
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating isomer specific photoprotection in a model plant sunscreen†
M. D.
Horbury
a,
A. L.
Flourat
b,
S. E.
Greenough
a,
F.
Allais
 b and
V. G.
Stavros
*a
b and
V. G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bChaire ABI–AgroParisTech, CEBB, 3 rue des Rouges Terres, 51110 Pomacle, France
First published on 2nd January 2018
Abstract
Sinapate esters are used throughout the plant kingdom, for example in photoprotection from ultraviolet radiation. Sinapate esters are naturally produced in their E-isomeric form; however, upon exposure to ultraviolet radiation, photoisomerization drives Z-isomer formation. To elucidate the photoprotection capacity of E vs. Z forms of sinapate esters, we explore the photochemistry of the model system, Z-ethyl sinapate. Following a novel Z-ethyl sinapate synthesis, we demonstrate that photoprotection is isomer independent. This suggests that, regarding photoprotection, there were no evolutionary pressures for biosynthesis of either isomer.
Sinapate esters are used in a multitude of processes within the plant kingdom, such as lignin synthesis, disease resistance and plant growth regulation.1,2 They are produced via the phenylpropanoid pathway1 in their thermodynamically more stable E-isomer.3,4 Recently, the photochemistry of sinapate esters has received considerable attention, in particular, sinapoyl malate ((2S)-2-{[(2E)-3-(4-hydroxy-3,5-dimethoxyphenyl)-2-propenoyl]oxy}succinic acid).3,5–7 Sinapoyl malate is a sunscreening agent used by plants in response to overexposure to ultraviolet (UV) radiation,1,8 in a manner akin to the melanin pigments in human skin.9–11 In an effort to elucidate the photoprotection mechanism of sinapoyl malate, its photochemistry has been previously investigated, following exposure to UVA (400–315 nm) and UVB (315–280 nm) radiation. These studies demonstrated that sinapoyl malate undergoes ultrafast (femtosecond (fs) to picosecond (ps), 10−15 and 10−12 seconds respectively) photoisomerization via an E–Z (trans–cis) isomerization pathway, thus reconciling its capacity for photoprotection.3,5–7 These studies also showed that the ester functional group has little effect to the overall photochemistry of the sinapate.5,6
While these sinapate esters have been studied previously, only the photochemistry of their naturally occurring E-isomeric forms have been obtainable.3,5,6 In general, when E-sinapates are exposed to UV, a high photoisomerization quantum yield leads to significant formation of the Z-isomer.3,5,6,12,13 Furthermore, a recent study on the related 2-ethylhexyl-4-methoxycinnamate, an EU approved sunscreening agent, has demonstrated that the Z-isomer is significantly more genotoxic than the E-isomer.14 Hence, an understanding into the photochemistry of the Z-isomer is highly pertinent, which hitherto has not been possible due to the difficulties in synthesizing pure Z-isomers of these esters. Therefore, we have developed a new synthetic method (Scheme 1) to produce a model system of sinapoyl malate, Z-ethyl sinapate (ethyl-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylate, Z-ES), enabling us to execute isomer specific fs time-resolved pump–probe spectroscopy (see ESI† for details), and thus compare the photochemistry of the E-ESversus Z-ES isomers.
 | ||
| Scheme 1 Z-Ethyl sinapate (Z-ES) synthesis. | ||
The novel synthetic approach developed to access the Z-α,β-unsaturated ester Z-ES from syringaldehyde (1) relied on the use of a Still–Gennari Z-selective olefination15 on ethyl P,P-bis(2,2,2-trifluoroethyl)phosphonoacetate, potassium hexamethyldisilazide, 18-crown-6 and THF at −78 °C (Scheme 1). To avoid the consumption of the phosphonoacetate anion by the phenol moiety, we first protected 1 using an acetate before performing the Still–Gennari olefination. Although this reaction provided the expected Z-ES isomer (80%), the latter was rapidly isomerized into the E-ES isomer upon acetate cleavage either in basic or acidic conditions. Indeed, as described in Scheme 1, the phenolate (2) obtained through the deprotection rapidly isomerizes to give the corresponding methylene quinone (3). The latter can then undergo a rotation around the Cβ–Cγ bond, leading to the E-ES isomer after re-aromatization. It thus seemed necessary to avoid any treatment that may lead to the phenolate formation. Under this consideration, 1 was submitted directly to Still–Gennari olefination. The reaction led to the formation of a separable Z-ES and E-ES isomer mixture, however it is noteworthy to mention that the Z-ES isomer co-eluted with phosphonate by-products, rendering the purification of the former quite difficult (for further synthetic and characterization details please see ESI†).
