
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the photobehavior of a 2,1,3-benzothiadiazole-based HOF and its molecular units: experimental and theoretical insights into their spectroscopic properties in solution and in the solid state†
Mario
de la Hoz Tomás
 a,
Juan Ángel
Organero
a,
Maria Rosaria
di Nunzio
a,
Taito
Hashimoto
b,
Ichiro
Hisaki
b and
Abderrazzak
Douhal
*a
a,
Juan Ángel
Organero
a,
Maria Rosaria
di Nunzio
a,
Taito
Hashimoto
b,
Ichiro
Hisaki
b and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, and INAMOL, Universidad de Castilla-La Mancha, Avenida Carlos III, S/N, 45071 Toledo, Spain. E-mail: abderrazzak.douhal@uclm.es
bGraduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 29th May 2024
Abstract
2,1,3-Benzothiadiazole (BTD) acid derivatives have emerged as versatile new building blocks for the fabrication of smart porous materials. Herein, we report experimental and theoretical studies on a BTD-based H-bonded organic frameworks (HOFs) and its methylated ester and carboxylic acid molecular units in solutions and in solid state. BTIA-ester and BTIA-COOH in solutions display a large Stokes-shifted emission (∼6000 cm−1) as a result of an intramolecular charge-transfer (ICT) reaction in the excited species, followed by a twisting of phenyl moieties. Femtosecond (fs) experiments on the ester and acid derivatives in N,N-dimethylformamide (DMF) reveal that an ICT reaction takes place in ∼300 fs while the phenyl twisting occurs in ∼6 ps. BTIA-ester shows a single emission lifetime of 7.52 ns, while BTIA-COOH displays lifetimes of 390 ps and 1.15 and 7.75 ns assigned to different emitting species. Theoretical calculations on the ester compound agree with experimental observations. Flash photolysis experiments on both samples in DMF and under ambient conditions show a triplet state population living for ∼500 ns and a charge-separated species living for ∼2 μs. In solid state, BTIA-ester shows an abnormally slow ICT event (80 ps) leading to ICT aggregates with lifetimes of 1.20 and 2.87 ns, whereas BTIA-HOF exhibits fast ICT and intermolecular proton-transfer (PT) reactions (<15 ps), producing ICT and ionic species with lifetimes of 0.52 and 1.23 ns, respectively. At the single-crystal level, BTIA-HOF displays fluorescence properties not dependent on the interrogated point under fluorescence microscopy, suggesting a homogeneous distribution of the molecular units of the HOF. These results provide new findings for a better understanding of the photobehavior of BTD derivatives and related HOFs and will help in the development of new HOFs for photonic applications.
1. Introduction
The design and fabrication of new materials with luminescence properties are one of the main research interests in the field of smart materials for advanced optoelectronics.1–4 In particular, luminescent porous materials have attracted considerable attention owing to their large surface area, high porosity, thermal stability, and chemical tunability, allowing their possible use in a wide range of applications.5–12 Among these porous materials, luminescent metal–organic frameworks (MOFs) have been extensively studied over the last two decades.5–7,13,14 However, the spectroscopic properties of other porous materials, such as covalent organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs), are not yet fully understood.10–12,15–17In the last decade, HOFs have emerged as new porous materials with diverse possible applications, such as in gas storage and separation, catalysis, luminescent sensing, and optoelectronic devices.10–12,16,18–23 HOFs are assembled from organic units through intermolecular hydrogen (H) bonding interactions, and the resulting frameworks can be further strengthened via other interactions, such as π–π stacking, van der Waals, dipole–dipole and/or C–H–π interactions.11,12,17,24,25 The inherent features of H-bond interactions, such as reversibility and directionality, endow these materials with unique properties, including crystallinity, flexibility, structural diversity, and easy recovery via recrystallization.11,12,16–18,24,25 These properties, together with a large π-conjugated system of the involved aromatic moieties, H-bonding interactions, and π–π stacking, provide HOFs with interesting luminescent properties, which may include proton- and charge-transfer reactions for different photoinduced applications, from sensing to lighting.10,11,26–30
Thus, the design of HOFs for fine tuning their optical properties has been the focus of many researchers. In recent years, 2,1,3-benzothiadiazole (BTD) derivatives have emerged as a versatile building block due to their easy functionalization and remarkable fluorescence properties. BTD is a typical electron-withdrawing aromatic group which when combined with an electron-donating one forms a “push–pull” system.31–34 BTD derivatives are characterized by a high molar extinction coefficient, high fluorescence quantum yield, large Stokes shift, and the feasibility to tune the absorption/emission wavelength to the near-infrared region.34–36 In addition to that, it is also well documented that these “push–pull” chromophores undergo intramolecular charge-transfer (ICT) reactions upon photoexcitation.37–40 When functionalized with thiophene groups, the ICT reaction in non-planar conformers is followed by a twisting/rotation motion of the involved moieties, leading to a planar conformation known as a planarized ICT (PLICT) structure.35,36,41,42 Thus, these photoinduced properties make these systems very attractive materials to investigate for their possible applications in organic light-emitting diodes (OLEDs)43–45 and organic photovoltaic cells46,47 and for use in the construction of novel porous materials with enhanced luminescent properties.48–53
HOFs with possible applications based on light (e.g., lighting, photosensing, photochromism, photocatalysis, to name a few)11 offer several advantages over normal chromophores. For example, the presence of π–π, dipole–dipole, and H-bonds interactions in the HOF network leads to the observation of different photophysical processes (e.g., intermolecular charge- and proton-transfer reactions) that might not be observed with molecular chromophores. These photoinduced reactions lead to significant changes in the luminescence properties, prompting the search for new HOFs for specific applications.10–12,16,18–23 Also, HOFs can serve as a host for guest molecules (drugs, ions, chromophores) with specific capabilities. This multifunctionality expands the potential applications of HOFs in a wide variety of fields, not limited to photonics, such as sensing, catalysis, and drug delivery. Among the organic linkers used to synthesize luminescent HOFs, to the best of our knowledge, there are not many examples in which the fundamental unit contains donor–acceptor (D–A) groups. Indeed, only a few HOFs based on hexaazatriphenylene28,29,54 and dehydrobenzoannulenes55,56 with ICT reactions have been reported. Recently, a two-component HOF based on BTD and an electron-donor group was synthetized, showing an efficient energy-transfer process, which allowed modulating the fluorescence emission colour by modifying the content of BTD.57 Furthermore, two new two-dimensional HOFs constructed from BTD derivatives have been reported, although their photophysical properties have not been extensively explored.52,53 Thus, elucidating their photophysics would be of great interest to deeply understand their photobehavior for possible applications in lighting or photosensing.
Here, we report on detailed studies of a HOF based on 5,5′-(benzo[c][1,2,5]thiadiazole-4,7-diyl) diisophthalic acid (BTIA-HOF) and its molecular units (BTIA-ester and BTIA-COOH) (Fig. 1A). The emission spectra of the ester and acid compounds in N,N-dimethylformamide (DMF) are characterized by large Stokes-shifted bands (∼6000 cm−1) due to the occurrence of a photoinduced ICT reaction in ∼300 fs coupled to the twisting motions of the phenyl rings that happen in ∼6 ps. These observations are in agreement with the theoretical calculations on BTIA-ester suggesting the formation of a planar conformation at S1. The ICT species of BTIA-ester in DMF has a lifetime of ∼7.5 ns, while those of BTIA-COOH are 350 ps (H-bonding complex), 1.15 ns (anionic species), and 7.75 ns (planar ICT species). Flash photolysis experiments on both samples in DMF reveal the existence of triplet and charge-separated states with lifetimes of 500 ns and 2 μs, respectively. In solid state, BTIA-ester exhibits a slow ICT reaction (80 ps) producing emissive aggregates with lifetimes of 1.20 and 2.87 ns, while BTIA-HOF undergoes ultrafast ICT and intermolecular proton-transfer reactions (<15 ps), giving rise to ICT and ionic species with lifetimes of 0.52 and 1.23 ns, respectively. Moreover, fluorescence microscopy experiments on BTIA-HOF single crystals suggest a homogeneous distribution of the molecular acid units forming the network of the HOF as a result of the growth in the direction of π-stacking and H-bonds between the BTD moieties. These results provide new findings for a deeper understanding of the photobehavior of BTD derivatives and related HOFs and will help for developing new HOF materials based on BTD with improved emission properties for application in lighting and sensing devices.
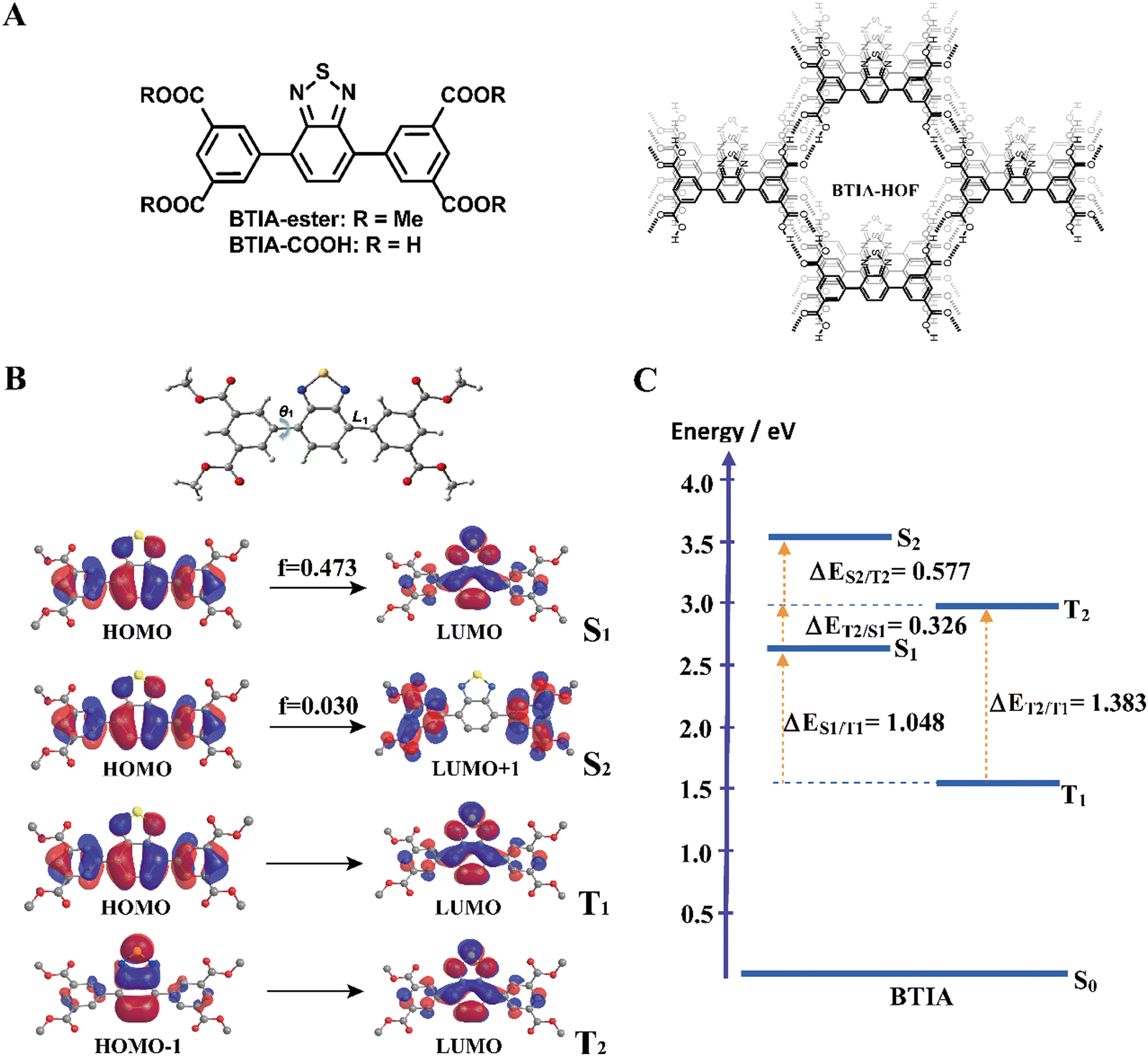 | ||
| Fig. 1 (A) Molecular structures of BTIA-ester, BTIA-COOH, and BTIA-HOF. (B) DFT optimized structure of BTIA-ester at S0 state, where θ1 and L1 are the twisting angle and rotatable bonds connecting aromatic moieties. Frontier molecular orbitals involved in the S1, S2, T1 and T2 electronic excited states of BTIA-ester molecule; f corresponds to the oscillator strength. (C) Schematic representation of energy levels, in electron volts (eV), of the different electronic states of the BTIA-ester molecule when using the continuum method with DMF. The dashed levels correspond to Franck–Condon excitation. Energy gaps between S1/T1 (ΔES1/T1), S1/T2 (ΔES1/T2), S2/T2 (ΔES2/T2), and T1/T2 (ΔET1/T2) states are also given. | ||
2. Materials and methods
The synthesis of BTIA-ester, BTIA-COOH, and BTIA-HOF compounds have been already described.52 Fig. S1 and S2 (ESI†) show the 1H-NMR spectra of BTIA-ester and BTIA-COOH in CDCl3 and DMSO-d6, respectively. The computational details are given in the ESI.† The photophysical properties of the three systems were characterized by a combination of complementary spectroscopic techniques, including steady-state absorption and emission, time-correlated single-photon counting, flash photolysis, fs up-conversion and fluorescence microscopy systems. Information about the used setups is provided in the ESI.†3. Results and discussion
3.1 Theoretical calculations
For the theoretical study, we performed calculations on the BTIA-ester molecule in the gas phase, and in DCM and DMF as solvents (Fig. 1). Since we did not find a big differences between the results in the gas phase and in DCM and DMF solvents, we only discuss the results obtained in DMF.The full optimization of BTIA-ester in DCM and DMF (continuum model) was carried out at the S0, S1, S2, T1 and T2 states. The main structural difference observed in the different states corresponds to the twisting angle (θ1) which defines the planarity between the BTD core and the two dimethylbenzoate (MBZ) rings in BTIA-ester (Fig. 1B). The obtained values of θ1 are 41.3°, 18.9°, 31.4°, 23.4° and 36.4° for S0, S1, S2, T1 and T2, respectively. No significant differences were found in the gas phase, and in DCM and DMF solvents (Table S1, ESI†). It is noteworthy that there is a decrease in the torsional angles of the BTIA-ester in both the excited singlet and triplet states. This leads to an enhanced electronic coupling and conjugation gain, especially in the S1 state, where the BTD and phenyl branches act as a significant π-conjugation skeleton. This phenomenon promotes the existence of the locally excited (LE) and charge-transfer (CT) states in molecules.58 This fact is also manifested in the decrease in the bond lengths connecting the phenyl branches to BTD at their S1 (1.447 Å) and T1 (1.452 Å) states with respect to those at the S0 (1.479 Å) state (Table S2, ESI†).
