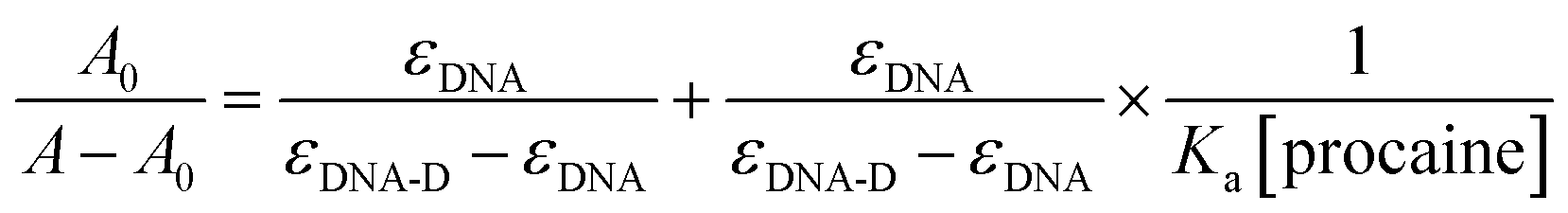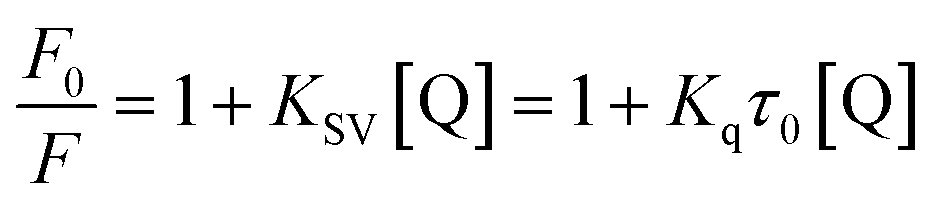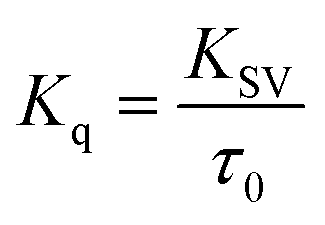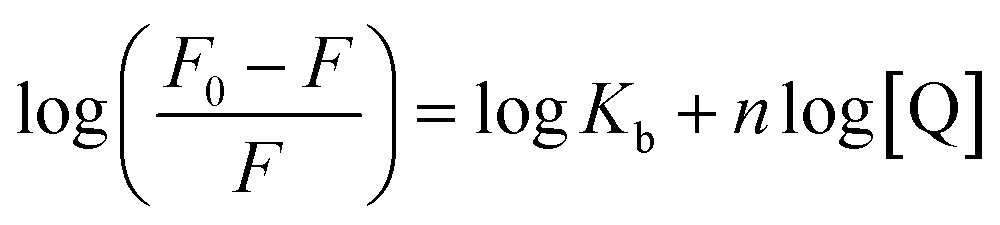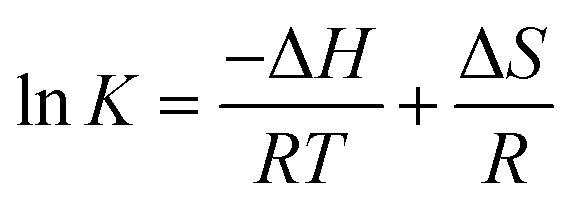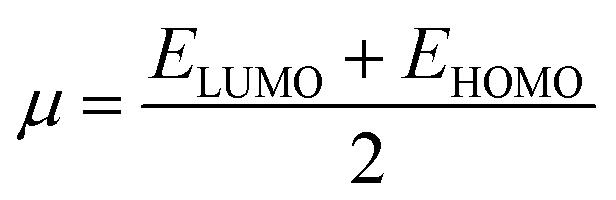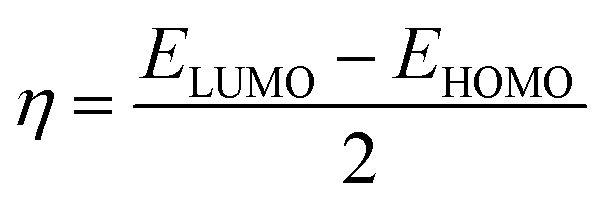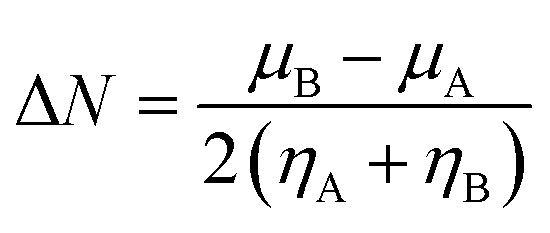DOI:
10.1039/C7RA13647A
(Paper)
RSC Adv., 2018,
8, 9083-9093
Comprehensive exploration of the anticancer activities of procaine and its binding with calf thymus DNA: a multi spectroscopic and molecular modelling study†
Received
26th December 2017
, Accepted 21st February 2018
First published on 1st March 2018
Abstract
Procaine is an anesthetic drug commonly administrated topically or intravenously for use in local anesthesia. Promisingly, some anticancer activities of procaine have also been reported. Therefore, the mechanism of interaction between anesthetic drug procaine with ct-DNA was determined collectively by means of various spectroscopic and molecular docking methods. Minor groove 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding of procaine to the ct-DNA was evidenced from absorption spectroscopy, fluorescence quenching, DNA melting, competitive binding measurements with EB and DAPI dyes, viscosity and CD spectroscopy together with molecular docking simulations and DFT calculations. Molecular docking on five different B-DNA structures (taken from the Protein Data Bank) shows that procaine binds in the AT rich region of all five B-DNA structures. Thermodynamic parameters, evaluated using van't Hoff's isotherm, shown that the interaction was feasible and the binding forces involved were hydrophobic as well as hydrogen bonding which were, further, confirmed by molecular docking. The frontier molecular orbitals (HOMO and LUMO) of procaine and DNA bases have been calculated by DFT method and the chemical potential (μ), chemical hardness (η) and fraction number of electrons (ΔN) from procaine to DNA bases were evaluated, which have shown that procaine acts as an electron donor to the DNA bases. Simultaneously, anticancer activities of procaine alone and in combination with doxorubicin were observed on the MCF-7 breast cancer cell line. The results showed that the combined treatment with both procaine and doxorubicin enhanced the cytotoxic and apoptotic inducing potential of doxorubicin.
:1 binding of procaine to the ct-DNA was evidenced from absorption spectroscopy, fluorescence quenching, DNA melting, competitive binding measurements with EB and DAPI dyes, viscosity and CD spectroscopy together with molecular docking simulations and DFT calculations. Molecular docking on five different B-DNA structures (taken from the Protein Data Bank) shows that procaine binds in the AT rich region of all five B-DNA structures. Thermodynamic parameters, evaluated using van't Hoff's isotherm, shown that the interaction was feasible and the binding forces involved were hydrophobic as well as hydrogen bonding which were, further, confirmed by molecular docking. The frontier molecular orbitals (HOMO and LUMO) of procaine and DNA bases have been calculated by DFT method and the chemical potential (μ), chemical hardness (η) and fraction number of electrons (ΔN) from procaine to DNA bases were evaluated, which have shown that procaine acts as an electron donor to the DNA bases. Simultaneously, anticancer activities of procaine alone and in combination with doxorubicin were observed on the MCF-7 breast cancer cell line. The results showed that the combined treatment with both procaine and doxorubicin enhanced the cytotoxic and apoptotic inducing potential of doxorubicin.
1. Introduction
Procaine, an anesthetic drug, has been used in dentistry for a long time. Several other uses of procaine have also been reported, such as for the cure of depression, inflammation, obesity, etc.1–5 Apart from these uses, procaine has an enormous range of applications, particularly, in cancer related research. It has been found to inhibit the triggering as well as the induction of thermotolerance in mouse fibroblast LM cells.6 Furthermore, it was found to improve the therapeutic index of chemotherapy agent cisplatin around two-fold.7 Procaine in a small dose of 2 mM has shown cytotoxicity to leukemic cells with further enhancement in the cytotoxicity at 42 °C.8 The most noticeable role of procaine related to the cancer chemotherapy is the demethylation of DNA.9,10 DNA methylation has enormous applications in gene regulation. A hallmark of cancer is the deregulation of the DNA methylation machinery and aberrant DNA methylation patterns. “DNA methyltransferases (DNMTs) catalyzes the transfer of a methyl group from S-adenosyl-L-methionine to the carbon-5 position of cytosine residues that results in an epigenetic change”.11 “These enzymes regulate gene expression, for example, hypermethylation of the promoter lead to transcriptional silencing of tumor suppressor genes. Therefore, DNMT inhibitors or demethylating agents are promising new drugs for the treatment of diseases such as cancer and brain disorders”.11,12 DNA demethylation is a process in which inhibitor suppresses the activity of DNA methyltransferases.13
Owing to the promising roles of the procaine in molecular biology it is important to understand the interaction of this pharmacologically important drug with nucleic acid. DNA is a significant genomic constituent of life that transports most of the hereditary data and enables the biological synthesis of proteins and enzymes through the replication and transcription.14 The investigation of binding of DNA with small molecules is also of importance among the researchers to investigate its modes of binding. Small molecules, such as drugs, can binds to the DNA either with covalent mode of binding or non-covalently. Covalent binding mode is ascribed to the formation of covalent adducts between DNA and ligand which generally takes place via alkylation or inter- and intrastrand crosslinking and is irreversible in nature which can restrict the activity, such as, transcription and replication of DNA and a consequent cell death.15,16 Non-covalent binding includes three different modes which are (1) intercalative binding, which comprises of the insertion of the ligand inside the DNA helix which may cause in the discrepancy in the DNA structure and functions; (2), electrostatic binding or external binding between the negatively charged DNA phosphate backbone and positive end of the molecules; and (3) groove binding that occurs either in major groove or minor groove depending on the size of the ligand as the bigger molecules tend to bind at major groove and smaller molecules generally occupies the minor groove.17 Intercalative and groove binding occur near or inside the DNA double helix, while the electrostatic binding takes place outside the groove.18,19
As procaine is an established anesthetic drug with many other uses including anticancer activities, therefore, in this study we have seen the interaction of procaine with ct-DNA by using theoretical as well as experimental methods in order to understand the mechanism of the drug binding. The anticancer capability of procaine was also seen by elucidating its cytotoxic and apoptotic effects on human breast cancer (MCF-7) cells. Finally, an effort was made to determine the influence of procaine combined with doxorubicin, an anthracycline antibiotic which is one of the most effective anticancer agents used for the treatment of breast cancer.
