
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible and irreversible reaction mechanisms of Li–CO2 batteries†
Xinxin
Zhang
,
Yu
Wang
 * and
Yafei
Li
*
* and
Yafei
Li
*
Jiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: yu.wang@njnu.edu.cn; yafeili@njnu.edu.cn
First published on 16th February 2024
Abstract
Li–CO2 batteries are considered a versatile solution for CO2 utilization. However, their development, including reversibility and efficiency, is impeded by an inadequate understanding of Li–CO2 electrochemistry, particularly the decomposition of carbon and the generation of by-product O2. Here, using typical Ru(0001) (reversible) and Ir(111) (irreversible) as model catalysts and employing state-of-the-art first-principles calculations, the rechargeable/reversible reaction mechanisms of Li–CO2 batteries are disclosed. We find that electrolyte, often neglected or oversimplified in Li–CO2 modelling, plays an essential role in CO2 activation and C–C coupling affects the generation pathways of discharge intermediates due to the sluggish kinetics. The results rationalize experimental observations, which are also examined by constant-potential modelling. Specifically, by exploring the kinetics of the charging process, we discover that the reversibility of Ru(0001) is attributed to its ability to suppress O–O coupling while co-oxidizing Li2CO3 and carbon. In contrast, Li2CO3 decomposition on Ir(111) preferentially produces O2, during which carbon can only be partially decomposed. These findings solve long-standing questions and highlight the necessity of describing the explicit solvent effect in modelling, which can promote further studies on Li–CO2 batteries.
Introduction
Li–CO2 batteries have garnered extensive attention from the scientific community owing to their high potential for CO2 fixation while simultaneously enabling energy storage with a theoretical energy density of 1876 W h kg−1.1–3 In contrast to Li-ion batteries, their performance is governed by Li–CO2 electrochemistry, which operates according to the following reaction: 4Li + 3CO2 ↔ 2Li2CO3 + C (E0 = 2.8 V vs. Li/Li+).4–7 However, CO2 reduction during the discharging process is kinetically sluggish, which results in large discharging overpotentials, and a high charge voltage is also required to decompose the discharge product, Li2CO3, due to its wide bandgap of 5.03 eV and high thermodynamic stability (ΔGf = −1132.1 kJ mol−1).8,9 The inadequate decomposition of the discharge product carbon limits energy efficiency and leads to an irreversible reaction (e.g., 2Li2CO3 → 4Li+ + 4e− + 2CO2 + O2).10–12 Moreover, the passivation of catalyst surfaces and instability of electrolytes affect the cycle performance of Li–CO2 batteries. Such issues pose massive challenges to the practical applications of Li–CO2 batteries, thereby inspiring the rational design of catalysts that demonstrate high reversibility and low overpotentials.13–15Uncovering the underlying mechanisms can facilitate the design of efficient catalysts.16–21 Based on experimental characterization and theoretical calculations, much effort has been expended to reveal the reaction pathway on the catalysts, which directly determines the reversibility of Li–CO2 electrochemistry.22–25 For the discharging process, C–C coupling, such as the dimerization of *CO2 (* indicates an adsorption site) and the reaction of *Li2CO2 and *CO2, was regarded as the essential steps for Li2CO3 formation.12,18,26 However, the dominant coupling step and reaction pathway are still controversial. Another important issue is that the oxidative decomposition mechanism of Li2CO3 and carbon remains quite ambiguous. For example, in principle, the as-formed O species can react with carbon, but in the case of most catalysts, such as Au(111) and Ir(111), only the decomposition of Li2CO3 has been observed; both O2 and CO2 were found to evolve concomitantly during the charging process.3,26–30 Note that the generated O2 may contain aggressive singlet oxygen (1O2), which can attack battery components and cause severe parasitic reactions.13,31 In the cases of Ru-based metal catalysts, Li2CO3 and carbon disappear together and only CO2 is released, corresponding to a reversible process.3 However, to date, no satisfactory explanation for such phenomena exists. It is of great significance to gain a mechanistic understanding of Li–CO2 electrochemistry.
Herein, we investigate the underlying mechanisms of Li–CO2 electrochemistry by conducting comprehensive first-principles calculations to provide fundamental insights into the reversibility with a focus on the initial charge and discharge processes. Ru(0001) and Ir(111) are selected as model catalysts because the former has been extensively reported to achieve good reversibility in Li–CO2 electrochemistry, while the latter suffers from irreversibility issues. The modelling results show that the electrolyte can facilitate the activation of CO2, and the disproportionation of *CO2 corresponds to the predominant kinetic barrier for the discharging process. The two catalysts exhibit different CO2 charging processes. Ru(0001) can suppress O–O coupling to form O2 while co-oxidizing carbon and Li2CO3 to generate CO2, thereby ending a complete battery cycle. By contrast, in the case of Ir(111), Li2CO3 decomposes and releases both CO2 and O2, and carbon can only be partially decomposed into small fragments.
