
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrustrated Lewis pairs on pentacoordinated Al3+-enriched Al2O3 promote heterolytic hydrogen activation and hydrogenation†
Qingyuan
Wu‡
ab,
Ruixuan
Qin‡
 ac,
Mengsi
Zhu
b,
Hui
Shen
a,
Shenshui
Yu
a,
Yuanyuan
Zhong
a,
Gang
Fu
a,
Xiaodong
Yi
*a and
Nanfeng
Zheng
*ab
ac,
Mengsi
Zhu
b,
Hui
Shen
a,
Shenshui
Yu
a,
Yuanyuan
Zhong
a,
Gang
Fu
a,
Xiaodong
Yi
*a and
Nanfeng
Zheng
*ab
aNew Cornerstone Science Laboratory, State Key Laboratory for Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, National & Local Joint Engineering Research Center of Preparation Technology of Nanomaterials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: nfzheng@xmu.edu.cn; xdyi@xmu.edu.cn
bInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361102, China
cFujian Key Laboratory of Rare-Earth Functional Materials, Fujian Shanhai Collaborative Innovation Center of Rare-Earth Functional Materials, Longyan 366300, China
First published on 16th January 2024
Abstract
As an emerging class of metal-free catalysts, frustrated Lewis pairs (FLPs) catalysts have been greatly constructed and applied in many fields. Homogeneous FLPs have witnessed significant development, while limited heterogeneous FLPs catalysts are available. Herein, we report that heterogeneous FLPs on pentacoordinated Al3+-enriched Al2O3 readily promote the heterolytic activation of H2 and thus hydrogenation catalysis. The defect-rich Al2O3 was prepared by simple calcination of a carboxylate-containing Al precursor. Combinatorial studies confirmed the presence of rich FLPs on the surface of the defective Al2O3. In contrast to conventional alumina (γ-Al2O3), the FLP-containing Al2O3 can activate H2 in the absence of any transition metal species. More importantly, H2 was activated by surface FLPs in a heterolytic pathway, leading to the hydrogenation of styrene in a stepwise process. This work paves the way for the exploration of more underlying heterogeneous FLPs catalysts and further understanding of accurate active sites and catalytic mechanisms of heterogeneous FLPs at the molecular level.
1 Introduction
In catalytic hydrogenation of both chemical transformations and biological functions, most of the hydrogen (H2) activation processes occurred at the transition metal center.1–7 The exploration of metal-free systems for H2 activation is quite appealing while rather challenging.8 Until 2006, Stephan and co-workers first reported one such unique system, the metal-free zwitterionic phosphoniumborate, which could reversibly activate H2 under mild conditions. The proposed activation center of hydrogen was frustrated Lewis acid–base pairs (FLPs), which were defined as Lewis acids and bases sterically prevented from interaction to form Lewis acid–base adjuncts.9 Since then, the traditional paradigm of Lewis acid–base chemistry was changed and development of new FLPs for promoting chemical reactivity was heatedly pursued.10–13 During the past few decades, a large number of homogeneous FLPs catalysts have been synthesized, which have found wide application in the fields of hydrogenation catalysis, small-molecule activation, organic chemistry, radical chemistry, transition metal chemistry, and enzyme models, as well as polymers and materials.14–16Despite the significant progress, the studies on heterogeneous (semi-solid and solid) FLPs catalysts were rather limited.17–30 The prominent examples included semi-solid (two-phase) FLPs catalysts of imine or nitrile modified gold nanoparticles,18 as well as solid FLPs catalysts that covered atypical oxides such as In2O3−x(OH)y and CeO2−x.22,31 The abundant homogeneous while rare heterogeneous FLP catalysts indicated the difficulty in the design and preparation of heterogeneous FLP catalysts, which was attributed to the poor understanding of the structure–property relationship of solid FLPs. As such, the development of simple and practical synthetic strategies, detailed characterization of active sites, and insight into the reaction mechanism of heterogeneous FLPs are of high importance.
Among oxide supports used in industry, alumina is generally considered to be inert to H2 and thus make no contribution to the hydrogenation process by supported metal catalysts. Close examination of the literature revealed that, however, alumina after dehydration treatment at 450–650 °C was able to hydrogenate ethylene, in which the active sites were believed to chemisorb and polymerize olefins on alumina.32,33 Nevertheless, it remained elusive for the true active sites and the mechanism of the hydrogenation process.