Fig. 1B depicts the shape of the orbitals involved in the electronic transitions and the vertical (Franck–Condon) excitations for the S1, S2, T1 and T2 states. The results also show that all the orbitals implied in these electronic excitations are π-type orbitals. The symmetry of the BTIA-ester molecule corresponded to the C2 group, where the C2 axe passes through the sulphur atom parallel to molecular plane of the BTD core in all the studied electronic states. This is reflected on the fact that the shape of their molecular orbitals spread symmetrically over the full structure. The vertical Franck–Condon excitation reveals a significant oscillator strength for S0 → S1 transition at 405 nm (f = 0.473). This indicates a pronounced photoactive nature of the S1 state which, along with the T1 one, predominantly originates from HOMO → LUMO excitations, suggesting a π–π* character for both states. The large oscillator strength values of the vertical S0 → S1 transition is due to the noteworthy spatial overlap between the HOMO and LUMO orbitals, respectively, as well as the change in electron density distribution across the molecular framework upon the electronic transitions. In addition to that, the population analysis of these orbitals reveals the occurrence of a π–π* charge-transfer process upon excitation (Fig. 1B). This analysis shows that the corresponding fragmental contributions for MBZ, the benzene ring (BZ) belonging to the benzothiadiazole moiety, nitrogen atoms (N) and sulphur atom (S) account for 37%, 51%, 11% and 1% in the HOMO orbital, and 11%, 41%, 27% and 21% in the LUMO orbital, respectively (Table 1). As a result, the observed spatial separation between the wave functions of the frontier orbitals, along with a certain degree of overlap, indicate a hybridized local and charge-transfer excited state (HLCT) nature for the S1 state, which shows a mixed locally excited (LE) and charge-transfer (CT) character.59 The greater value of the dipole moments in the excited states compared to the ground state also indicates polarized S1 and S2 states resulting from a CT process after photoexcitation in DMF (Table 2). Similar results were found in DCM, while the dipole moments found in the gas phase are significantly lower. Thus, the excited states are characterized by a significant redistribution of the electron density, leading to a more polar state compared to the ground state. This increased polarity of the excited state results in a stronger interaction with the solvent, which further increases the dipole moment value in polar solvents. On the other hand, the S0 → S2 Frank–Condon transition (322 nm, f = 0.030) predominantly arises from HOMO → LUMO+1. The small orbital overlap between these pairs of orbitals provokes a low oscillator strength of this transition. This suggests that the probability of populating the S2 state is very low. Notably, the LUMO+1 orbital is predominantly localized in the MBZ moieties (98%), indicating the CT character of S2, which is also reflected as a large dipole moment value (Table 2). Finally, the T2 state mainly comes from HOMO−1→ LUMO excitation.
| Orbital | Energy/eV | Fragmental contributions/% |
|---|---|---|
| LUMO+1 | −1.99 | MBZ (98) |
| LUMO | −2.81 | MBZ (11) BZ (41) N (27) S (21) |
| HOMO | −6.44 | MBZ (37) BZ (51) N (11) S (1) |
| HOMO−1 | −7.51 | MBZ (7) BZ (63) N (1) S (29) |
| HOMO−2 | −7.61 | MBZ (85) BZ (6) N (4) S (5) |
| Molecule | Electronic states | μ/D | ||
|---|---|---|---|---|
| GAS | DCM | DMF | ||
| BTIA-ester | S0 | 1.20 | 1.91 | 2.13 |
| S1 | 4.24 | 5.72 | 6.07 | |
| S2 | 15.79 | 19.65 | 20.17 | |
| T1 | 2.83 | 4.29 | 4.64 | |
| T2 | 1.49 | 2.59 | 2.93 | |
Fig. 1C shows the energy levels of the stationary points of BTIA-ester at the S1, S2, T1 and T2 states, while Table S3 (ESI†) displays the energy gaps between the involved states. The calculations of the optimized low-lying excited states show low energy values for T1 and a large energy gap between T2 and T1 (ΔET1/T2 = 1.383 eV), which may suppress the internal conversion (IC) decay from T2 to T1 as the rate of IC is inversely related with the energy gap.60 In addition to that, the large energy gap between S1 and T1 states (ΔES1/T1 = 1.048 eV) does not favour the occurrence of intersystem crossing (ISC) and reverse intersystem crossing (RISC) processes from T1 to S1. On contrary, the significant smaller energy gap between T2 and S1 states (ΔES1/T2 = 0.326 eV) may induce an ISC process and enhance the RISC between these states, as predicted by the Fermi golden rule.61 Comparable energy gap values (ΔES1/T2 = 0.277 eV and ΔET1/T2 = 0.972 eV) have been observed in other BTD derivatives, which undergo a RISC process.62 The HOMO−1 transition involved in the T2 state exhibits significantly higher contributions from the BZ fragment and sulphur atom, with a lesser contribution from the MBZ moiety compared to the HOMO transition associated with the S1 state. This results in considerable differences in the CT character of the S1 and T2 states. Such distinction facilitates the ISC process between the two states. This phenomenon is in agreement with El Sayed's rule, which predicts highly efficient spin–flip processes between two states involving a change in orbital components, thereby resulting in strong spin–orbit coupling.63 Furthermore, the nearly negligible energy difference between the S0 → S1 vertical transition energy (3.06 eV) and the diabatic excitation energy of the T2 state (2.98 eV) indicate the potential for overlap between the vibrational states of both singlet excited states and T2 states, thereby increasing the probability of the occurrence of an ISC process.
In brief, the theoretical calculations of BTIA-ester in DMF suggest that: (i) the optimized geometry of the molecule in the ground state is not planar, while it is more planar in the electronically excited state with a larger electronic conjugation; (ii) a photoinduced ICT reaction occurs in the S1 state from the peripheral phenyl rings to the BTD core; (iii) the existence of an HLCT nature at the S1 state, which is a combination of LE and CT states; (iv) the energy gaps between T2 and S1, along with the vertical transition energy value of the latter, may contribute to ISC and RISC processes; (v) the notable differences observed between the character of the transition orbitals involved in S1 and T2 favour the existence of strong spin–orbit coupling between these states. These theoretical calculations will help for a better understanding of the experimental findings that will be shown and discussed in the following section.
3.2 Steady-state absorption and emission studies in DMF solutions
To get a preliminary understanding of the photobehavior of BTIA-HOF, we first studied the spectroscopic properties of the fundamental units that constitute the HOF (BTIA-ester and BTIA-COOH). To begin with, we selected DMF as a solvent to investigate the photophysical behaviour of the molecular units and the HOF, since BTIA-HOF was crystallized using a mixed solution of DMF and methyl benzoate. In this way, we aimed to compare the photophysical processes that occur in the linkers and the HOF in the same solvent. BTIA-ester and BTIA-COOH samples were prepared in DMF solutions, while BTIA-HOF was studied as synthesized, removing part of the solvent containing the dissolved BTIA-COOH molecules and taking the suspended HOF crystals (see the ESI†). Fig. 2A shows the UV-visible absorption and emission spectra, while the relevant information is given in Table 3.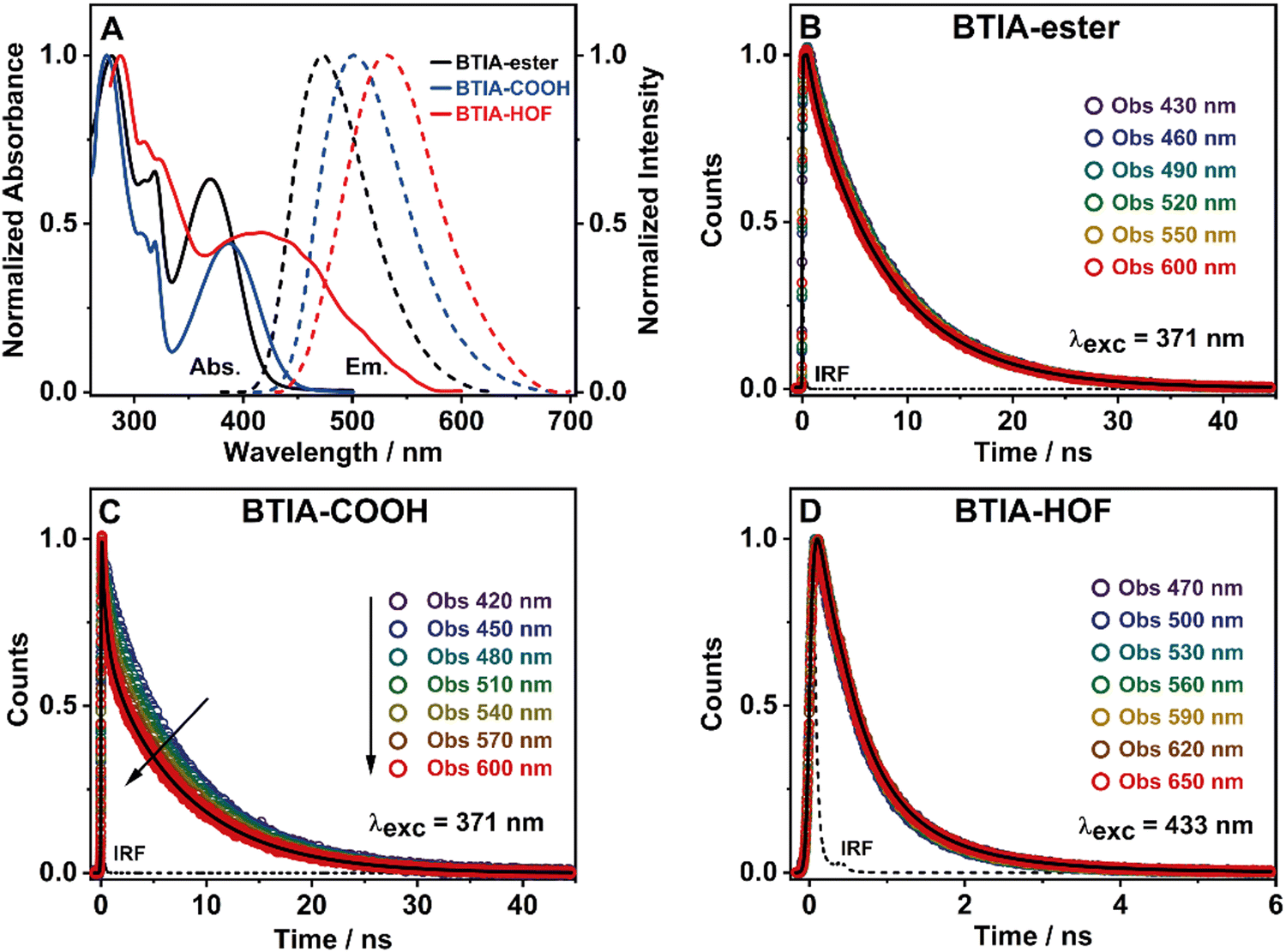 | ||
| Fig. 2 (A) Normalized UV-visible absorption (solid line) and emission (dashed line) spectra of BTIA-ester (black), BTIA-COOH (blue) and BTIA-HOF (red) in a DMF solution or suspension. For the emission spectra, the excitation wavelength was 370 nm. Magic-angle ps-emission decays of BTIA-ester (B), concentrated sample of BTIA-COOH (C) and BTIA-HOF (D) in a DMF solution or suspension, upon excitation and observation as indicated in the inset. The solid lines are from the best multiexponential fits, and the IRF is the instrumental response function (∼70 ps). | ||
| Medium | Sample | λ abs/nm | λ em/nm | Δνss/cm−1 | FWHM/cm−1 | Φ F/% |
|---|---|---|---|---|---|---|
| DMF solution | BTIA-ester | 278, 318, 370 | 477 | 6060 | 3259 | 63 |
| BTIA-COOH | 275, 318, 385 | 502 | 6050 | 3533 | 11 | |
| BTIA-HOF | 286, 323, 415 | 530 | 5230 | 3511 | — | |
| Solid state | BTIA-ester | 294, 377 | 504 | 6680 | 3456 | 1 |
| BTIA-HOF | 302, 380 | 540 | 7800 | 3810 | — | |
To begin with, the absorption spectrum of BTIA-ester in DMF solution shows two intense broad bands centred at ∼280 and ∼370 nm, and a well-defined narrow band at ∼320 nm, all assigned to π → π* transitions of the BTD moiety (Fig. 2A).64,65 As we discussed in the theoretical part, BTIA-ester displays a high oscillator strength for the S0 → S1 transition. Therefore, we assign the lower energy absorption band (370 nm) to the S0 (π) → S1 (π*) transition, which should have an ICT character as suggested by the theoretical calculations (Fig. 1B), and as previously reported for other BTD systems.65–69 To further investigate the photophysical properties of BTIA-ester, we also measured the absorption spectra in methylcyclohexane (MCH), dichloromethane (DCM), triacetin (TAC), and in a rigid matrix (poly(methyl methacrylate): PMMA) (Fig. S3 and Table S4, ESI†). The absorption spectra in the selected media are similar and comparable to the one observed in DMF, indicating that the ground-state species are not influenced by the solvent properties. The absorption spectrum of the acid, BTIA-COOH, in DMF shows a shape similar to that of the ester derivative, while that of BTIA-HOF exhibits a broad lower energy band (Fig. 2A). We assign the observed bands to the S0 (π) → S1 (π*) transition. Compared to BTIA-ester, the lower energy band is shifted to longer wavelengths (by 15 and 45 nm, respectively, Table 3), which suggests the presence of different species in the ground state (S0). The presence of carboxylic acid groups in the latter can lead to the formation of anionic species or H-bonded complexes by interaction with DMF molecules. To investigate this possibility, we recorded the UV-visible absorption spectra of BTIA-COOH in DMF solution at different concentrations [optical densities (ODs) in a 1 cm path at 380 nm were 0.8, 0.3, and 0.02] (Fig. S4, ESI†). For the concentrated and intermediate samples, the absorption spectra are very similar in terms of their shape and position (±3 nm), while upon dilution we observe a red-shift of the 370 nm band by 10 nm. BTIA-COOH behaves like a weak acid, increasing its dissociation with dilution (Ostwald's law) and leading to the formation of anionic species. A similar photobehaviour has been previously reported for other molecules with carboxylic acid groups, which can generate H-bonded complexes and anionic species with the used solvent.28,55,56