Results and discussion
To explore rechargeable/reversible mechanisms of Li–CO2 batteries, the Ru(0001) and Ir(111) surfaces were constructed (Fig. 1a). We first investigated the discharge process with a focus on the CO2 activation (forming *CO2) and subsequent C–C coupling that are crucial steps in the Li–CO2 electrochemistry. Note that the interactions between reaction intermediates and solvent molecules can be strong, thereby affecting the discharge pathways; for example, prior experiments have indicated that the addition of DMSO can induce the generation of oxalate intermediates.32,33 However, the solvent effect is often neglected or oversimplified in Li–CO2 electrochemistry modelling. Hence, an explicit description of the solvent effect was conducted, where DMSO was chosen as a demonstration.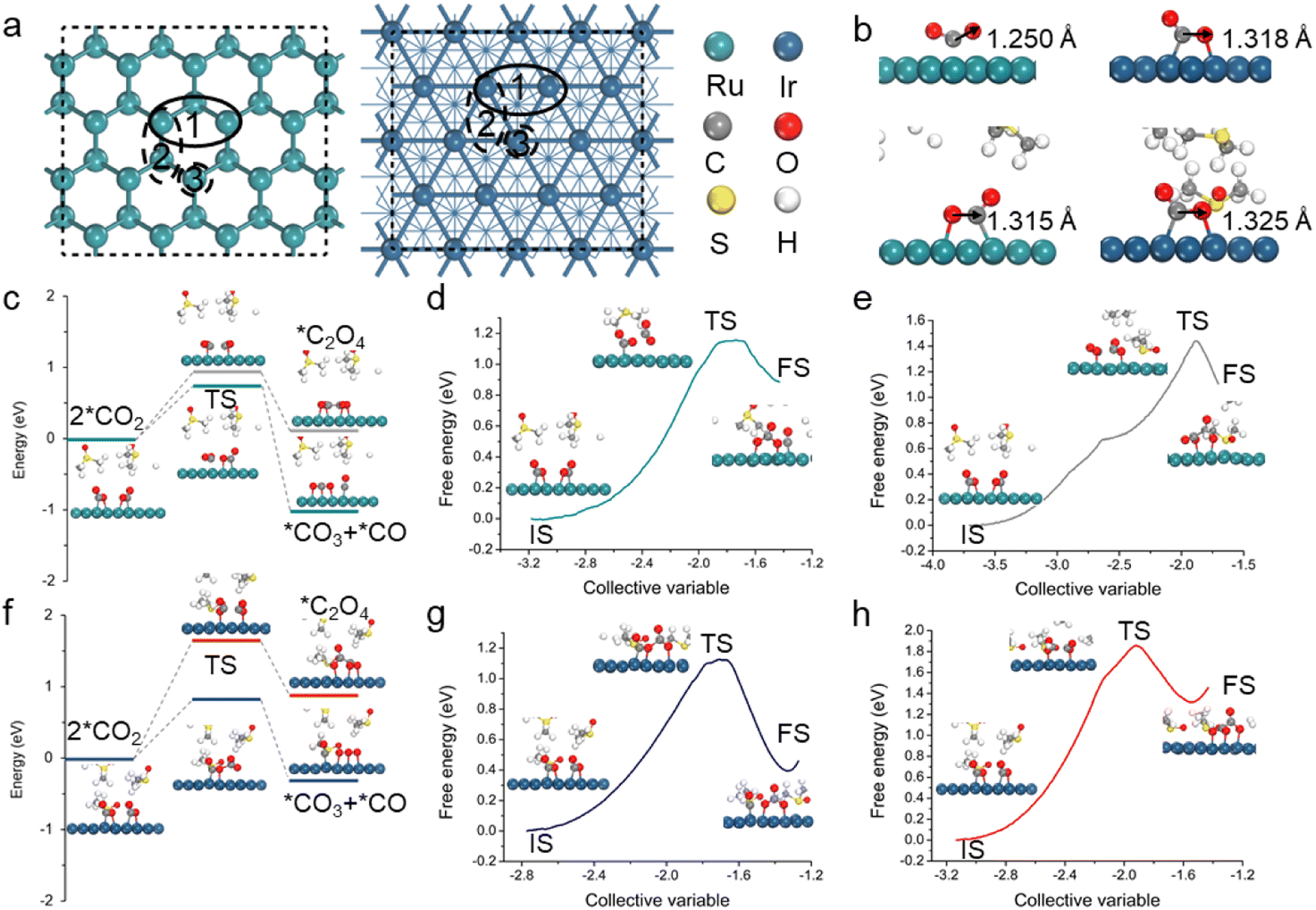 | ||
| Fig. 1 (a) Structures and possible adsorption sites of the Ru(0001) (left) and Ir(111) (right) surfaces. (b) The most stable CO2 adsorption configurations of Ru(0001) and Ir(111) in a vacuum (upper) and DMSO solvent (bottom). C, O, S, H, Ru, and Ir are represented by gray, red, yellow, white, green, and dark cyan spheres, respectively. (c and f) Energy barriers of C–C coupling on (c) Ru(0001) and (f) Ir(111), in which the DESW and CBD methods are employed. The insets show geometric structures of the initial state (IS), transition state (TS), and final state (FS). (d and e) Free energy profiles of the (d) CO2 disproportionation and (e) dimerization pathways on Ru(0001). (g and h) Free energy profiles of the (g) CO2 disproportionation and (h) dimerization pathways on Ir(111). | ||
Different CO2 adsorption sites on the two surfaces were investigated. Fig. 1b shows the lowest-energy structure of *CO2 in which the CO2 molecule is chemically adsorbed onto Ru(0001) and Ir(111) with a dual-site adsorption configuration. The adsorption free energies are −0.35 eV for Ru(0001) and −0.30 eV for Ir(111). It is worth mentioning that the solvent effect has resulted in an extension of the C–O bond length with respect to both the Ru(0001) and Ir(111) surfaces, with an increase from 1.250 Å to 1.315 Å for the former and 1.318 Å to 1.325 Å for the latter, indicating an overall stronger CO2 adsorption in both cases (Fig. 1b). The dual-site adsorption presents a longer C–O bond length, which may facilitate bond breaking in the subsequent disproportionation reaction. Please also note that in the case of Ru, the configuration of *CO2 changes from single-site adsorption (forming a Ru–C bond) to dual-site adsorption (forming Ru–O and Ru–C bonds) after considering the solvent effect. These results highlight the essential role of electrolytes in the activation of CO2 and demonstrate the necessity of describing the explicit solvent effect in Li–CO2 electrochemistry modelling.