2. Experimental
Sodium salt of ct-DNA (D1501, type I, fibers, 41.9 mol% G–C and 58.1 mol% A–T) and procaine hydrochloride (99%) (Scheme S1†) were purchased from Sigma, USA. Rests of the chemicals were of high purity grades or molecular biology grades and the details of all the materials used are given in ESI.† Stock solutions of ct-DNA were prepared by reported methods and the ratio of absorbance at 260 nm and 280 nm was in the range of 1.8–1.9. For experimental binding studies, we have used UV-visible, fluorescence, circular dichroism spectroscopies and viscometry.20 For molecular modelling the geometries of procaine and DNA bases were optimized at DFT/BP RI by ORCA.21 Autodock 4.2.3 program was used to perform docking calculations of B-DNAs with procaine.22 Anticancer activities of procaine alone and in combination of doxorubicin on MCF-7 breast cancer cell lines were performed using well established methods. Detailed information of all experiments and computational studies is given in ESI.†
3. Results and discussions
3.1. Experimental investigation of interaction of procaine with ct-DNA
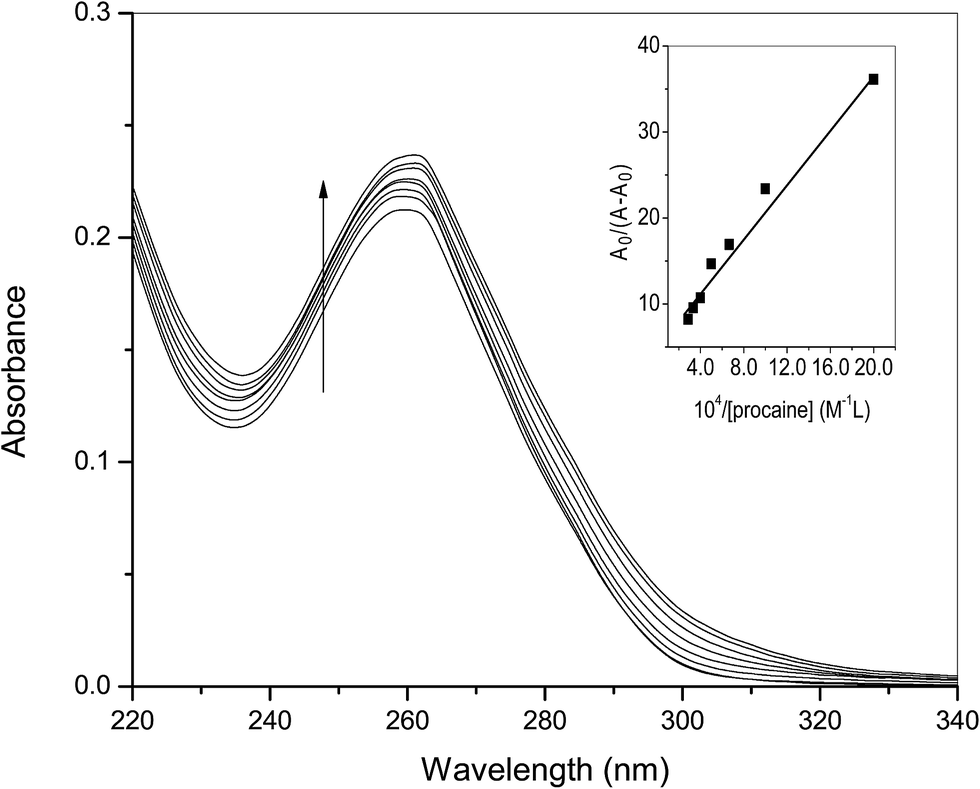
3.1.1. UV-visible spectroscopic studies. Interaction of ct-DNA with procaine was seen experimentally by means of a number of physico-chemical techniques already given in the Introduction section. Absorption spectroscopy is a basic technique used to determine the binding/association strength of small molecules with biomolecules, such as, proteins and nucleic acids.16,23 The UV spectra of ct-DNA in absence and presence of various amounts of procaine are given in Fig. 1. The UV spectrum of ct-DNA exhibits characteristic absorption peak at 260 nm. On the progressive addition of procaine, gradual increase in the absorbance (hyperchromic shift) of ct-DNA absorption was observed with a slight red-shift (∼1 nm) in the wavelength of maximum absorption. When a molecule intercalates to the ct-DNA base pairs a coupling of the π-antibonding orbital of former takes place with π-bonding orbital of latter16 which causes a decrease in the π–π* transition energy and a consequent red shift of absorption band. It is generally observed that in case of classical intercalators the UV absorption spectra show a significant red shift. However, no or slight change in the position of absorption band with hyperchromic effect can be due to the minor groove binding which is associated to the overlapping of the electronic states of the chromophore of the molecule with the nitrogenous bases in the grooves of DNA.24 Therefore, results of absorption spectroscopy give a primary idea that binding of procaine is more likely at minor groove of ct-DNA.
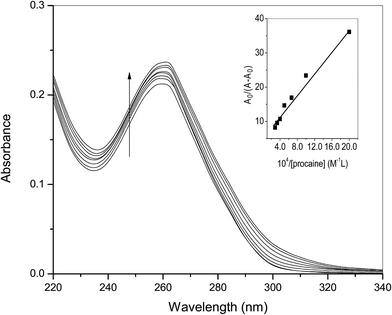 |
| | Fig. 1 Difference absorption spectra of ct-DNA (30 × 10−6 M) in the presence of increasing amount of procaine (0, 5, 10, 15, 20, 25, 30, 35 × 10−6 M) at 25 °C. Inset: plot of A0/(A − A0) versus 1/[procaine]. | |
The association constant (Ka) between ct-DNA and procaine can be calculated by using Benesi–Hildebrand equation16,24 which is given as:
| |
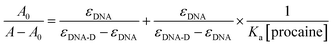 | (1) |
where
A0 and
A are the respective absorbances of ct-DNA in absence and presence of procaine at 260 nm and in a similar way
εDNA and
εDNA-D are the molar extinction coefficients of free DNA and procaine–ct-DNA complex, respectively. The association constant (
Ka) can be obtained from the linear regression of plot of
A0/(
A −
A0) and 1/[procaine] (inset of
Fig. 1). The analyzed value of
Ka at 25 °C was found to be 3.22 × 10
4 M
−1.
3.1.2. Fluorescence quenching studies. Procaine when excited at 290 nm shows an emission peak at 355 nm (Fig. S1–S3†), though; its fluorescence is negligible while compared with intrinsic fluorescence of bovine serum albumin at the excitation wavelength of 280 nm as we have reported in our recent publication,25 however, on increasing the slit width of the instrument (10 nm for both excitation and emission slits) we have got the substantial fluorescence emission spectra of procaine while ct-DNA didn't show any significant emission under identical conditions. Increasing amount of ct-DNA results in the quenching of the fluorescence emission of procaine but the position of the emission maximum remains unchanged. This is a clear indication that procaine could interact with ct-DNA. Due to the absorbance of ct-DNA at 290 and 355 nm, though very small, which are the excitation and emission wavelengths for procaine, respectively, we have corrected the fluorescence data for the inner filter effect using the method described in ESI (eqn (S1))† and given in established literature.26 The corrected fluorescence data were than quantified with the help of Stern–Volmer equation which is given as:| |
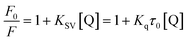 | (2) |
| |
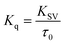 | (3) |
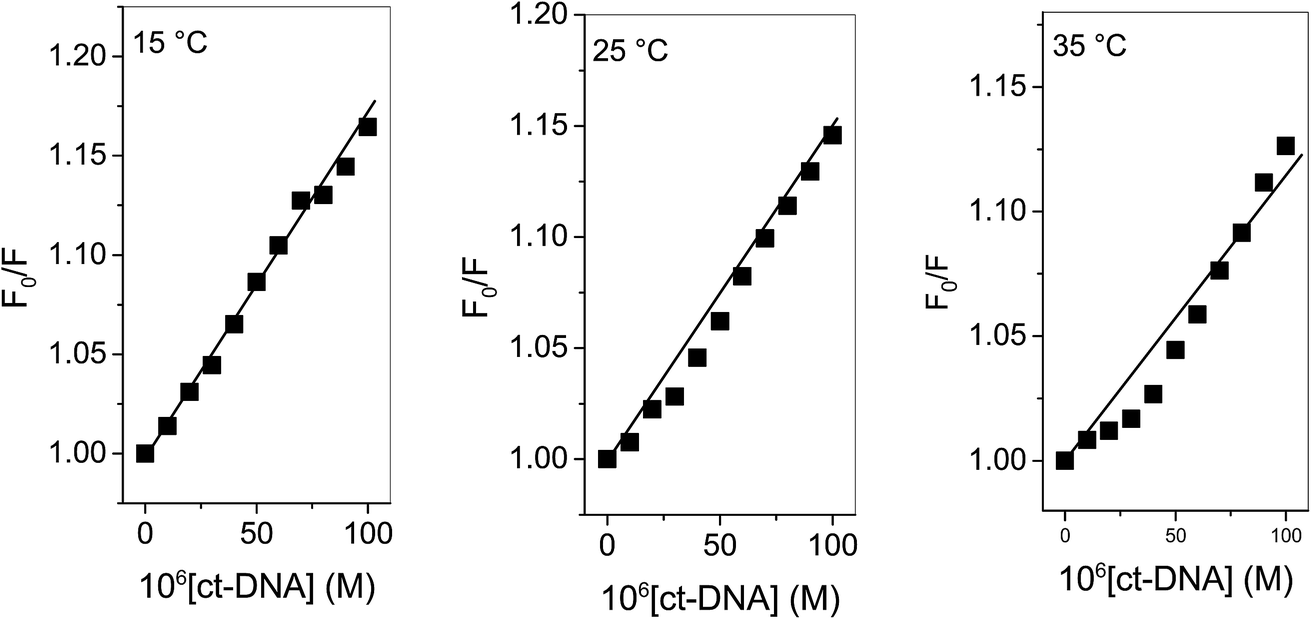
“where F0 and F are corrected fluorescence intensities in absence and presence of quencher (procaine), respectively, and KSV is Stern–Volmer constant and [Q] is the concentration of quencher, Kq is the bimolecular quenching constant and τ0 is life time of fluorophore in the absence of quencher which is equal to the 5.7 × 10−9 s−1”.26,27 When quenching of fluorophore occurs by a quencher the mechanism is either static or dynamic. Static quenching leads to the formation of the ground state complex formation while dynamic quenching comprises of the collision between the fluorophore and the quencher.28 Dynamic quenching depends on molecular diffusion. Therefore, increase in the temperature results in the increase in the diffusion which may directs to the increase in dynamic quenching constant. In contrary to that rise in temperature makes the complex less stable which ensue a decrease in static quenching constant. Thus, effect of varying temperature can distinguish between the static quenching and dynamic quenching. For that reason, we have conducted our studies at three temperatures, viz., 15, 25 and 35 °C (Fig. S1–S3†) to differentiate between these two types of quenchings.KSV can be obtained from the linear regression of eqn (2) for which the plots of F0/F versus [Q] at three temperatures are given in Fig. 2. Calculated values of KSV, which are given in Table 1, show a decreasing trend on increasing the temperature conclusive of involvement of static quenching mechanism in procaine–ct-DNA interaction.