Herein, we report the simple synthesis of a new type of pentacoordinated Al3+-enriched Al2O3, in which a large number of Lewis acidic and basic sites are co-present on the surface. The rigidity of the solid lattice prevents them from interacting to be acid–base adducts, thus giving access to heterogeneous FLPs catalysts. Interestingly, the as-obtained Al2O3 catalyst can activate hydrogen in the heterolytic pathway to produce equal amounts of O–Hδ+ and Al–Hδ− in the absence of transition metals. The heterogeneous FLPs exhibited nice catalytic activity toward styrene hydrogenation in a stepwise mechanism.
2 Results and discussion
2.1 Preparation and characterization of the pentacoordinated Al3+-enriched Al2O3 catalyst
The pentacoordinated Al3+-enriched defective Al2O3 (denoted as d-Al2O3) catalyst was prepared by calcining aluminum hydroxide acetate, [Al(OH)(CH3COO)2], at 500 °C in an inert atmosphere (N2 or Ar). The catalyst was first characterized by solid state 27Al magic-angle spinning nuclear magnetic resonance (27Al MAS-NMR) spectroscopy (Fig. 1a).34 Three peaks at chemical shifts of ∼64, 33 and 6 ppm were assigned to Al3+ in tetrahedral (Alt), pentahedral (Alp) and octahedral (Alo) coordination environments, respectively. Unexpectedly, a large proportion of Alp was observed in the as-prepared d-Al2O3,35 while only Alt and Alo were present in the lattice of commercial γ-Al2O3 (Fig. 1a).36 As surface atoms play a key role in both substrate interaction and chemical transformation, we then compared the relative content of surface Alp by using gaseous Lewis base molecules of NH3.37–40 As shown in Fig. 1b, the coordination of NH3 to surface Alp led to a decrease in the intensity of the peak at ∼33 ppm and correspondingly a distinct increase of the peak at ∼6 ppm, suggesting the conversion of Alp to Alo after the treatment and thus the presence of rich Lewis acidic sites (Alp) on the surface of d-Al2O3.36 The content of Alp decreased from 39.1% in d-Al2O3 to 23.8% in NH3-d-Al2O3 (Fig. S1 and Table S1†). Thus, the portion of surface Alp was estimated to be approximately 15.3% of the total Al3+ sites. The presence of surface Alp on d-Al2O3 was also confirmed by in situ Fourier transform infrared spectroscopy of adsorbed NH3 (in situ NH3-FTIR) and temperature-programmed desorption coupled with mass spectrometry using NH3 (NH3-TPD-MS) (Fig. S2†). We also revealed that the d-Al2O3 surface featured richer and stronger Lewis