
Catalytic asymmetric formal total syntheses of (+)- and (−)-cycloclavine†
Saikat
Chaudhuri
,
Santanu
Ghosh
,
Subhajit
Bhunia
and
Alakesh
Bisai
 *
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal – 462 066, Madhya Pradesh, India. E-mail: alakesh@iiserb.ac.in
First published on 2nd January 2018
Abstract
We report an expeditious catalytic asymmetric approach to clavine alkaloids via a key Heck cyclization. This reaction sets the formation of vicinal stereocenters with excellent diastereoselectivity. Utilizing the aforementioned strategy, the formal total synthesis of cycloclavine (1) has been achieved via another key late-stage ester-aminolysis of 6.
Clavine alkaloids (1 and 2; Fig. 1) are a subclass of the ergot family of indole-containing alkaloids produced by several members of the Clavicipitaceae and Trichocomaceae families of filamentous fungi.1,2 They have also been identified in plants of the families Convolvulaceae, Poaceae and Polygalaceae.3 Ergot alkaloids (1–4; Fig. 1) primarily target serotonin (5-HT) receptors4a and α-adrenergic and dopamine receptors. Reportedly, some natural or semisynthetic ergoline derivatives are used as drugs, such as pergolide (2d) used as an anti-prolactin and anti-Parkinson's disease drug.4,5
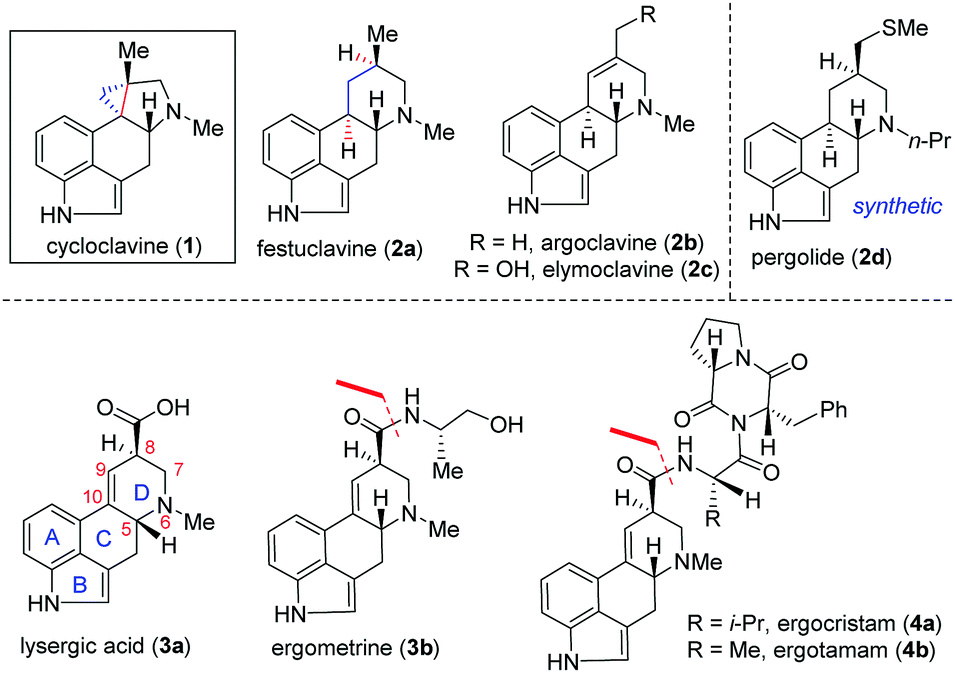 | ||
| Fig. 1 Clavine alkaloids (1 and 2) and lysergic acid (3 and 4) family. | ||
Therefore, significant progress has been made in the identification and characterization of genes responsible for the biosynthesis of clavine alkaloids (Fig. 1).6 Structurally, clavine alkaloids can exist in pentacyclic [such as cycloclavine (1)] and tetracyclic [such as festuclavine (2a–c)] forms.4b Lysergic acid (3a) (and its derivatives such as ergometrine 3b and ergopeptam alkaloids 4a–b) differs from clavine alkaloids 2a–c only in the oxidation state [see, 3a].7a,b
Cycloclavine (1) was isolated from the seeds of the African morning glory shrub Ipomoea hildebrandtii, and later from a species of filamentous fungus, Aspergillus japonicus.8a,b Although smaller in size, structurally cycloclavine (1) poses a formidable challenge because of its complex architecture with a pyrrolidine ring linked with a strained cyclopropane ring with three contiguous stereocenters, out of which two are vicinal all-carbon quaternary stereocenters.9 Despite the encouraging medicinal value of select clavine congeners, a comprehensive biological evaluation for the majority of these naturally occurring alkaloids has yet to be undertaken. From 2008 till 2016, only racemic syntheses of cycloclavine (1) have been reported, out of which three total syntheses10–12 and two formal total syntheses13,14 are reported. Interestingly, two consecutive coupling reactions such as selective alkylation of a dienolate and an intramolecular Heck reaction are utilized by Opatz and Netz for a racemic formal synthesis of cycloclavine (1).14
Recently, the first catalytic enantioselective total synthesis of unnatural (−)-cycloclavine (ent-1) has been achieved by Wipf and McCabe15avia Rh-catalyzed enantioselective cyclopropanation (up to 74% ee) of an unsubstituted allene to access a methylenecyclopropane derivative. Very recently, Cao and co-workers have reported an elegant formal total synthesis of naturally occurring (+)-cycloclavine (1)15b while our manuscript was under preparation. This synthesis features a Zn-mediated asymmetrical nucleophilic addition of N-tert-butanesulfinimine, an intramolecular ester-aminolysis reaction followed by isomerization of an exocyclic double bond and a late-stage intramolecular Heck coupling reaction.15b In this context, a unified strategy for the synthesis of 1 and 2 in enantioenriched form would present opportunities to provide access to significant quantities of the natural products and related analogues.