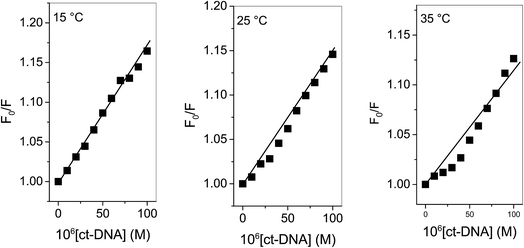 |
| | Fig. 2 Stern–Volmer plots of ct-DNA interaction with procaine at various temperatures. [procaine] = 30 × 10−6 M. | |
Table 1 Stern–Volmer quenching constants, binding parameters and thermodynamic parameters for the interaction of ct-DNA with procaine at various temperatures
| T (K) |
Stern–Volmer quenching constants |
Binding parameters |
Thermodynamic parameters |
| KSV (M−1) |
Kq (M−1 s−1) |
n |
Kb (M−1) |
ΔG (kJ M−1) |
ΔH (kJ M−1) |
ΔS (J M−1 K−1) |
| 288 |
3.3 × 103 |
5.8 × 1011 |
1.03 |
5.1 × 103 |
−20.5 |
−6.4 |
48.9 |
| 298 |
2.8 × 103 |
4.9 × 1011 |
1.04 |
4.8 × 103 |
−21.0 |
|
|
| 308 |
2.2 × 103 |
3.9 × 1011 |
1.04 |
4.3 × 103 |
−21.5 |
|
|
3.1.3. Determination of binding and thermodynamic parameters. Modified Stern–Volmer equation (eqn (4)) can be used to determine the binding constant (Kb) as well as number of binding sites (n).| |
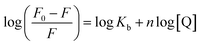 | (4) |
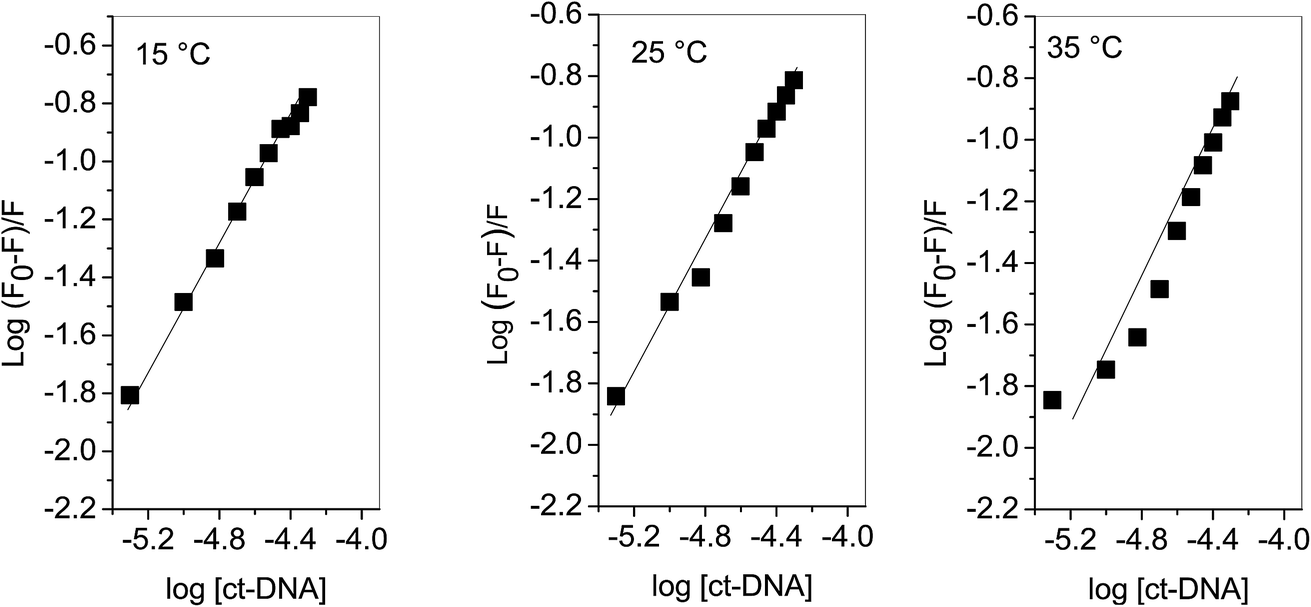
Plots of log(F0 − F)/F versus log[Q] (Fig. 3) were used to evaluate the binding constant and number of binding sites which are given in Table 1. The Kb obtained at 25 °C is in very close agreement from the one obtained using absorption spectroscopy. The value of n also validates that there is 1:1 binding between procaine and ct-DNA.
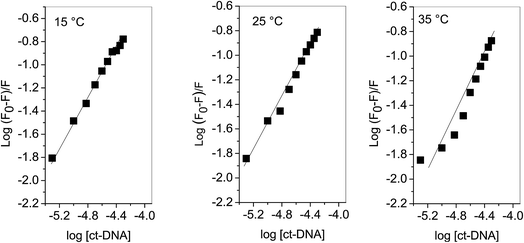 |
| | Fig. 3 Plot of log(F0 − F)/F as a function of log[ct-DNA]. | |
The involvement of various forces (hydrophobic, van der Waals, electrostatic and hydrogen bonding) between a ligand and macromolecule can be understood by assessing the thermodynamic parameters. Change in enthalpy (ΔH) and entropy (ΔS) of interaction process along with free energy change (ΔG) are promising factors to define the binding modes involved in the interaction.