basic sites than that of γ-Al2O3 by in situ CO2-FTIR and CO2-TPD-MS (Fig. S3†), although the specific surface area of γ-Al2O3 (176 m2 g−1) was higher than that of d-Al2O3 (78 m2 g−1) based on N2 adsorption/desorption isotherms (Fig. S4†). With the Alp sites serving as the Lewis acid sites and the surface oxygen species acting as the Lewis base sites, the rigid structure of d-Al2O3 prevented them from interacting to form acid–base adducts, so obtained d-Al2O3 was rich in FLPs. In addition, X-ray powder diffraction (XRD) characterization revealed that in comparison to well-crystallized γ-Al2O3, d-Al2O3 was much less crystallized, indicating that Al3+ ions in the alumina lattice of d-Al2O3 were highly disordered and in diverse coordination modes (Fig. S5†).35,41 Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and fast Fourier transform (FFT) measurement also confirmed the amorphous characteristic of d-Al2O3 (Fig. S6†). These results match well with those of the solid state 27Al MAS-NMR.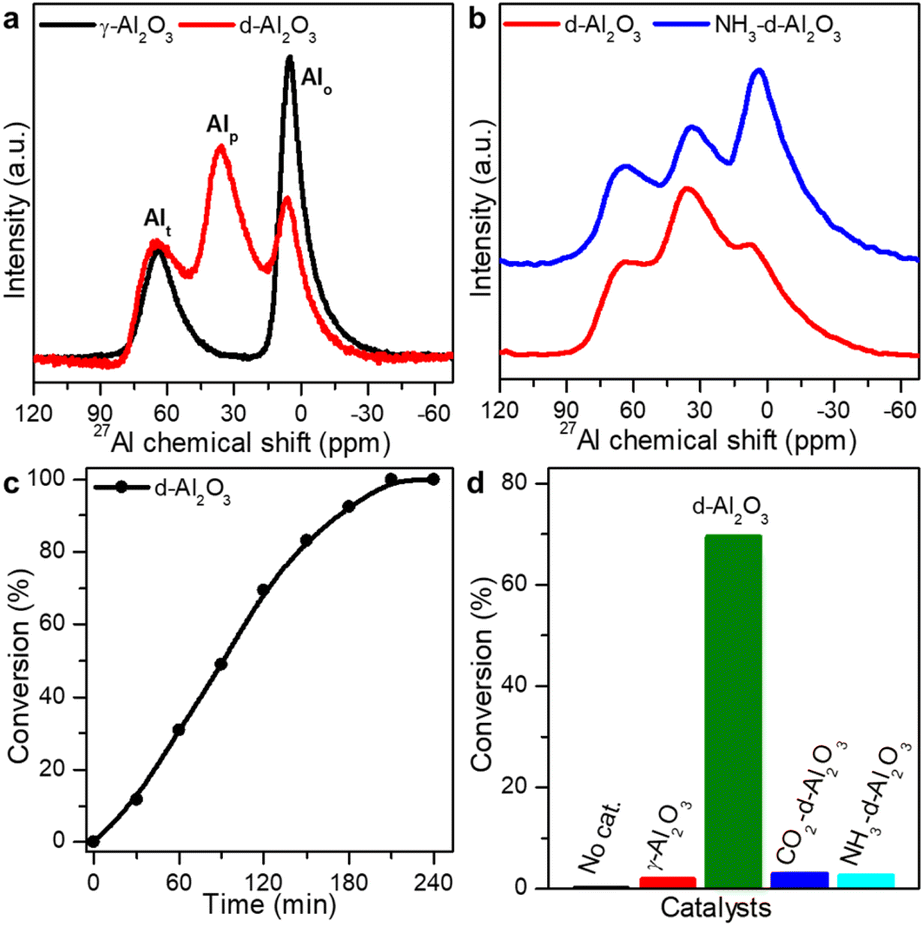 | ||
| Fig. 1 (a) Solid state 27Al MAS-NMR spectra of γ-Al2O3 and d-Al2O3. (b) Solid state 27Al MAS-NMR spectra of d-Al2O3 and NH3 treated d-Al2O3. (c) Catalytic hydrogenation of styrene by d-Al2O3. (d) Catalytic performance comparison of no catalyst, γ-Al2O3, d-Al2O3 and d-Al2O3-treated with gaseous Lewis acid or base molecules of CO2 and NH3, respectively. Reaction time: 120 min. | ||
2.2 FLPs as active sites for the catalytic hydrogenation of styrene
With the presence of a large amount of FLPs on the as-obtained d-Al2O3, we then employed the hydrogenation of styrene as a model reaction to demonstrate the surface reactivity of d-Al2O3. 50 mg catalyst of d-Al2O3 and 20 μL styrene were added to 5 mL anhydrous toluene within a well-stirred autoclave, which was then charged with H2 to 1 MPa and kept at 100 °C. As shown in Fig. 1c, d-Al2O3 exhibited nice reactivity toward hydrogenation, as ∼100% styrene conversion was achieved within 4 h, and the complete conversion was confirmed using 1H NMR spectra (Fig. S7†). Moreover, recycling tests revealed that the d-Al2O3 catalyst did not deactivate after 8 cycles, suggesting high robustness of FLP structures on the d-Al2O3 surface (Fig. S8†). We noted that the presence of FLPs on d-Al2O3 was the key to achieving the catalytic activity. In the control experiment where no catalyst was used or γ-Al2O3 without surface FLPs was employed, no reactivity was observed (Fig. 1d). In addition, we also used a gaseous probe to validate the necessity of FLPs for the hydrogenation. When NH3 or CO2 was used to neutralize surface Alp or basic sites, the catalytic activity of the d-Al2O3 was significantly declined (Fig. 1d). Based on the above observations, we considered that FLPs were the real active centers for the catalytic hydrogenation of styrene.2.3 Heterolytic activation of H2 on FLPs on d-Al2O3
The unexpected hydrogenation activity motivated us to deeply understand the H2 activation and hydrogenation mechanism over the heterogeneous FLPs. The feature of activated hydrogen species on the solid product was first characterized directly by two-dimensional (2D) solid state heteronuclear correlation (HETCOR) NMR spectroscopy.42,43 Compared to H2-treated γ-Al2O3, we detected an additionally intense cross peak linking the Alp site and Hδ− products in H2-treated d-Al2O3 with a short CP contact time (Fig. 2a and S9†). This suggests that H species are adjacent to Alp sites.44 Additionally, we confirmed the existence of Alp-bonded hydrogen species (Alp–Hδ−) through in situ H2-FTIR spectroscopy. An obvious peak in the range of 1730–1970 cm−1 confirmed the presence of Alp–Hδ− in H2-treated d-Al2O3, and abundant O–Hδ+ species were observed on the surface of H2-treated d-Al2O3 (Fig. 2b). The above results convincingly revealed that H2 was activated at the FLP sites via the heterolytic activation pathway with the formation of Alp–Hδ− and O–Hδ+.45–48 Furthermore, considering the complicated chemical environment of hydrogen species in the sample,49,50 some bound to FLPs while others not, we employed 2H to probe the hydrogen species on the FLPs to exclude other influencing factors.51 As shown in Fig. 2c (red trace), the solid state 2H MAS-NMR spectra of D2-treated d-Al2O3 showed a peak at 4.2 ppm corresponding to the 2H NMR signal of D2O (Fig. S10†),52 which represented the activated hydrogen species on the FLPs. In contrast to d-Al2O3, no 2H signal was detected when γ-Al2O3 was treated with D2 under the same conditions (Fig. 3c, black trace). These results clearly indicate that d-Al2O3 with FLPs readily activates H2, while γ-Al2O3 without FLPs did not. Moreover, in situ D2-FTIR spectroscopy demonstrated that d-Al2O3 activated D2 rather than γ-Al2O3, as indicated by the distinct signal of the O–D bond during the treatment of d-Al2O3 with D2 (Fig. S11†). The activated hydrogen species (O–Hδ+ and