To investigate the photochemistry of the synthesized Z-ES, we utilized the fs time-resolved solution-phase technique, transient electronic (UV/visible) absorption spectroscopy (TEAS). These measurements were performed on 1 mM solutions of Z- and E-ES in cyclohexane photoexcited at 319 nm (UVA region absorption maxima, see Fig. 1) and the resulting transient absorption spectrum/spectra (TAS) are shown in Fig. 2a and b. After initial photoexcitation, the TAS of both isomers are dominated by two features, an excited state absorption (ESA) centred around 425 nm and a ground state bleach (GSB), centred at ∼335 nm. As the time-delay between the pump and probe pulses (Δt) is increased, these spectral features begin to decay back to the baseline. At Δt >20 ps, the ESA has completely decayed away, revealing a pair of spectral features in the probe region of 330–350 nm consisting of the GSB and a photoproduct absorption.
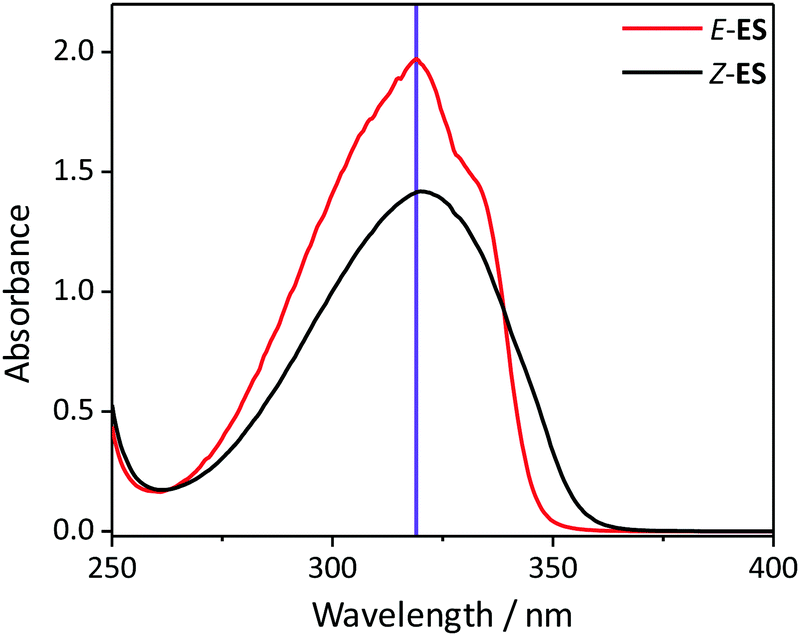 | ||
| Fig. 1 The UV/visible absorption spectra of 1 μM solutions Z- and E-ES in cyclohexane (black and red respectively). The excitation wavelength (319 nm) is shown by the purple line. | ||
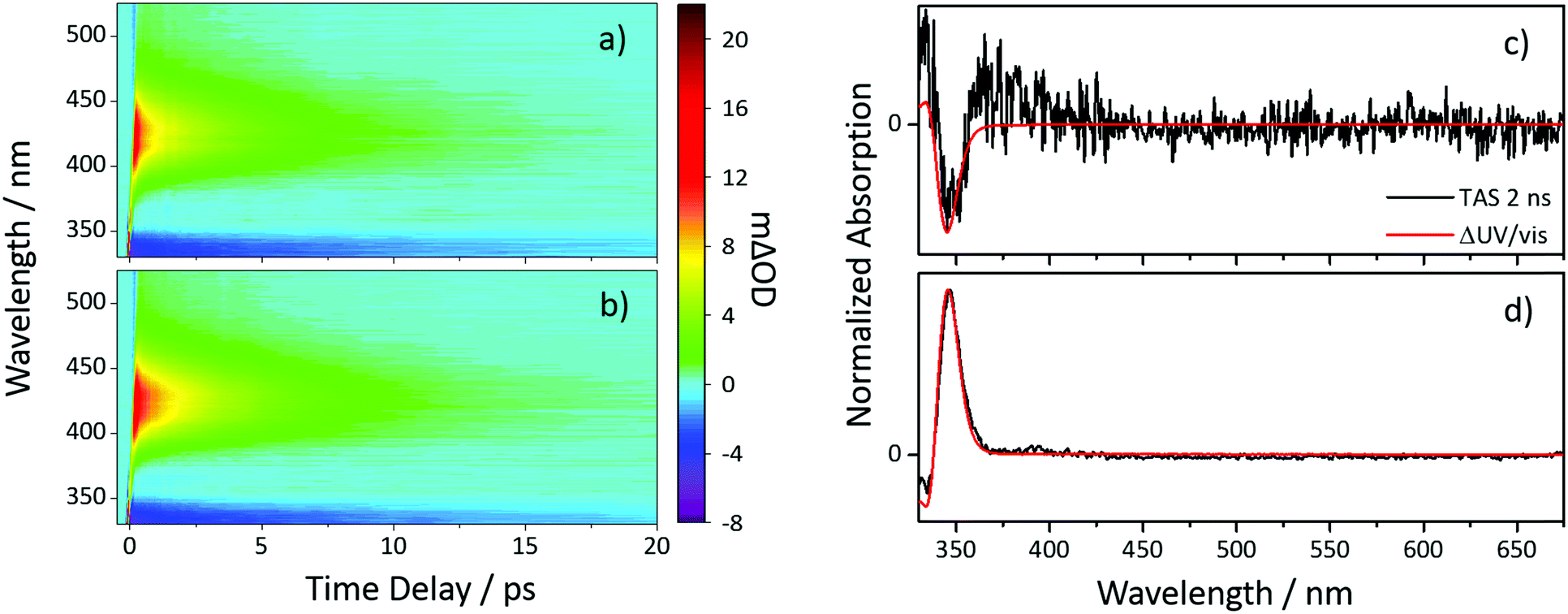 | ||
| Fig. 2 TAS of (Z/E)-ES shown as false colourmaps representing the change in optical density (ΔOD) in cyclohexane (a) Z-ES and (b) E-ES photoexcited at 319 nm. TAS taken at a Δt of 2 ns of (c) Z-ES and (d) E-ES are shown and overlaid with ΔUV/vis spectra. | ||
This pair of spectral features differ between the two isomers; this is highlighted by TAS at Δt = 2 ns shown in Fig. 2c and d. In the E-ES TAS the GSB is centred at ∼335 nm, with a photoproduct absorption at ∼345 nm, while in the Z-ES TAS the features are reversed, with the GSB at ∼345 nm and the photoproduct absorption at ∼335 nm. Note the increased