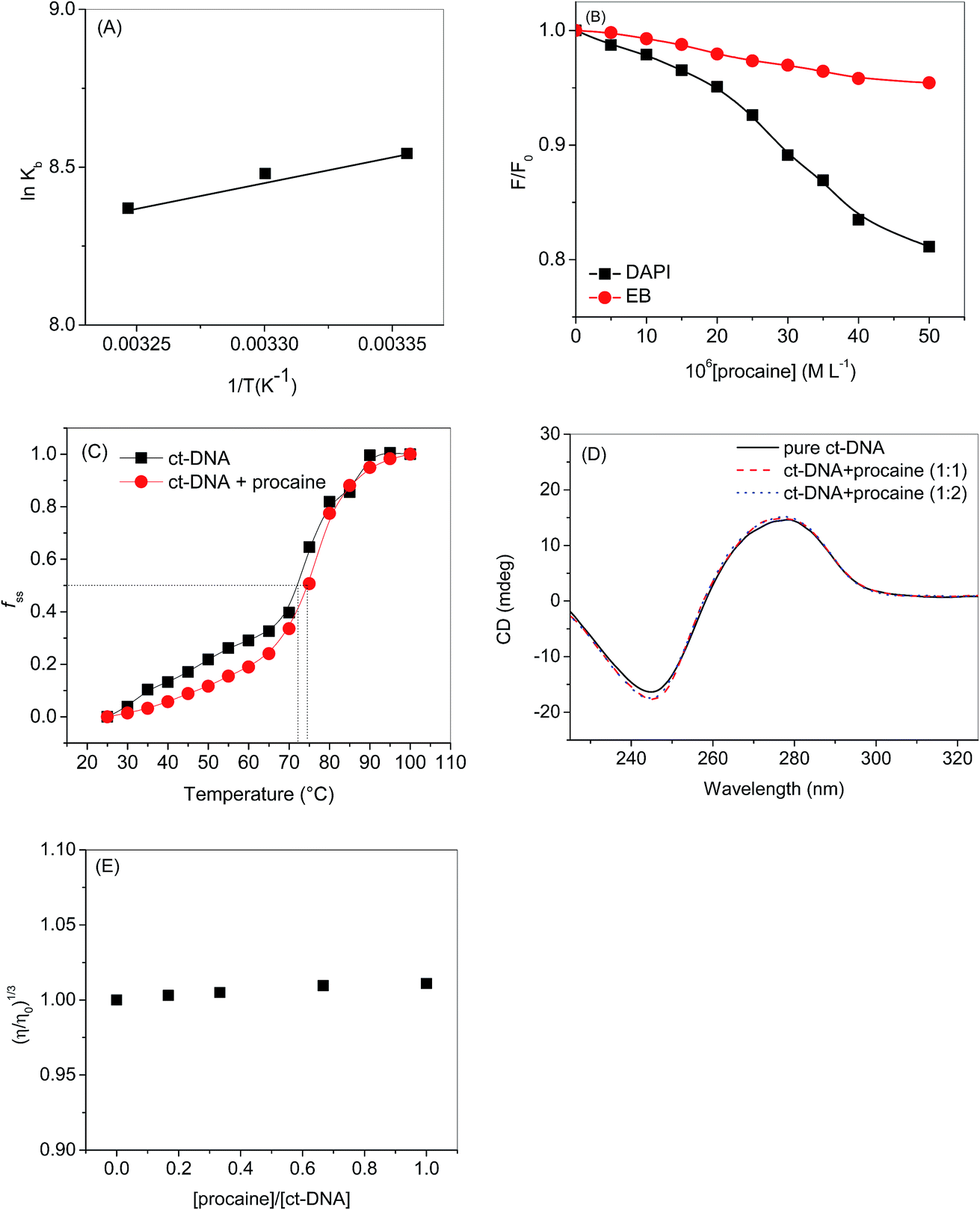
If the ΔH does not change too much on the temperature variation than both ΔH and ΔS can be evaluated from the van't Hoff equation (eqn (5)):
| |
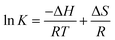 | (5) |
van't Hoff plot of interaction of ct-DNA with procaine are given in Fig. 4(A). The values of ΔG were found to be negative at all temperatures envisage the feasibility of interaction. From enthalpy and entropy point of views the binding process was exothermic in nature i.e., negative ΔH with positive ΔS. It is well understood that hydrophobic interaction is characterized by the positive values of both ΔH and ΔS. Involvement of van der Waals forces and hydrogen bonding results in the negative values of both ΔH and ΔS whereas in case of electrostatic interaction the value of ΔH is either very less or zero.29 In present case the negative value of ΔH and positive value of ΔS suggests that the main forces involved in the binding of procaine with ct-DNA are hydrophobic forces and hydrogen bonding that may play major roles in the interaction.30 The presence of other interaction forces cannot be ruled out, though, it can be said that the interaction forces are dominated by hydrophobic forces and hydrogen bonding.31
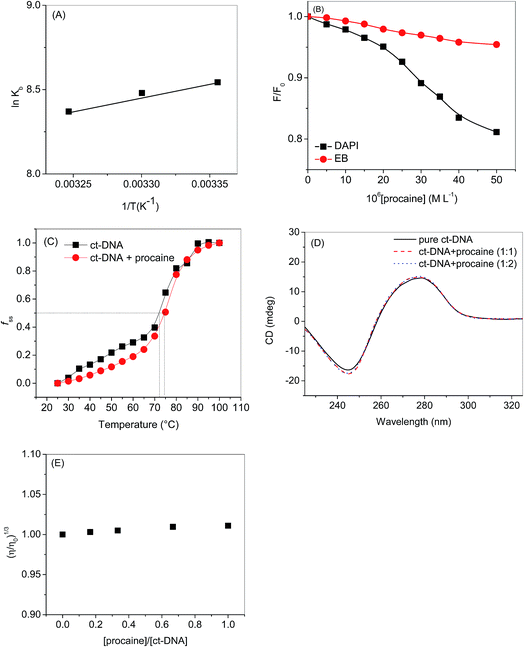 |
| | Fig. 4 (A) van't Hoff plot of ct-DNA–procaine interaction. (B) Relative fluorescence intensity of titration of ct-DNA and DAPI/EB with [procaine]. The fluorescence intensities at wavelength of maximum emission have been selected for both dyes which are 490 and 590 for DAPI and EB respectively. (C) Melting curves of ct-DNA in the absence and presence of procaine at pH 7.4. [procaine] = 30 × 10−6 mol L−1, and [ct-DNA] = 30 × 10−6 M. (D) CD spectra of ct-DNA in absence and presence of procaine. The concentration of ct-DNA was taken as 50.0 × 10−6 M. (E) Effect of increasing amounts of procaine on the relative viscosity of ct-DNA at pH 7.4. [ct-DNA] = 30 × 10−6 M. | |
3.1.4. Dye displacement assay using EB and DAPI dyes. When a molecule binds to the DNA, non-covalently, the binding modes may be one or more among intercalation, groove binding and external binding.16 Mode of DNA binding can be studied by using DNA binding dye as there are several dyes which have been well studied and their binding modes are well established. If any molecule that competitively replaces a DNA bound dye is likely to bind the DNA at that particular dye binding site, for instance, molecule with affinity towards minor groove will compete and replace the dyes which are minor groove binders whereas an intercalator will compete with the dyes which are established intercalation dyes.16 DAPI is an important dye which is useful in DNA staining studies. Though, DAPI generally binds at the minor groove of double stranded DNA20,32,33 but it also has been reported to intercalate between the DNA strands.34 The binding mode of the double stranded DNA is dependent on the sequence of the base pairs, for instance, in case of G–C rich DNA, prefers to bind at intercalating site while in A–T rich DNAs it prefers to bind at minor groove region.20,35–39 Under our present experimental conditions the DAPI is supposed to bind at minor groove region of ct-DNA, because it is A–T rich DNA (58.1% as given in specifications from Sigma-Aldrich company), and can be used as a probe to identify the minor groove interaction between ct-DNA and procaine.20,35–39 The fluorescence of DAPI enhances intensely in presence of DNA which is approximately twenty times greater than the DAPI without ct-DNA.40 Minor groove binding agent could affect the DAPI–ct-DNA binding and consequently can cause the decrease in the fluorescence intensity of DAPI. On the other side if there is very small or no change in the fluorescence of DNA bound DAPI, the molecule will preferably bind to the other side. In a similar way EB is strongly intercalates into the ct-DNA and its fluorescence also increases after ct-DNA binding. If a molecule intercalates into the ct-DNA helix, it can compete and displace EB from the EB–ct-DNA complex which may cause in the decrease of fluorescence intensity of EB–ct-DNA complex. Henceforth, the effect of procaine on the DAPI–ct-DNA and EB–ct-DNA complex was studied using fluorescence spectroscopy. The results corresponding to DAPI and EB have been displayed in Fig. S4(a) and (b),† respectively, and the relative fluorescence intensity as a function of [procaine] is given in Fig. 4(B). From the results it can be seen that successive addition of procaine affects the fluorescence intensity of DAPI while there was negligible effect of procaine on the fluorescence intensity of EB (Fig. 4(B)). The decrement of fluorescence intensity of DAPI in presence of procaine is due to the replacement of some of DAPI molecules from its binding site, i.e., minor groove. There was very small change in the intensity of EB that means there is no displacement of the dye from the intercalation region. Therefore, the preferable binding site of procaine in ct-DNA is minor groove.
3.1.5. DNA melting studies. Melting curves of ct-DNA in absence and presence of procaine are given in Fig. 4(C). The midpoints of curves based on fss = (A − A0)/(Af − A0) versus temperature were defined as melting temperatures (Tm), where A0 and Af are the initial and final absorbance intensities at 260 nm, respectively, and A is the absorbance intensity corresponding to its temperature.41 The intercalation of small molecules into the double helix of DNA has remarkable effect on the Tm of DNA, but, there is very small change in Tm in case of non-covalent groove binding.42 In general an intercalating molecule could increase the Tm from 5 to 8 °C (ref. 43) but the Tm of ct-DNA increases from 72.4 to 75 °C in presence of procaine which also supports our conclusions that the binding of procaine with ct-DNA is taking place in minor groove region.
3.1.6. Circular dichroism spectroscopy. CD spectroscopy can also be used to understand the binding of small molecules to the DNA. The CD spectrum of ct-DNA gives a negative peak at 245 nm due to the stacking of the base pairs and a positive peak at 275 nm which corresponds to the polynucleotide helicity (Fig. 4(D)). Both these peaks are the characteristics of B-DNA.44 The intercalating molecule, generally, grounds to the increase in the positive band due to its stacking interaction between the base pairs of DNA.45 Contrariwise, groove binding agents do not amend the CD spectrum of DNA. Procaine didn't cause any significant change in the CD spectrum of ct-DNA point towards the groove binding between ct-DNA and procaine.
