007, 2897–2908 CrossRef.
- Y.-T. Bi, L. Li, Y.-R. Guo and Q.-J. Pan, Inorg. Chem., 2019, 2, 1290–1300 CrossRef PubMed.
- C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14–38 CrossRef CAS.
- J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115–5127 RSC.
- W. Zhou, N. I. Saper, J. P. Krogman, B. M. Foxman and C. M. Thomas, Dalton Trans., 2014, 43, 1984–1989 RSC.
- N. I. Saper, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Polyhedron, 2016, 114, 88–95 CrossRef CAS.
- H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS.
- K. J. Blakeney, P. D. Martin and C. H. Winter, Dalton Trans., 2018, 47, 10897–10905 RSC.
- H. S. Zijlstra, J. Pahl, J. Penafiel and S. Harder, Dalton Trans., 2017, 46, 3601–3610 RSC.
- T. Agou, T. Wasano, T. Sasamori and N. Tokitoh, Organometallics, 2014, 33, 6963–6966 CrossRef CAS.
- S. M. Mansell, C. A. Russell and D. F. Wass, Inorg. Chem., 2008, 47, 11367–11375 CrossRef CAS PubMed.
- J. P. Charmant, M. F. Haddow, F. E. Hahn, D. Heitmann, R. Fröhlich, S. M. Mansell, C. A. Russell and D. F. Wass, Dalton Trans., 2008, 6055–6059 RSC.
- J. A. Cabeza, J. M. Fernández-Colinas, P. García-Álvarez and D. Polo, Inorg. Chem., 2012, 51, 3896–3903 CrossRef CAS.
- S. I. Al-Rafia, P. A. Lummis, M. J. Ferguson, R. McDonald and E. Rivard, Inorg. Chem., 2010, 49, 9709–9717 CrossRef CAS PubMed.
- M. Chen, J. R. Fulton, P. B. Hitchcock, N. C. Johnstone, M. F. Lappert and A. V. Protchenko, Dalton Trans., 2007, 2770–2778 RSC.
- T. J. Hadlington, J. A. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC.
- T. Fjeldberg, H. Hope, M. F. Lappert, P. P. Power and A. J. Thorne, J. Chem. Soc., Chem. Commun., 1983, 639–641 RSC.
- M. Aman, O. Mrózek, L. Dostál, Z. Růžičková and R. Jambor, J. Organomet. Chem., 2018, 872, 1–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and IR spectra and crystallographic studies. CCDC 1949410–1949417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9DT03472B |
| This journal is © The Royal Society of Chemistry 2019 |