The activated *CO2 species can undergo direct dimerization to form oxalate or carbonate intermediates, and they are also available to be coupled with the lithiated intermediates, including *LiCO2 and *Li2CO2. After adsorbing the first CO2 molecule, the second CO2 molecule is also activated; the adsorption energies are −0.31 and −0.20 eV for Ru(0001) and Ir(111), respectively. To find the most likely reaction path, we performed a kinetic analysis using the double-ended surface walking (DESW) and constrained Broyden dimer (CBD) approaches.34,35 Note that the methods can determine a low-energy pathway linking two minima even without iterative optimization of the path, from which the transition state (TS) can be located readily. As shown in Fig. S1 and S2,† *CO2 is more favourable to disproportionate into *CO3 and *CO for both Ru(0001) and Ir(111). However, the coupling of *Li2CO2 and *CO2 on Ru(0001) tends to generate oxalates, while in the case of Ir(111), the formation of carbonates is still predominant. As mentioned above, polar electrolytes have a significant impact on the formation of discharge intermediates. Therefore, ab initio molecular dynamics (AIMD) simulations were performed to obtain explicit DMSO structures for the Ru(0001) and Ir(111) surfaces (Fig. S3†),36 and both reaction pathways under solvent conditions were further analysed.
The kinetic results involving the solvation effect shown in Fig. 1c and f indicate that the disproportionation pathway (2*CO2 → *CO3 + *CO) is more energetically favourable than the dimerization pathway (2*CO2 → *C2O4). For the disproportionation pathway, the energy barriers of Ru(0001) and Ir(111) are 0.76 and 0.83 eV, respectively, which are lower than those of the dimerization pathway (0.96 and 1.65 eV). In addition, to further catch the solvent network, the slow-growth method was also employed to sample the energy change of the aforementioned optimal reaction pathway; multiple independent slow-growth samplings for each step were conducted (Fig. 1d, e, g, h and S4†). The results prove that *CO2 on the catalysts has a higher propensity towards undergoing disproportionation to form *CO3 and *CO, which is consistent with the above kinetic results (Fig. 1c and f). Note that we also conducted slow-growth simulations up to 50 ps and found similar results. This is not surprising as highly polar electrolytes do not directly participate in the Li–CO2 reactions, which is different from typical aqueous electrochemical reactions (e.g., the hydrogen evolution reaction and the reduction of CO2 and H2O to CH3OH). On a different note, the competing coupling of *Li2CO2 and *CO2 to form *Li2C2O4 in DMSO also exhibits a high energy barrier on the Ru(0001) surface (Fig. S5†). Therefore, the disproportionation of *CO2 to form *CO3 and *CO is the dominant pathway for Ru(0001) and Ir(111).
The Li2CO3 and carbon formation processes were subsequently investigated using the constant charge method (Fig. 2).2,12,24 The relative free energy changes (ΔG) are summarized in Table S1.† Please also note that the modelling of the Li–CO2 reaction still faces many challenges, such as accurately describing the formation process of Li2CO3; currently, one common strategy is to simplify it to a Li2CO3 moiety, which successfully explains the reaction processes and operation potentials.2,11,12,22,24,25 This strategy was adopted in this work. It was found that after the disproportionation of *CO2, *CO3 can convert to *Li2CO3via two-step lithiation reactions, which are both thermodynamically spontaneous for the Ru(0001) and Ir(111) surfaces (Fig. 2). Subsequently, *Li2CO3 desorbs to produce the first Li2CO3 species, whereas the previously generated *CO remains on the catalyst surface.