3.1.7. Viscosity measurements. In the Experimental part of binding studies, we have performed viscosity measurements, too, in order to further confirm the mode of binding (Fig. 4(E)). Viscosity is an important hydrodynamic property which, simply, can offer augmented hint on the binding modes of small molecules to the DNA. In general, classical intercalator results in the lengthening/enlargement of the DNA helix due to the split-up of base pairs at the intercalation site which results in the concomitant increase in the relative specific viscosity. Minor groove binders don't show any significant increase in the viscosity of the solution comprising DNA. Therefore, viscosity measurement can be regarded as least vague assessment for understanding the groove and intercalation mode of binding.18,46–48 As can be seen from Fig. 4(E) that there was very small change in the relative viscosity of ct-DNA on the addition of procaine which is accredited to the minor groove binding.49
3.2. Computational analyses of interaction of procaine with DNA
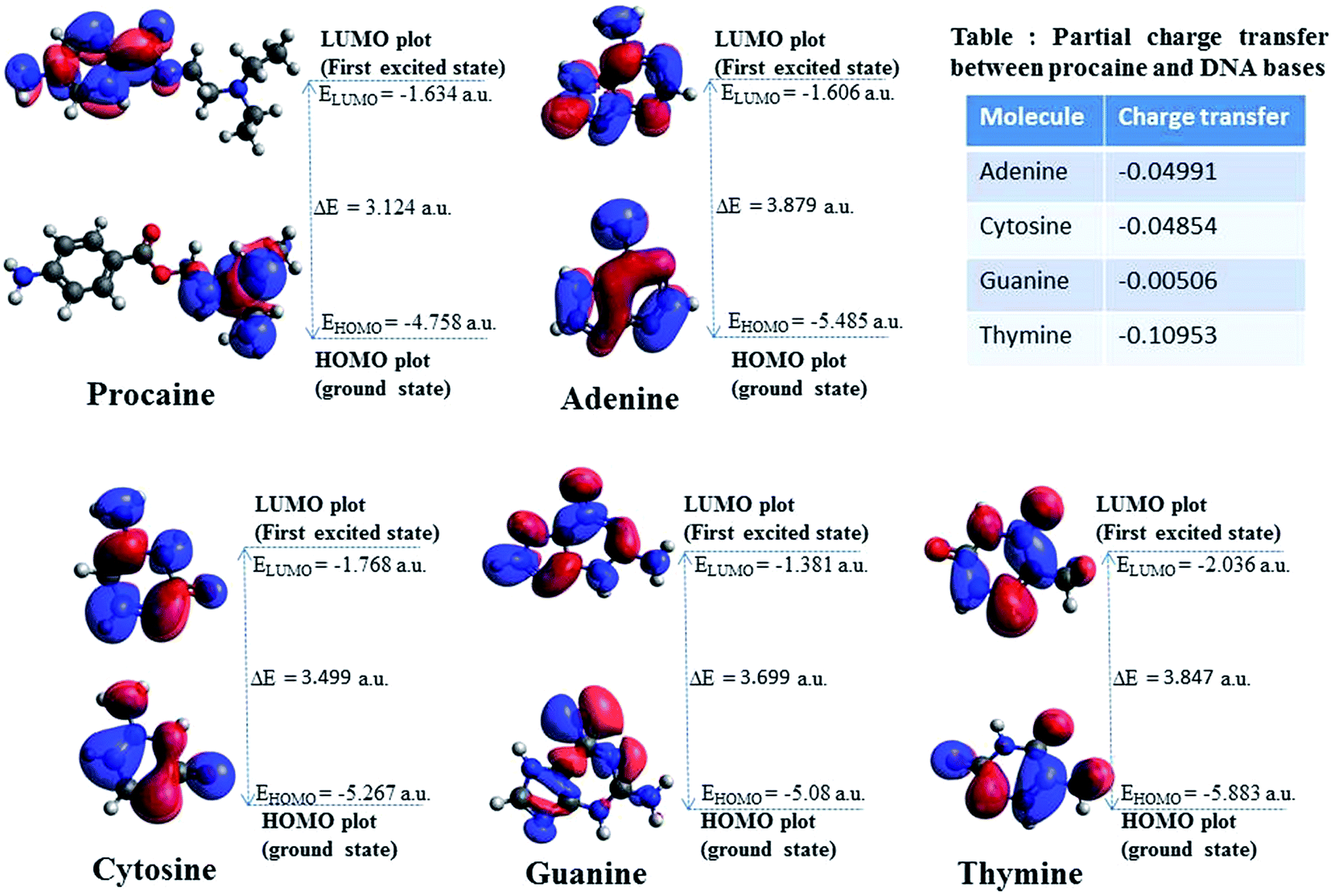
3.2.1. Molecular docking. Computational studies which can generate the specific sequences and interactions of nucleic acids with small molecules are very helpful in designing and development of chemotherapeutic agents. One such computational framework is molecular docking which, currently, is very useful to understand the drug–biomolecules interactions for the rational drug design and discovery. The mechanism of the drug–biomolecules interaction can easily be understood by molecular docking methods and validation of experimental results can also be achieved by these computational methods.50 As discussed, there are two modes of non-covalent interactions between DNA and small molecules which are intercalation or groove binding. Further, the interactions taking place in major or minor groove are also differing in nature in terms of the electrostatic potential and steric effects because of the narrow shape of minor groove. Small molecules tend to bind at narrow minor groove while large molecules find its binding site at major groove.51 Therefore, molecular docking calculations have been performed on the procaine–DNA system. Since we have used ct-DNA in our studies which is a B-DNA we have taken five protein data bank crystals structures of B-DNAs (https://www.rcsb.org/) in order to certify the mode of binding of procaine on it. The conformer with minimum binding energy was selected from the various conformers which are given in Fig. S5.† Interestingly, all confirmers show the minor groove binding as preferential binding. The nucleotides involved in the binding mechanism for all types of B-DNA–procaine complexes are given in Fig. 5 and it is evident from the figure that procaine binds mainly in the AT rich region of all types of B-DNAs. Flexibility of procaine also plays an important role in its binding with DNA as displayed by the Fig. S6† which describe the conformations of procaine in absence and presence of various DNA structures. Altered spatial conformation of procaine also confirms the binding between DNA and procaine. From the collective information of the types of interactions belongs to all types of DNA conformers (Table S1†) a supposition can be drawn that interaction of procaine with B-DNA involves hydrophobic, hydrogen bonding and electrostatic interactions. When a ligand interacts with biomolecules, there is a chance for many types of interactions between them (for instance, hydrophobic, electrostatic, van der Waals, hydrogen bonding, etc.), but due to the predominant nature of one or two, which depends on the structure of the ligand, the understanding of others is difficult.52 Though, from theoretical models it is easier to understand all types of interactions involved despite of various shortcomings involved in these modellings.53 The PDB crystal structures of B-DNA used for molecular docking in this study have been developed by various authors and that's why there might be a possibility of some variations (bond length, bond angle, polarity, etc.) among them. Therefore, we have obtained various informations and possibilities of interaction of procaine with DNA. These results are in good agreement with the one obtained through experimental techniques.
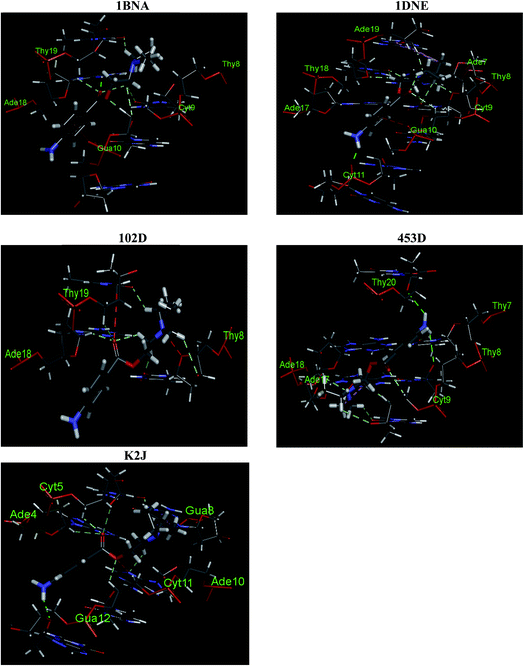 |
| | Fig. 5 Predicted binding mode of procaine docked into DNA. | |
3.3. Anticancer and anti-proliferative activities of procaine
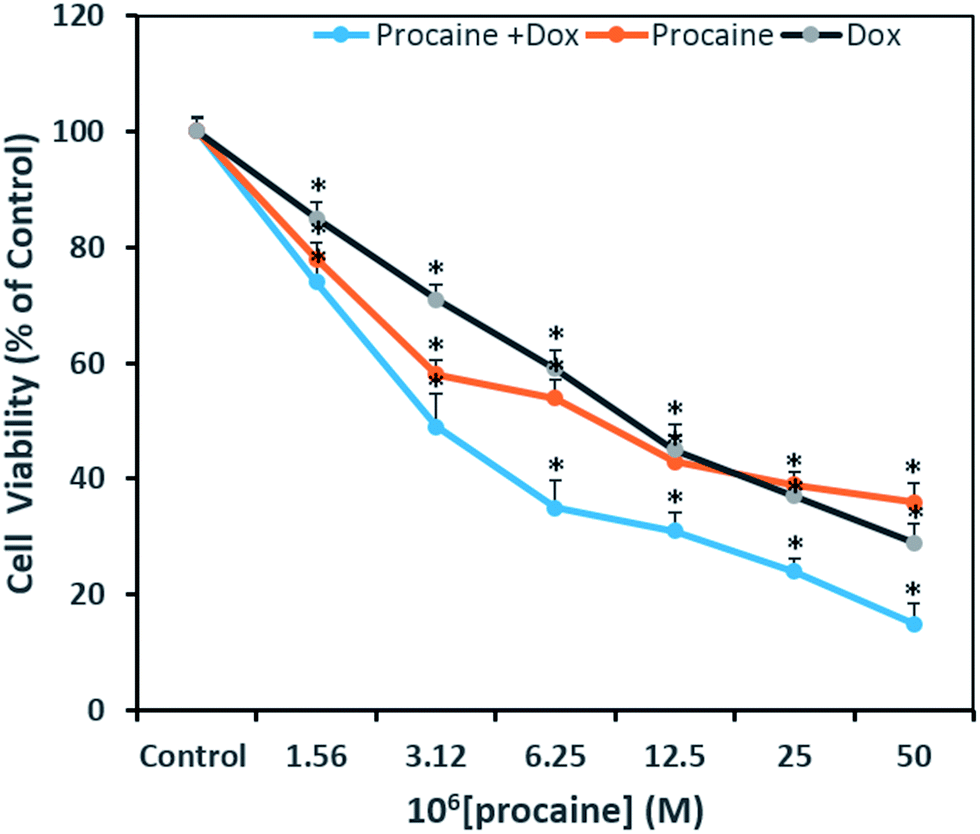
3.3.1. Cytotoxicity assay. The MTT assay was performed to evaluate the cytotoxic activity of procaine and compare its efficacy with that of anti-tumor agent, doxorubicin. This assay is useful in measuring the IC50 value, which is the concentration of the test compound that can inhibit or kill 50% of the total cells. Fig. 7 shows the percent viability of cells exposed to different concentrations of procaine, doxorubicin alone or in combination. The IC50 values estimated at 24 h post-treatment in MCF-7 for procaine is 9 μM, doxorubicin is 10 μM and for combined treatment it was observed at 3 μM. Procaine exhibited a marginally higher inhibition as compared to doxorubicin whereas, both procaine and doxorubicin treated in combination exhibited higher cytotoxicity in MCF-7 calls.
