
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIronIII-catalyzed asymmetric inverse-electron-demand hetero-Diels–Alder reaction of dioxopyrrolidines with simple olefins†
Tangyu
Zhan
,
Liang
Zhou
,
Yuqiao
Zhou
 ,
Bingqian
Yang
,
Xiaoming
Feng
* and
Xiaohua
Liu
*
,
Bingqian
Yang
,
Xiaoming
Feng
* and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 29th February 2024
Abstract
The asymmetric catalytic inverse-electron-demand hetero-Diels–Alder reaction of dioxopyrrolidines with a variety of simple olefins has been accomplished, significantly expanding the applicability of this cyclization to both cyclic hetero-dienes and dienophiles. A new type of strong Lewis acid catalyst of ferric salt enables the LUMO activation of dioxopyrrolidines via formation of cationic species, this method yields a range of bicyclic dihydropyran derivatives with exceptional outcomes, including high yields (up to 99%), diastereoselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and enantioselectivity (up to 99% ee) under mild conditions. This facile protocol was available for the late-stage modification of several bioactive molecules and transformation into macrocycle molecules as well. The origins of enantioselectivity were elucidated based on control experiments.
:1) and enantioselectivity (up to 99% ee) under mild conditions. This facile protocol was available for the late-stage modification of several bioactive molecules and transformation into macrocycle molecules as well. The origins of enantioselectivity were elucidated based on control experiments.
Introduction
The synthesis of novel heterocyclic scaffolds continues to capture the attention of the organic chemistry community owing to their potential pharmaceutical activities.1 The inverse-electron-demand hetero-Diels–Alder (IEDHDA) reaction, a significant variant of Diels–Alder reactions, proves adept at incorporating functional groups into hetero-rings (Scheme 1a). Particularly noteworthy is its asymmetric catalytic version, enabling the construction of optically enriched six-membered rings.2 Mechanistically, this reaction unfolds through the interaction between the LUMO of an electron-deficient hetero-diene and the HOMO of electron-rich dienophiles. In this context, the use of chiral Lewis acids or organocatalysts becomes instrumental in expediting enantioselective transformations, employing three primary strategies: elevating the HOMO of dienophiles, lowering the LUMO energy of dienes, or concurrently activating both through dual modulation.3 As evidenced in Scheme 1a, the asymmetric catalytic IEDHDA reactions involving α,β-unsaturated carbonyls with electron-rich dienophiles, such as cyclic or acyclic 1,3-dienes,4 vinyl ethers5 and the related,6 or hydrazone conjugated carbon–carbon double bond,7 have been well demonstrated. HOMO-raising strategies, employing enamine8 or enolate activation9 with organocatalysts, further broaden the scope of dienophiles. The generation of active dienophiles in situ, exemplified by the conversion of 3-cyclopropylideneprop-2-enone into cyclobutene-fused furans10 or the oxidation of ethers,11 facilitates rapid and alternative access to cycloaddition products. In contrast, IEDHDA reaction of simple alkenes is generally challenging12,14 and achievable only by lowering the LUMO of hetero-dienes. Two successful examples involve the asymmetric [4 + 2] cycloaddition of β,γ-unsaturated α-ketoesters into chiral oxanes (Scheme 1b), as illustrated in Scheme 1b. Luo's approach employs a chiral binary acid complex that synergistically combines a chiral phosphoric acid with a metal salt (InIII or ScIII).13 Sakakura and Ishihara's report features an n-cation copper complex.14 Critically, both processes aim to enhance the cationic nature of the metal centers for the LUMO activation of dienes. Despite these advancements, further exploration of diene species, such as cyclic enones instead of linear unsaturated α-ketoesters, for the asymmetric catalytic IEDHDA reaction of unactivated alkenes, which may suffer partial polymerization, remains a challenging frontier in current research.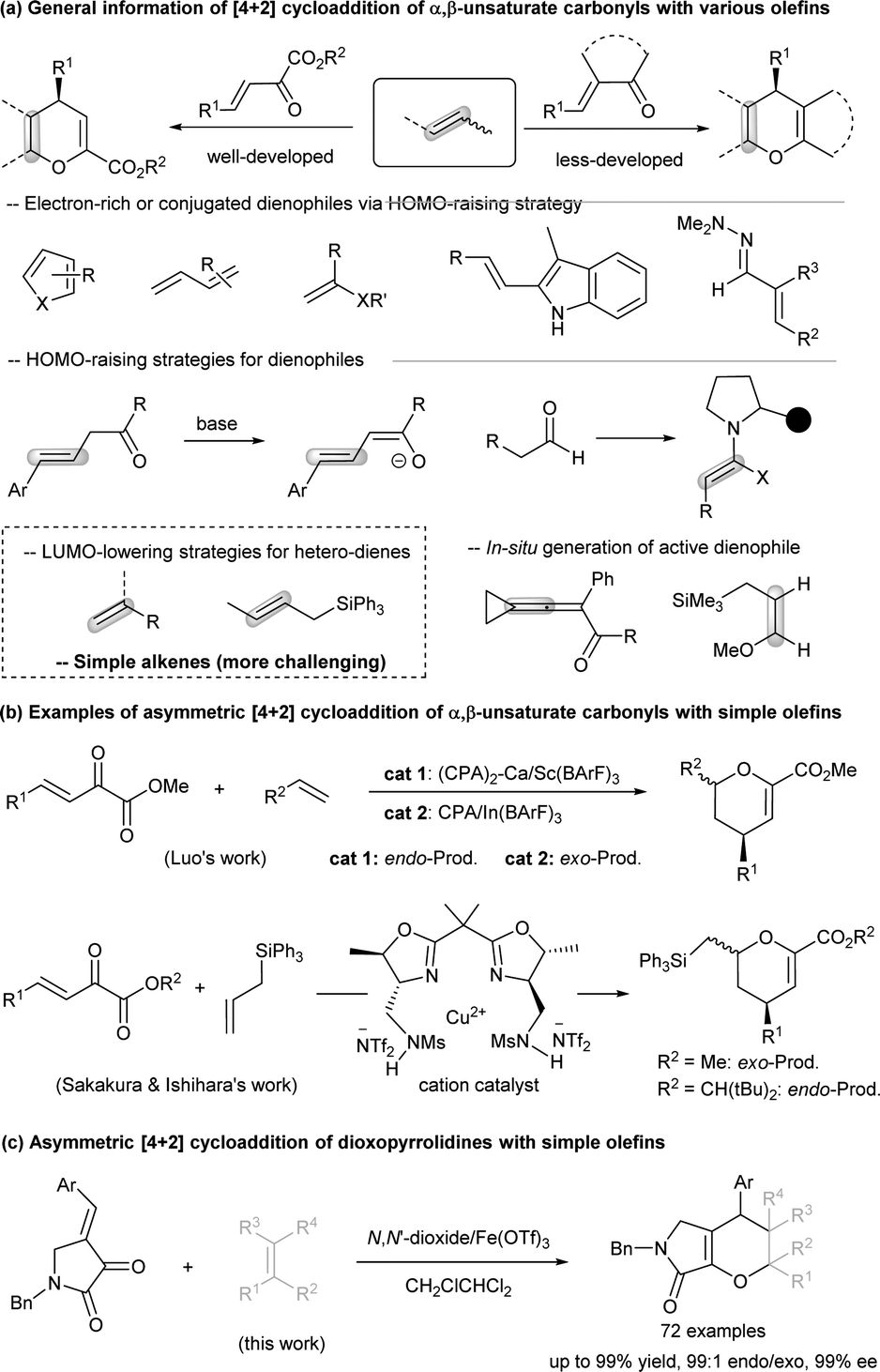 | ||