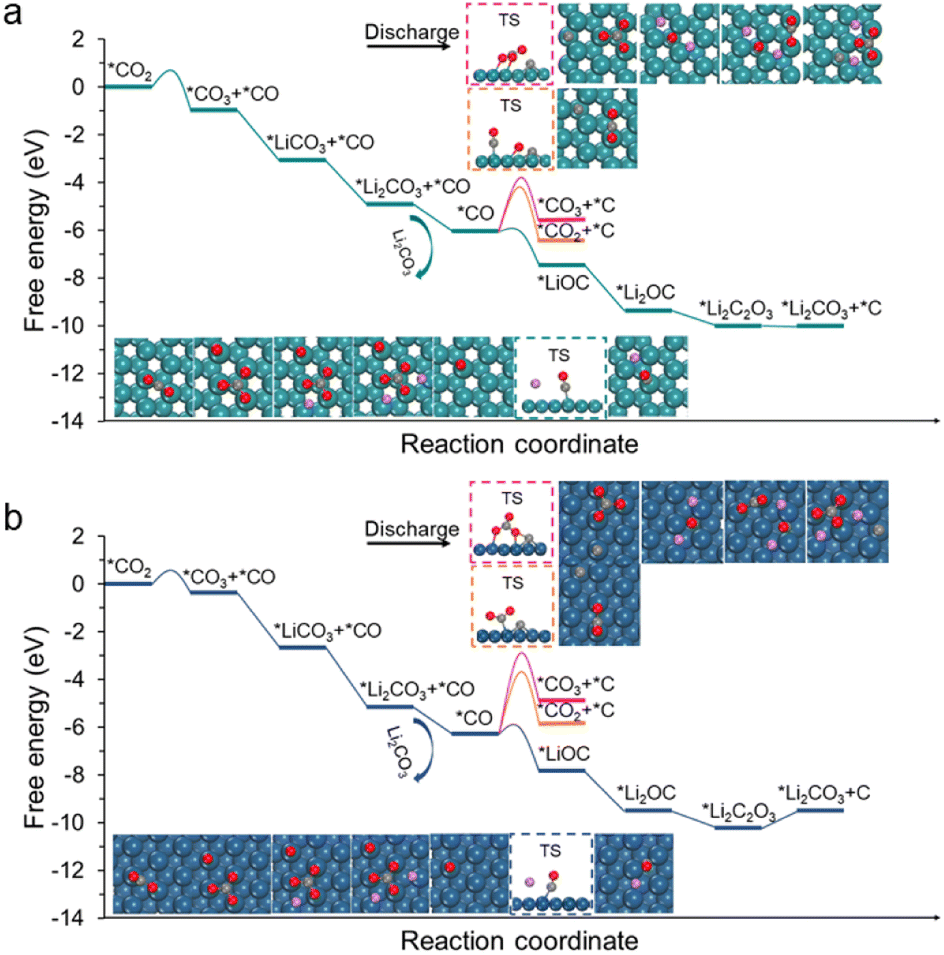 | ||
| Fig. 2 Free energy diagrams of CO2 reduction (discharging process) on the (a) Ru(0001) and (b) Ir(111) surfaces. Insets display geometric structures of free slabs, adsorbates, and key transition states that are highlighted by dashed boxes. The structures of transition states are shown in Fig. S6 and S7.† | ||
The remaining question is whether CO undergoes disproportionation or participates in subsequent lithiation reactions. According to our calculations, *CO lithiation to form *LiOC is an exothermic reaction, with respective ΔG values of −1.43 eV for Ru(0001) and −1.54 eV for Ir(111); in accordance with our expectations, the kinetic barriers of such a lithiation step are small (less than 0.3 eV). By contrast, the disproportionation of *CO to form *C and *CO2 is considerably endothermic, and the kinetic barriers are as high as 1.92 and 2.98 eV for Ru(0001) and Ir(111), respectively. We also calculated the coupling of *CO and *CO2 to form *CO3 and *C but found that the relative kinetic barriers are very high (2.31 and 3.28 eV). These results suggest that the two reactions are difficult to occur. Moreover, the lithiation of *LiOC to *Li2OC is exothermic, and the as-formed *Li2OC can react with CO2 to form *Li2C2O3 and then decompose into *Li2CO3 and *C species. Such a decomposition step is slightly endothermic by 0.07 eV for Ru(0001) and 0.66 eV for Ir(111).
The theoretical limiting potential (UL) is an important indicator to bridge the experimental activity, such as discharge potential. Based on the free-energy diagrams in Fig. 2, UL was identified to be 1.43 V for Ru(0001) and 1.54 V for Ir(111). To better simulate electrochemical environments, we performed constant potential modelling, which can describe the effects of the surface charge of catalysts on electrochemical reactions.37–40 The total energy of each reaction intermediate as a function of potential is fitted (Figs. S8 and S9†), and Fig. 3a and b show the fitting results for *LiCO3 and *CO intermediates on Ru(0001) and Ir(111), respectively. We found that the UL values of Ru(0001) and Ir(111) obtained based on the constant potential method are close to the above constant charge results (Fig. 3c and d), and these values are in agreement with experimental observations.41 These results demonstrate the rationality of our proposed discharge mechanism. Considering the computational cost, the constant charge method was used in subsequent calculations.