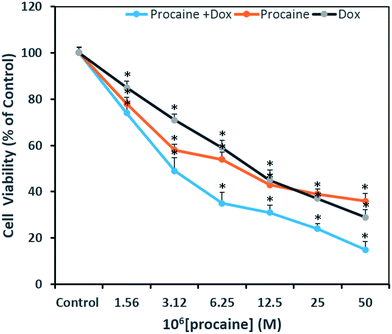 |
| | Fig. 7 Antiproliferative effects on MCF-7 cells treated as indicated with two-fold dilutions of procaine and doxorubicin alone or in combinations. All data are expressed as mean ± SE. * Significant (p < 0.05) compared with corresponding controls. | |
A concentration dependent decrease in the cell viability was evident. Procaine and doxorubicin inhibited cell proliferation by 71% and 64%, respectively at highest concentration of 50 μM. While, combined treatment reduced cell growth by 85% and higher concentration. These data suggest that procaine enhanced the antiproliferative property of doxorubicin.
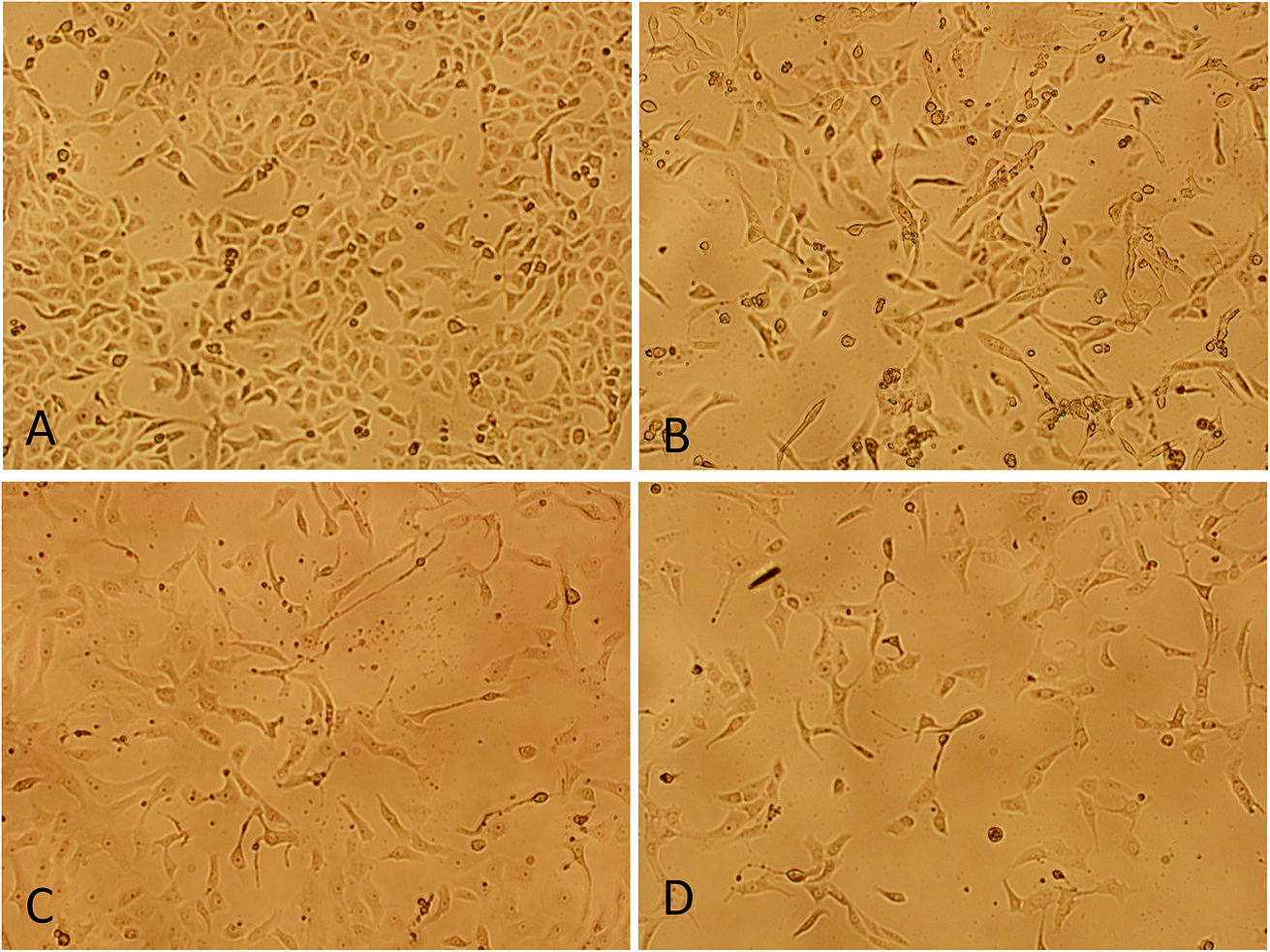
3.3.2. Morphological changes analysis. To evaluate the cytotoxicity at morphological level, cells were treated with below IC50 concentration (5 μM) of procaine and doxorubicin alone or in combination (5 μM dox + 5 μM procaine) for 24 h. The representative image of morphological changes observed under phase contrast inverted microscopy in MCF-7 cells is shown in Fig. 8. No significant changes in the morphology were observed in control cells (Fig. 8(A)). The cells appeared to have a normal shape, were attached to the surface and reached about 95–100% confluence. Conversely, the cells lost their normal epithelial cell morphology, becoming longer, and swelled. A decreased cell density was observed in treated MCF-7 cells (Fig. 8(B–D)).
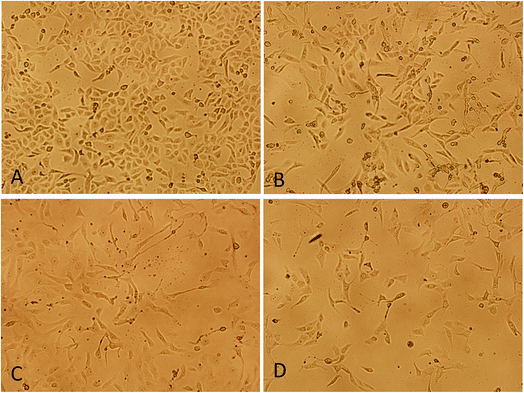 |
| | Fig. 8 Morphological changes analysis of MCF-7 cells by phase contrast inverted microscopy. (A) Control, (B) 5 × 10−6 M procaine, (C) 5 × 10−6 M doxorubicin, (D) 5 × 10−6 M procaine + doxorubicin. Magnification: 100×. | |
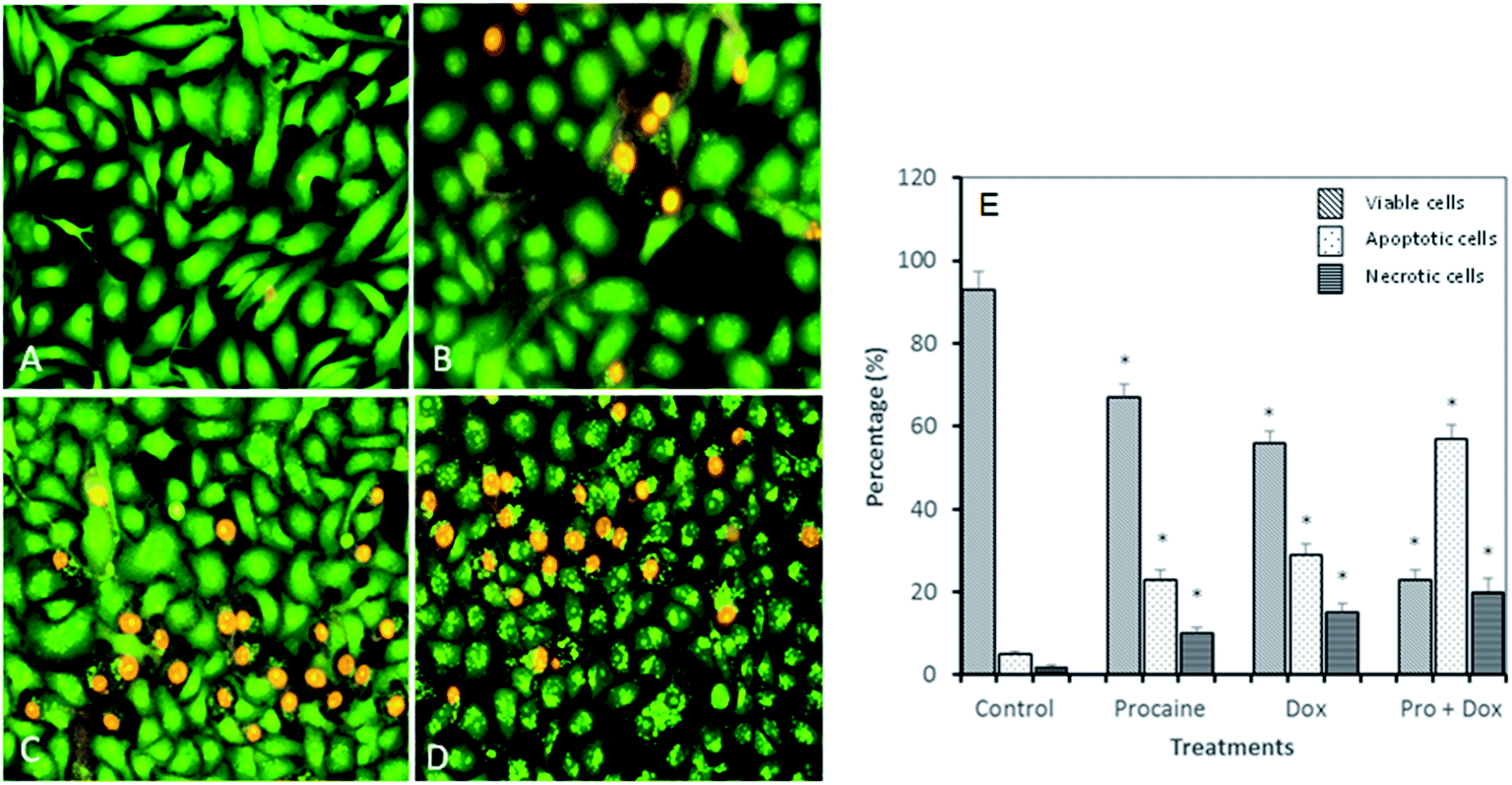
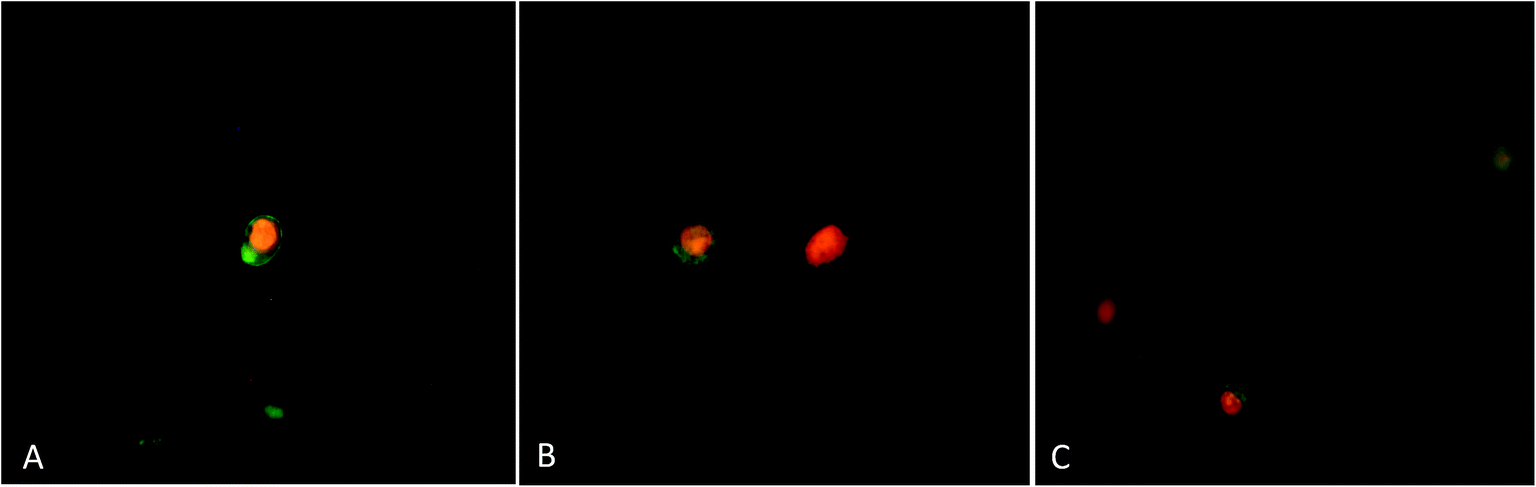
3.3.3. Apoptotic morphological changes in MCF-7 cells. To test whether the decrease in cell viability observed after treatment with procaine and doxorubicin is due to apoptosis, MCF-7 cells were stained with acridine orange/ethidium bromide (AO/EB) dye (Fig. 9). AO is able to permeate into both live and dead cells to stain all nucleated cells and excite green florescence, while EB only enters dead cells with damaged membranes and generates red florescence. Around 93% of viable cells were prominently evident in untreated MCF-7 cells. Control cells showed evenly distributed AO stain (green fluorescence) with no morphological changes (Fig. 9(A) and (E)). The percentage of viable cells however, decreased significantly (p < 0.05) in all treated samples. As shown in Fig. 9(B)–(E), AO/EB staining which correlated with the presence of cells with typical apoptotic nuclear morphology (nuclear shrinkage, DNA condensation and fragmentation). Quantification of apoptotic and necrotic cells revealed that combined treatment of procaine and doxorubicin induced highest percentage of apoptotic cells (57%) and necrotic cells (20%) (Fig. 9(E)).
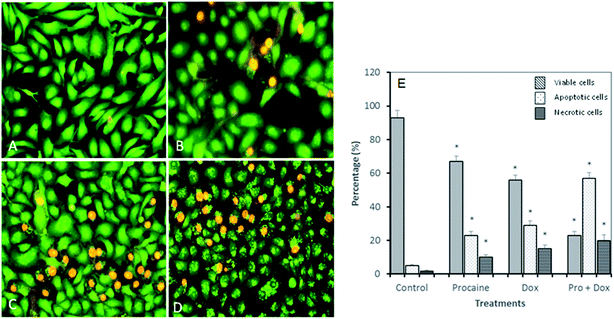 |
| | Fig. 9 Apoptotic morphological changes in MCF-7 cells observed under fluorescence microscopy. (A) Control, (B) 5 × 10−6 M procaine, (C) 5 × 10−6 M doxorubicin, (D) 5 × 10−6 M procaine + doxorubicin. Magnification: 200×. (E) Quantification of apoptotic and necrotic cells based on the uptake of acridine orange and propidium iodide in more than 300 cells. All data are expressed as mean ± SE. * Significant (p < 0.05) compared with corresponding controls. | |