Fig. 2A shows the emission spectra of BTIA-ester, BTIA-COOH and BTIA-HOF in a DMF solution, while Table 3 gives the values of the parameters related to the absorption and emission spectra. The three samples display a single fluorescence band, with the maximum intensity at 477, 502, and 530 nm, respectively. The fluorescence spectra are characterized by a large Stokes shift (6060, 6050, and 5230 cm−1, respectively). We anticipate that this shift is the result of the occurrence of an ICT process (from the peripheral phenyl rings to the BTD core), probably coupled to twisting motions of the phenyl rings in the electronically first excited state (S1). The theoretical calculations at the S0 and S1 states indicate the occurrence of an ICT event and twisting of the phenyl moieties (Tables S1 and S2, ESI†). It is well known that BTD systems undergo PLICT reactions resulting in a large electronic coupling and conjugation gain, and thus exhibiting high fluorescence quantum yields (ΦF).34–36 The obtained ΦF value for BTIA-ester in DMF solution is high, i.e. ΦF = 0.63, following the behaviour of BTD derivatives. To further investigate the ICT process, we also recorded the emission spectra of BTIA-ester in different media with changing the polarity and viscosity (MCH, DCM, TAC, and PMMA film). Fig. S3 (ESI†) shows that the absorption spectra do not show significant changes, while the positions for the emission ones are influenced by the properties of the solvent. In apolar (MCH) and rigid media (PMMA), the emission spectra maxima are at shorter wavelengths (456 and 446 nm, respectively), while we observe comparable spectral positions in DCM and TAC solutions (465 and 463 nm, respectively) and a red-shift in DMF (477 nm) (Table S4, ESI†). Compared to MCH, the red-shift in DMF (∼20 nm) indicates a polarity effect on the relaxed emitting species, in agreement with the theoretical calculations showing a change of the dipole moment value from 2.0 to 6.4 D upon excitation to S1 and reflecting a clear ICT process. It is well known that upon increasing the environment polarity around a polar fluorophore, the emission band will shift to longer wavelengths. The observed blue-shift in TAC [polar and highly viscous solvent, η (25 °C) =17.4 cP] and in the PMMA film could be explained in terms of a restriction of the twisting motion of the phenyl rings, which prevents the molecule relaxing to more stable levels at S1. A similar photobehaviour has been observed in many BTD derivatives encapsulated in polymer matrices.35,70,71
In agreement with the absorption spectra, the red-shifted emission bands of BTIA-COOH and BTIA-HOF in DMF, when compared to the ester one, indicates the presence of several emitters, most likely anionic species and/or H-bonded complexes with DMF molecules, as discussed above. The large value of the full width at half maximum (FWHM) of the emission intensity (∼3530 cm−1) suggests that the emission originate from more than one species (Table 3). To further investigate the emission properties of BTIA-COOH, we also recorded the emission spectra in DMF solution at different concentrations (ODs at 380 nm were 0.8, 0.3, and 0.02) (Fig. S4, ESI†). For the concentrated and intermediate samples, the emission spectra display their intensity maxima at ∼480 nm, while that of diluted one is at ∼500 nm. The obtained spectra for the former are very similar to that of the methylated derivative (477 nm), indicating that the electronic structures of the emitting species are not very different, with an ICT character. The red-shifted emission of BTIA-COOH observed upon dilution indicates the emission of different structures, most likely anionic species that should emit at longer wavelengths than the neutral one, as observed in other molecules having carboxylic acid groups.28,55,56 The fluorescence quantum yield of BTIA-COOH (ΦF = 0.11) is lower than that obtained for its methylated ester derivative (ΦF = 0.63), as a consequence of the H-bond interactions with DMF molecules enhancing the non-radiative transitions. Finally, BTIA-HOF in DMF shows an intense yellow emission (535 nm), which is slightly red-shifted when compared to BTIA-COOH. In addition to the existence of anionic species, the structural arrangements and the resulting intermolecular interactions (H-bonds and dipole–dipole) present in the HOF network led to changes in their photobehaviour, resulting in a shift of the emission spectra towards longer wavelengths.11,12,52
To further characterize the spectroscopic properties of BTIA-ester, BTIA-COOH and BTIA-HOF in DMF, we recorded their excitation spectra (Fig. S5, ESI†). The spectra of BTIA-ester do not depend on the observation wavelength, and display bands similar to those of the absorption one, indicating a common ground-state for the emissive species. However, the excitation spectra of BTIA-COOH and BTIA-HOF show a clear difference in spectral positions, in which the lower energy band is shifted to shorter wavelengths with respect to the absorption one. This difference suggests that the visible part of the absorption spectra contains absorbing species that are not emissive, possibly due to the establishment of H-bonds with DMF molecules or the presence of anions. To get more details on the photobehaviour of BTIA-ester, BTIA-COOH and BTIA-HOF, we performed picosecond (ps) time-resolved emission experiments in DMF solutions.
3.3 Picosecond time-resolved emission experiments in solutions
| Sample | λ obs/nm | τ 1/ns | a 1 | c 1 | τ 2/ns | a 2 | c 2 | τ 3/ns | a 3 | c 3 |
|---|---|---|---|---|---|---|---|---|---|---|
| BTIA-ester λ exc = 371 nm | 430 | — | — | 7.52 | 100 | 100 | ||||
| 460 | 100 | 100 | ||||||||
| 550 | 100 | 100 | ||||||||
| 600 | 100 | 100 | ||||||||
| BTIA-COOH λ exc = 371 nm | 420 | 0.39 | 13 | 1 | — | 7.75 | 87 | 99 | ||
| 450 | 15 | 1 | 85 | 99 | ||||||
| 570 | 37 | 3 | 63 | 97 | ||||||
| 600 | 41 | 3 | 59 | 97 | ||||||
| BTIA-HOF λ exc = 433 nm | 470 | 0.38 | 67 | 31 | 1.12 | 33 | 69 | — | ||
| 500 | 69 | 46 | 31 | 54 | ||||||
| 620 | 68 | 44 | 32 | 56 | ||||||
| 650 | 70 | 48 | 30 | 52 | ||||||
To begin with, the emission decays of BTIA-ester in MCH, DCM, and DMF solutions were well-fitted (single wavelength and global fits) by a monoexponential function over the entire spectral range with time constants of 5.32, 6.87, and 7.52 ns, respectively (Fig. 2B and Fig. S6, Table 4, and Tables S5–S7, ESI†). It is clear that its value increases with the solvent polarity. According to the theoretical calculations, which suggest the occurrence of an ICT process coupled to twisting motions, and considering our previous results and discussion in the steady-state experiments, we suggest the occurrence of an ultrafast ICT process leading to the formation of long-living planar (less twisted) species. Therefore, we assigned this time constant to the lifetime of these PLICT species (Scheme 1A). With a ∼15 ps time resolution of the used set-up (see ESI†), we could not observe any rising component in the red part of the emission spectrum, suggesting that the ICT is an ultrafast process. However, we could resolve the issue using a fs-set-up (vide infra). To explore to what extent the viscosity and rigidity of the medium affect the emission decays, we also collected the decays in TAC and PMMA film upon excitation at 371 nm (Fig. S6, ESI†). The photobehaviour of BTIA-ester in TAC, a highly viscous solvent (η = 17.4 cP at 298 K), is more complex and the emission decays were well fitted by a biexponential function with time constants of 0.24 ns and 7.34 ns (Table S8, ESI†). The longest component decays over the entire spectral range, reaching its maximum contribution at 490 nm. Considering the value of the longest component, which is similar to that observed in DCM and DMF solutions, we assign its value to the lifetime of planar ICT species. On the other hand, the shortest component (240 ps) decays at the blue part of the emission spectrum and rises at the reddest one (Fig. S7, ESI†). This is an indication that a photoinduced process occurs at S1, and according to our previous discussion, we assign it to a PLICT event that is slow due to the high viscosity of the medium. Last and least, the emission decays of BTIA-ester in a rigid medium (PMMA film) show a biexponential behaviour with time constants of τ1 = 3.19 ns and τ2 = 6.73 ns (Table S9, ESI†). Both components are decaying throughout the entire spectrum, with the shortest one displaying its highest contribution in the bluest part of the emission spectrum and the longest one in the reddest region. Knowing that PMMA cavities can trap molecules, we propose that the short and long components are due to conformations having restricted and not restricted motions, respectively.