Retrosynthetically, we envisioned that cycloclavine (1) can be synthesized from an advanced enantiopure intermediate α,β-unsaturated amide 5 (Scheme 1) via isomerization followed by reduction of the amide functionality and cyclopropanation.10 Compound 5 can be accessed from α,β-unsaturated ester 6, which could in fact be the advanced intermediate for clavine alkaloids 2a–d sharing vicinal stereocenters (Fig. 1). We reasoned that ester 6 has the potential to afford two different tetracyclic intermediates such as 5 and 13 (Scheme 2). An ester-aminolysis of 6 can provide access to 5, on the other hand 6 can also afford ester 13 following a 5-endo-trig cyclization (Scheme 2). We argued that as the secondary amine (HOMO) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O π* (LUMO) are in proper alignment (see the orbital representation in 11), an ester-aminolysis of 6 would be facile to afford tetracyclic amide 5. However, a 5-endo-trig cyclization of 6 would not be possible because of bad alignment of the secondary amine (HOMO) and CC π* (LUMO) (see the orbital representation of intermediates 12a and 12b).
O π* (LUMO) are in proper alignment (see the orbital representation in 11), an ester-aminolysis of 6 would be facile to afford tetracyclic amide 5. However, a 5-endo-trig cyclization of 6 would not be possible because of bad alignment of the secondary amine (HOMO) and CC π* (LUMO) (see the orbital representation of intermediates 12a and 12b).
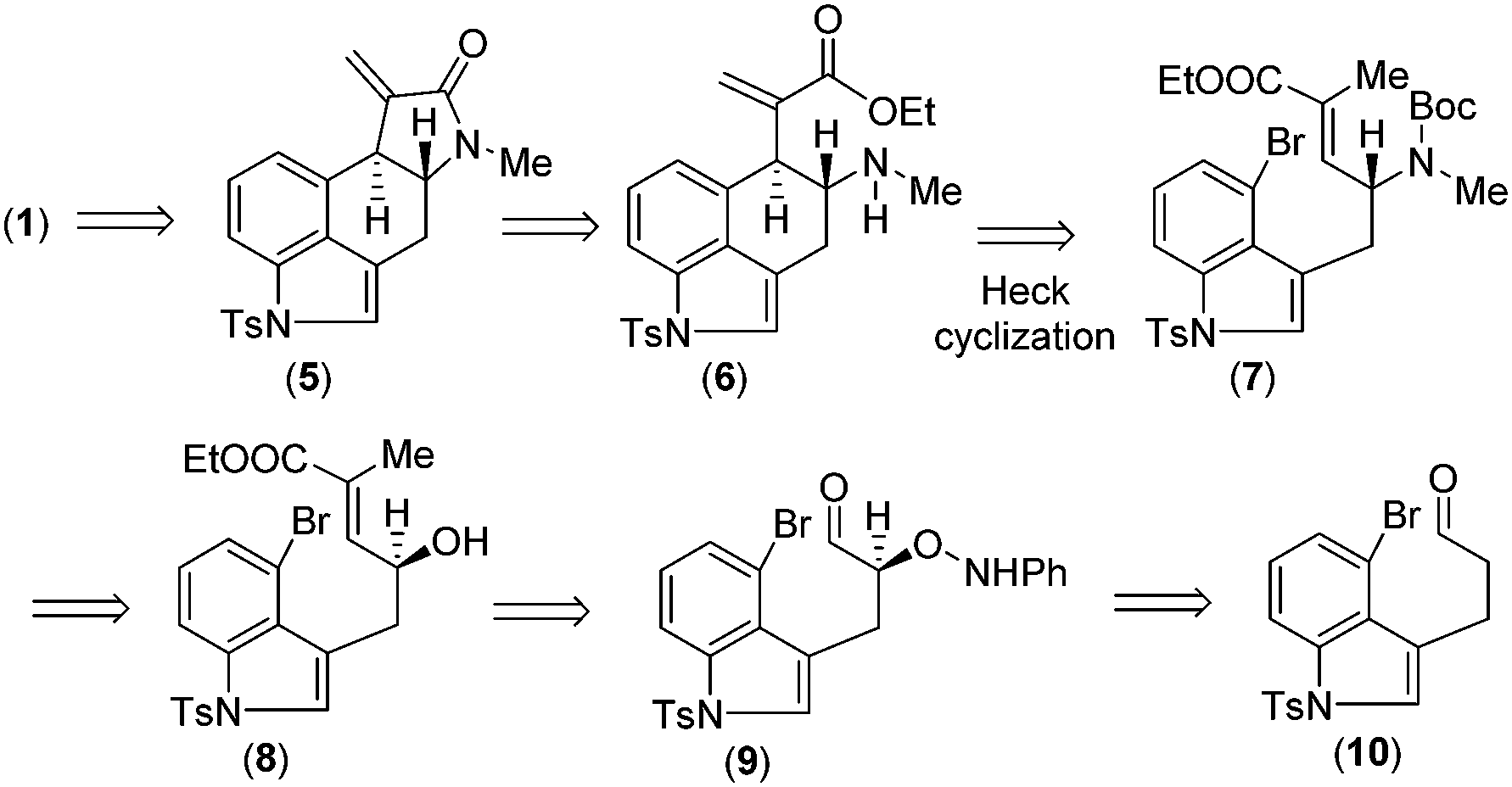 | ||
| Scheme 1 Retrosynthetic analysis of cycloclavine (1). | ||
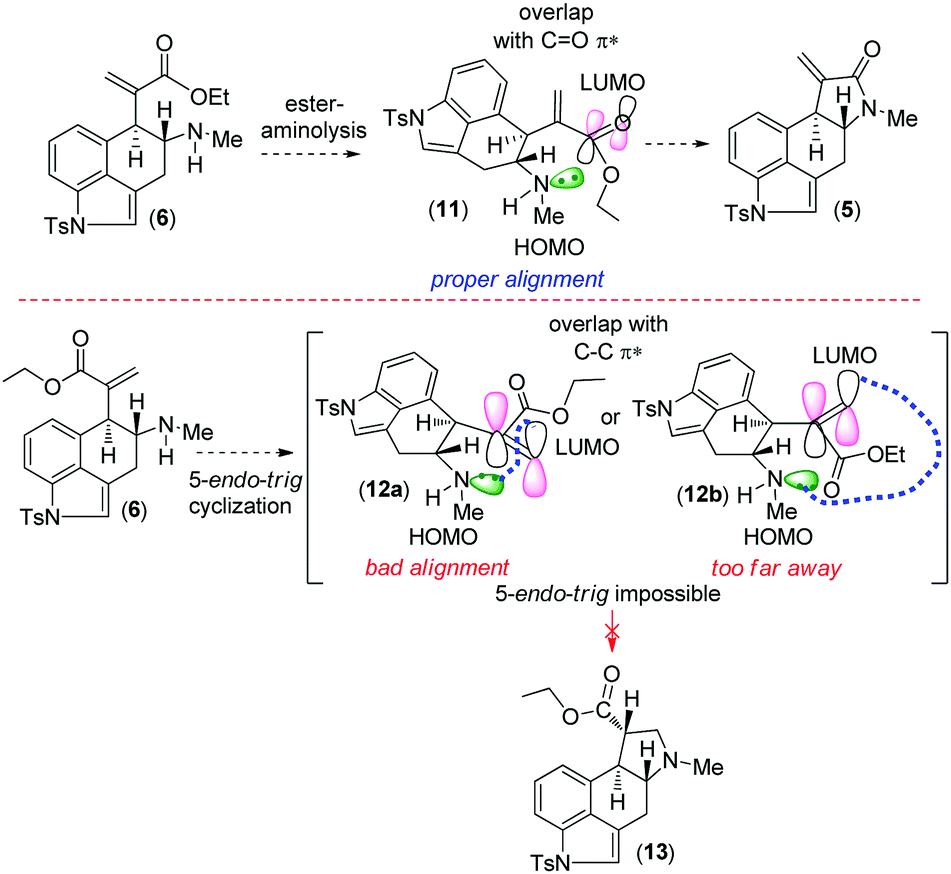 | ||
| Scheme 2 An ester-aminolysis versus 5-endo-trig cyclization of α,β-unsaturated ester 6. | ||
Further, we thought that α,β-unsaturated ester 6 with an exo-double bond can be synthesized via a key intramolecular Heck cyclization of allylamine 7 (Scheme 1). Enantioenriched allylic amine 7 can be synthesized from allylic alcohol 8via Mitsunobu type inversion using an azide nucleophile followed by synthetic manipulations. Non-racemic allyl alcohol 8 can be accessed from aldehyde 10via a D-proline catalysed α-aminoxylation reaction with nitrosobenzene through the intermediate aldehyde 9 (Scheme 1). Importantly, since both enantiomers of proline are commercially available, one can synthesize both antipodes of allylic alcohols, i.e.8 and ent-8.