Al–Hδ−) formed on the FLPs of d-Al2O3 reacted readily with styrene. As shown by solid state 2H MAS-NMR, the 2H signal of the deuterated species from the d-Al2O3 treated with D2 disappeared upon the introduction of styrene (Fig. 2c, blue trace). When the d-Al2O3 catalyst was treated with D2 again after the reaction with styrene, the 2H signal reappeared (Fig. 2c, green trace).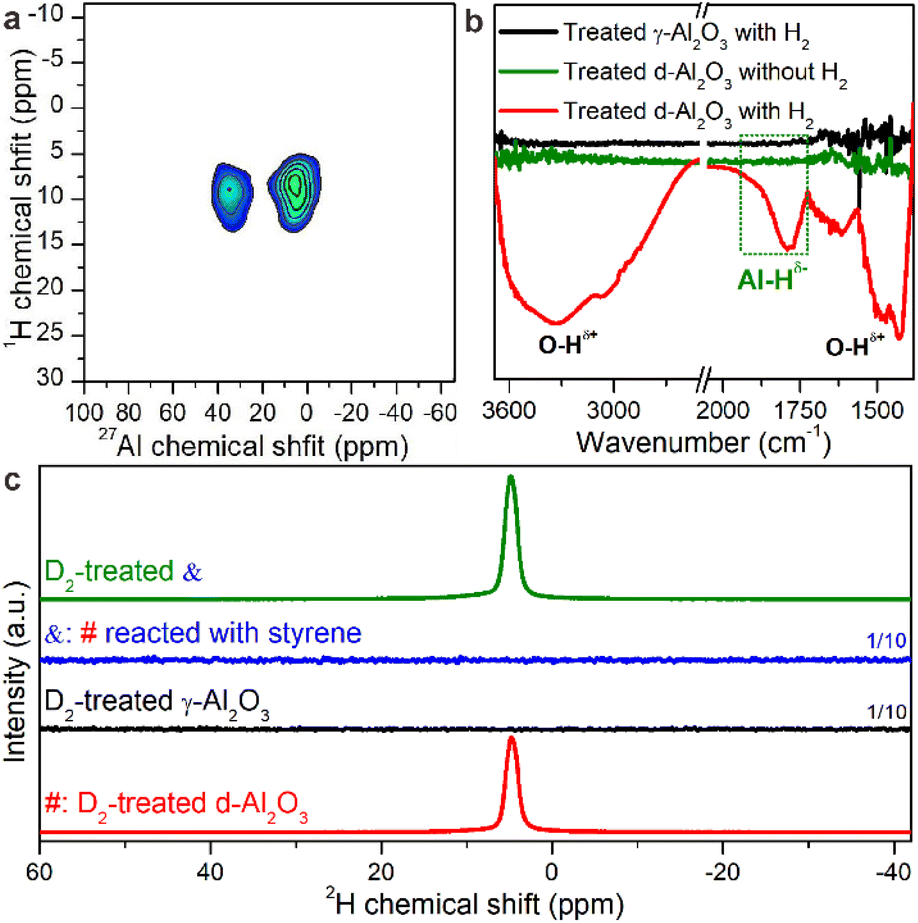 | ||
| Fig. 2 (a) 2D solid state 1H–27Al HETCOR spectrum of H2-treated d-Al2O3. (b) In situ H2-FTIR of γ-Al2O3, d-Al2O3 treated without H2 and d-Al2O3 treated with H2. (c) Solid state 2H MAS-NMR spectra of D2-treated d-Al2O3 (denoted as #), D2-treated γ-Al2O3, # reacted with styrene (denoted as &), and D2-treated &. | ||
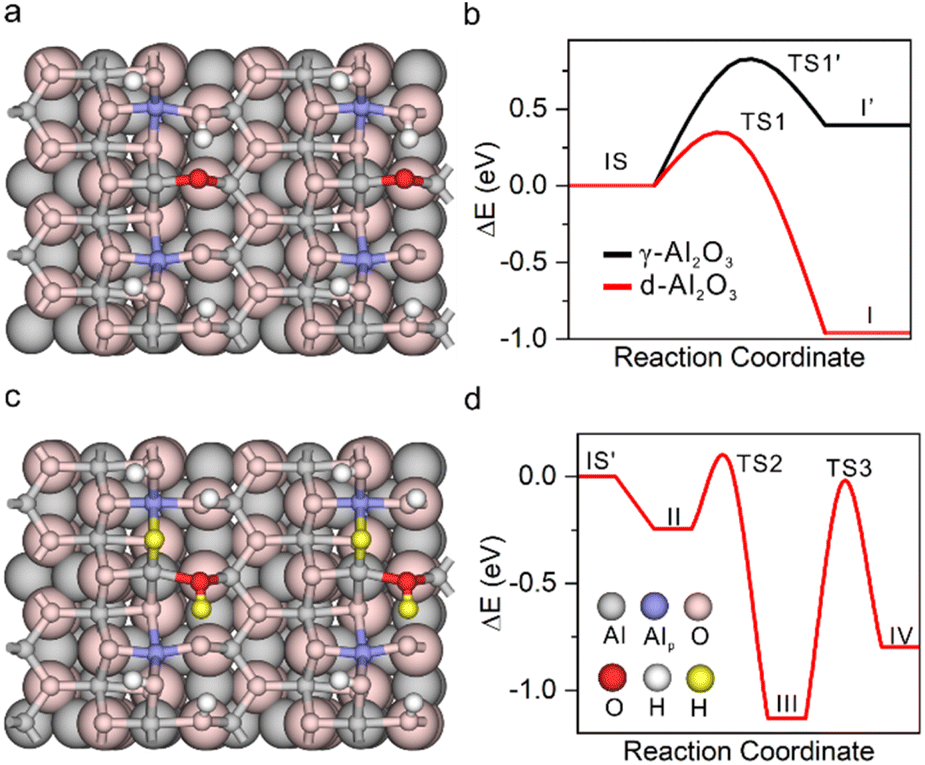 | ||
| Fig. 3 Structure of (a) d-Al2O3 and (c) d-Al2O3 with heterolytically dissociated H2. The colour code is shown in panel (d). Energetic profiles of (b) H2 activation over γ-Al2O3 and d-Al2O3, and (d) following stepwise hydrogenation of C2H4 over d-Al2O3. | ||
DFT calculation was further applied to understand the hydrogenation process.53–55 The structure of γ-Al2O3 was constructed following the same method in our previous study.56 The d-Al2O3 model with surface Alp was constructed from γ-Al2O3 following a dehydration process. Considering the high surface concentration of Alp on the surface of d-Al2O3 reported in this work, 1/2 surface hydroxyl group was removed. The resulting coordinatively unsaturated Alp site and the deprotonated –O– site serve as the FLP center for the H2 activation and hydrogenation (Fig. 3a). By overcoming a barrier of 0.30 eV (TS1), the heterolytic dissociation of H2 at the FLP site was exothermic by ∼0.96 eV over d-Al2O3 (Fig. 3b), resulting in O–Hδ+ and Alp–Hδ− (Fig. 3c). By contrast, the dissociation of H2 on γ-Al2O3 can be only realized on the surface with reconstruction of the surface Alt with a barrier of 0.82 eV (TS1′) and endothermcity of 0.40 eV (Fig. S12†), implying that the activation of H2 on the pristine γ-Al2O3 was restricted. The hydrogenation of ethylene was further calculated to confirm that the heterolytically dissociated hydrogen species were responsible for the catalytic hydrogenation. As shown in Fig. 3d and S13,† the barriers for the stepwise hydride and proton transfer from Alp–Hδ− and O–Hδ+ to produce ethane were 0.31 (TS2) and 1.11 eV (TS3), respectively, which was affordable under the catalytic conditions.