 | ||
| Scheme 1 Proposed photodynamic schemes for BTIA-ester (A), BTIA-COOH (B) and BTIA-HOF (C) in a DMF solution or suspension, in which PLICT is the planarized intramolecular charge-transfer process, τICT is the ICT reaction time, τPh-Ph is the time of the twisting motions of the phenyl rings, and τPT is the time of the intermolecular PT process. The scheme is not to scale. | ||
The photodynamics of BTIA-COOH in DMF is more complex and depended on the concentration of the sample, in agreement with the steady-state observation. Fig. 2C displays the emission decays of a relatively highly concentrated sample of BTIA-COOH [ODs (380 nm) = 0.8], while those of the intermediate and diluted samples are shown in Fig. S8 (ESI†). The data obtained from the best multiexponential fit are given in Table 4 and Tables S10–S12 (ESI†). We first focus the discussion on the results obtained for the concentrated sample, in which anionic species were not observed in the steady-state spectrum. The emission decays exhibit a biexponential behaviour with time constants of 0.39 and 7.75 ns. Both components decay throughout the entire spectral range (420–600 nm) with the shortest component displaying its highest contribution in the reddest part of the emission spectrum and the longest one in the bluest region (Table S10, ESI†). Based on the steady-state explanation where no anionic species were found at this concentration, we suggest the presence of neutral BTIA-COOH species and BTIA-COOH molecules having H-bonded interactions with DMF molecules. The longer component (7.75 ns), which is very similar to that of BTIA-ester (7.52 ns), reflects the emission lifetime of planar ICT species that do not interact or are less interacting with DMF molecules, while the shortest component is assigned to the emission lifetime of BTIA-COOH⋯DMF complex. With a ∼15 ps time resolution of the set-up, we did not observe any rising component in the red part of the emission spectrum, suggesting that the ICT process takes place on a shorter time scale. Next, we discuss the photobehaviour of the intermediate and diluted samples (Fig. S8, ESI†). The emission decays show comparable behaviours and depend on the observation wavelength, being shorter when observed at longer wavelengths. The emission decays of both samples were fitted with a multiexponential function, giving three time constants: τ1 = 0.27, τ2 = 1.29, and τ3 = 7.72 ns for the intermediate solution, and τ1 = 0.35, τ2 = 1.15, and τ3 = 7.64 ns for the diluted one (Tables S11 and S12, ESI†). Although the time constants are very similar for both samples, the contributions of each component to the global signal depend on the sample concentration. All the components decay over the entire spectral range, with the longest one displaying its maximum contribution in the blue part of the emission spectra, and the shortest ones in the red one. The shortest and longest components (∼0.3 and ∼7.7 ns) are comparable to those observed for the concentrated sample (0.4 and 7.8 ns), and which we assigned to the lifetimes of H-bonded complexes and PLICT species, respectively. Therefore, we assign these components to similar species of BTIA-COOH in DMF. However, we obtained an additional component (1.15 ns), which was not observed in the concentrated sample. This component displays its maximum contribution in the reddest part of the emission spectrum and reaches its higher contribution in the diluted sample. Anionic species of this kind of molecules display their emission at longer wavelengths.28,55,56 Based on the literature and on the discussion in the steady-state section, we assign this component to the lifetime of anionic species, which may have ICT character. Scheme 1B summarizes the proposed photodynamical behaviour of BTIA-COOH in DMF.
Last but not least, we investigated the photodynamic behaviour of BTIA-HOF in a DMF suspension, exciting at 433 nm where the visible absorption band shows its maximum of intensity, and recording the signal at different wavelengths (from 470 to 650 nm) (Fig. 2D). Surprisingly, the emission decays do not depend on the observation wavelength and show a biexponential behaviour with time constants of 0.38 and 1.12 ns (Table 4 and Table S13, ESI†). Both components are decaying throughout the emission spectrum without any variation in their contribution, being ∼70% that of the shortest component. The presence of H-bond interactions in the HOF plays a key role in the stability and photobehaviour of the network. The interactions of two carboxylic acid groups of neighbouring BTD molecules might lead to the occurrence of intermolecular proton-transfer (PT) reactions at the electronically first excited state. As a result, it is possible to form ionic structures that may have protonated carbonyls and deprotonated OH groups, and thus the formation of anionic, cationic, and zwitterionic species. On the other hand, the possible presence of “free” COOH groups in the network may lead to the formation of intermolecular H-bonds with DMF molecules, and therefore open the door to forming anionic species. Based on the previous discussion of the results for BTIA-ester and BTIA-COOH, where ICT and anionic species were observed, we suggest that the shorter component reflects the lifetime of the initially excited species, which are susceptible to undergo an ultrafast ICT reaction (below instrumental resolution, <15 ps), while the longest one, which is similar to that observed for anionic species of BTIA-COOH (1.15 ns), corresponds to the lifetime of anionic species with an ICT character (Scheme 1C). Similar photobehaviour have been previously reported for other HOFs based on hexaazatriphenylene and dehydrobenzoannulenes derivatives containing carboxylic acid groups.28,55
To conclude the ps part, the initially electronically excited species of BTIA-ester in DMF undergo an ultrafast ICT reaction (<15 ps) leading to long-living planar species (7.52 ns). However, BTIA-COOH shows a complex behaviour due to the existence of different species coexisting in the ground state (neutral, H-bonded complexes, and anionic species). The neutral BTIA-COOH species undergo a photoinduced ICT reaction (<15 ps) leading to planar structures with a lifetime of 7.75 ns. On the other hand, the excited H-bonded complexes (BTIA-COOH⋯DMF) can relax to the S0 state with a lifetime of 390 ps, while the excited anionic molecules emit at longer wavelengths with a lifetime of 1.15 ns. The photobehaviour of BTIA-HOF reflects the presence of two different emitters: excited species which undergo an ICT process and relax with a lifetime of 0.38 ns, and anionic ones with a lifetime of 1.12 ns. Scheme 1 illustrates the discussed photobehaviours of BTIA-ester, BTIA-COOH and BTIA-HOF in DMF.
3.4 Femtosecond dynamics of the BTIA-ester and acid in DMF solutions
To explore the ultrafast photoevents in BTIA-ester and BTIA-COOH, we performed fs-emission experiments on both molecules in DMF solution, upon excitation at 360 nm and probing at different emission wavelengths (from 420 to 580 nm). Fig. 3 shows the representative transient emission decays in a short time scale, while Fig. S10 (ESI†) exhibits the selected emission decays in a longer window. The obtained time constants (τi) and pre-exponential factors (ai) normalized to 100 are shown in Table 5.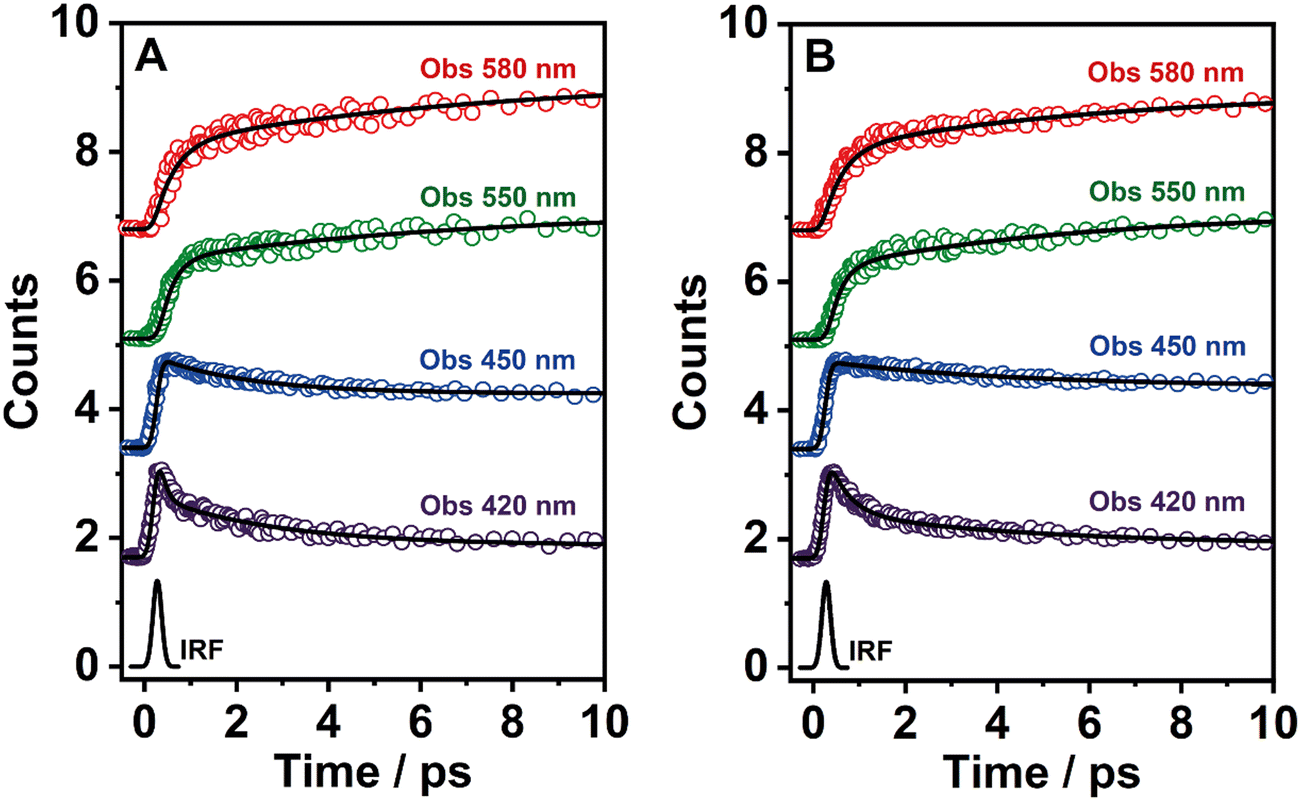 | ||
| Fig. 3 Representative fs-emission transients of (A) BTIA-ester and (B) BTIA-COOH in a DMF solution. The samples were excited at 360 nm and recorded at the indicated wavelengths. The solid lines are from the best multiexponential fit and the IRF is the instrumental response function (220 fs). | ||
| Sample | λ obs/nm | τ 1/ps | a 1 | τ 2/ps | a 2 | τ 3/ns | a 3 |
|---|---|---|---|---|---|---|---|
| a Fixed values in the fitting considering the TCSPC experiments. | |||||||
| BTIA-ester | 420 | 0.26 | 68 | 6.26 | 25 | 7a | 7 |
| 450 | — | 5.94 | 35 | 65 | |||
| 550 | 0.29 | −62 | 6.43 | −38 | 100 | ||
| 580 | 0.31 | −54 | 6.48 | −46 | 100 | ||
| BTIA-COOH | 420 | 0.44 | 55 | 4.57 | 32 | 7a | 13 |
| 450 | — | 5.12 | 28 | 72 | |||
| 550 | 0.35 | −57 | 5.93 | −43 | 100 | ||
| 580 | 0.42 | −55 | 6.07 | −45 | 100 | ||
To record the fs-transient decays, we used concentrated samples of BTIA-ester and BTIA-COOH, thus excluding or lowering the possibility of having anionic species. The collected signals of both molecules show similar complex photobehaviours. The best fit of the data using a multiexponential analysis needs three components (Table 5): two short time constants (∼300 fs and ∼6.5 ps for BTIA-ester, and ∼400 fs and ∼6.0 ps for BTIA-COOH) and an offset component of 7 ns (fixed from the values obtained from the TCSPC experiments), which correspond to the lifetime of ICT planar species. The two shortest components decay at the bluest part (420–450 nm) and rise at the reddest part of the emission spectrum (from 490 nm) (Fig. 3 and Table 5). This photobehaviour is a clear indication of the occurrence of an ultrafast event in the S1 state. Hence, following our previous discussion (steady-state and picosecond sections), we ascribed this process to a PLICT process. Previous studies on BTD derivatives functionalized with thiophen groups have reported that these molecules undergo an ICT process followed by twisting motions of the phenyl rings, occurring both in coupled processes in the picosecond timescale.35,36 It should be expected that the ICT event takes place faster than the twisting motions of the phenyl rings, with the former occurring in the fs time scale. Therefore, we assign the ∼300 fs component to the ICT process, while the ∼6 ps component reflects the twisting motion of the phenyl rings (Scheme 1A).
We also recorded the ultrafast photodynamics of BTIA-ester in DCM solution upon excitation at 360 nm. Fig. S11 (ESI†) displays the representative transient decays, and Table S16 (ESI†) gives the time constants and pre-exponential factors obtained from the best multiexponential fitting. The fs-emission transients in DCM solution behaves as we observed in DMF, with time constants of τ1 = ∼300 fs, τ2 = ∼2 ps and an offset component of 6 ns. The similarity of the time constants and the behaviour (decay in blue and rise in red) allowed assigning these components to the processes described above in DMF, with the only difference in the value of the time constant assigned to the twisting motions of the phenyl rings, which is shorter (∼2 ps in DCM vs. ∼6 ps in DMF) due to the lower viscosity of DCM solvent compared to DMF. Note that, this time becomes longer up to 240 ps in highly viscous solvent (TAC, η298K = 17.4 cP) (Table S8, ESI†). The assignment of the obtained components in DCM solution is the same as that given previously for DMF solutions.