On the basis of previous studies on the proline catalysed α-aminoxylation reaction of aliphatic aldehydes with nitrosobenzene,16 we decided to investigate the potential of this process in the catalytic asymmetric total synthesis of clavine alkaloids (Fig. 1). Towards this direction, we synthesized 3-allyl-4-bromoindole 15 from the Pd(0)-catalyzed reaction of 4-bromoindole 14 with allyl alcohol in the presence of triethylborane using Tamaru's report.17 This was then reacted with borane followed by oxidation with H2O2 in the presence of NaOH to afford a primary alcohol, which was then oxidized to obtain aldehyde 10 under Swern oxidation (Scheme 3). Having aldehyde 10 in hand, we then conducted a catalytic enantioselective α-aminoxylation reaction with nitrosobenzene in the presence of 10 mol% D-proline (Scheme 3). This reaction afforded an α-aminoxylated aldehyde, which was immediately reacted with a stabilized Wittig reagent prepared from 2-bromo ethylpropionate to afford compound E-ester 16 as the sole isomer in 85% yield over 2 steps with 96% enantioselectivity.18
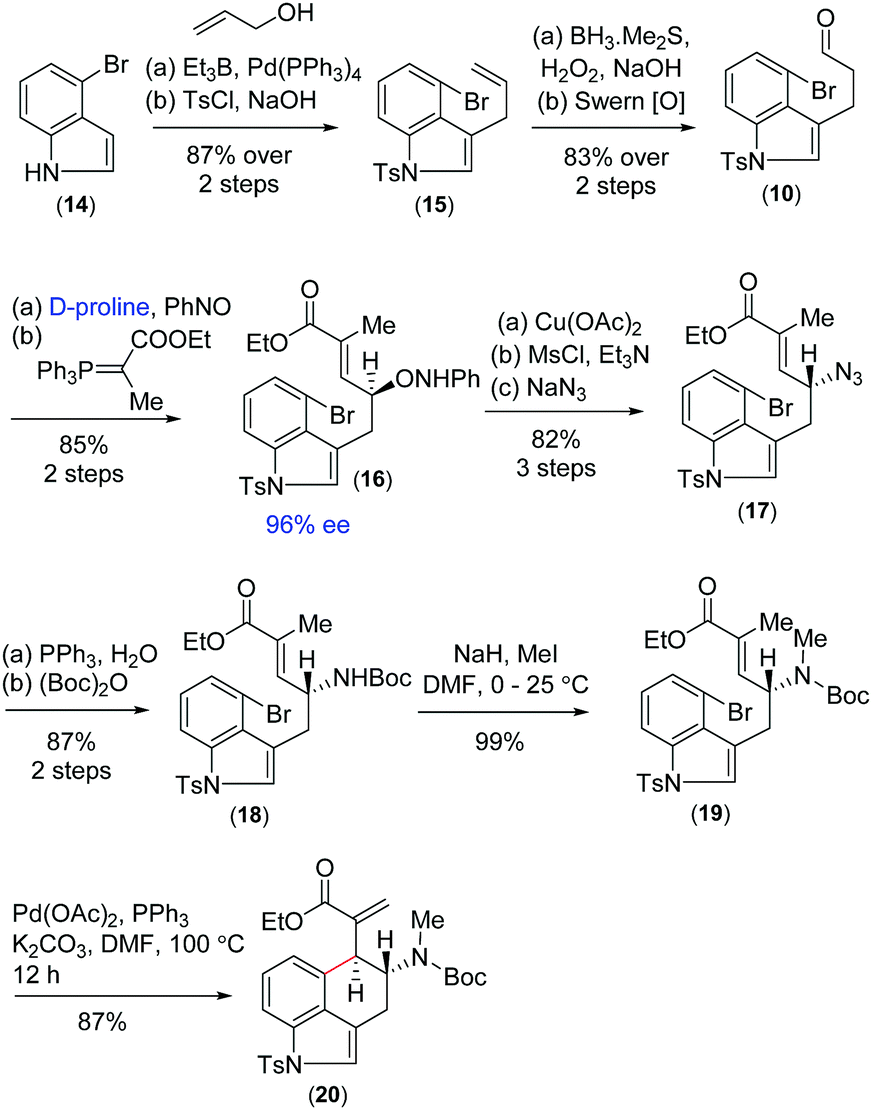 | ||
| Scheme 3 Asymmetric synthesis of key α,β-unsaturated ester 20. | ||
With compound 16 in hand, our effort was thereafter to elaborate to allylic amine 19 for key Heck cyclization (Scheme 3). Towards this, N–O bond cleavage was performed with anhydrous Cu(OAc)2,19,20 followed by mesylation and azide formation, affording 17 in 82% yield over 3 steps. The azide functionality was reduced under Staudinger