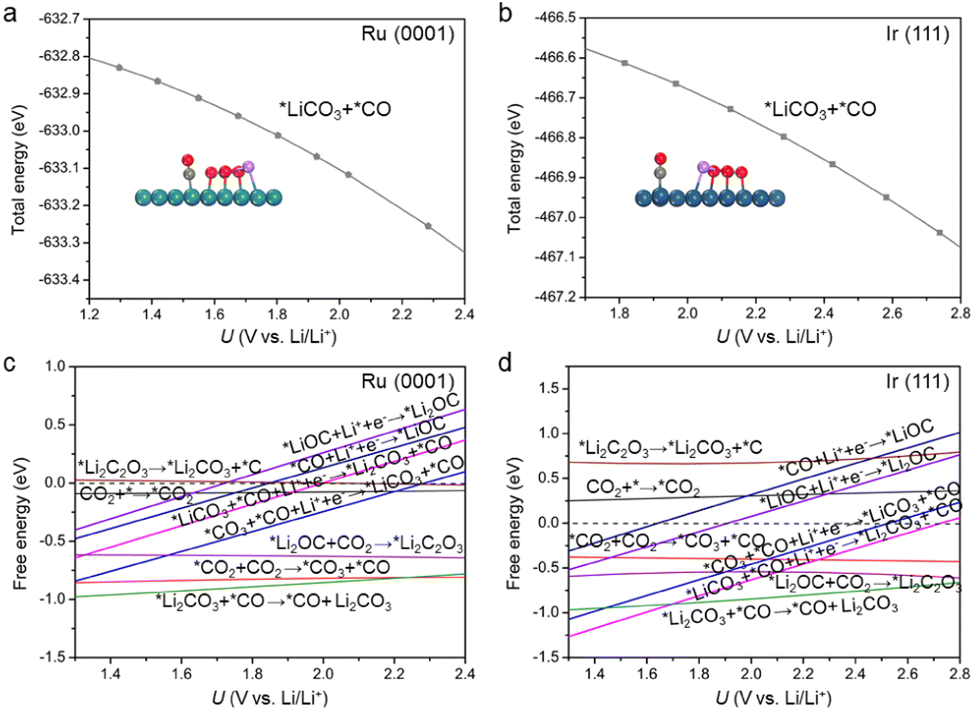 | ||
| Fig. 3 (a and b) Total energy of the absorbed *LiCO3 and *CO species of (a) Ru(0001) and (b) Ir(111) as a function of potential. (c and d) Free energy changes of discharge elementary reactions of (c) Ru(0001) and (d) Ir(111) based on the constant potential method. | ||
Compared with the discharge reaction, the charging mechanism is relatively complex and ambiguous. According to whether the discharge products Li2CO3 and carbon can be completely decomposed, the charging process can be divided into reversible and irreversible reactions.3,25,27–29 Please note that the reversibility can be influenced by many aspects, such as discharge product-caused passivation of catalyst surfaces, instability of electrolytes, and the incomplete decomposition of discharge products that directly determines the reversibility of Li–CO2 reactions. In this modelling work, we focused on the decomposition of discharge products during the charging process.
It was previously reported that both chemical and electrochemical pathways may exist for the decomposition of Li2CO3.42,43 For most reported catalysts, only the decomposition of Li2CO3 was observed during the charging process while carbon was partially decomposed.3,12–15 Meanwhile, for exceptional catalysts such as Ru, no satisfactory explanation of the underlying mechanism of the oxidation of Li2CO3 and carbon has been provided. In this regard, we proposed two possible mechanisms and performed corresponding calculations, as discussed later.
The discharge product carbon is a complex system; in principle, its decomposition is the gradual transformation of large fragments into small fragments. According to our calculations, the decomposition of carbon on Ru(0001) is much more energetically favourable than that on Ir(111), and as the number of C atoms in the carbon fragments decreases, the energy barrier for removing C atoms typically increases. For example, we investigated the kinetics of removing a C atom from C13 and C6 clusters (Fig. S10†). The results show that the decomposition barrier of the C13 cluster is 0.38 and 0.94 eV for Ru(0001) and Ir(111), respectively. In the case of the C6 cluster, the barriers are significantly higher, being 1.45 eV for Ru(0001) and 1.98 eV for Ir(111). Note that after the carbon six-membered ring is broken, the subsequent barrier is moderate; for instance, the barrier of removing a C atom from a C5 cluster is 1.05 eV for Ru(0001) and 1.21 eV for Ir(111).
However, the decomposition of carbon and Li2CO3 might not be simply oxidized separately but is a co-oxidation process.27 Thus, we further include the contributions from Li2CO3 in carbon decomposition. Herein, the C6 cluster is adopted as the representative model because its decomposition exhibits the largest energy barrier compared with other carbon fragments (Fig. S10†). For the charging process, *Li2CO3 first loses two Li and then generates *CO3via two delithiation steps (Fig. 4a and b), and the kinetic barriers are 0.67 and 0.83 eV for Ru(0001) and Ir(111), respectively (Fig. S11†). The relative free energy changes are summarized in Tables S2 and S3.† These steps are relatively easy to occur on both Ru(0001) and Ir(111) by applying moderate charging voltage. The further decomposition of *CO3 to *CO2 and *O is also energetically favourable, displaying a moderate barrier of 0.51 eV and 0.50 eV on Ru(0001) and Ir(111), respectively.