| Scheme 1 General information about the catalytic asymmetric IEDHDA reaction. | ||
Bicyclic dihydropyrans fused with a γ-lactam or pyrrolidine moiety constitute core structures in bioactive molecules and natural products.15 The asymmetric IEDHDA reaction of γ-lactam-derived cyclic enones offers an efficient pathway for constructing these valuable backbones (Scheme 1c).16 We propose that chiral Lewis acid catalysts, specifically those employing N,N′-dioxide ligands, may expedite the cycloaddition by LUMO-activating dioxopyrrolidine17 for cyclization with simple olefins. This rationale is grounded in the observation that chelation of these tetra-oxygen ligands allows the counterion of the metal precursor to delocalize from the metal center to extend, generating n-cation-characterized stronger Lewis acid catalysts.18 Concerning stereoselectivity, challenges extend beyond enantioselectivity to include the endo/exo ratio influenced by orbital-favored transition states and a stepwise 1,4-addition/cyclization process.19 In this context, we present a chiral iron-complex catalyzed asymmetric IEDHDA reaction of dioxopyrrolidines with simple alkenes, yielding various optically active bicyclic dihydropyran derivatives with outstanding results: up to 99% yield, 99:1 diastereoselectivity, and up to 99% enantioselectivity, all achieved under mild conditions.
Results and discussion
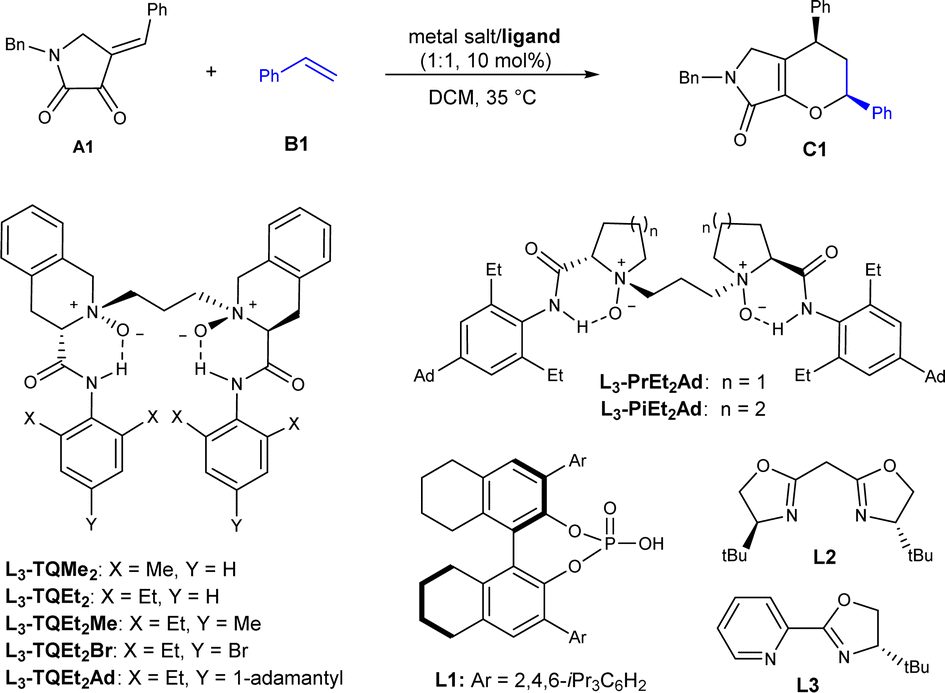
Our investigation of the IEDHDA reaction began with dioxopyrrolidine A1 and styrene B1 as model substrates to optimize the reaction conditions (Table 1). We identified the critical parameters to the reactivity and found that Lewis acidity of the metal salts played a decisive role in whether the reaction occurs (see ESI† for details), which is in consistent with the mechanism of LUMO-activation of hetero-diene. The reaction performed only in the presence of stronger Lewis acids, such as In(OTf)3, Fe(OTf)3, or Al(OTf)3, to give the desired product C1 in low diastereoselectivity. The comparison with the reaction of heterosubstituted alkenes which could be performed with NiII complexes manifested critical of the Lewis acidity of the catalyst.20 Using 10 mol% of In(OTf)3 in CH2Cl2 at 35 °C, the cycloaddition product was isolated in 84% yield and 48:52 endo:exo, which is selected to identify optimal chiral ligands to control the stereoselectivity. Noteworthily, the diastereoselectivity is poor, differing from the preferred endo-selectivity of achiral Lewis acid-promoted IEDHDA of β,γ-unsaturated α-ketoesters.21 Investigating asymmetric catalytic reaction showed that the substituents of chiral N,N′-dioxide ligands at the anilines affected the diastereo- and enantioselectivity a lot (Table 1, entries 1–3). The reaction catalyzed by In(OTf)3/L3-TQEt2Me delivered the product in 91% yield with 73:27 endo:exo, and 84% ee for endo-isomer when 1,1,2-trichloroethane was used as the solvent (entry 4). The use of Fe(OTf)3 instead led to slightly increased endo-diastereoselectivity (entry 5). Further modification of para-substitutions at the aniline units of the ligands was carried out (entries 5–7), and it was observed that by introduction of steric hindered 1-adamantyl group into the para-position (L3-TQEt2Ad) lead to a slightly higher diastereoselectivity and enantioselectivity, and the product C1 was isolated in 95% ee with 90:10 endo:exo ratio and 97% ee (entry 7). Reducing the usage of styrene to 5 equivalents led to no disadvantage to the outcomes (entry 8). Reinvestigation of the amino backbone of N,N′-dioxide ligands showed that the reactivity dropped a lot when L-proline or L-pipeclic acid based ones were employed (entries 9 and 10). Comparatively, other representative chiral ligands, such as chiral phosphoric acid L1, bisoxazoline L2 and pyridine-oxazoline L3 resulted in poor reactivity (entries 11–13), which manifests the importance of ligand-acceleration in asymmetric Lewis acid catalysis.22 In these cases, the minor exo-product was obtained in lower ee value than the endo-product. The absolute configuration of endo-product C1 was determined to be (2S, 4R) by X-ray crystallography analysis23 (see ESI† for details).