According to the results described above, we can present a global and also a detailed picture of the fs-ns photodynamics of BTIA-ester and BTIA-COOH in DMF. Upon photoexcitation to S1, the initially excited species undergo an ultrafast ICT reaction (∼300–400 fs) accompanied by a twisting motion of the phenyl rings, which depends on the solvent viscosity: ∼2, ∼6, and 240 ps in DCM, DMF, and TAC, respectively. The results of both processes lead to the formation of a long-lived planar ICT species (∼7 ns), emitting with a large Stokes-shifted emission. Scheme 1 presents an illustration of the photobehaviour of BTIA-ester and BTIA-COOH in DMF solutions.
3.5 Flash photolysis transient absorption studies
BTD derivatives also display slow photodynamics due to the existence of accessible triplet states.62,66,67,74 Our results from the theoretical calculations on BTIA-ester in DMF (Section 3.1) show the possibility of an ISC process between the S1 and T2 states (ΔES1/T2 = 0.326 eV), leading to species which may populate the T2 state. To explore the slow relaxation of BTIA-ester and BTIA-COOH in DMF solutions, we carried out ns–ms flash photolysis experiments upon excitation at 355 nm in presence and absence of molecular oxygen. Fig. 4(A) and (C) show the transient absorption spectra (TAS) at different delay times, while Fig. 4(B) and (D) display the related decays at the maximum intensity of the transient absorption band, and collected under different atmospheric conditions (air, nitrogen, and oxygen). Table 6 gives the values of the obtained time constants from the best fits of the decays.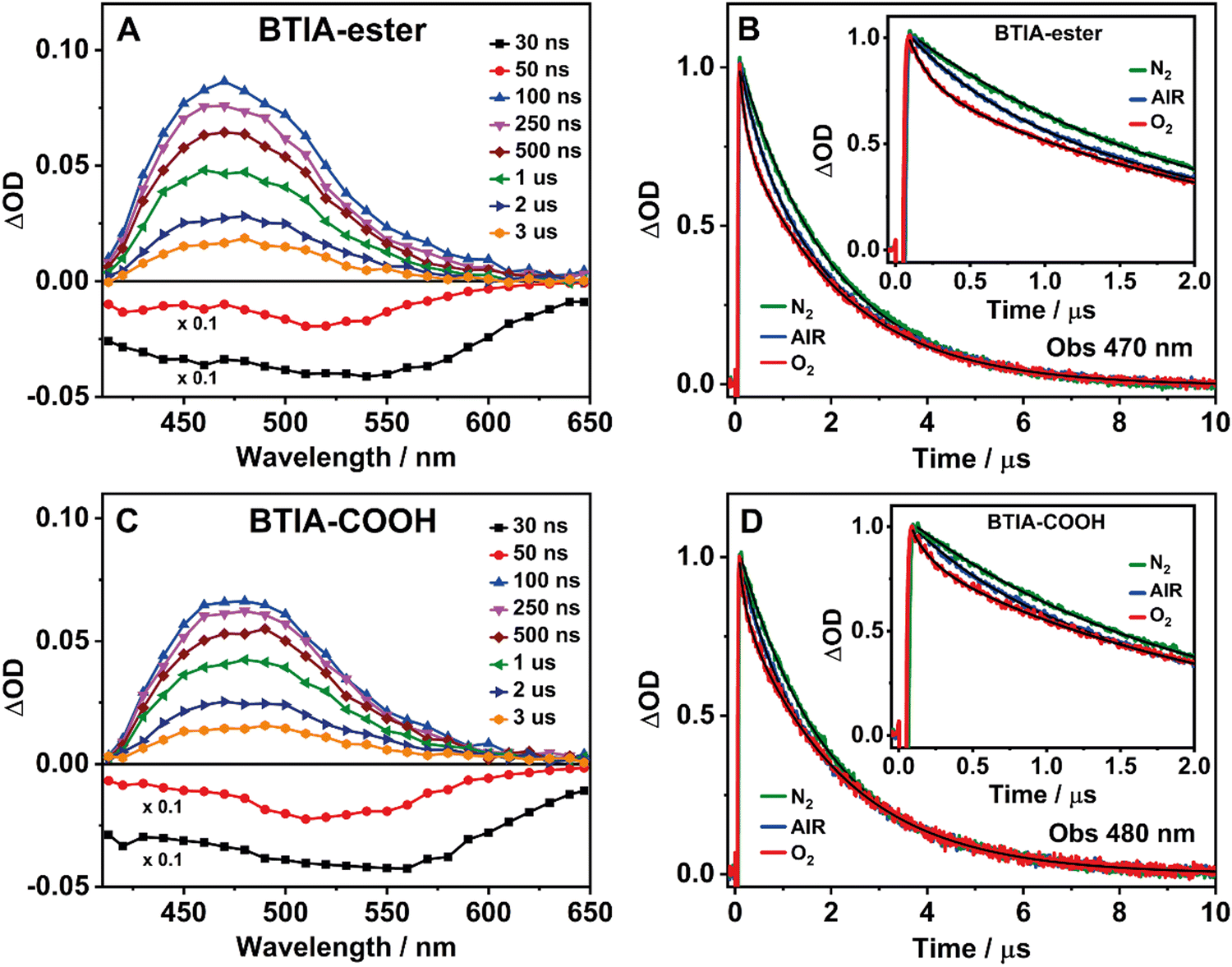 | ||
| Fig. 4 Time-resolved transient absorption spectra (TAS) of BTIA-ester (A) and BTIA-COOH (C) in a DMF solution at different delay times. Transient absorption decays of BTIA-ester (B) and BTIA-COOH (D) in DMF solutions probed at 470 nm and 480 nm under different atmospheric conditions: air (blue), oxygen (red), and nitrogen (green). The excitation wavelength was 355 nm. | ||
| Sample | Conditions | τ 1/ns | a 1 | τ 2/μs | a 2 |
|---|---|---|---|---|---|
| BTIA-ester/DMF λ obs = 470 nm | Air | 490 | 21 | 2.10 | 79 |
| O2 | 140 | 31 | 2.08 | 69 | |
| N2 | 840 | 12 | 1.97 | 88 | |
| BTIA-COOH/DMF λ obs = 480 nm | Air | 520 | 19 | 2.12 | 81 |
| O2 | 135 | 23 | 2.12 | 77 | |
| N2 | 790 | 13 | 2.07 | 87 | |
At delay times longer than 50 ns, the TAS of BTIA-ester and BTIA-COOH display a single positive band covering a spectral range from 410 to 650 nm with the maximum intensity around 475 nm, while we record a negative band at shorter delay times (Fig. 4). Based on the spectral position of the latter, which coincides with the emission spectra of both molecules (Fig. 2), we assign it to a stimulated emission from the S1 state. The positive band arises from the transient absorption of triplet states as their decays are sensitive to the presence or absence of molecular oxygen, as shown below. The transient absorption decays of both BTIA-ester and BTIA-COOH molecules in DMF equilibrated with air exhibit a biexponential behaviour with time constants of ∼500 ns and ∼2.1 μs (Table 6). To support the explanation of the existence of triplet states, we recorded the transient decays in DMF, saturated with O2 and N2 gases. At first glance, the transient decays become shorter and longer under oxygen and nitrogen conditions, respectively, confirming the existence of a triplet state (Fig. 4). The recorded transient decays in the presence and absence of molecular oxygen exhibit a biexponential behaviour, and the analysis gives time constants of ∼150 ns and ∼2.1 μs, and ∼800 ns and ∼2.0 μs, respectively (Table 6). Interestingly, only the shorter component is influenced by the presence of oxygen, while the longer one remains unchanged. Based on this observation, we suggest that the shorter component (∼500 ns under air conditions) reflects the lifetime of a triplet state. Our theoretical calculations suggest that an ISC process may occur between T2 and S1 states, and the calculated large energy gap between T2 and T1 (ΔET1/T2 = 1.383 eV) may suppress the interconversion process from T2 to T1, as reported for other BTD systems.66,67 Therefore, we assign the shortest component to the lifetime of a non-emissive T2 state. On the other hand, the longest component is not influenced by the presence of oxygen and has a similar value in all the studied environments (∼2 μs). It is well known that a push–pull system (such as the one studied here) can generate long-lived charge-separated (CS) states after photoexcitation.75–77 Based on that, it is possible that the ∼2 μs component reflects the lifetime of a long-lived CS state, which is produced after the photoexcitation and as a consequence of the ultrafast ICT. Finally, we also recorded the TAS and the transient decays of BTIA-ester in DCM solution under different atmospheric conditions (Fig. S12 and Table S17, ESI†). The obtained results are similar to those obtained in DMF, indicating that the solvent properties do not affect the slow photodynamics of BTIA-ester.
3.6 Steady-state and picosecond time-resolved emission studies in solid state
To elucidate the photophysical properties of the present HOF in solid state, relevant to its possible applications in light-based devices, we also investigated the photobehaviours of BTIA-ester and BTIA-HOF in solid state (Fig. 5). While BTIA-ester forms an amorphous solid, BTIA-HOF has an ordered fibrous crystalline structure due to the inter-unit H-bonds, and dipole–dipole interactions in the network.52 | ||
| Fig. 5 (A) Normalized diffuse reflectance (converted to K–M function, solid line) and emission (dashed line) spectra of BTIA-ester (black) and BTIA-HOF (red) in solid state. For the emission spectra, the excitation wavelength was 370. Magic-angle ps-emission decays of BTIA-ester (B) and BTIA-HOF (C) in solid state, upon excitation at 371 nm and observation at 430, 460, 490, 520, 550, 580, 610, and 640 nm. The solid lines are from the best multiexponential fits, and IRF is the instrumental response function (70 ps). | ||
To begin with, the diffuse reflectance (transformed to a K–M function) spectra of both samples consist of a broad spectrum with two clear bands with maximum intensities at ∼295 and ∼380 nm, which reflects the presence of several absorbing structures (Fig. 5A and Table 3). We also recorded the emission spectra of BTIA-ester and BTIA-HOF in solid state (Fig. 5A). BTIA-ester displays a green emission with a maximum at 504 nm, while BTIA-HOF shows a yellow one (540 nm). The emission spectra in solid state are red-shifted compared to those in DMF solution. The structural arrangements and interactions (dipole–dipole and H-bonds in the HOF) between neighbouring BTD molecules in solid state result in the formation of aggregated or crystal structures that emit at longer wavelengths.78 This behaviour is also reflected in the lower ΦF value in solid state for BTIA-ester (ΦF = 0.01 vs. ΦF = 0.63 in DMF, Table 3). In both cases, the emission results in a large Stokes shift value, which reflects the occurrence of an ICT reaction, as previously discussed in DMF solution. The larger value of Stokes shift obtained for BTIA-HOF (∼7800 cm−1), compared to methylated ester derivative (6680 cm−1), indicates that the former can undergo additional photoinduced processes, most likely intermolecular PT reactions between the acid units. The H-bond interactions between neighbouring molecules may originate the formation of ionic species, which emitts at longer wavelengths.28,29,55 Note that the interactions between the neighbouring carboxylic acid groups may produce structures with protonated carbonyls, deprotonated OH groups, and the possible formation of zwitterionic species.79 The larger FWHM value (3800 cm−1) of the emission spectrum of BTIA-HOF also confirms the presence of several emitting species.
The excitation spectra of BTIA-ester show a clear different spectral shape between 450 and 550 nm compared to that of the absorption one (Fig. S13, ESI†). The additional band observed (∼475 nm) in the excitation spectra increases in intensity when gating the emission at longer wavelengths. This agrees with the slight red-shift observed in the emission spectrum upon excitation at longer wavelengths (Fig. S14, ESI†). This difference could be due to the presence of a small fraction of emissive aggregates with different conformations that absorb in this region and emit at longer wavelengths, most likely aggregates with an ICT character. In contrast to what is observed in the amorphous BTIA-ester sample, the excitation spectra of BTIA-HOF display similar bands and shapes to those obtained in the absorption one, indicating a common ground-state for the emissive species (Fig. S13, ESI†). The directionality of the H-bonds between the carboxyl groups and the dipole–dipole interactions between the BTD moieties lead to the formation of a crystalline structure, excluding the formation of aggregated structures.52
To further investigate the photobehaviour in solid state, we carried out ps-experiments exciting at 371 nm. Fig. 5 shows the emission decays at different wavelengths (from 430 to 640 nm). While Fig. 6 displays the time-resolved emission spectra (TRES) at different delay times. Table 7 gives the obtained data from the best fit of the most representative wavelengths, while those at all observed wavelengths are shown in Tables S18 and S19 (ESI†).