noise present in Fig. 2c is due to weaker photoproduct signal in the TAS (we return to discuss this below). To identify the species responsible for the photoproduct absorption, the TAS at Δt = 2 ns were compared to steady-state difference absorption spectra (‘ΔUV/vis’, see ESI† for details). Since E- and Z-ES have differences in their respective ground state absorptions (Fig. 1), with Z-ES having a slightly broader and weaker absorption than the E-ES, a distinct difference absorption signature is observed. The TAS and ΔUV/vis spectra for initially photoexcited E-ES are near matches of each other (Fig. 2d). However, while the TAS and ΔUV/vis spectra for photoexcited Z-ES are similar (Fig. 2c) providing assurance that the E-ES photoproduct is being formed, a difference is present nonetheless. We attribute this difference to the presence of the phenolic radical ES species generated via two-photon ionization, as seen in previous studies.5,6,16 We believe that this is only present in Z-ES as its ionization potential is likely to be low enough for two-photon ionization (with the 319 nm photoexcitation pulse) to occur, while in E-ES the ionization potential appears to be higher and therefore two-photon ionization is precluded. To confirm this, additional TEAS measurements on E-ES in cyclohexane were carried out using an excitation wavelength of 305 nm (see ESI† Fig. S5). At this excitation wavelength, a broadening of the absorption peak present at a Δt = 2 ns was observed akin the Z-ES absorption peak. This supports the idea that the ionization potential of E-ES is higher in energy than Z-ES. It is worth noting that this is an artefact of the high photon densities present in fs laser pulses and is highly unlikely to occur in nature, where single-photon (solar) excitation processes dominate.