2.4 Stepwise hydrogenation mechanism
The catalytic hydrogenation mechanism utilizing O–Hδ+ and Al–Hδ− species on FLPs was also verified by isotope-labelling experiments. First, when D2-treated d-Al2O3 was used to react with styrene in anhydrous toluene, the characteristic peaks of deuterated ethylbenzene were observed in 2H NMR spectra (Fig. 4a), indicating that the D2 dissociated on FLPs of d-Al2O3 can deuterate styrene and thus verifying the results observed in Fig. 2c. Correspondingly, when D2 was used as the hydrogen source and D2 treated d-Al2O3 as the catalyst, a very strong signal of deuterated ethylbenzene without the presence of other impurities was observed in the 2H NMR spectrum, further confirming that d-Al2O3 can catalyze hydrogenation of styrene to afford ethylbenzene in ∼100% purity (Fig. 4b). Interestingly, we found that the number of D atoms on the alpha carbon atom (Cα) was significantly less than that on the beta carbon atom (Cβ). The peak area ratio, αD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :βD, was calculated to be 1:2.6 (Fig. 4c). The results suggested the existence of the hydrogen–deuterium exchange in the hydrogenation process, which was an indication that the hydrogenation reaction was performed in two steps rather than one step.57 As expected, when D2 and H2 were used as hydrogen sources respectively, a typical primary kinetic isotope effect (KIE, kH2/kD2 > 5) was observed (Fig. 4d), so the rate-determining step was the cleavage of the O–D bond.58,59
:βD, was calculated to be 1:2.6 (Fig. 4c). The results suggested the existence of the hydrogen–deuterium exchange in the hydrogenation process, which was an indication that the hydrogenation reaction was performed in two steps rather than one step.57 As expected, when D2 and H2 were used as hydrogen sources respectively, a typical primary kinetic isotope effect (KIE, kH2/kD2 > 5) was observed (Fig. 4d), so the rate-determining step was the cleavage of the O–D bond.58,59
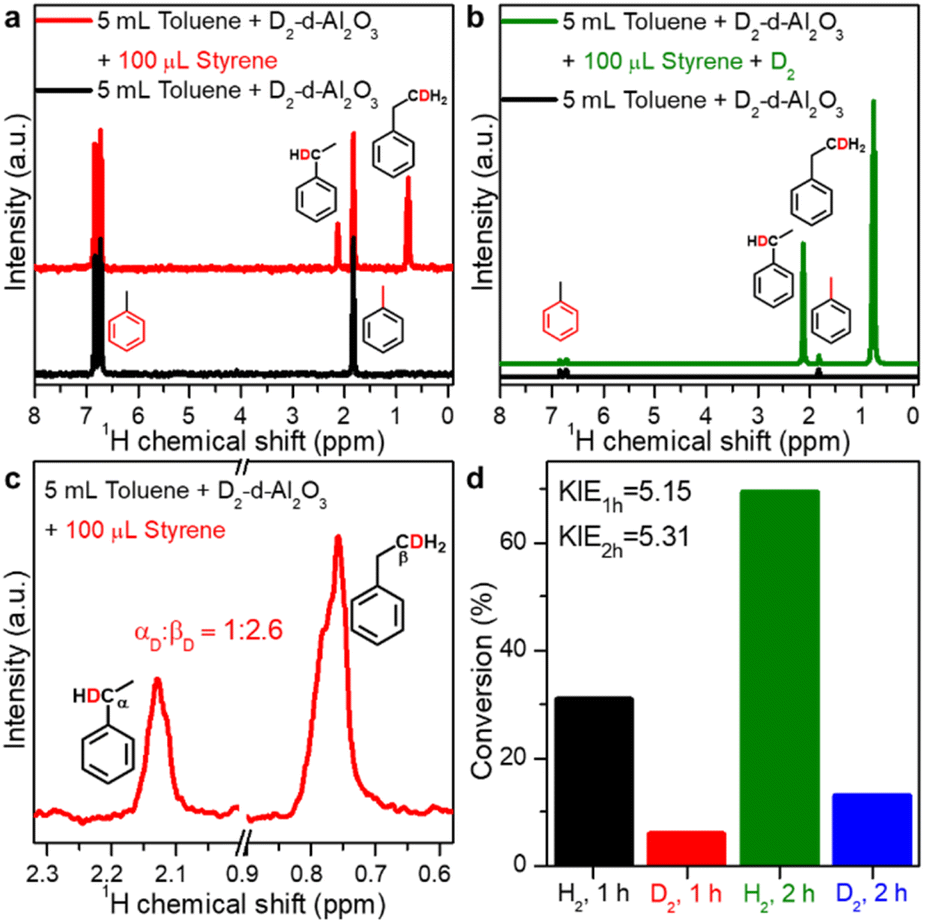 | ||
| Fig. 4 (a) 2H NMR spectra of toluene mixed with D2-treated d-Al2O3 and D-ethylbenzene from reacting styrene with D2-treated d-Al2O3 in toluene. (b) 2H NMR spectra of toluene mixed with D2-treated d-Al2O3 and D-ethylbenzene from reacting styrene with D2 and D2-treated d-Al2O3 in toluene. (c) Enlargement of the two peaks of αD and βD in Fig. 4a. (d) Primary kinetic isotope effect (PKIE) of styrene hydrogenation. | ||
Based on the above systematic studies, the catalytic pathway of the reaction was proposed (Fig. S14†). The reaction begins with the heterolytic activation of H2 by FLPs of d-Al2O3 to produce O–Hδ+ and Al–Hδ−. Next, the active hydride species react with the adsorbed styrene following an Eley–Rideal-like mechanism, attacking βC, and then the protons at the O atom interact with αC. Finally, the obtained ethylbenzene was released and the catalytically active sites were recovered.