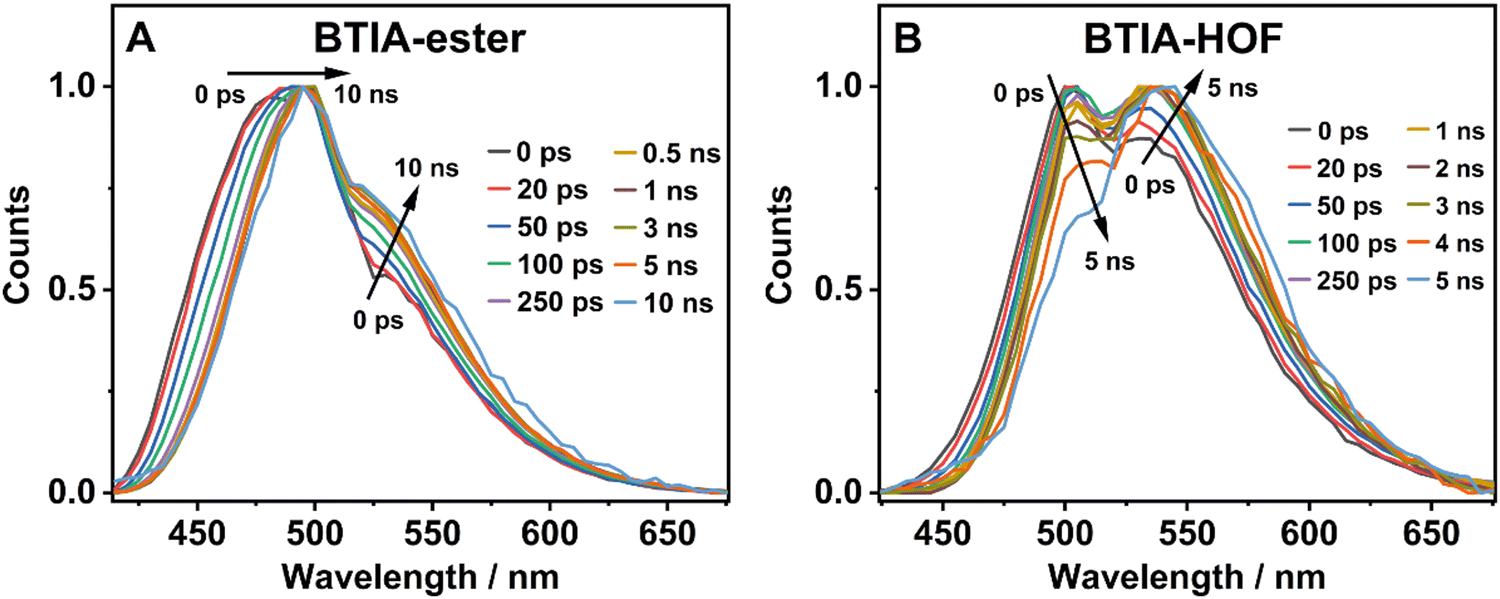 | ||
| Fig. 6 Normalized time-resolved emission spectra (TRES) of BTIA-ester (A) and BTIA-HOF (B) in solid state, upon excitation at 371 nm and gating at the indicated delay times. | ||
| Sample | λ obs/nm | τ 1/ns | a 1 | c 1 | τ 2/ns | a 2 | c 2 | τ 3/ns | a 3 | c 3 | τ 4/ns | a 4 | c 4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BTIA-ester | 430 | 0.08 | 64 | 10 | 0.38 | 16 | 12 | 1.20 | 11 | 27 | 2.87 | 9 | 51 |
| 460 | 53 | 7 | 20 | 13 | 17 | 35 | 10 | 45 | |||||
| 610 | −100 | −100 | 16 | 4 | 62 | 51 | 22 | 45 | |||||
| 640 | −100 | −100 | 16 | 5 | 69 | 62 | 15 | 33 | |||||
| BTIA-HOF | 460 | 0.14 | 64 | 22 | 0.52 | 18 | 23 | 1.23 | 18 | 55 | — | ||
| 490 | 34 | 8 | 37 | 32 | 29 | 60 | |||||||
| 610 | 1 | 1 | 45 | 26 | 54 | 73 | |||||||
| 640 | — | 46 | 26 | 54 | 74 | ||||||||
In contrast to the monoexponential decay observed in DMF solutions, the emission decays of BTIA-ester in solid state show a complex behaviour that depends on the observation wavelength (Fig. 5B). The decays were fitted using a multiexponential function, obtaining time constants of 80 ps, 0.38, 1.20, and 2.87 ns (Table 7 and Table S18, ESI†). We could not get an accurate fit using less components. The shortest component decays at the bluest part and rises at the reddest one (from 510 nm), while the other components are decaying throughout the entire spectral range. The 0.38 ns component displays its maximum contribution at the blue part of the emission spectrum, while it almost disappears from 510 nm. The 1.20 ns component scarcely contributes to the bluest part and reaches its maximum contribution at the reddest region of the emission spectra. Finally, the longest component (2.87 ns) displays its maximum contribution at 510 nm, contributing less to the global signal at the beginning and end of the spectrum (Table 7 and Table S18, ESI†). To get more insights into the photodynamics of BTIA-ester, we recorded the TRES in solid state (Fig. 6A and Fig. S15, ESI†). The TRES displays a clear spectral shift to longer wavelengths when the time increases. At t = 0 ps, the TRES shows an emission band with the intensity maximum at 480 nm and a shoulder around 530 nm. As the gating time increases (t = 250 ps), the initial band shifts to longer wavelengths (∼500 nm) and the shoulder located at 530 nm increases in intensity. At longer delay times (>250 ps), the TRES evolves towards longer wavelengths and the contribution of the 530 nm band increases. Based on these observations, the contributions obtained from the fit of the ps-emission decays and considering the solution results and explanation, we propose the following picture to explain the photobehaviour of BTIA-ester in solid state. The initial emission band recorded at early times reflects the emission of the initially excited species, which emit at 480 nm with a lifetime of 380 ps. These species undergo an ICT reaction from the peripheric phenyl groups to the BTD core, which takes place in 80 ps, producing ICT structures which emit at longer wavelengths with lifetimes of 1.20 and 2.87 ns. The heterogeneity of the sample (amorphous solid) gives rise to different types of aggregates of different conformations, and thus emits at different lifetimes.28,55 The 500 nm emission band recorded in the TRES corresponded to ICT species emitting with a lifetime of 2.87 ns, while those emitting at longer wavelengths (∼530 nm) had a lifetime of 1.20 ns.
Fig. 5C exhibits the collected ps-emission decays of BTIA-HOF, while Table 7 gives the obtained values from the best global fits. The decays also exhibit a complex behaviour, being shorter at the blue side of the emission spectrum. However, we do not observe a ps-rising component. The emission decays were well fitted using lifetimes of 0.14 ps, 0.52 and 1.23 ns. The components are decaying over the entire spectral range, with the shortest component displaying its maximum contribution at the bluest part and disappearing at the reddest, while the longer ones exhibit the opposite behaviour. The 0.52 ns component reaches its maximum contribution at ∼500 nm and remains unchanged at longer wavelengths. On the other hand, the 1.23 ns component displays its maximum contribution at the reddest part of the emission spectrum (Table 7 and Table S19, ESI†).
We also recoded the TRES of BTIA-HOF in solid state (Fig. 6B and Fig. S15, ESI†), which shows differences with respect to that of BTIA-ester. At t = 0 ps, we observe an emission spectrum with two bands located at ∼500 and ∼550 nm, with the intensity of the former decreasing at longer gating times (5 ns) and the latter increasing. The 500 nm band recorded for BTIA-HOF is similar to that previously observed for its ester derivative, and which we assigned to ICT species; whereas the 550 nm band is red-shifted (∼15 nm) compared to the one observed in BTIA-ester, indicating the presence of different emitting species. Interestingly, we do not observe the 480 nm band that was recorded in the BTIA-ester. This is an indication that the initial species evolves very fast to ICT structures (sub-picosecond time scale), a result consistent with the absence of a ps-rising component in the emission decays (Table 7 and Table S19, ESI†).
Based on these observations, we propose the following picture for BTIA-HOF in solid state. The shortest component, which displays its maximum contribution at the bluest part of the emission spectra (450–480 nm), reflects the lifetime of the initially excited species (0.14 ns). These undergo an ultrafast ICT reaction, which takes place below the instrument resolution (<15 ps), giving rise to ICT structures with a lifetime of 0.52 ns. In addition to the ICT process, the presence of carboxylic acid groups in the structure may lead to the observation of intermolecular PT reactions between neighbouring and H-bonded molecules, producing ionic species that cannot be formed in BTIA-ester. Therefore, we suggest that the longest component (1.23 ns), which reaches its maximum contribution at the reddest side of the emission spectrum, corresponds to the lifetime of these ionic species in the HOF structure. We cannot exclude the possibility that ionic species also have an ICT character in their structure. Scheme 2 illustrates the photophysical processes occurring in the S1 state of the amorphous BTIA-ester solid and the ordered crystalline structure of BTIA-HOF.