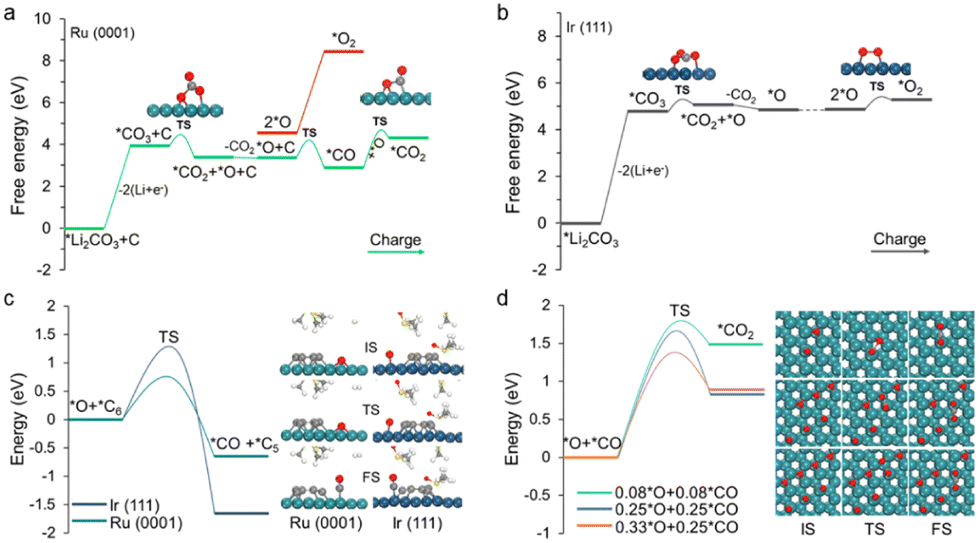 | ||
| Fig. 4 (a and b) Calculated energetic profiles of the charging process of (a) Ru(0001) and (b) Ir(111). (c) Energy barriers for the oxidative decomposition of a C6 cluster to form *C5 and *CO on Ru(0001) and Ir(111). (d) Energy barriers for *CO oxidation under different *CO and *O coverages. | ||
The remaining question is how *O is oxidized. Two possible reaction pathways for *O oxidation were studied. In the first pathway, *O species couple with one another to generate O2, corresponding to the irreversible charge; in the second pathway, *O attacks carbon to form carbon oxides, which enables a complete battery cycle. It was found that the as-formed *O species can facilitate the carbon decomposition on both the Ru(0001) and Ir(111) surfaces (Fig. S12†). Meanwhile, in accordance with our expectations, the reaction of *O with the C6 cluster to form *CO and *C5 on Ru(0001) exhibits a moderate energy barrier (0.86 eV), whereas in the case of Ir(111), the value is as high as 1.60 eV (Fig. S12†). To further investigate the oxidative decomposition of carbon, the reaction energy barriers with respect to the DMSO solvent were also computed (Fig. 4c). A slight decline in the respective energy barriers was observed, validating the feasibility (infeasibility) of oxidative decomposition of carbon on Ru (Ir).
Finally, the adsorbed CO species over Ru(0001) can react with another *O to form *CO2. Although the energy barrier of the *CO oxidation reaction is slightly high, it becomes more favourable as the coverage of *O and *CO increases (Fig. 4d). This is also consistent with previous reports that CO oxidation on Ru will be more active under high gas pressure.44,45 On a different note, the dimerization of *O to form O2 on Ru(0001) is virtually impossible on account of the massive changes in a free energy of 3.90 eV (Fig. 4a). In the case of Ir(111), the *O intermediate preferentially couples with another *O species to generate O2, which is attributed to a moderate kinetic barrier (0.53 eV). Note that high O coverage reduces the energy barrier of O–O coupling on Ir(111), and while a similar situation occurs in Ru(0001), the energy barrier is still very high (Fig. S13†). Overall, the above modelling results demonstrate that Li2CO3 and carbon can be co-oxidized on Ru to conclude a complete battery cycle, while Ir(111) can only partially decompose carbon into small fragments together with oxidizing Li2CO3 into CO2 and O2. Therefore, our observations are in good agreement with the aforementioned experimental phenomena.3,28
To gain a fundamental understanding of the difference in the oxidation of carbon on Ru(0001) and Ir(111), the surface charge distributions of *C adsorbed on the two metal surfaces were assessed through a Bader charge analysis. As shown in Fig. S14,† a considerable amount of electronic charge is transferred from both metal surfaces to the *C species. The electron transfer value of the Ru(0001) surface (1.39e) significantly exceeds that of the Ir(111) surface (1.02e), corresponding to a substantially stronger Coulomb interaction between C and Ru(0001). In addition, the continuous formation of the *O intermediate on the Ru(0001) surface results in the former being widely distributed on the latter, which also facilitates reactions between *O and *C to generate CO2.
Conclusions
In conclusion, using comprehensive first-principles calculations, the complete reversible and irreversible mechanism pathways of Li–CO2 batteries were disclosed for the first time. The modelling results highlighted that the electrolyte may play an essential role in the activation of CO2; both Ru(0001) and Ir(111) prefer to undergo the direct disproportionation of *CO2 to *CO3 and *CO during the discharge process. The constant-potential calculations verified the rationality of our reaction mechanism. Moreover, we elucidated the reversible co-oxidation process of the discharge products carbon and Li2CO3 on Ru(0001) as well as the rationale for the partial decomposition of carbon and the generation of O2 on Ir(111). Our findings provide a solution to long-standing questions and impart ample guidance in the development of reversible Li–CO2 batteries.Methods
First-principles calculations
All first-principles calculations were performed using the Vienna ab initio simulation package (VASP) with the projector augmented wave method and Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation exchange-correlation functional.46–48 The cutoff energy was set to 400 eV after a series of tests, and the criteria were set as 10−5 eV for energy and 0.05 eV Å−1 for force. The Brillouin zone was sampled using a Monkhorst–Pack 3 × 3 × 1 k-point grid, and the Grimme-D3 approach was applied to describe the dispersion interactions.49 The reaction pathways and transition states were located using the double-ended surface walking (DESW) and constrained Broyden dimer (CBD) methods, as implemented in the LASP software.34,35 All transition states have been verified by using vibrational frequency calculations (only one imaginary frequency). Note that these methods can determine a low-energy pathway linking two minima even without iterative optimization of the path, from which the transition state (TS) can be located readily.Structure models