|
|
|||||
|---|---|---|---|---|---|
| Entry | M(OTf)3 | Ligand | Yieldb (%) | endo/exoc | eec (%) |
|
a The reactions were carried out with A1 (0.1 mmol), B1 (1.0 mmol) and ligand/metal (1:1, 10 mol%) in CH2Cl2 (1.0 mL) at 35 °C for 24 h.
b Yield of isolated product.
c Determined by chiral UPC2 on a chiral stationary phase.
d CH2ClCHCl2 as solvent.
e With 0.5 mmol B1 for 48 h. NR, no reaction.
|
|||||
| 1 | In(OTf)3 | L3-TQMe2 | 84 | 56:44 |
43/44 |
| 2 | In(OTf)3 | L3-TQEt2 | 99 | 68:32 |
59/40 |
| 3 | In(OTf)3 | L3-TQEt2Me | 98 | 73:27 |
75/53 |
| 4d | In(OTf)3 | L3-TQEt2Me | 91 | 73:27 |
84/59 |
| 5d | Fe(OTf)3 | L3-TQEt2Me | 87 | 83:17 |
93/19 |
| 6d | Fe(OTf)3 | L3-TQEt2Br | 29 | 78:22 |
65/0 |
| 7d | Fe(OTf)3 | L3-TQEt2Ad | 95 | 90:10 |
97/72 |
| 8d,e | Fe(OTf)3 | L3-TQEt2Ad | 93 | 90:10 |
97/73 |
| 9d,e | Fe(OTf)3 | L3-PrEt2Ad | 44 | 77:23 |
39/31 |
| 10d,e | Fe(OTf)3 | L3-PiEt2Ad | 36 | 87/13 | 92/69 |
| 11d,e | Fe(OTf)3 | L1 | NR | — | — |
| 12d,e | Fe(OTf)3 | L2 | Trace | — | — |
| 13d,e | Fe(OTf)3 | L3 | Trace | — | — |
Under the optimal conditions, the substrate scope of IEDHDA reaction of dioxopyrrolidines with styrene were evaluated as listed in Scheme 2. Most of the desired products could be obtained in excellent total yields (84–98%) and enantioselectivities (86–99% ees) for the endo-isomers (C2–C14, C16, and C17). But those with ortho-substituted phenyl groups (25–80% yields; C18–C21) and meta-methoxyl substituted C15 (34% yield) were exceptions, especially 2-Cl and 2-Br-containing ones whose endo-selectivity dropped a lot, and ee value of the exo-isomers are higher than endo-isomers (C19–C20). It is also interesting to found that the endo:exo ratio of the tetrahydropyranopyrrolones with electron-donating aryls (95:5 to 97:3) is higher than the electron-deficient ones (87:13 to 92:8), manifesting the influence of a concert or stepwise pathway. The phenyl ring could be replaced by 2-thiophene (C22), 2-naphthyl (C23), 2-benzothiophene (C24), and phenylallyl substituents (C25), which were also suitable.
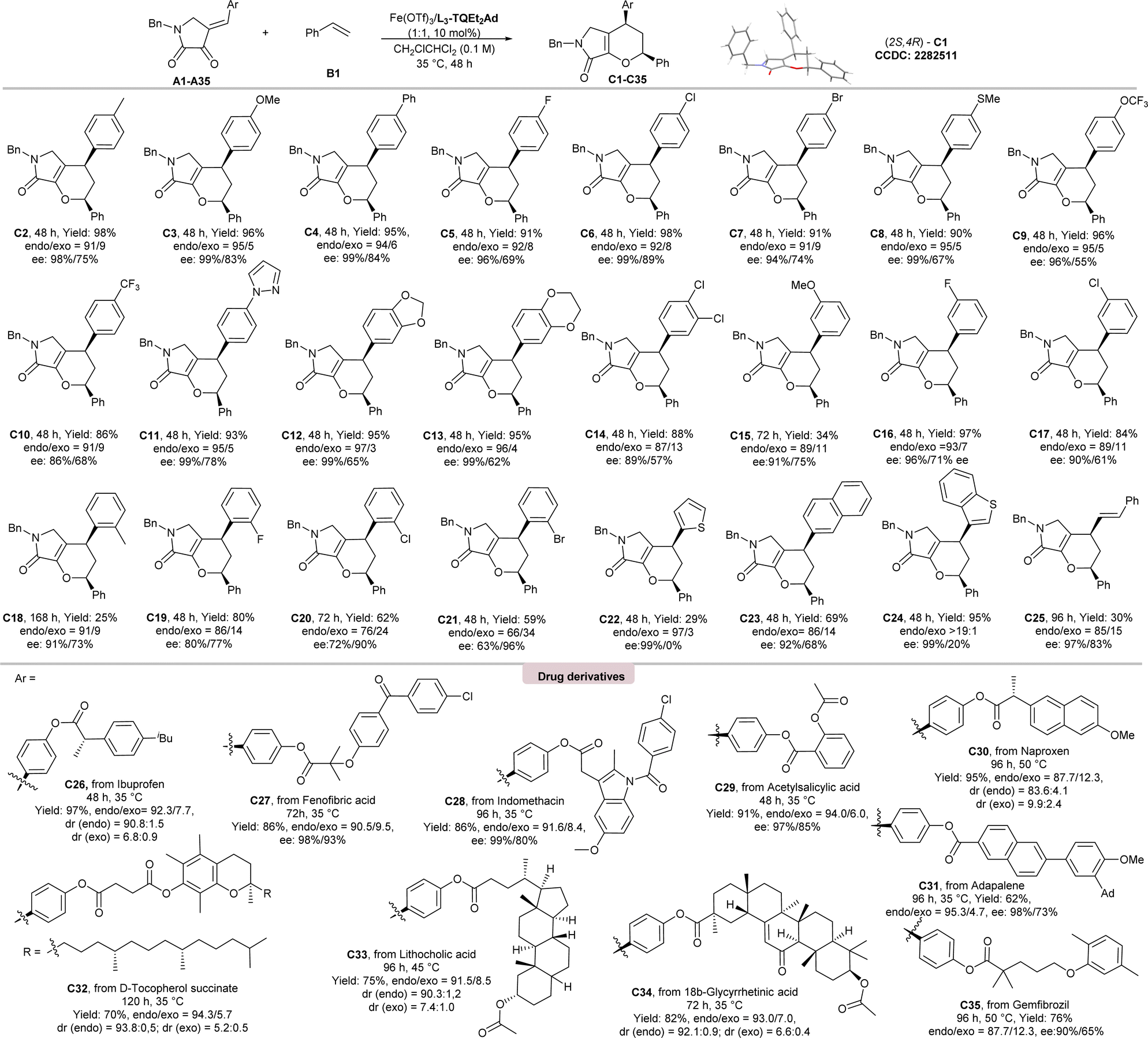 | ||
| Scheme 2 The substrate scope of dioxopyrrolidines. a Unless otherwise noted, all reactions were performed with A (0.1 mmol), styrene B1 (0.5 mmol) and Fe(OTf)3/L3-TQEt2Ad (1:1, 10 mol%) in CH2ClCHCl2 (1.0 mL) at 35 °C. The yield is isolated yield. The endo:exo ratio was determined by 1H NMR analysis and the ee value was determined by UPC2 or HPLC on a chiral stationary phase. | ||
To our delight, the current method could be used in the late-stage modification of complex and bioactive compounds. As illustrated in Scheme 2, the reaction of ibuprofen, fenofibric acid, indomethacin, acetylsalicylic acid, naproxen, adapalene, D-tocopherol succinate, as well as gemfibrozil derived dioxopyrrolidines with styrene B1 underwent smoothly, affording the desired bicyclic dihydropyranes (C26–C35) in excellent yields (62–97%), endo:exo ratio (88:12–94:6) and enantioselectivities (90–99% ees). Complex terpenoid derivatives underwent smoothly, affording the desired products (C33–C34) in good yields (75–82%), and excellent enantioselectivities (98% ee). Nevertheless, the current catalytic system is not fit for the cyclization of acyclic hetero-diene (see ESI† for details).