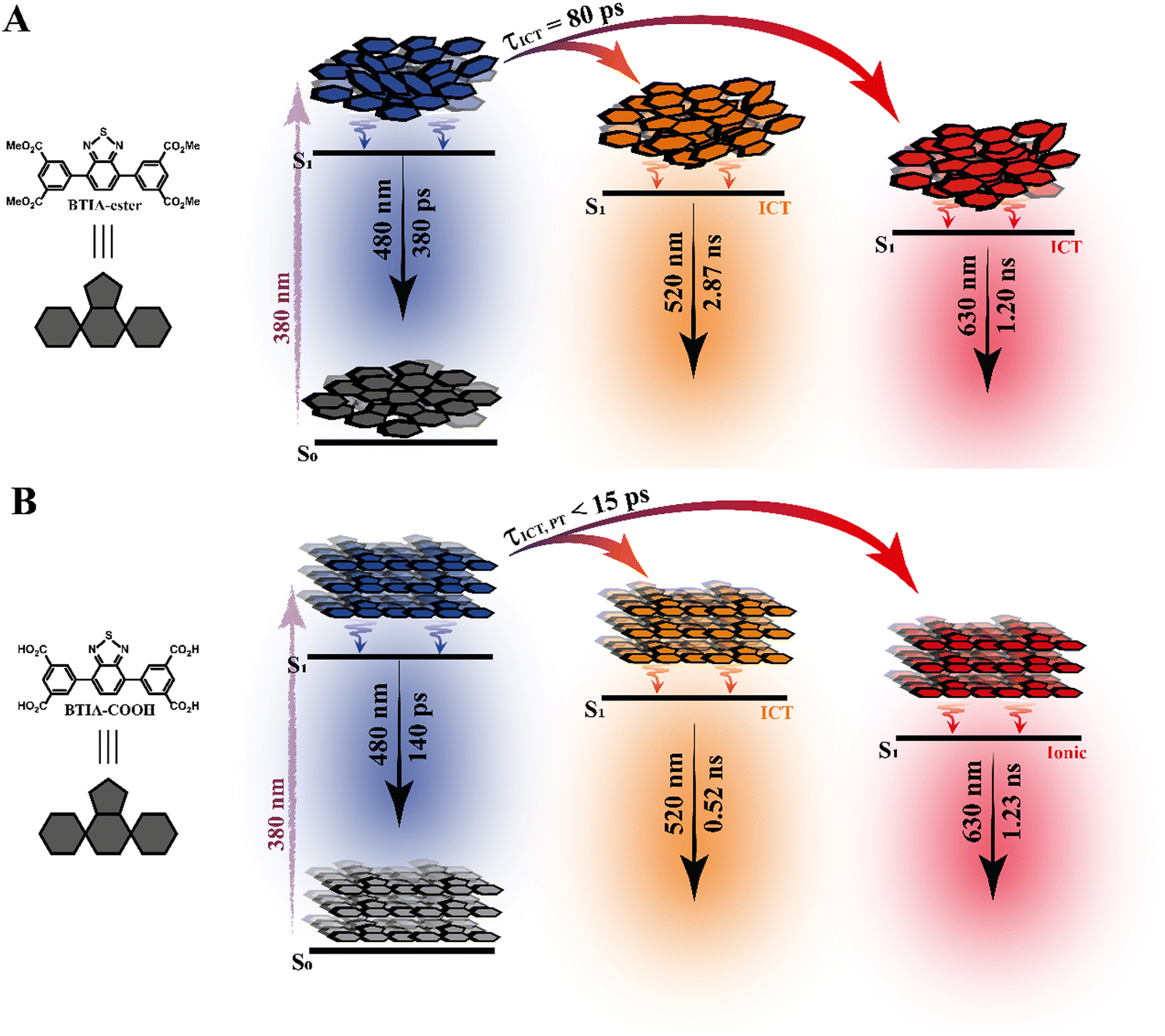 | ||
| Scheme 2 Proposed photodynamic schemes for BTIA-ester (A) and BTIA-HOF (B) in solid state, in which ICT is the intramolecular charge-transfer process and PT is the intermolecular proton-transfer reaction. The emission wavelengths were chosen based on the time-resolved spectra and the contributions obtained in the fit of the picosecond emission decays. The schemes are not to scale. | ||
3.7 Single-crystal fluorescence microscopy of BTIA-HOF
To obtain further information on the solid-state fluorescence of the HOF and explore the effect of the crystals size and possible defects in the crystalline structure, we carried out time and space fluorescence microscopy experiments on BTIA-HOF single crystals (Fig. 7).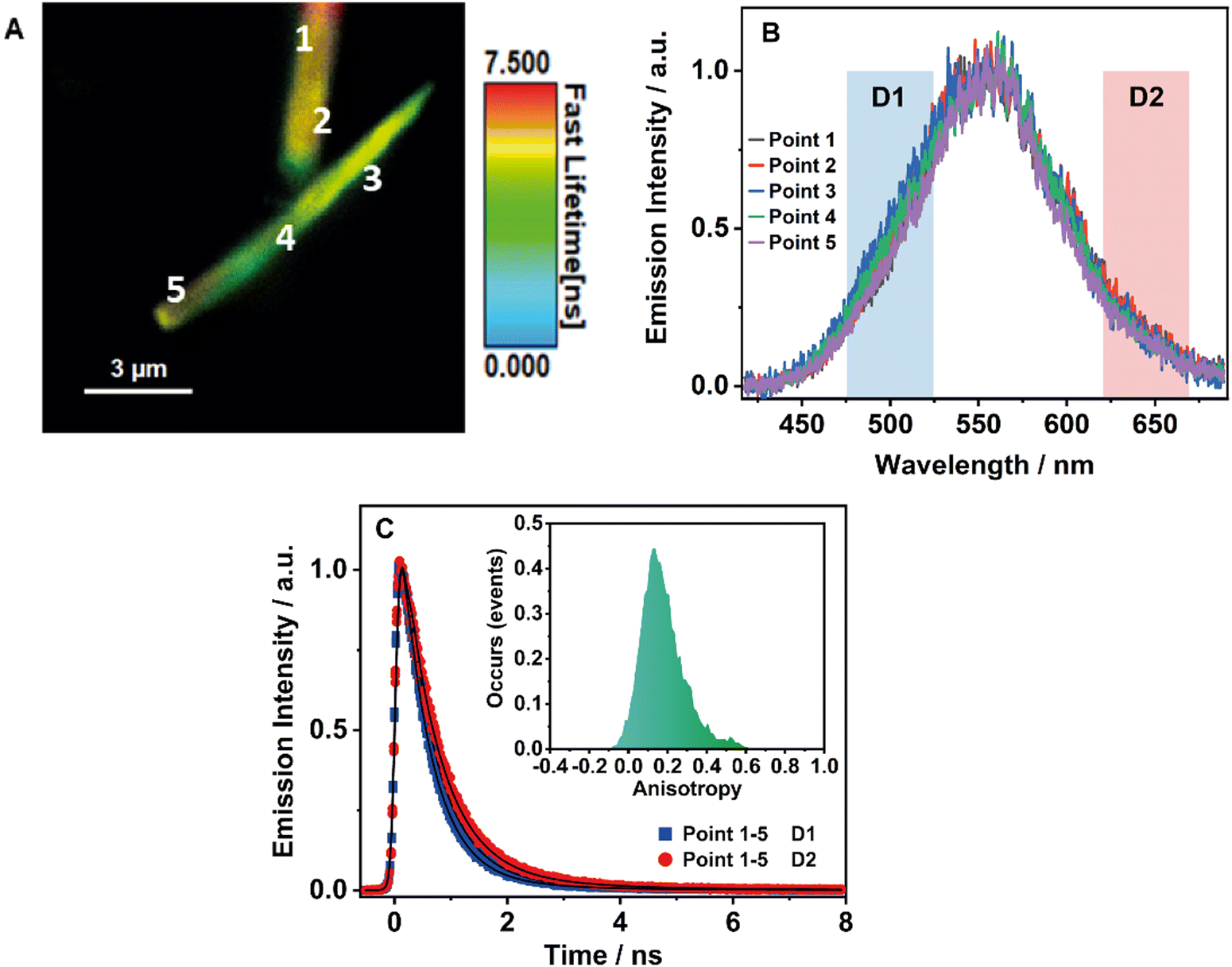 | ||
| Fig. 7 (A) FLIM of BTIA-HOF crystals. (B) Normalized emission spectra of BTIA-HOF at different points of the crystals. (C) Fluorescence emission decays of BTIA-HOF crystals in the selected spectral range using two different filters (D1 and D2 in (B)). The solid lines are from the best fit using a multiexponential function. Inset shows the histogram of the emission anisotropy for BTIA-HOF crystals. The excitation wavelength was 371 nm. | ||
To begin with, Fig. 7A displays the fluorescence image (FLIM) of two crystals of BTIA-HOF. All the examined crystals have a needle-shaped structure and are 5–10 μm in length and 0.5–1 μm in thickness (see also ESI†). From the lifetime distribution (mostly green with a large fraction of yellow colour), we suggest that the fluorescence lifetimes are the same over the crystals. However, we observe an intense red colour distribution in some crystals, which may indicate the presence of possible molecules adsorbed or adhered on the surface of the single crystals (Fig. S16, ESI†). Aiming for more information on the single crystals photophysical properties, we also recorded the fluorescence spectra at different points for the same crystal (Fig. 7B). The spectra display a broad band with intensity maxima at ∼550 nm regardless of the examined point. The FWHM of the emission band (∼3300 cm−1) scarcely changed with the recording position, indicating that the fluorescence emission comes from the same emitting species. We also studied other crystals with different sizes and thicknesses, and they show similar photobehaviours (Fig. S16, ESI†). The fact that the emission spectra and FHWM do not change with the interrogated position (middle or corner) or crystals indicates that the defects, if they exist, do not affect the photophysical properties of the crystalline structure, at least under the used experimental and resolution conditions.
To shed more light on the single crystals photobehaviour, we also collected the ps-emission decays in two different regions of the emission spectrum using two different filters to select the signal: detector 1 (D1) for the blue–green emission region (475–525 nm) and detector 2 (D2) for the red one (620–670 nm) (Fig. 7C and Fig. S16, ESI†). The results show a small difference between the interrogated points in the crystal, with the emission decays showing a biexponential behaviour with time constants of ∼0.6 and ∼1.7 ns (Table 8 and Table S20, ESI†). Both components are decaying over the whole spectral range, with the shorter component displaying its maximum contribution at the blue part and the longer one at the red side of the emission spectrum. The obtained values are comparable to those observed for the crystals ensemble (0.14, 0.52, and 1.23 ns). Note that we do not observe the shortest component (0.14 ns) as it is within the instrumental response function of the set-up (IRF = ∼200 ps). Therefore, we assign the ∼0.6 ps component to species with an ICT character and the ∼1.7 ns one to the lifetime of ionic species formed after an intermolecular PT reaction in the crystal.
| Point | Detector | τ 1/ns | a 1 | τ 2/ns | a 2 |
|---|---|---|---|---|---|
| 1 | 1 | 0.59 | 95 | 1.65 | 5 |
| 2 | 0.63 | 92 | 1.80 | 8 | |
| 2 | 1 | 0.59 | 95 | 1.72 | 5 |
| 2 | 0.61 | 91 | 1.70 | 9 | |
| 3 | 1 | 0.56 | 96 | 1.64 | 4 |
| 2 | 0.56 | 90 | 1.78 | 10 | |
| 4 | 1 | 0.58 | 95 | 1.75 | 5 |
| 2 | 0.59 | 92 | 1.67 | 8 | |
| 5 | 1 | 0.63 | 95 | 1.84 | 5 |
| 2 | 0.62 | 91 | 1.73 | 9 | |
Finally, to get information on the alignment and molecular order in the HOF, we studied the anisotropy properties of single crystals using fluorescence microscopy (Fig. 7C, Inset). BTIA-HOF shows a high anisotropic emission behaviour caused by growth in a direction of π-stacking due to dipole–dipole interactions between the BTD moieties. The emission anisotropy distribution displays one broad band with the maximum intensity at 0.2, suggesting that the crystal structure of BTIA-HOF do not have a preferential orientation for the emitters. It is worth to mentioning that the anisotropy distribution histogram is very broad. When crystallizing the HOF, we used methyl benzoate (MB) as a solvent, and thus the obtained HOF crystals contained a few MB molecules, disturbing the dipole-dipole interactions of the HOF molecular units and causing an anisotropic crystal growth.52 This leads to a heterogeneous distribution of the molecular interactions between the molecular building units. We also determined the anisotropy properties of crystals of different sizes, and we observe the same anisotropic behaviour (Fig. S16, ESI†), with a broad anisotropy distribution located at 0.2. To conclude this part, fluorescence microscopy experiments on BTIA-HOF single crystals indicate that the photobehaviour does not depend on the interrogated point reflecting a comparable homogeneity, and they show a high anisotropic behaviour as a result of the growth in the direction of π-stacking due to the dipole-dipole interactions and intermolecular H-bonds between the BTD moieties.
Based on the above results, we can provide a comparison of the spectroscopic properties of the molecular units and the HOFs in solution and in the solid state. The main spectral difference between the emission spectra of the ester and HOF in the solid state is a red-shift in the emission of the latter (∼1100 cm−1) as a result of ICT and PT events. The presence of H-bond interactions between the neighbouring molecules of the HOF leads to the formation of ionic species with longer lifetimes. Furthermore, the structural arrangement and the outcome of the main intermolecular interactions (π–π, dipole-dipole, and H-bonds) between the units forming the network are behind the changes in the optical properties and lifetimes of the HOF when compared to their molecular units in solution. In solid state, we show that both ICT/PT processes in BTIA-HOF are ultrafast (<15 ps), while the ICT event observed in the amorphous solid BTIA-ester is slow (∼80 ps). This indicates that the ordered structure in the network and the above interactions favour the occurrence of both processes and make them faster events. Both ICT/PT photoreactions together with the ordered structural arrangement are the origin of the changes in the luminescence properties of the HOF when compared to the molecular units. The combination of these characteristics present in BTIA-HOF gives this network different luminescent properties and will serve as guiding knowledge for the development of smart HOFs with improved optical properties, allowing the use of these for specific applications, such as lighting and sensing.