conditions, followed by Boc-protection leading to intermediate 18 in 87% yield over 2 steps. The latter was N-methylated using methyl iodide to afford allyl amine 19 (Scheme 3). The intramolecular Heck cyclization of 19 was performed with 5 mol% Pd(OAc)2 and 10 mol% PPh3. Gratifyingly, this reaction afforded a single diastereomer of 20 in 87% yield (Scheme 3).21
We urged that the Heck cyclization of 19 can proceed through the intermediate Pd(II)-species 21a (Scheme 4). However, in order to minimize the steric clash, 21a could immediately form 21bvia a C–C bond rotation. The formation of the tetra-substituted α,β-unsaturated ester 22 from this intermediate is not possible since Pd(II) and β-hydride are anti-position to each other (Scheme 4). At this situation, a β-hydride transfer from an adjacent methyl group in 21c could afford 20 having vicinal stereogenic centers (Scheme 4).
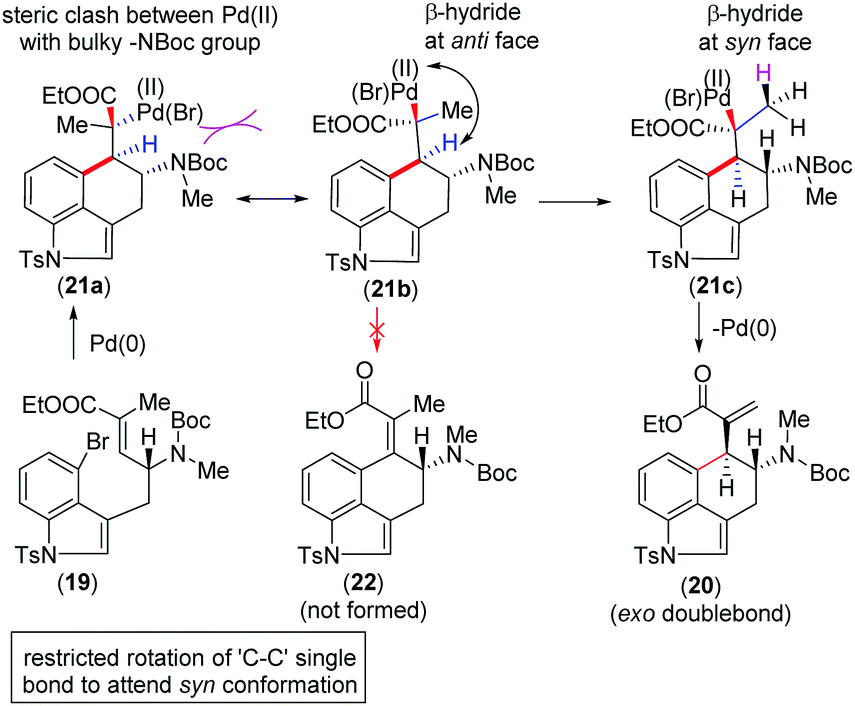 | ||
| Scheme 4 Rationale of highly diastereoselective Heck cyclization. | ||
Further, compound 20 was elaborated under a key cyclization in order to get the tetracyclic core of cycloclavine (1). Towards this, we deprotected the Boc group in the presence of trifluoroacetic acid at 25 °C to afford 6, which under refluxing toluene afforded the ester-aminolysis product 5 with an exocyclic double bond in 88% isolated yield over 2 steps (Scheme 5).22 To our delight, no trace of the 5-endo-trig cyclization (aza-Michael reaction) product was observed, as confirmed from 1H-NMR analysis of the crude reaction mixture.