Subsequently, we turned our attention to the substrate scope of alkenes (Scheme 3) to illustrate the IEDHDA reaction. A series of styrenes B (B2–B37), regardless of the substituent pattern and the electronic property of the aryl moiety, reacted with dioxopyrrolidine A1 smoothly, providing the desired bicyclic dihydropyranes D1–D36 in moderate to good yields and high enantioselectivities. The position of substitution at phenyl group displayed a significant effect on the stereocontrol of [4 + 2] cycloaddition. Styrenes with different functional groups at para-position were tolerated well to deliver the corresponding products. Generally, electron-withdrawing groups, as 4-fluror (D7) or 4-chloro (D8) substitutions, gave better diastereo- and enantioselectivity than electron-donating groups (D1–D6). However, if the steric hindrance of para-position of the aryl group increased, the reactivity dropped obviously and displayed a negligible effect on the stereocontrol (D7–D9). Lower diastereo- and enantioselectivity were given for 4-methoxyphenyl substituted D4 (86% yield, endo:exo = 65:35, 31%/27% ee), implying the disfavored competition of stepwise 1,4-addition/cyclization process. In contrast, electron-withdrawing groups or weak electron-donating groups at 3- or 3,4- substituted substrates were found to be suitable in the reaction (D10–D23; 46–99% yields, 76:24–93:7 endo:exo, 81–99% ees). The absolute configuration of product D13 was determined to be (2S, 4R) by X-ray crystallography analysis (see ESI† for details).23 3,4-Dicholor, and 2-fluoro substituted styrenes could be achieved in good selectivity, unfortunately, the reactivity is lower (C24–C25). Moreover, the phenyl ring could be replaced by 2-benzofuran (D26), 2-benzothiophene (D27), 2-naphthyl (D28), but the reaction temperature had to be rationalized in order to balance the enantioselectivity and yield. Propen-2-ylbenzene participated in the cyclization in moderate yield and dropped stereoselectivity (D29). Both trans- and cis-propen-1-ylbenzene could perform the reaction in good endo-selectivity and excellent enantioselectivity but the yields were moderate (D30–D31) after the addition of NaBArF4. Similarly, when trans-methylstyrene bearing 3,4-dimethoxyl substituents was subjected, only moderate stereoselectivity was obtained (see ESI† for details). Although the trans-configuration of olefine delivered into the product without