4. Conclusions
In this contribution, we report on studies of a new HOF based on a 2,1,3-benzothiadiazole (BTD) derivative and their molecular units in DMF solutions and in solid state. The absorption spectra of BTIA-HOF and its molecular units in DMF exhibit an intense broad band located at ∼400 nm assigned to S0 (π) → S1 (π*) transition with an ICT character. The emission spectra of both BTIA-ester and BTIA-COOH in DMF are characterized by a large Stokes shift due to the occurrence of an ICT reaction followed by twisting of the phenyl moieties, which is in agreement with the theoretical calculation results. The fs-experiments on BTIA-ester and BTIA-COOH in DMF solutions show an ICT process taking place at ∼300 fs, and twisting motions of the phenyl rings in ∼6 ps. The photophysical properties of BTIA-ester in DMF reflect the occurrence of a single planarized ICT (PLICT) process giving rise to a long-lived species (7.52 ns). However, the photobehaviour of BTIA-COOH in DMF is more complex, producing different emitting species: H-bonded complexes (390 ps), planar ICT species (7.75 ns), and anion structures (1.15 ns). Flash photolysis experiments in DMF reveal a population of a triplet state with a lifetime of ∼500 ns, and the formation of a long-lived charge-separated state living for ∼2 μs. In solid state, BTIA-ester shows an ICT reaction occurring in 80 ps and producing ICT aggregate structures with lifetimes of 1.20 and 2.87 ns. On the other hand, BTIA HOF displays a rich photochemistry due to ICT and intermolecular PT reactions (<15 ps), giving rise to ICT and ionic species with lifetimes of 0.52 and 1.23 ns, respectively. Fluorescence microscopy experiments on BTIA-HOF single crystals show that their photobehaviour does not depend on the interrogated point, reflecting a homogeneity of the crystal formation. We believe that the present work provides new findings on HOFs photochemistry for the development of new HOFs based on benzothiadiazole derivatives with improved fluorescence properties for application in lighting and sensor devices.Author contributions
I. Hisaki designed, I. Hisaki and T. Hashimoto synthesized and characterized the materials. J. A. Organero carried out the theoretical calculations. M. de la Hoz Tomás and M. R. di Nunzio performed, analyzed, and discussed the experimental results. A. Douhal designed, supervised, and discussed the experimental and theoretical work. M. de la Hoz Tomás, J.A. Organero, M. R. di Nunzio and A. Douhal wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the following grants: PID2020-116519RB-I00 and TED2021-131650B-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union, EU”; PRTR-C17.I1 funded by European Union NextGenerationEU; SBPLY/21/180501/000108 funded by JCCM and by the EU through “Fondo Europeo de Desarollo Regional” (FEDER) and 2022-GRIN-34325 funded by UCLM (FEDER). MH thanks MCIN for the FPI fellowship PRE2021-099064 financed by MCIN/AEI/10.13059/501100011033 and by FSE+. IH thanks KAKENHI (JP21H01919) from MEXT/JSPS Japan, and Multidisciplinary Research Laboratory System for Future Developments (MRL), Graduate School of Engineering Science, Osaka University.References
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed
.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef
- J. Zhang, X. Zhao, H. Shen, J. W. Y. Lam, H. Zhang and B. Z. Tang, Adv. Photonics, 2021, 4(1), 014001 Search PubMed
- J. Zhang, H. Shen, X. Liu, X. Yang, S. L. Broman, H. Wang, Q. Li, J. W. Y. Lam, H. Zhang, M. Cacciarini, M. B. Nielsen and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202208460 CrossRef CAS PubMed
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC
- S. Dalapati, C. Gu and D. Jiang, Small, 2016, 12, 6513–6527 CrossRef CAS PubMed
- J. Ma, T. Shu, Y. Sun, X. Zhou, C. Ren, L. Su and X. Zhang, Small, 2022, 18, 2103516 CrossRef CAS PubMed
- S. Cai, Z. An and W. Huang, Adv. Funct. Mater., 2022, 32, 2207145 CrossRef CAS
- M. R. di Nunzio, I. Hisaki and A. Douhal, J. Photochem. Photobiol., C, 2021, 47, 100418 CrossRef CAS
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC
- M. Gutiérrez, Y. Zhang and J.-C. Tan, Chem. Rev., 2022, 122, 10438–10483 CrossRef PubMed
- M. Gutiérrez, U. Díaz, B. Cohen and A. Douhal, J. Mater. Chem. C, 2023, 11, 14043–14069 RSC
- L. Zhang, L. Yi, Z.-J. Sun and H. Deng, Aggregate, 2021, 2, e24 CrossRef CAS
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed
- L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Mater. Adv., 2022, 3, 3680–3708 RSC
- J. Yang, J. Wang, B. Hou, X. Huang, T. Wang, Y. Bao and H. Hao, Chem. Eng. J., 2020, 399, 125873 CrossRef CAS
- Y. Cai, J. Gao, J.-H. Li, P. Liu, Y. Zheng, W. Zhou, H. Wu, L. Li, R.-B. Lin and B. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308579 CrossRef CAS PubMed
- C. Chen, L. Shen, H. Lin, D. Zhao, B. Li and B. Chen, Chem. Soc. Rev., 2024, 53, 2738–2760 RSC
- X.-J. Xi, Y. Li, F. Lang, J. Pang and X.-H. Bu, Chem. Sci., 2024, 15, 4529–4537 RSC
- X. Liu, G. Liu, T. Fu, K. Ding, J. Guo, Z. Wang, W. Xia and H. Shangguan, Adv. Sci., 2024, n/a, 2400101 CrossRef PubMed
- Z. Xiong, S. Xiang, Y. Lv, B. Chen and Z. Zhang, Adv. Funct. Mater., 2024, n/a, 2403635 CrossRef
- Y.-F. Han, Y.-X. Yuan and H.-B. Wang, Molecules, 2017, 22, 266 CrossRef PubMed
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS PubMed
- Z. Sun, Y. Li, L. Chen, X. Jing and Z. Xie, Cryst. Growth Des., 2015, 15, 542–545 CrossRef CAS
- E. Gomez, M. Gutiérrez, B. Cohen, I. Hisaki and A. Douhal, J. Mater. Chem. C, 2018, 6, 6929–6939 RSC
- E. Gomez, Y. Suzuki, I. Hisaki, M. Moreno and A. Douhal, J. Mater. Chem. C, 2019, 7, 10818–10832 RSC
- I. Hisaki, Y. Suzuki, E. Gomez, Q. Ji, N. Tohnai, T. Nakamura and A. Douhal, J. Am. Chem. Soc., 2019, 141, 2111–2121 CrossRef CAS PubMed
- Y. Suzuki, M. Gutiérrez, S. Tanaka, E. Gomez, N. Tohnai, N. Yasuda, N. Matubayasi, A. Douhal and I. Hisaki, Chem. Sci., 2022, 13, 14410 RSC
- Y. Farré, M. Raissi, A. Fihey, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, Dyes Pigm., 2018, 148, 154–166 CrossRef
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS
- A. M. Thooft, K. Cassaidy and B. VanVeller, J. Org. Chem., 2017, 82, 8842–8847 CrossRef CAS PubMed
- F. Ricci, F. Elisei, P. Foggi, A. Marrocchi, A. Spalletti and B. Carlotti, J. Phys. Chem. C, 2016, 120, 23726–23739 CrossRef CAS
- A. Iagatti, B. Patrizi, A. Basagni, A. Marcelli, A. Alessi, S. Zanardi, R. Fusco, M. Salvalaggio, L. Bussotti and P. Foggi, Phys. Chem. Chem. Phys., 2017, 19, 13604–13613 RSC
- B. Patrizi, A. Iagatti, L. Abbondanza, L. Bussotti, S. Zanardi, M. Salvalaggio, R. Fusco and P. Foggi, J. Phys. Chem. C, 2019, 123, 5840–5852 CrossRef CAS
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed
- S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845–851 CrossRef CAS
- S. Lee, M. Jen, T. Jang, G. Lee and Y. Pang, Sci. Rep., 2022, 12, 6557 CrossRef CAS PubMed
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC
- G. Haberhauer, R. Gleiter and C. Burkhart, Chem. – Eur. J., 2016, 22, 971–978 CrossRef CAS PubMed
- G. Haberhauer, Chem. – Eur. J., 2017, 23, 9288–9296 CrossRef CAS PubMed
- H. Wu, J. Luo, Z. Xu, Z. Wang, D. Ma, A. Qin and B. Z. Tang, Chem. Res. Chin. Univ., 2020, 36, 61–67 CrossRef CAS
- H. Wu, Y. Pan, J. Zeng, L. Du, W. Luo, H. Zhang, K. Xue, P. Chen, D. L. Phillips, Z. Wang, A. Qin and B. Z. Tang, Adv. Opt. Mater., 2019, 7, 1900283 CrossRef
- B. Zhang, H. Wu, Z. Wang, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2020, 8, 4754–4762 RSC
- V. Malytskyi, J.-J. Simon, L. Patrone and J.-M. Raimundo, RSC Adv., 2015, 5, 354–397 RSC
- N. Yeh and P. Yeh, Renewable Sustainable Energy Rev., 2013, 21, 421–431 CrossRef CAS
- J. Li, Y. Zhu, H. Xu, T.-F. Zheng, S.-J. Liu, Y. Wu, J.-L. Chen, Y.-Q. Chen and H.-R. Wen, Inorg. Chem., 2022, 61, 3607–3615 CrossRef CAS PubMed
- Q.-Q. Li, P.-H. Pan, H. Liu, L. Zhou, S.-Y. Zhao, B. Deng, Y.-J. He, J.-X. Song, P. Liu, Y.-Y. Wang and J.-L. Li, Inorg. Chem., 2023, 62, 17182–17190 CrossRef CAS PubMed
- J.-Y. Zeng, X.-S. Wang, B.-R. Xie, M.-J. Li and X.-Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10087–10094 CrossRef CAS PubMed
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., 2020, 132, 17050–17057 CrossRef
- Z. Yang, A. Moriyama, R. Oketani, T. Nakamura and I. Hisaki, Chem. Lett., 2021, 50, 1909–1912 CrossRef CAS
- Z. Yang, A. Saeki, A. Inoue, R. Oketani, K. Kamiya, S. Nakanishi, T. Nakamura and I. Hisaki, Cryst. Growth Des., 2022, 22, 4472–4479 CrossRef CAS
- I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Chem. – Eur. J., 2017, 23, 11611–11619 CrossRef CAS PubMed
- E. Gomez, M. R. di Nunzio, M. Moreno, I. Hisaki and A. Douhal, J. Phys. Chem. C, 2020, 124, 6938–6951 CrossRef CAS
- E. Gomez, M. Gutiérrez, M. Moreno, I. Hisaki, S. Nakagawa and A. Douhal, Phys. Chem. Chem. Phys., 2018, 20, 7415–7427 RSC
- Q. Chen, T. Zhang, X. Chen, M. Liang, H. Zhao, P. Yuan, Y. Han, C.-P. Li, J. Hao and P. Xue, ACS Appl. Mater. Interfaces, 2022, 14, 24509–24517 CrossRef CAS PubMed
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS
- S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC
- Y. Xu, C. Wang, X. Zhou, J. Zhou, X. Guo, X. Liang, D. Hu, F. Li, D. Ma and Y. Ma, J. Phys. Chem. Lett., 2019, 10, 6878–6884 CrossRef CAS PubMed
- J. U. Kim, I. S. Park, C.-Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765 CrossRef CAS PubMed
- X. Dong, S. Shen, Y. Qin, X. Xie, Z. Pang, H. Gao, G. Liu, C.-S. Lee, P. Wang and Y. Wang, J. Phys. Chem. C, 2024, 128, 974–983 CrossRef CAS
- C. M. Marian, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed
- E. Xu, H. Zhong, J. Du, D. Zeng, S. Ren, J. Sun and Q. Fang, Dyes Pigm., 2009, 80, 194–198 CrossRef CAS
- X.-Z. Zhu, H.-D. Hu, S.-H. Chen, Y.-M. Li, J.-F. Yan and Y.-F. Yuan, New J. Chem., 2023, 47, 4299–4305 RSC
- I. Bala, R. A. K. Yadav, M. Devi, J. De, N. Singh, K. Kailasam, J. Jayakumar, J.-H. Jou, C.-H. Cheng and S. K. Pal, J. Mater. Chem. C, 2020, 8, 17009–17015 RSC
- S. Kongsabay, T. Loythaworn, S. Petdee, P. Chasing, P. Funchien, W. Waengdongbung, P. Wongkaew, T. Sudyodsuk and V. Promarak, ChemPhotoChem, 2022, 6, e202200154 CrossRef CAS
- P. Gautam, R. Maragani, S. M. Mobin and R. Misra, RSC Adv., 2014, 4, 52526–52529 RSC
- D. Jiang, S. Chen, Z. Xue, Y. Li, H. Liu, W. Yang and Y. Li, Dyes Pigm., 2016, 125, 100–105 CrossRef CAS
- R. Karpicz, S. Puzinas, S. Krotkus, K. Kazlauskas, S. Jursenas, J. V. Grazulevicius, S. Grigalevicius and V. Gulbinas, J. Chem. Phys., 2011, 134, 204508 CrossRef CAS PubMed
- Z.-L. Qiu, C. Tang, X.-R. Wang, Y.-Y. Ju, K.-S. Chu, Z.-Y. Deng, H. Hou, Y.-M. Liu and Y.-Z. Tan, Angew. Chem., Int. Ed., 2020, 59, 20868–20872 CrossRef CAS PubMed
- C. M. Hu and R. Zwanzig, J. Chem. Phys., 1974, 60, 4354–4357 CrossRef CAS
- J. S. Baskin and A. H. Zewail, J. Phys. Chem. A, 2001, 105, 3680–3692 CrossRef CAS
- D. G. Patel, F. Feng, Y.-Y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599–2612 CrossRef CAS PubMed
- M. E. El-Khouly, Y. Chen, X. Zhuang and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 6370–6371 CrossRef CAS PubMed
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed
- M. Gutiérrez, L. Duplouy-Armani, L. Angiolini, M. Pintado-Sierra, F. Sánchez and A. Douhal, Int. J. Mol. Sci., 2020, 21, 4366 CrossRef PubMed
- S. Banerjee, A. K. Both and M. Sarkar, ACS Omega, 2018, 3, 15709–15724 CrossRef CAS PubMed
- M. Yamaguchi, M. D. L. H. Tomás, A. Fujiwara, R. Oketani, K. Okubo, K. Oka, N. Tohnai, A. Douhal and I. Hisaki, Bull. Chem. Soc. Jpn., 2024, 97, uoae004 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc01368a |
| This journal is © The Royal Society of Chemistry 2024 |