2.5 d-Al2O3 as the catalyst for hydrogenation of other substrates
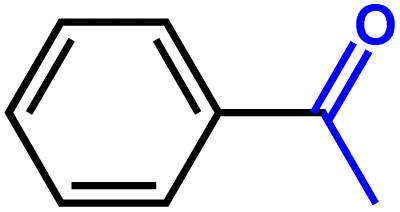
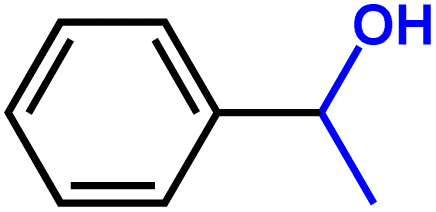
Furthermore, insight into the mechanism also motivated us to explore the general applicability of the d-Al2O3 catalyst. Table 1 shows the results of these hydrogenation reactions. First, d-Al2O3 exhibited nice activity in the hydrogenation of various olefins including aromatic and aliphatic olefins (Table 1, entries 1–4). Moreover, the hydrogenation of aromatic and aliphatic alkynes over d-Al2O3 gave the corresponding alkanes with excellent yields too (Table 1, entries 5–9). These results strongly suggested that FLPs enabled the hydrogenation of both terminal and internal carbon–carbon multiple bonds. Since heterolytic activation of H2 yielded both O–Hδ+ and Al–Hδ− on the d-Al2O3 surface, d-Al2O3 should allow the hydrogenation of polar unsaturated bonds.60–62 As expected, we observed good catalytic performance in the hydrogenation of benzaldehyde or acetophenone by d-Al2O3 (Table 1, entries 10–11).




3 Conclusions
In summary, amorphous Al2O3 enriched with surface pentacoordinated Al3+ species has been synthesized via a simple synthesis method. The co-presence of a large amount of Lewis acidic and basic sites in the same rigid phase thus gave rise to a new type of heterogeneous transition metal-free FLP catalyst. The d-Al2O3 catalyst is highly reactive to enable the heterolytic activation of molecular hydrogen. In the hydrogenation of styrene, the FLP-enriched d-Al2O3 exhibited nice activity and robustness, and more importantly, a stepwise hydrogenation mechanism. Since heterolytic activation of H2 yielded both O–Hδ+ and Al–Hδ− on the d-Al2O3 surface, d-Al2O3 catalyzed the hydrogenation of polar unsaturated bonds in aldehydes or ketones. More work on employing FLP-rich oxides for the discovery of other exciting catalysts should be further explored.4 Experimental methods
4.1 General procedure for synthesis of d-Al2O3
For a typical synthesis process of d-Al2O3, 500 mg Al(OH)(CH3COO)2 was transferred into a glass reaction tube (Φ = 1 cm). The sample was heated to 500 °C at a rate of temperature increase of 1 °C min−1 for 4 h under N2 or Ar protection. After cooling to room temperature, the white products were quickly transferred to a dry centrifuge tube, sealed and stored in a desiccator, and used for further characterization.4.2 General procedure for treatment of d-Al2O3 with gaseous Lewis base molecules
For a typical treatment process, d-Al2O3 was first synthesized by the above method in a glass reaction tube. After cooling to room temperature, high-purity gases (5 vol% NH3/Ar) at a rate of 20 mL min−1 were passed with mass flow for 12 h. The products were quickly transferred to a dry centrifuge tube, sealed and stored in a desiccator, as well as used for further characterization. The samples were marked as NH3-d-Al2O3.4.3 General procedure for treatment of d-Al2O3 with H2 or D2
For a typical treatment process, d-Al2O3 was first synthesized by the above method in a glass reaction tube. After cooling to room temperature, the sample was heated to 100 °C at a rate of temperature increase of 1 °C min−1 for 2 h under H2 or D2 protection. After cooling to room temperature, the products were quickly transferred to a dry centrifuge tube, sealed and stored in a desiccator, which was used for further characterization and applications.4.4 General procedure for catalysis tests
For a typical styrene hydrogenation reaction, a freshly prepared 50 mg catalyst of d-Al2O3 was dispersed in 5 mL anhydrous toluene, and then 20 μL styrene was added in an autoclave under stirring. Next, the reactor was charged with H2 to 1 MPa and kept at 100 °C in an oil bath during stirring. The products were analyzed by gas chromatography (GC9720Plus, Fuli Instruments) for conversion determination. For other catalysts, the reaction conditions were the same as above. In the catalyst recycling test, after the first cycle was carried out for 150 min, the same amount of styrene was added to the reactor and the reaction was allowed to take place. As mentioned above, the recycling was repeated another six times.4.5 Solid state 27Al MAS NMR characterization