The lattice parameters of bulk Ru and Ir are optimized to be a = b = 2.719 Å, c = 4.297 Å, and a = b = c = 3.872 Å, respectively, which are consistent with experimental values. Two typical metal surfaces, i.e., Ru(0001) and Ir(111), were constructed using 2 × 4 × 1 supercells. A vacuum region of 15 Å thickness along the z direction was chosen to eliminate the interactions between neighbouring surfaces. Specifically, to better describe the reactions in the electrode interface, we considered the solvent effect and potential effect, which are often neglected or oversimplified in the Li–CO2 electrochemistry modelling. A typical electrolyte, DMSO, was adopted to describe the solvent effect on reaction pathways as a demonstration; five explicit DMSO molecules fill the vacuum gap to provide an experimental density of ρ = 1.1 g cm−3.36 Note here that other electrolytes (e.g., TEGDME) were not considered because they have negligible influence on reversibility. The top layer, the adsorbates, and all DMSO molecules are allowed to relax while the remaining atoms are fixed during the calculations. For the Ru(0001) and Ir(111) surfaces, we first performed 10 ps AIMD simulations to obtain explicit DMSO structures. Then, adsorbates were placed on the metal surfaces for further study of the CO2 electrochemical processes.Slow-growth simulations
The slow-growth method was used to sample the free-energy change during the disproportionation reaction.50,51 Nose–Hoover thermostat was adopted to keep temperature at 300 K.52 The corresponding increment speed of the CV was within the range between 0.0003 and 0.0004 CV per step, and the simulation time ranged from 5 to 10 ps depending on the length of the reaction pathways. We performed multiple independent slow-growth simulations for each step, and all results revealed that the disproportionation pathway (2*CO2 → *CO3 + *CO) is more energetically favourable than the dimerization pathway (2*CO2 → *C2O4). In addition, we also conducted slow-growth simulations up to 50 ps and found no impact on determining the dominant pathway.Free energy calculations
The free energy change (ΔG) for each elemental step was calculated using| ΔG = ΔE + ΔEZPE - TΔS | (1) |
| Eads = Etotal − Eslab − Eadsorbate | (2) |
Data availability
The computational data supporting the findings can be found in the article and ESI,† and are available from the authors upon reasonable request.Author contributions
Y. L. designed the research, X. Z. and Y. W. demonstrated the initial idea and collected all the data. X. Z., Y. W. and Y. L. wrote the paper and all authors commented on it.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We are grateful for funding support from the National Key R&D Program of China (2019YFA0308000), the Natural Science Foundation of China (No. 22173048 and 22203044), the Jiangsu Specially Appointed Professor Plan and the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- J. Zou, G. Liang, F. Zhang, S. Zhang, K. Davey and Z. Guo, Adv. Mater., 2023, 35, 2210671 CrossRef CAS PubMed.
- B. Chen, D. Wang, J. Tan, Y. Liu, M. Jiao, B. Liu, N. Zhao, X. Zou, G. Zhou and H.-M. Cheng, J. Am. Chem. Soc., 2022, 144, 3106–3116 CrossRef CAS PubMed.
- Y. Qiao, J. Yi, S. Wu, Y. Liu, S. Yang, P. He and H. Zhou, Joule, 2017, 1, 359–370 CrossRef CAS.
- A. Ahmadiparidari, R. E. Warburton, L. Majidi, M. Asadi, A. Chamaani, J. R. Jokisaari, S. Rastegar, Z. Hemmat, B. Sayahpour, R. S. Assary, B. Narayanan, P. Abbasi, P. C. Redfern, A. Ngo, M. Voros, J. Greeley, R. Klie, L. A. Curtiss and A. Salehi-Khojin, Adv. Mater., 2019, 31, 1902518 CrossRef CAS PubMed.
- Z. Zhang, Q. Zhang, Y. Chen, J. Bao, X. Zhou, Z. Xie, J. Wei and Z. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6550–6553 CrossRef CAS PubMed.
- X. Zhang, Q. Zhang, Z. Zhang, Y. Chen, Z. Xie, J. Wei and Z. Zhou, Chem. Commun., 2015, 51, 14636–14639 RSC.
- Y. Jiao, J. Qin, H. M. K. Sari, D. Li, X. Li and X. Sun, Energy Storage Mater., 2021, 34, 148–170 CrossRef.
- L. Qie, Y. Lin, J. W. Connell, J. Xu and L. Dai, Angew. Chem., Int. Ed., 2017, 56, 6970–6974 CrossRef CAS PubMed.
- P. D. James Speight, Lange's Handbook of Chemistry, Mcgraw-Hill Education, New York, 16th edn, 2005 Search PubMed.
- X. Sun, X. Mu, W. Zheng, L. Wang, S. Yang, C. Sheng, H. Pang, W. Li, C.-H. Li, P. He and H. Zhou, Nat. Commun., 2023, 14, 536 CrossRef CAS PubMed.
- C. Guo, F. Zhang, X. Han, L. Zhang, Q. Hou, L. Gong, J. Wang, Z. Xia, J. Hao and K. Xie, Adv. Mater., 2023, 35, 2302325 CrossRef CAS PubMed.
- Z. Zhao, L. Pang, Y. Su, T. Liu, G. Wang, C. Liu, J. Wang and Z. Peng, ACS Energy Lett., 2022, 7, 624–631 CrossRef CAS.
- X. Mu, H. Pan, P. He and H. Zhou, Adv. Mater., 2020, 1903790 CrossRef CAS PubMed.
- B. Liu, Y. Sun, L. Liu, J. Chen, B. Yang, S. Xu and X. Yan, Energy Environ. Sci., 2019, 12, 887–922 RSC.
- Z. Zhang, W.-L. Bai, K.-X. Wang and J.-S. Chen, Energy Environ. Sci., 2020, 13, 4717–4737 RSC.