While the TAS of both Z-ES and E-ES appear qualitatively similar, we can gain quantitative insight into their respective photochemistry by extracting the kinetics contained within the TAS. To do this, a sequential (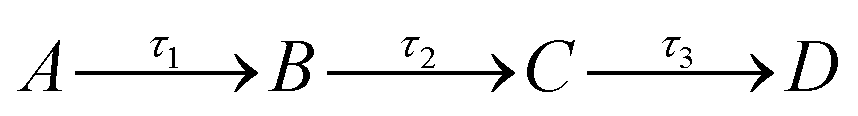 ) global fitting technique was performed, using the software package Glotaran.17 The resulting time-constants (τn), which describe the timescale for each step in the relaxation pathway, are shown in Table 1, (see ESI† for evolution associated difference spectra).
) global fitting technique was performed, using the software package Glotaran.17 The resulting time-constants (τn), which describe the timescale for each step in the relaxation pathway, are shown in Table 1, (see ESI† for evolution associated difference spectra).
| τ ivr (fs) | τ iso (ps) | τ pp (ns) | |
|---|---|---|---|
| Z-ES | 330 ± 40 | 5.05 ± 0.06 | ≫2 |
| E-ES | 290 ± 40 | 4.60 ± 0.04 | ≫2 |
The dynamics responsible for the time-constants produced in our sequential global fits have been extensively discussed in previous literature for the E-isomer;5,6,18 we shall provide a quick overview here. After the initial photoexcitation, E-ES undergoes a rapid geometry relaxation, in the form of intramolecular vibrational redistribution (IVR), which we assign the time-constant τivr. From this relaxed geometry, E-ES proceeds to undergo a photoisomerization, on the timescale of τiso, along the E to Z isomerization coordinate. This results in the reformation of the E-ES reactant in its electronic ground state, along with the formation of Z-ES in its electronic ground state as a photoproduct, with a lifetime represented by the time-constant τpp.
Regarding the photochemistry of Z-ES, the TAS and the results produced by the global fit for Z-ES match very closely with the TAS and global fit results of E-ES. Due to this, we are confident that after photoexcitation, Z-ES undergoes the same (or at least very similar) photochemical processes as E-ES, with the photoisomerization being Z to E instead. A caveat however should be noted: while the photochemistry of Z- and E-ES appears to be similar, the photoisomerization of E-ES occurs 10% faster; this is most likely due to the electronic structure of the E-ES compared to Z-ES; slight differences in the topologies of the respective potential energy surfaces can noticeably impact these timescales. Furthermore, from the TAS taken at a Δt = 2 ns, the GSB of Z-ES recovers more than the GSB of E-ES, suggesting that the isomerization is likely biased towards the Z-isomer, reconciled by the (apparent) poor signal-to-noise from the weaker photoproduct absorbance. This is further supported by the extracted ratio of Z- to E-ES following steady state irradiation at 319 nm (see ESI† Fig. S8 for details). From these measurements, the ratio was found to bias towards Z-ES (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 of Z- to E-isomer). This result implies that the resultant ratio is kinetically, rather than thermodynamically, driven (cf. E-ES is thermodynamically the more stable isomer).
:30 of Z- to E-isomer). This result implies that the resultant ratio is kinetically, rather than thermodynamically, driven (cf. E-ES is thermodynamically the more stable isomer).
Additional TEAS measurements of Z- and E-ES in ethanol have been performed (see ESI† for results), to observe the impact of a more strongly interacting solvent environment on the photoproduct formation. We avoid detailed discussion of the results herein, owing to the excessive contamination of the phenoxyl radical, which is more prone to generation in polar solvents,5,16 masking observation of the Z- or E-photoproduct. However, it is still clear the excited state relaxation dynamics occur on the ps timescale. Importantly we see that there is no observable difference in the photoisomerization time-constants for each isomer (cf. 10% different for Z/E-ES in cyclohexane), implying that E-ES may experience a greater perturbation (to its dynamics) from the increased solvent interaction, when compared to Z-ES.