change, and no obverse intermediate was detected, stepwise pathway or a concerted asynchronous could not be ruled out. Moreover, when α,α-dialkyl olefins or 2,3-dimethylbut-2-ene were used as the dienophiles (D32–D35), good enantioselectivity could be given after reinvestigation of the chiral catalyst as In(OTf)3/L3-TQEt2 combination. The reaction of tetrahydronaphthalene bearing an exocyclic double bond worked in the presence of Fe(OTf)3/L2-TQEt2Ad to afford only endo-D36 in 52% yield with 67% ee.
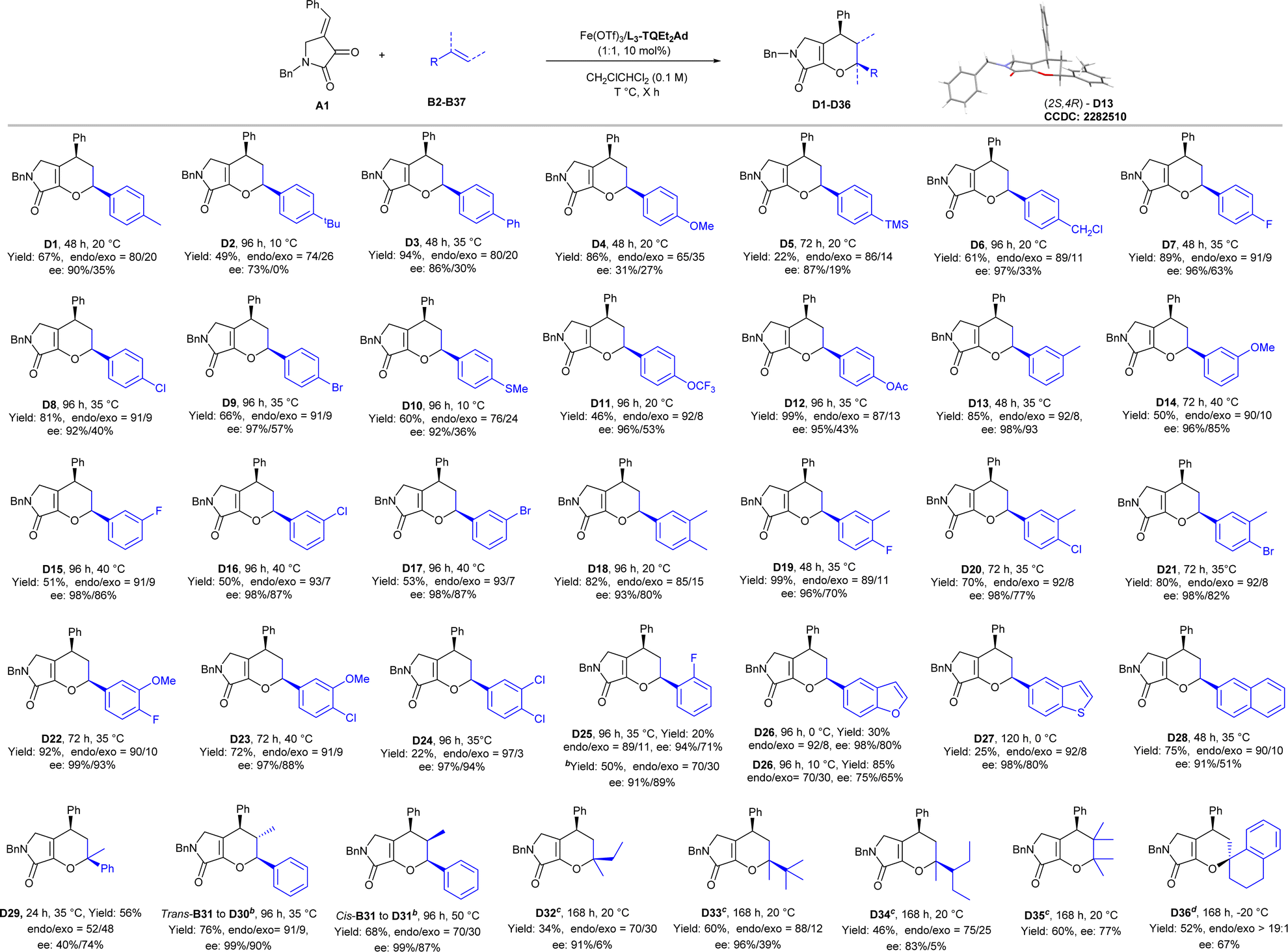 | ||
| Scheme 3 The substrate scope of olefins.a Unless otherwise noted, all reactions were performed with dioxopyrrolidine A1 (0.1 mmol), olefin (0.5 mmol) and Fe(OTf)3/L3-TQEt2Ad (1:1, 10 mol%) in CH2ClCHCl2 (1.0 mL). Isolated yield. The endo:exo ratio was determined by 1H NMR analysis and the ee value was determined by UPC2 or HPLC on a chiral stationary phase. b NaBArF4 (50 mol%). c In CH3CN with In(OTf)3/L3-TQEt2 (1:1, 20 mol%). d Fe(OTf)3/L2-TQEt2Ad (1:1, 10 mol%). | ||
To illustrate the potential synthetic utility of the current catalytic system, a scale-up synthesis of C1 were performed. As shown in Scheme 4a, under the optimized reaction condition, dioxopyrrolidine A1 (3.2 mmol) reacted smoothly with styrene B1 (5.0 equiv.), affording the desired product C1 in 94% yield (1.15 g) with 90:10 endo/exo ratio and 99% and 71% ee separately. Under the oxidation of RuCl3 with NaIO4, the double bond of dihydropyrrolone ring of endo-C1 broke to give O,N-based macrocycle E1 in good yield with slight loss of stereoselectivity.23 Alternatively, reduction reaction mediated by H2 in the presence of Pd/C in methanol led to the ring-opening reaction to yield E2 in nearly completely yield but the optical purity decreased a little (Scheme 4b).
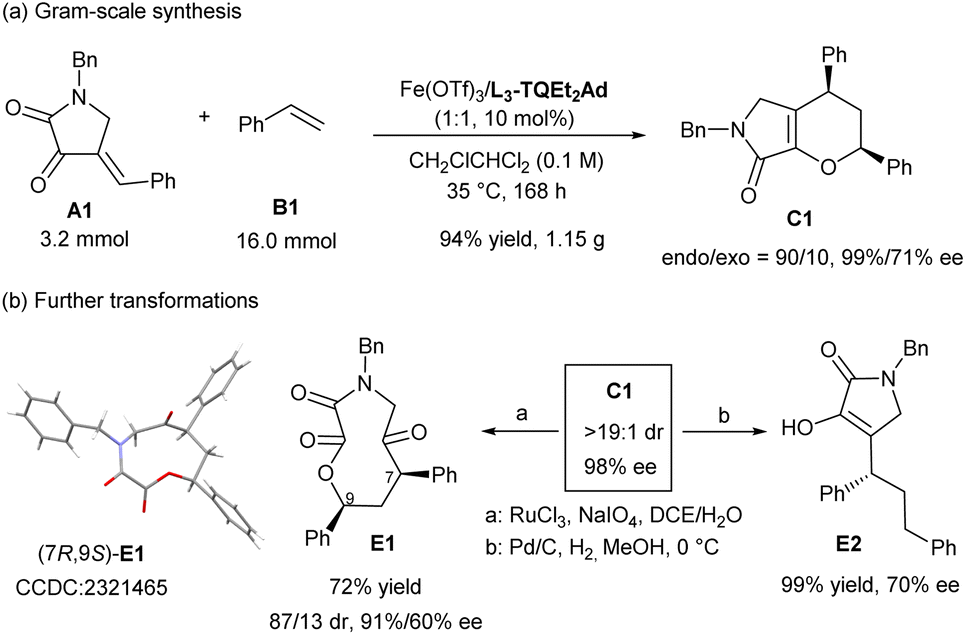 | ||
| Scheme 4 Gram-scale synthesis and further transformations. | ||
UV-vis absorption spectra were carried out to show the interaction of chiral ferric iron catalyst with the diene (Fig. 1a). There was obvious hyperchromic effect upon mixing Fe(OTf)3 with dioxopyrrolidine A1, especially in the presence of the L3-TQEt2Ad ligand. In addition, the investigation of relationship between ee value of L3-TQEt2Ad and that of C1 showed a self-evident linear effect,24 implying catalytically active species was likely to be the monomeric complex of Fe(OTf)3 and L3-TQEt2Ad (Fig. 1b).