3.3.4. Annexin V-apoptosis assay by fluorescence microscopy. To further confirm that procaine and doxorubicin induces cell apoptosis, annexin V-FITC and PI double staining method was used to detect the translocation of phosphatidylserine on the outer membrane of apoptotic cells. MCF-7 cells underwent apoptosis after exposure to procaine and doxorubicin as evident in Fig. 10(A–C). The annexin V binding to PS (green membrane fluorescence) at outer plasma membrane shows early apoptosis while, PI staining (red fluorescence) of nucleus with membrane green fluorescence shows late apoptosis. Moreover, only red fluorescence of nucleus indicates necrosis. Procaine treated MCF-7 cells showed appearance of early and late apoptosis. Doxorubicin exposure induced apoptotic and necrotic cells while the number of late apoptotic and necrotic cells increased in combined treatment.
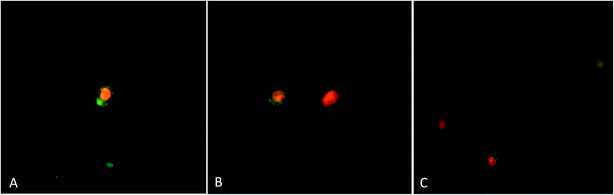 |
| | Fig. 10 Annexin-V/PI apoptosis assay. After treatment, cells were stained with annexin V-FITC and propidium iodide and observed by fluorescence microscopy for the detection of phosphatidylserine translocation to the plasma membrane. (A) 5 × 10−6 M procaine, (B) 5 × 10−6 M doxorubicin, (C) 5 × 10−6 M procaine + doxorubicin. Magnification: 400×. | |
Our results proved that procaine inhibits the proliferation of MCF-7 cells alone or in the presence of doxorubicin, thereby increasing its cytotoxicity. Previously, procaine has been reported to exert DNA demethylation and inhibits the growth of MCF-7 cells.9 Recently, procaine has been shown to induce mitochondrial dysfunction, elevated ROS level and apoptosis in human neuroblastoma cell line.56 On the other hand, doxorubicin was reported to exhibit its cytotoxic effect by intercalating between DNA base pairs and by inhibition of TOPO II enzyme, protein synthesis and overproduction of ROS.57 Unfortunately, doxorubicin is associated with several serious adverse effects such as myelosuppression, mucositis, alopecia, chronic cardiotoxicity that is limiting factor of the chemotherapeutic use of doxorubicin.58 Therefore, a wide variety of approaches has been investigated to minimize the side effects of doxorubicin and to improve its therapeutic use. Some reports emphasize that combining more than one anticancer drug can minimize the side effects of anticancer agent used alone. In this context, Li et al., demonstrated that lidocaine sensitizes the cytotoxicity of cisplatin against MCF-7 cells.59 However, in the present study, it is not clear by what mechanism procaine enhances the effects of doxorubicin, but these results support the possible use of procaine and its derivatives in cancer therapy with some other anticancer agents.
4. Conclusions
Assessment of ct-DNA binding properties (experimental and theoretical) of procaine along with its anticancer activities has been carried out in this study. Analyses of experimental investigations inferred that there was 1:1 binding between ct-DNA and procaine and the preferred binding mode was minor groove binding which involves hydrogen bonding, and hydrophobic forces. Molecular docking on five different structures of B-DNA and procaine interaction also favors the experimental observations and in all five B-DNA structures, minor groove, which is AT rich region, was the principle binding site, however, from molecular docking the involvement of electrostatic forces in the interaction was also observed. Molecular orbital calculations using DFT methods were used to calculate the chemical potentials of procaine and DNA bases which stated that procaine acts as electron donor to the DNA bases. Procaine was also found to have cytotoxic effects towards MCF-7 breast cancer cell lines. The results also showed that the combined treatment with both procaine and doxorubicin enhanced the cytotoxic and apoptosis inducing potential of doxorubicin. The results support the promising use of procaine and its derivatives in cancer therapy with some other anticancer agents.