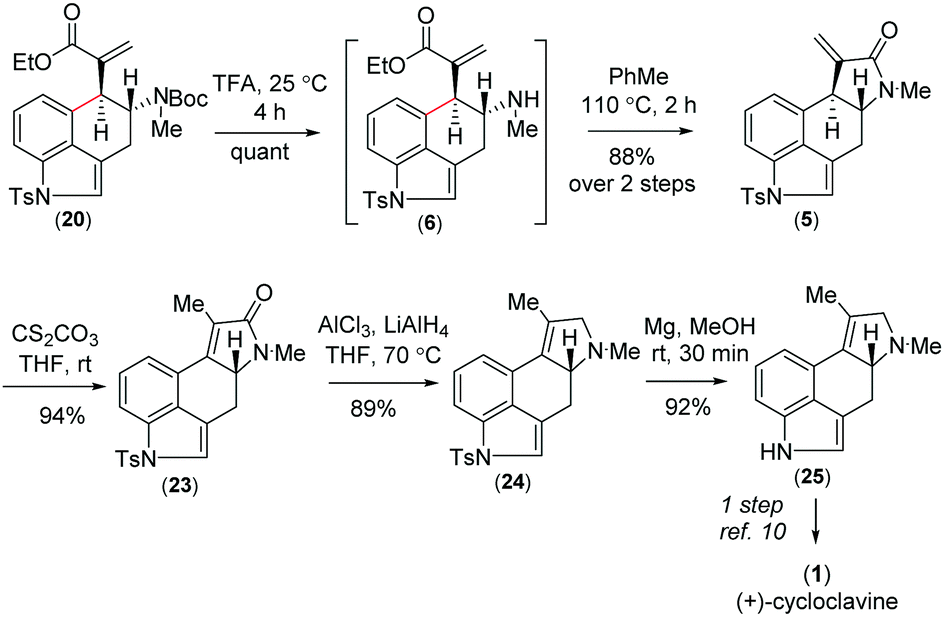 | ||
| Scheme 5 Asymmetric synthesis of (+)-cycloclavine (1). | ||
With the enantioenriched tetracyclic 5 in hand, we then isomerized to γ-lactam using cesium carbonate in THF to afford 23 in 94% yield (Scheme 5).23 The latter was reduced using LiALH4 in the presence of AlCl3 to furnish the electron-rich tetrasubstituted double bonded product 24 in 89% yield.24 Further, in order to access the antipode of 25, we performed the catalytic enantioselective α-aminoxylation reaction of 10 with nitrosobenzene in the presence of 10 mol% L-proline, which afforded the product ent-16 in 97% ee after a Wittig reaction (Scheme 6). This enantioenriched material was elaborated to ent-25via a similar reaction sequence as shown in Schemes 3 and 5. As the total synthesis of cycloclavine (1) from 25 is known, our effort culminated in the formal total synthesis of this alkaloid.