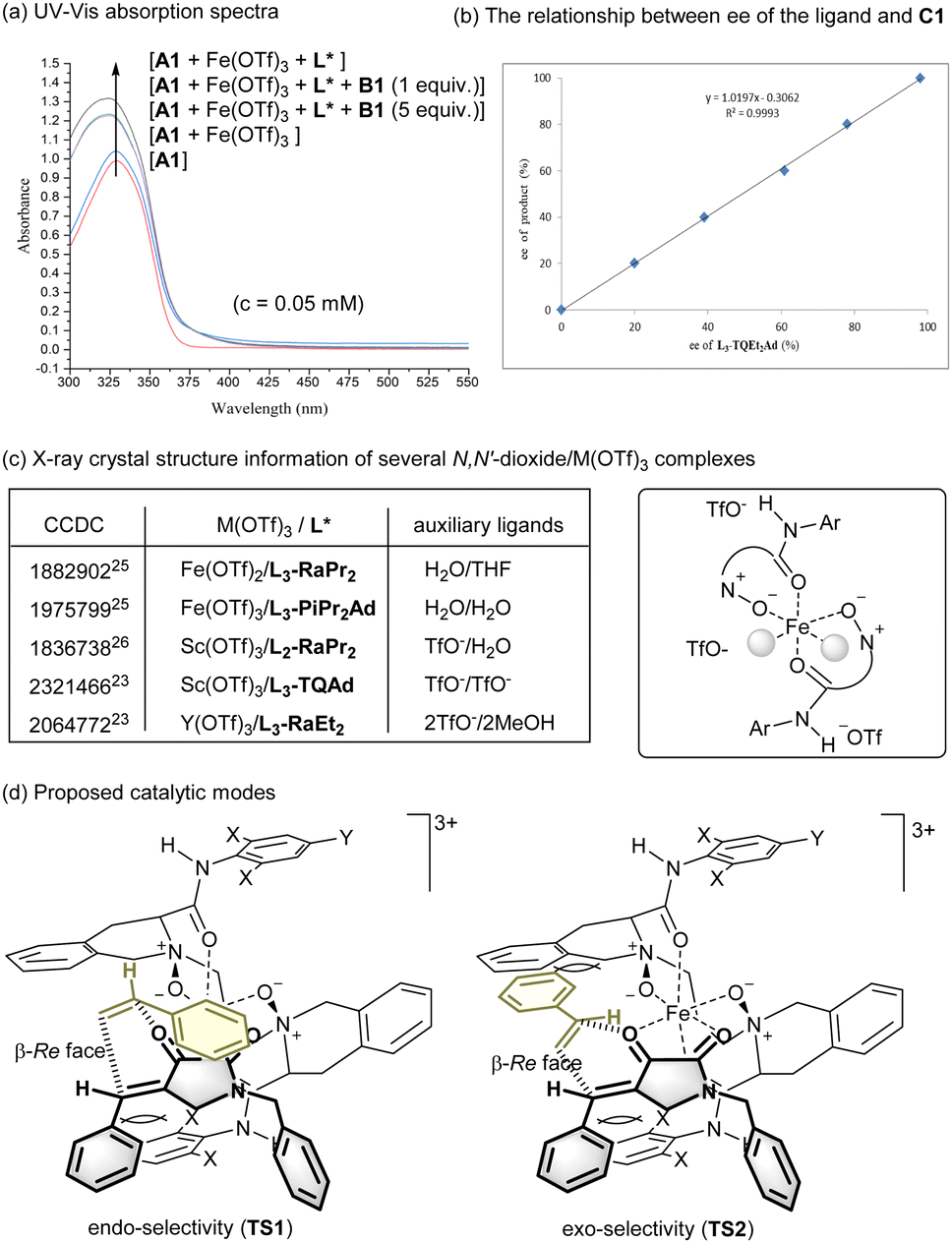 | ||
| Fig. 1 Reaction mechanism consideration. | ||
Chiral N,N′-dioxide rare-earth metal complexes, such as Sc(OTf)3 or Y(OTf)3, which are usually rationalized as good Lewis acid candidates, were found to sluggish in accelerating the cycloaddition (see ESI† for details). Analysis of several crystal structures of N,N′-dioxide-M(OTf)3 complexes provide interesting cooperation priority (Fig. 1c). It was found that the Fe(OTf)3 complexes of N,N′-dioxides were more likely to form trication characterized complexes although the whole structures are neutral overall.25 However, there are at least one anion coordinating to the Sc(III) center as showing in the related structures.26 In most cases, the counter ions locate around the complexes, having interaction via H-bonds with the outwards amide subunits.14 In addition, other rare-earth metal complexes with larger ion radii are prone to form metal complexes with coordination number beyond six,18,27 which exhibited different configuration geometry from the octahedral complexes of iron. Thus, the chiral ferric iron complexes are capable to efficiently lower the LUMO energy of the dienophile upon coordination.
Based on the observed stereoselectivity and the previous works,20 possible catalytic modes were proposed (Fig. 1d). Initially, the tetradentate L3-TQEt2Ad coordinates to Fe(III) to form an octahedral trication species. The substrate A1 bonds to the ferric iron center with the two carbonyls via bidentate manner, which leads to its β-Si face blocking by one of the amide subunits of the ligand. The free styrene B1 prefers to undergo cyclization from β-Re face of A1 in endo- or exo-selectivity (TS1 or TS2). The exo-product is the minor one which is in line with the disfavoured steric hindrance between the aryl substituent of olefin and tetrahydroisoquinoline backbone of the ligand as shown in TS2. As a result, the preferred endo-β-Re face selective cyclization gives raise to the formation of (2S,4R)-C1 as the major one. Additionally, the exo-cyclization may occur in a step-wise conjugate addition manner.
Conclusions
In summary, we have developed a highly enantioselective [4 + 2] cycloaddition of a number of simple olefins with cyclic hetero-diene of dioxopyrrolidines. The reaction proceeded well in the assistance of chiral N,N′-dioxide/FeIII complex catalysts, which could form cationic Lewis acid species to lower the LUMO energy of dioxopyrrolidines. It effectively delivered various optically active bicyclic dihydropyranes derivatives with excellent yield (up to 99%), diastereoselectivity (up to 99:1) and enantioselectivity (up to 99% ee) under mild conditions, including of late-stage modification of drug-molecular-based dienes. Mechanistic studies support the strategy and transition states were proposed to elucidate stereo-induction. Further investigations of asymmetric transformations of simple olefins are currently ongoing in our laboratory.
Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F {1H} NMR, HPLC spectra, X-ray crystallographic data for C1, D13 and E1 are available in the ESI.†Author contributions
T. Y. Z. performed experiments and prepared the ESI† and paper. L. Z repeated some experiments. Y. Q. Z for X-ray crystal analysis. B. Q. Y participated in structure characterization and discussion. X. H. L. and helped with modifying the paper and ESI.† X. H. L. and X. M. F. conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 22188101), and the Science & Technology Department of Sichuan Province (No. 2021YJ0561) for financial support.Notes and references
- (a) J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS; (b) A. Mermer, T. Keles and Y. Sirin, Bioorg. Chem., 2021, 114, 105076–110623 CrossRef CAS; (c) C. Cabrele and O. Reiser, J. Org. Chem., 2016, 81, 10109–10125 CrossRef CAS; (d) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- For selected examples of asymmetric IEDHDA reaction, see: (a) G. Huang, C. Kouklovsky and A. de la Torre, Chem.–Eur. J., 2021, 27, 4760–4788 CrossRef CAS; (b) V. Laina-Martín, J. A. Fernández-Salas and J. Alemán, Chem.–Eur. J., 2021, 27, 12509–12520 CrossRef; (c) M. S. Xie, L. L. Lin and X. M. Feng, Chem. Rec., 2017, 17, 1184–1202 CrossRef CAS; (d) Z. M. Png, H. N. Zeng, Q. Ye and J. W. Xu, Chem.–Asian J., 2017, 12, 2142–2159 CrossRef CAS; (e) X. Y. Hao, L. L. Lin, F. Tan, C. K. Yin, X. H. Liu and X. M. Feng, ACS Catal., 2015, 5, 6052–6056 CrossRef CAS; (f) W. Yao, X. Dou and Y. Lu, J. Am. Chem. Soc., 2015, 137, 54–57 CrossRef CAS; (g) Y. H. Miao, Y. Z. Hua, S. K. Jia, X. Xiao, M. C. Wang and G. J. Mei, Chem. Commun., 2023, 59, 6929–6932 RSC.