- S. Yang, Y. Qiao, P. He, Y. Liu, Z. Cheng, J.-J. Zhu and H. Zhou, Energy Environ. Sci., 2017, 10, 972–978 RSC.
- C. Ling, Y. Ouyang, L. Shi, S. Yuan, Q. Chen and J. Wang, ACS Catal., 2017, 7, 5097 CrossRef CAS.
- Y. Wang, T. Liu and Y. Li, Chem. Sci., 2022, 13, 6366–6372 RSC.
- J. Zhou, T. Wang, L. Chend, L. Liao, Y. Wang, S. Xie, B. Chen, T. Lin, Q. Zhang, C. Ye, X. Zhou, Z. Guan, L. Zhai, Z. He, G. Wang, J. Wang, J. Yua, Y. Ma, P. Lu, Y. Xiong, S. Lu, Y. Chen, B. Wang, C.-S. Leea, J. Cheng, L. Gu, T. Zhao and Z. Fan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204666119 CrossRef CAS PubMed.
- J. Li, L. Wang, Y. Zhao, S. Li, X. Fu, B. Wang and H. Peng, Adv. Funct. Mater., 2020, 30, 2001619 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng, A. Vasileff and S. Z. Qiao, Nano Energy, 2020, 71, 104601 CrossRef CAS.
- Y. Liu, S. Zhao, D. Wang, B. Chen, Z. Zhang, J. Sheng, X. Zhong, X. Zou, S. P. Jiang, G. Zhou and H.-M. Cheng, ACS Nano, 2022, 16, 1523–1532 CrossRef CAS PubMed.
- D. Guan, X. Wang, M. Li, F. Li, L. Zheng, X. Huang and J. Xu, Angew. Chem., Int. Ed., 2020, 59, 19518–19524 CrossRef CAS PubMed.
- C. Yang, K. Guo, D. Yuan, J. Cheng and B. Wang, J. Am. Chem. Soc., 2020, 142, 6983–6990 CrossRef CAS PubMed.
- J. Hu, C. Yang and K. Guo, J. Mater. Chem. A, 2022, 10, 14028–14040 RSC.
- L. Fan, H. Shen, D. Ji, Y. Xing, L. Tao, Q. Sun and S. Guo, Adv. Mater., 2022, 34, 2204134 CrossRef CAS PubMed.
- D. Cao, C. Tan and Y. Chen, Nat. Commun., 2022, 13, 4908 CrossRef CAS PubMed.
- C. Wang, Q. Zhang, X. Zhang, X. Wang, Z. Xie and Z. Zhou, Small, 2018, 14, 1800641 CrossRef PubMed.
- Y. Xing, Y. Yang, D. Li, M. Luo, N. Chen, Y. Ye, J. Qian, L. Li, D. Yang, F. Wu, R. Chen and S. Guo, Adv. Mater., 2018, 30, 1803124 CrossRef PubMed.
- C. Tan, A. Wang, D. Cao, F. Yu, Y. Wu, X. He and Y. Chen, Adv. Energy Mater., 2023, 13, 2204191 CrossRef CAS.
- N. Mahne, S. E. Renfrew, B. D. Mccloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529 CrossRef CAS PubMed.
- Y.-F. Wang, G.-J. Ji, L.-N. Song, X.-X. Wang and J.-J. Xu, ACS Energy Lett., 2023, 8, 1026–1034 CrossRef CAS.
- Z. Zhao, E. Wang, J. Wang, C. Liu and Z. Peng, J. Mater. Chem. A, 2021, 9, 3290–3296 RSC.
- C. Shang and Z.-P. Liu, J. Chem. Theory Comput., 2010, 6, 1136–1144 CrossRef CAS.
- X. Zhang, C. Shang and Z.-P. Liu, J. Chem. Theory Comput., 2013, 9, 5745–5753 CrossRef CAS PubMed.
- Z. Jiang and A. M. Rappe, J. Am. Chem. Soc., 2022, 144, 22150–22158 CrossRef CAS PubMed.
- J.-S. Filhol and M. Neurock, Angew. Chem., Int. Ed., 2006, 45, 402–406 CrossRef CAS PubMed.
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef CAS PubMed.
- D. Kim, J. Shi and Y. Liu, J. Am. Chem. Soc., 2018, 140, 9127–9131 CrossRef CAS PubMed.
- Y. Wang, L. You and K. Zhou, Chem. Sci., 2021, 12, 14065–14073 RSC.
- X. Xiao, Z. Zhang and P. Tan, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217454120 CrossRef CAS PubMed.
- L. A. Kaufman and B. D. McCloskey, Chem. Mater., 2021, 33, 4170–4176 CrossRef CAS.
- A. T. S. Freiberg, J. Sicklinger, S. Solchenbach and H. A. Gasteiger, Electrochim. Acta, 2020, 346, 136271 CrossRef CAS.
- A. Tetenoire, J. I. Juaristi and M. Alducin, J. Phys. Chem. C, 2021, 125, 12614–12627 CrossRef CAS.
- C. H. F. Peden, D. W. Goodman, M. D. Weisel and F. M. Hoffmann, Surf. Sci., 1991, 253, 44–58 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. P. Straatsma, H. Berendsen and J. Postma, J. Chem. Phys., 1986, 85, 6720–6727 CrossRef CAS.
- E. A. Carter, G. Ciccotti, J. T. Hynes and R. Kapral, Chem. Phys. Lett., 1989, 156, 472–477 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00383g |
| This journal is © The Royal Society of Chemistry 2024 |