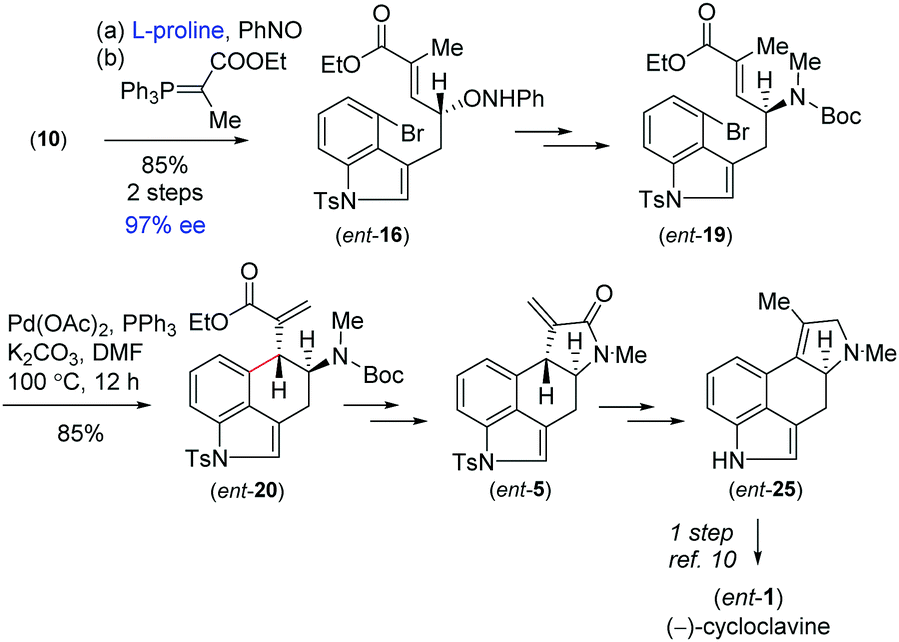 | ||
| Scheme 6 Asymmetric synthesis of (−)-cycloclavine (ent-1). | ||
In summary, the catalytic enantioselective formal total synthesis of both antipodes of cycloclavine (1) has been achieved via a late stage ester-aminolysis of an α,β-unsaturated ester intermediate 6. The vicinal stereocenters of this advanced intermediate were established following an intramolecular Heck cyclization of an enantioenriched α,β-unsaturated ester having allylamine 19. Since both enantiomers of proline are inexpensive and commercially available, our strategy offers an expeditious approach to either enantiomer of cycloclavine (1). Further efforts for a rational extension of the strategy to other congeners of clavine alkaloids are underway and will be reported in due course.25
Financial support from the SERB, DST [EMR/2016/000214] and CSIR [02(0295)/17/EMR-II], Govt. of India, is gratefully acknowledged. S. C. and S. B. thank the UGC and CSIR, respectively, for predoctoral fellowships. S. G. thanks IISER Bhopal for a postdoctoral fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Hofmann, in Plants in the developments of modern medicine, ed. T. Swain, Harvard University Press, Cambridge, Mass, 1972, pp. 235–260 Search PubMed; (b) Review: S. R. McCabe and P. Wipf, Org. Biomol. Chem., 2016, 14, 5894 RSC.
- (a) L. V. Boichenko, D. M. Boichenko, N. G. Vinokurova, T. A. Reshetilova and M. U. Arinbasarov, Microbiology, 2001, 71, 306 CrossRef; (b) P. L. Schiff, Am. J. Pharm. Educ., 2006, 70, 98 CrossRef PubMed.
- (a) D. Gröger and H. G. Floss, Alkaloids: Chem. Biol., 1998, 50, 171 Search PubMed . Review; (b) C. Wallwey and S.-M. Li, Nat. Prod. Rep., 2011, 28, 496 RSC.
- (a) A. Sinz, Pharm. Unserer Zeit, 2008, 37, 306 CrossRef CAS PubMed . Review; (b) H. Liu and Y. Jia, Nat. Prod. Rep., 2017, 34, 411 RSC.
- (a) I. Ninomiya and T. Kiguchi, in The Alkaloids, ed. A. Brossi, Academic Press, San Diego, CA, 1990, vol. 38, pp 1–156 Search PubMed; (b) M. Somei, Y. Yokoyama, Y. Murakami, I. Ninomiya, T. Kiguchi and T. Naito, in The Alkaloids, ed. G. A. Cordell, Academic Press, San Diego, CA, 2000, vol. 54, pp. 191–257 Search PubMed.
- (a) D. Jakubczyk, J. Z. Cheng and S. E. O’Connor, Nat. Prod. Rep., 2014, 31, 1328 RSC; (b) C. A. Young, C. L. Schardl, D. G. Panaccione, S. Florea, J. E. Takach, N. D. Charlton, N. Moore, J. S. Webb and J. Jaromczyk, Toxins, 2015, 7, 1273 CrossRef CAS PubMed.
- (a) C. L. Schardl, D. G. Panaccione and P. Tudzynski, Alkaloids: Chem. Biol., 2006, 63, 45 CAS; (b) T. Haarmann, et al. , Mol. Plant Pathol., 2009, 10, 563 CrossRef CAS PubMed and references cited therein.
- (a) D. Stauffacher, P. Niklaus, H. Tscherter, H. P. Weber and A. Hofmann, Tetrahedron, 1969, 25, 5879 CrossRef CAS PubMed; (b) T. Furuta, M. Koike and M. Abe, Agric. Biol. Chem., 1982, 46, 1921 CAS.
- Biosynthesis of cycloclavine (1), see; (a) D. Jakubczyk, L. Caputi, A. Hatsch, C. A. F. Nielson, M. Diefenbacher, J. Klein, A. Molt, H. Schröder, J. Z. Cheng, M. Naesby and S. E. O’Conner, Angew. Chem., Int. Ed., 2015, 54, 5117 CrossRef CAS PubMed; (b) D. Jakubczyk, L. Caputi, C. E. M. Stevenson, D. M. Lawson and S. E. O’Conner, Chem. Commun., 2016, 52, 14306 RSC.