- X. X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS.
- (a) Y. Zhu, X. H. Chen, M. S. Xie, S. X. Dong, Z. Qiao, L. L. Lin, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2010, 16, 11963–11968 CrossRef CAS; (b) J. Lv, L. Zhang, S. S. Hu, J. P. Cheng and S. Z. Luo, Chem.–Eur. J., 2012, 18, 799–803 CrossRef CAS; (c) H. D. Yang, J. B. Chen, C. Peng, W. K. Liu, S. S. Zhou, J. X. Song, Z. H. Qi, Y. Wang and X. W. Wang, Adv. Synth. Catal., 2022, 364, 4347–4362 CrossRef CAS; (d) S. S. Zhao, K. L. Li, X. Su, Z. G. Zha and Z. Y. Wang, Org. Lett., 2022, 24, 4224–4228 CrossRef CAS; (e) J. L. Zheng, F. Liu, X. Song, Z. Zhao, W. Du and Y. C. Chen, J. Org. Chem., 2023, 88, 7800–7809 CrossRef CAS.
- (a) Y. Zhu, M. S. Xie, S. X. Dong, X. H. Zhao, L. L. Lin, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2011, 17, 8202–8208 CrossRef CAS; (b) Y. B. Hong, T. J. Cui, L. Sergei, X. L. Xie and E. Meggers, Chem.–Eur. J., 2021, 27, 8557–8563 CrossRef CAS; (c) J. B. Chen, M. Xu, J. Q. Zhang, B. B. Sun, J. M. Hu, J. Q. Yu, X. W. Wang, Y. Z. Xia and Z. Wang, ACS Catal., 2020, 10, 3556–3563 CrossRef CAS; (d) D. A. Evans and J. S. Johnson, J. Am. Chem. Soc., 1998, 120, 4895–4896 CrossRef CAS; (e) S. Cao, Z. Q. Ye, Y. H. Chen, Y. M. Lin, J. H. Fang, K. J. Wang, B. X. Yang and L. Gong, CCS Chem., 2022, 4, 3122–3133 CrossRef CAS; (f) N. K. Li, B. B. Sun, J. B. Chen, H. D. Yang, B. L. Wang, J. Q. Yu, X. W. Wang and Z. Wang, Org. Chem. Front., 2021, 8, 2009–2018 RSC.
- (a) H. M. Huang, X. Y. Wu, B. R. Leng, Y. L. Zhu, X. C. Meng, Y. Hong, B. Jiang and D. C. Wang, Org. Chem. Front., 2020, 7, 414–419 RSC; (b) N. Thirupathi, F. Wei, C. H. Tung and Z. H. Xu, Nat. Commun., 2019, 10, 3158–3165 CrossRef; (c) S. j. Li, J. Lv and S. Z. Luo, Org. Chem. Front., 2018, 5, 1787–1791 RSC; (d) J. J. Zhao, S. B. Sun, S. H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS.
- Y. Hashimoto, T. Ikeda, A. Ida, N. Morita and O. Tamura, Org. Lett., 2019, 21, 4245–4249 CrossRef CAS.
- (a) Q. J. Liu, L. Wang, Q. K. Kang, Z. Peter and Y. Tang, Angew. Chem., Int. Ed., 2016, 55, 9220–9223 CrossRef CAS; (b) Y. H. Zhou, Y. Zhu, L. L. Lin, Y. L. Zhang, J. F. Zheng, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2014, 20, 16753–16758 CrossRef CAS.
- (a) S. X. Dong, X. H. Liu, Y. Zhu, P. He, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2013, 135, 10026–10029 CrossRef CAS; (b) M. He, G. J. Uc and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 15088–15089 CrossRef CAS; (c) K. X. Yang, D. S. Ji, H. X. Zheng, Y. C. Gu and P. F. Xu, Chem. Commun., 2022, 58, 4227–4230 RSC; (d) Ł. Albrecht, G. Dickmeiss, C. F. Weise, C. Rodríguez-Escrich and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 13109–13113 CrossRef; (e) B. W. Huang and J. L. Han, J. Org. Chem., 2023, 88, 16376–16390 CrossRef CAS; (f) L. L. Zhang, Y. Liu, K. Liu, Z. T. Liu, N. N. He and W. J. Li, Org. Biomol. Chem., 2017, 15, 8743–8747 RSC.
- J. He, W. Liu, J. Zhang, Z. W. Zhong, L. L. Lin and X. M. Feng, Sci. China: Chem., 2023, 66, 2803–2809 CrossRef CAS.
- A. Yesilcimen, N. C. Jiang, F. H. Gottlieb and M. Wasa, J. Am. Chem. Soc., 2022, 144, 6173–6179 CrossRef CAS.
- (a) J. Liu, X. Wang, L. Xu, Z. Hao, L. Wang and J. Xiao, Tetrahedron, 2016, 72, 7642–7649 CrossRef CAS; (b) Y. Zheng, S. C. Qian, P. C. Xu, T. T. Ma and S. L. Huang, Adv. Synth. Catal., 2022, 364, 3800–3804 CrossRef CAS.
- J. Lv, L. Zhang, S. Z. Luo and J. P. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9786–9790 CrossRef CAS.
- Y. Matsumura, T. Suzuki, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6131–6134 CrossRef CAS.
- (a) R. Sarabu, US Pat., 20110313002A1, 2011 Search PubMed; (b) J. H. Rigby, A. Cavezza and M. J. Heeg, J. Am. Chem. Soc., 1998, 120, 3664–3670 CrossRef CAS; (c) T. S. Kam, K. Yoganathan and C. Wei, Tetrahedron Lett., 1996, 37, 3603–3606 CrossRef CAS.