In conclusion, we have developed a novel, isomer specific, synthesis of a sinapate ester. This has enabled us to carry out isomer specific, time-resolved spectroscopic measurements on (E/Z)-ethyl sinapate, a model of the ubiquitous sinapoyl malate, which is a photoprotection molecule found in plant leaves. These measurements have revealed that the photochemistry of the Z-isomer is very likely comparable to the photochemistry of the naturally produced E-isomer in terms of rapidly disposing dangerous excess energy imparted due to potentially phototoxic UV absorption. Therefore, the capacity for photoprotection provided by the Z-isomer is similar to the E-isomer. This indicates that the intrinsic photochemistries of the two isomers do not induce a selective pressure upon which isomer the plant must produce. It is convenient that the plant can use the thermodynamic product from the phenylpropanoid pathway rather than having to evolve a more sophisticated synthesis.
We acknowledge that our model system has not taken the ester functional group of sinapoyl malate into account, however previous studies have shown such sinapates to be sufficient models for the more complex sinapoyl malate, in non-polar, polar and viscous environments.5,6 Furthermore, these studies have shown that photoisomerization of E-sinapates is achieved through large amplitude motion;6 an environment that impedes this motion (e.g. viscous) might retard the isomerisation and thus lead to competing and potentially detrimental side reactions. Whether the difference in geometric structure of the Z-isomer (versus E) can noticeably impede the photoisomerization, could be a contributing factor in the biosynthesis of the E-isomer. Additional work is currently underway (both synthesis and spectroscopy), towards more realistic scenarios.
The authors would like to acknowledge the Warwick Centre for Ultrafast Spectroscopy (WCUS) for the use of the Lambda 850 and Fluorolog 3. M. D. H. thanks the Leverhulme Trust for postdoctoral funding. S. E. G. thanks the University of Warwick for postdoctoral funding. A. L. F. and F. A. thank the Grand Reims, the Conseil Départemental de la Marne and the Region Grand Est for financial support. Finally, V. G. S. thanks the EPSRC for an equipment grant (EP/J007153), the Leverhulme Trust for a research grant (RPG-2016-055) and the Royal Society and Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship.
Conflicts of interest
There are no conflicts to declare.References
- C. M. Fraser and C. Chapple, The Arabidopsis Book, 2011, p. e0152 Search PubMed.
- C. Chapple, T. Vogt, B. E. Ellis and C. R. Somerville, Plant Cell, 1992, 4, 1413–1424 CrossRef CAS.
- J. Luo, Y. Liu, S. Yang, A. L. Flourat, F. Allais and K. Han, J. Phys. Chem. Lett., 2017, 8, 1025–1030 CrossRef CAS PubMed.
- M. L. Sallum and R. Erra-Balsells, Environ. Control Biol., 2013, 51, 1–10 CrossRef.
- L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- M. Horbury, W.-D. Quan, A. Flourat, F. Allais and V. Stavros, Phys. Chem. Chem. Phys., 2017, 19, 21127–21131 RSC.
- F. Liu, L. Du, Z. Lan and J. Gao, Photochem. Photobiol. Sci., 2017, 16, 211–219 CAS.
- K. Osakabe and Y. Osakabe, Plant Light Stress, in eLS, John Wiley & Sons, Ltd, Chichester, 2012 Search PubMed.
- J. P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572–594 CrossRef CAS PubMed.
- A. Huijser, A. Pezzella and V. Sundström, Phys. Chem. Chem. Phys., 2011, 13, 9119–9127 RSC.
- G. Kahnt, Phytochemistry, 1967, 6, 755–758 CrossRef CAS.
- J. S. Challice and A. H. Williams, J. Chromatogr. A, 1966, 21, 357–362 CrossRef CAS PubMed.
- A. Sharma, K. Bányiová, P. Babica, N. El Yamani, A. R. Collins and P. Čupr, Sci. Total Environ., 2017, 593, 18–26 CrossRef PubMed.
- W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405–4408 CrossRef CAS.
- D. S. Larsen, I. H. M. van Stokkum, M. Vengris, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 87, 1858–1872 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, J. Stat. Softw., 2012, 49(3) Search PubMed.
- S. Wang, S. Schatz, M. C. Stuhldreier, H. Böhnke, J. Wiese, C. Schröder, T. Raeker, B. Hartke, J. K. Keppler, K. Schwarz, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2017, 19, 30683–30694 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, 1H and 13C NMR, additional TAS data, fitting details and additional ΔUV/vis spectra. See DOI: 10.1039/c7cc09061g |
| This journal is © The Royal Society of Chemistry 2018 |