- M. Incze, G. Dörnyei, I. Moldvai, E. Temesvári-Major, O. Egyed and C. Szánty, Tetrahedron, 2008, 64, 2924 CrossRef CAS.
- F. R. Petronijevic and P. Wipf, J. Am. Chem. Soc., 2011, 133, 7704 CrossRef CAS PubMed.
- N. D. Jabre, T. Watanabe and M. Brewer, Tetrahedron Lett., 2014, 55, 197 CrossRef CAS PubMed.
- W. Wang, J.-T. Lu, H.-L. Zhang, Z.-F. Shi, J. Wen and X.-P. Cao, J. Org. Chem., 2014, 79, 122 CrossRef CAS PubMed.
- N. Netz and T. Opatz, J. Org. Chem., 2016, 81, 1723 CrossRef CAS PubMed.
- (a) S. R. McCabe and P. Wipf, Angew. Chem., Int. Ed., 2017, 56, 324 CrossRef CAS PubMed; (b) J.-Q. Chen, L.-L. Song, F.-X. Li, Z.-F. Shi and X.-P. Cao, Chem. Commun., 2017, 53, 12902 RSC.
- (a) L-Proline catalyzed α-aminoxylation of aldehydes, see; G. Zhong, Angew. Chem., Int. Ed., 2003, 42, 4247 CrossRef CAS PubMed; (b) L-proline catalyzed α-amination of aldehydes, see; (c) B. List, J. Am. Chem. Soc., 2002, 124, 5656 CrossRef CAS PubMed; (d) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790 CrossRef.
- M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592 CrossRef CAS PubMed.
- For α-aminoxylation and Horner–Wadsworth–Emmons olefination of aldehydes, see, N. B. Kondekar and P. Kumar, Org. Lett., 2009, 11, 2611 CrossRef CAS PubMed.
- (a) Y. Hayashi, M. Shoji, H. Ishikawa, J. Yamaguchi, T. Tamura, H. Imai, Y. Nishigaya, K. Takabe, H. Kakeya and H. Osada, Angew. Chem., Int. Ed., 2008, 47, 6657 CrossRef CAS PubMed; (b) J. S. Yadav, K. Ramesh, U. V. S. Reddy, B. V. S. Reddy and A. A. K. A. Ghamdi, Tetrahedron Lett., 2011, 52, 2943 CrossRef CAS.
- N–O bond cleavage using Pd/C under 1 atm H2 led to a complex mixture of products.
- For leading references for Pd-catalyzed intramolecular Heck cyclization for the synthesis of natural products, see; (a) M. Zhang, X. Huang, L. Shen and Y. Qin, J. Am. Chem. Soc., 2009, 131, 6013 CrossRef CAS PubMed; (b) J. Wu, J. Becerril, Y. Lian, H. L. M. Davies, J. A. Porco Jr. and J. S. Panek, Angew. Chem., Int. Ed., 2011, 50, 5938 CrossRef CAS PubMed; (c) J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Org. Lett., 2012, 14, 2714 CrossRef CAS PubMed; (d) W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 1818 CrossRef CAS PubMed; (e) L.-Y. Pu, J.-Q. Chen, M.-L. Li, Y. Li, J.-H. Xie and Q.-L. Zhou, Adv. Synth. Catal., 2016, 358, 1229 CrossRef CAS.
- For references on intramolecular ester-aminolysis to from α-methylene-γ-lactam, see; (a) S. Bonazzi, B. Cheng, J. S. Wzorek and D. A. Evans, J. Am. Chem. Soc., 2013, 135, 9338 CrossRef CAS PubMed; (b) J. A. Sirvent, F. Foubelo and M. Yus, J. Org. Chem., 2014, 79, 1356 CrossRef CAS PubMed . Also, see ref. 14.
- Other bases such as K2CO3, KHMDS, and DBU afforded isomerization products in 59–72% isolated yields along with some decomposition under heating.
- Reduction using LiAlH4 or Red-Al afforded a mixture of products as seen from TLC.
- For a biomimetic approach to clavine alkaloids, see; S. Chaudhuri, S. Bhunia, A. Roy, M. K. Das and A. Bisai, Org. Lett., 2018, 20, 288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional reaction optimization, spectroscopic data for all new compounds. See DOI: 10.1039/c7cc09045e |
| This journal is © The Royal Society of Chemistry 2018 |