- (a) S. Zhang, Y. C. Luo, X. Q. Hu, Z. Y. Wang, Y. M. Liang and P. F. Xu, J. Org. Chem., 2015, 80, 7288–7294 CrossRef CAS; (b) J. L. Li, K. C. Yang, Y. Li, Q. Li, H. P. Zhu, B. Han, C. Peng, Y. G. Zhi and X. J. Gou, Chem. Commun., 2016, 52, 10617–10620 RSC; (c) L. L. Zhang, X. Lu, Y. C. Wang, H. Xu, R. T. Zhan, W. W. Chen and H. C. Huang, Org. Lett., 2019, 21, 10069–10074 CrossRef.
- (a) Y. Lu, Y. H. Zhou, L. L. Lin, H. F. Zheng, K. Fu, X. H. Liu and X. M. Feng, Chem. Commun., 2016, 52, 8255–8258 RSC; (b) Y. Luo, H. Zhang, S. Y. Wang, Y. Q. Zhou, S. X. Dong and X. M. Feng, Org. Lett., 2020, 22, 2645–2650 CrossRef CAS; (c) F. Q. Zhang, B. T. Ren, Y. Q. Zhou, Y. B. Liu and X. M. Feng, Chem. Sci., 2022, 13, 5562–5567 RSC; (d) C. Q. Ke, Q. H. Cao, Y. Luo, X. H. Liu and X. M. Feng, Chem. Commun., 2022, 58, 5881–5884 RSC; (e) H. Yu, T. Y. Zhan, Y. Q. Zhou, L. Chen, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 5136–5144 CrossRef CAS; (f) W. L. Xiao, L. C. Ning, S. Xin, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211596 CrossRef CAS; (g) Y. H. Mo, Q. Y. Chen, J. Z. Li, D. Ye, Y. Q. Zhou, S. X. Dong, X. H. Liu and X. M. Feng, ACS Catal., 2023, 13, 877–886 CrossRef CAS; (h) S. Y. Wang, Y. Q. Zhou, W. L. Xiao, Z. G. Li, X. H. Liu and X. M. Feng, Chem. Sci., 2023, 14, 1844–1851 RSC; (i) S. Kobayashi and H. Ishitani, J. Am. Chem. Soc., 1994, 116, 4083–4084 CrossRef CAS.
- For selected reviews and examples on N,N′-dioxide ligands, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS; (e) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS; (f) S. X. Dong, X. H. Liu and X. M. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS; (g) Y. H. Wen, X. Huang and X. M. Feng, Synlett, 2005, 16, 2445–2448 Search PubMed; (h) T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2022, 5, 2101–2110 CrossRef; (i) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423–1433 Search PubMed; (j) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1894–1902 Search PubMed; (k) G. H. Pan, C. L. He, M. Chen, Q. Xiong, W. D. Cao and X. M. Feng, CCS Chem., 2022, 4, 2000–2008 CrossRef CAS; (l) L. L. Lin, Y. Q. Zhou, W. D. Cao, S. X. Dong, X. H. Liu and X. M. Feng, Sci. Sin. Chim., 2023, 53, 246–258 CrossRef; (m) Y. M. He, Y. Z. Cheng, Y. D. Duan, Y. D. Zhang, Q. H. Fan, S. L. You, S. Z. Luo, S. F. Zhu, X. F. Fu and Q. L. Zhou, CCS Chem., 2023, 5, 2685–2716 CrossRef CAS; (n) Q. W. He, M. P. Pu, Z. Jiang, H. Y. Wang, X. M. Feng and X. H. Liu, J. Am. Chem. Soc., 2023, 145, 15611–15618 CrossRef CAS; (o) Z. Tan, L. Chen, L. Y. Li, Y. Z. Li, Y. Luo, F. Wang, S. X. Dong and X. M. Feng, Angew. Chem., Int. Ed., 2023, 62, e202306146 CrossRef CAS; (p) F. C. Zhang, Y. Q. Zhou, H. S. Zhao, L. Chen, W. D. Cao and X. M. Feng, Precis. Chem., 2023, 1, 423–428 CrossRef CAS.
- (a) X. F. Lin, Y. Liu and C. Li, Chem.–Eur. J., 2020, 26, 14173–14180 CrossRef CAS; (b) J. H. Zhou, B. Jiang, F. F. Meng, Y. H. Xu and T. P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS.
- X. Y. Hu, Y. H. Zhou, Y. Lu, S. J. Zou, L. L. Lin, X. H. Liu and X. M. Feng, J. Org. Chem., 2018, 83, 8679–8687 CrossRef CAS.
- (a) L. M. Stephenson, D. E. Smith and S. P. Current, J. Org. Chem., 1982, 47, 4170–4173 CrossRef CAS; (b) L. Maingot, S. Leconte, I. Chataigner, A. Martel and G. Dujardin, Org. Lett., 2009, 11, 1619–1622 CrossRef CAS.
- (a) S. X. Dong, W. D. Cao, M. P. Pu, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2717–2735 CrossRef CAS; (b) D. J. Berrisford, C. Bolm and K. B. Sharpless, Angew. Chem., Int. Ed., 1995, 34, 1059–1070 CrossRef CAS; (c) L. J. Li, Y. L. He, Y. H. Yang, J. Guo, Z. Lu, C. Y. Wang, S. L. Zhu and S. F. Zhu, CCS Chem., 2024, 6, 537–584 CrossRef.
- CCDC 2282511 (C1), CCDC 2282510 (D13), CCDC 2321465 (E1) contains the supplementary crystallographic data for this paper.
- X. T. Zhu, Y. J. Li and H. L. Bao, Chin. J. Chem., 2023, 41, 3097–3114 CrossRef CAS.
- (a) F. Wang, L. L. Feng, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Commun., 2020, 56, 3233–3236 RSC; (b) P. F. Zhou, X. H. Liu, W. B. Wu, C. R. Xu and X. M. Feng, Org. Lett., 2019, 21, 1170–1175 CrossRef CAS.
- F. Tan, M. P. Pu, J. He, J. Z. Li, J. Yang, S. X. Dong, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 2394–2402 CrossRef CAS.
- (a) Y. Zhang, F. C. Zhang, L. Chen, J. Xu, X. H. Liu and X. M. Feng, ACS Catal., 2019, 9, 4834–4840 CrossRef CAS; (b) Y. Luo, Q. Wei, L. K. Yang, Y. Q. Zhou, W. D. Cao, Z. S. Su, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 12984–12992 CrossRef CAS; (c) D. Zhang, M. P. Pu, Z. Z. Liu, Y. Q. Zhou, Z. D. Yang, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2023, 145, 4808–4818 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra. X-ray crystallographic data for C1 (CIF). CCDC 2282510, 2282511 and 2321465. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00078a |
| This journal is © The Royal Society of Chemistry 2024 |