Conflicts of interest
There are no conflicts to declare.
Abbreviations
| ct-DNA | Calf thymus DNA |
| CD | Circular dichroism |
| EB | Ethidium bromide |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DFT | Density functional theory |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project no. RGP-148.
References
- E. J. Olsen, L. Bank and L. F. Jarvik, J. Gerontol., 1978, 33, 514–520 CrossRef CAS PubMed
 .
. - R. Jakobs, M. U. Adamek, A. C. Von Bubnoff and J. F. Riemann, Scand. J. Gastroenterol., 2000, 35, 1319–1323 CrossRef CAS PubMed .
- L. J. Casarett and C. D. Klaassen, Casarett and Doull's Toxicology: The Basic Science of Poisons, McGraw-Hill, 2001 Search PubMed .
- W. W. Zung, D. Gianturco and E. Pfieffer, Psychopharmacol. Bull., 1976, 12, 50–51 CAS .
- M. D. A. R. P. Bulcao, M. Roehrs, C. Paniz, F. L. Cervi, F. V. Thiesen, M. B. Leal and S. C. Garcia, Rev. Cienc. Farm. Basica Apl., 2011, 32, 297–303 Search PubMed .
- A. W. T. Konings, Cancer Res., 1985, 45, 2016 CAS .
- M. Esposito, R. A. Fulco, P. Collecchi, A. Zicca, A. Cadoni, F. Merlo, R. Rosso and A. Sobrero, JNCI, J. Natl. Cancer Inst., 1990, 82, 677–684 CrossRef CAS PubMed .
- Y. Rong, X. Xia and B. Lin, Chin. J. Cancer Res., 1993, 5, 70–73 CrossRef .
- A. Villar-Garea, M. F. Fraga, J. Espada and M. Esteller, Cancer Res., 2003, 63, 4984–4989 CAS .
- M. Tada, F. Imazeki, K. Fukai, A. Sakamoto, M. Arai, R. Mikata, T. Tokuhisa and O. Yokosuka, Hepatol. Int., 2007, 1, 355–364 CrossRef PubMed .
- M. Rius and F. Lyko, Oncogene, 2012, 31, 4257–4265 CrossRef CAS PubMed .
- J. M. Foulks, K. M. Parnell, R. N. Nix, S. Chau, K. Swierczek, M. Saunders, K. Wright, T. F. Hendrickson, K. K. Ho, M. V. McCullar and S. B. Kanner, J. Biomol. Screening, 2012, 17, 2–17 CrossRef CAS PubMed .
- N. Martinet, B. Y. Michel, P. Bertrand and R. Benhida, MedChemComm, 2012, 3, 263–273 RSC .
- Y. Ni, M. Wei and S. Kokot, Int. J. Biol. Macromol., 2011, 49, 622–628 CrossRef CAS PubMed .
- C. Silvestri and J. S. Brodbelt, Mass Spectrom. Rev., 2013, 32, 247–266 CrossRef CAS PubMed .
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed .
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS PubMed .
- L. S. Lerman, J. Mol. Biol., 1961, 3, 18–30 CrossRef CAS PubMed .
- R. Vijayalakshmi, M. Kanthimathi, V. Subramanian and B. U. Nair, Biochem. Biophys. Res. Commun., 2000, 271, 731–734 CrossRef CAS PubMed .
- B.-M. Liu, C.-L. Bai, J. Zhang, Y. Liu, B.-Y. Dong, Y.-T. Zhang and B. Liu, J. Lumin., 2015, 166, 48–53 CrossRef CAS .
- F. Neese, Wiley Interdisciplinary Reviews: Computational Molecular Science, 2018, vol. 8, p. e1327 Search PubMed .
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed .
- J.-H. Shi, T.-T. Liu, M. Jiang, J. Chen and Q. Wang, J. Photochem. Photobiol., B, 2015, 147, 47–55 CrossRef CAS PubMed .
- M. Purcell, J. F. Neault and H. A. Tajmir-Riahi, Biochim. Biophys. Acta Protein Struct. Mol. Enzymol., 2000, 1478, 61–68 CrossRef CAS .
- M. S. Ali and H. A. Al-Lohedan, J. Mol. Liq., 2017, 236, 232–240 CrossRef CAS .
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, pp. 277–330, DOI:10.1007/978-0-387-46312-4_8 .
- U. Anand, C. Jash and S. Mukherjee, J. Phys. Chem. B, 2010, 114, 15839–15845 CrossRef CAS PubMed .
- S. M. Ng, M. Koneswaran and R. Narayanaswamy, RSC Adv., 2016, 6, 21624–21661 RSC .
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed .
- S. Naveenraj and S. Anandan, J. Photochem. Photobiol., C, 2013, 14, 53–71 CrossRef CAS .
- Y. Ma, G. Zhang and J. Pan, J. Agric. Food Chem., 2012, 60, 10867–10875 CrossRef CAS PubMed .
- F. A. Tanious, J. M. Veal, H. Buczak, L. S. Ratmeyer and W. D. Wilson, Biochemistry, 1992, 31, 3103–3112 CrossRef CAS PubMed .
- E. Trotta, E. D'Ambrosio, N. Del Grosso, G. Ravagnan, M. Cirilli and M. Paci, J. Biol. Chem., 1993, 268, 3944–3951 CAS .
- E. Trotta, E. D'Ambrosio, G. Ravagnan and M. Paci, Nucleic Acids Res., 1995, 23, 1333–1340 CrossRef CAS PubMed .
- D. Banerjee and S. K. Pal, J. Phys. Chem. B, 2008, 112, 1016–1021 CrossRef CAS PubMed .
- W. D. Wilson, F. A. Tanious, H. J. Barton, R. L. Jones, K. Fox, R. L. Wydra and L. Strekowski, Biochemistry, 1990, 29, 8452–8461 CrossRef CAS PubMed .
- J. Kapuscinski, Biotech. Histochem., 1995, 70, 220–233 CrossRef CAS PubMed .
- X. Cai, P. J. Gray Jr and D. D. Von Hoff, Cancer Treat. Rev., 2009, 35, 437–450 CrossRef CAS PubMed .
- B. S. P. Reddy, S. M. Sondhi and J. W. Lown, Pharmacol. Ther., 1999, 84, 1–111 CrossRef CAS PubMed .
- E. N. Zaitsev and S. C. Kowalczykowski, Nucleic Acids Res., 1998, 26, 650–654 CrossRef CAS PubMed .
- X. Zhou, G. Zhang and J. Pan, Int. J. Biol. Macromol., 2015, 74, 185–194 CrossRef CAS PubMed .
- B. Jana, S. Senapati, D. Ghosh, D. Bose and N. Chattopadhyay, J. Phys. Chem. B, 2012, 116, 639–645 CrossRef CAS PubMed .
- S. Bi, H. Zhang, C. Qiao, Y. Sun and C. Liu, Spectrochim. Acta, Part A, 2008, 69, 123–129 CrossRef PubMed .
- A. Rajendran and B. U. Nair, Biochim. Biophys. Acta Gen. Subj., 2006, 1760, 1794–1801 CrossRef CAS PubMed .
- V. Uma, M. Kanthimathi, T. Weyhermuller and B. U. Nair, J. Inorg. Biochem., 2005, 99, 2299–2307 CrossRef CAS PubMed .
- H. Yang, P. Tang, B. Tang, Y. Huang, X. Xiong and H. Li, RSC Adv., 2017, 7, 10242–10251 RSC .
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS PubMed .
- J. M. Kelly, A. B. Tossi, D. J. McConnell and C. OhUigin, Nucleic Acids Res., 1985, 13, 6017–6034 CrossRef CAS PubMed .
- C. Metcalfe, C. Rajput and J. A. Thomas, J. Inorg. Biochem., 2006, 100, 1314–1319 CrossRef CAS PubMed .
- R. Rohs, I. Bloch, H. Sklenar and Z. Shakked, Nucleic Acids Res., 2005, 33, 7048–7057 CrossRef CAS PubMed .
- R. Corradini, S. Sforza, T. Tedeschi and R. Marchelli, Chirality, 2007, 19, 269–294 CrossRef CAS PubMed .
- I. Haq, Arch. Biochem. Biophys., 2002, 403, 1–15 CrossRef CAS PubMed .
- X. Du, Y. Li, Y.-L. Xia, S.-M. Ai, J. Liang, P. Sang, X.-L. Ji and S.-Q. Liu, Int. J. Mol. Sci., 2016, 17, 144 CrossRef PubMed .
- J. Padmanabhan, R. Parthasarathi, V. Subramanian and P. K. Chattaraj, Chem. Res. Toxicol., 2006, 19, 356–364 CrossRef CAS PubMed .
- Y.-Q. Wang and H.-M. Zhang, J. Photochem. Photobiol., B, 2015, 149, 9–20 CrossRef CAS PubMed .
- X.-j. Yu, W. Zhao, Y.-j. Li, F.-x. Li, Z.-j. Liu, H.-l. Xu, L.-y. Lai, R. Xu and S.-y. Xu, Sci. Rep., 2017, 7, 45316 CrossRef CAS PubMed .
- L. Roca-Alonso, L. Pellegrino, L. Castellano and J. Stebbing, Cardiology, 2012, 122, 253–259 CrossRef CAS PubMed .
- C. Carvalho, R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS PubMed .
- K. Li, J. Yang and X. Han, Int. J. Mol. Sci., 2014, 15, 23519 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13647a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mohammad Abul Farahb,
Hamad A. Al-Lohedana and
Khalid Mashay Al-Anazib
*a,
Mohammad Abul Farahb,
Hamad A. Al-Lohedana and
Khalid Mashay Al-Anazib