Solid state 27Al MAS-NMR measurement (Fig. 1a) was performed using a Bruker Avance NEO 600 MHz spectrometer with a 3.2 mm probe head at a spinning rate of 20 kHz. Spectra were accumulated for 1000 scans with a cycle delay of 2.5 s, using a pulse width of π/6. Solid state 27Al MAS-NMR measurement (Fig. 1b) was performed using a Bruker Avance III 400 MHz spectrometer with a 4.0 mm probe head at a spinning rate of 12 kHz. Spectra were accumulated for 256 scans with a recycle delay of 2 s and a pulse width of π/2 was used. It is worth noting that the magnetic field strength and pulse width hardly affected the relative content of Al3+ ions in the different coordination environments in our test. The fitting of 27Al NMR spectra was conducted using the Dmfit program, which includes the quadrupolar interactions for 27Al MAS NMR. The CzSimple model was applied for the fitting, which implements a rapid version of the Czjzek distribution of quadrupolar interaction. The Gaussian isotropic model for d = 5 with an uncoupled distribution of isotropic chemical shift was included in the simulations. All the other parameters were freely variable during the fitting.634.6 2D solid state 1H–27Al heteronuclear correlation (HETCOR) experiment
The experiment was performed on a Bruker Avance NEO 600 MHz spectrometer with a 3.2 mm probe head under MAS conditions of 15 kHz with a very short CP contact time of 50 μs.4.7 Solid state 2H MAS NMR characterization
Solid state 2H MAS NMR spectra were recorded on a Bruker Avance III NEO 600 MHz spectrometer with a 3.2 mm probe head at a spinning rate of 15 kHz, and the spectra were accumulated for 1024 scans with a cycle delay of 1 s, and a pulse width of π/2 was used.4.8 Liquid state 1H NMR characterization
1H NMR spectra were recorded on an AVANCE III 500 MHz spectrometer.4.9 Liquid state 2H NMR characterization
2H NMR spectra were recorded on an AVANCE III 850 MHz spectrometer. All NMR data were processed on MestReNova software.4.10 In situ diffuse reflectance FTIR characterization
Typically, d-Al2O3 was first synthesized in an in situ chamber of FTIR (Thermo Fisher IS50) at 500 °C under Ar protection, and the sample was cooled to 30 °C to acquire background. For in situ NH3-FTIR, 5 vol% NH3/Ar was charged into the chamber at a rate of 30 mL min−1 while pure Ar was turned off, and spectra were collected until stable adsorption with NH3. In situ CO2-FTIR spectra were recorded in the same way. In situ D2-FTIR spectra were recorded in the same way except that the test temperature is 100 °C.4.11 TPD-MS characterization
Typically, NH3-TPD-MS was performed on a Micromeritics AutoChem II 2920 instrument equipped with a mass spectrometer. The 100 mg d-Al2O3 sample was first synthesized at 500 °C under Ar protection. The sample was then cooled to room temperature and treated under 5 vol% NH3/He gas flow for 30 min. The gaseous and weakly adsorbed NH3 was subsequently removed by purging with He for 60 min. Subsequently, the temperature was raised from room temperature to 700 °C at a rate of 10 °C min−1 and the NH3-TPD profile was recorded by using a mass spectrometer with a signal of m/z = 16. For CO2-TPD-MS, 5 vol% CO2/He gas was used, and the CO2-TPD profile was recorded by using a mass spectrometer with a signal of m/z = 44.4.12 N2 adsorption/desorption characterization
N2 adsorption/desorption measurements were conducted on a Micromeritics ASAP 2020 gas adsorption analyzer. The freshly prepared sample was first degassed at 300 °C for 8 h. Then, the specific surface areas were determined using the Brunauer–Emmett–Teller (BET) and Langmuir equations from the N2 sorption data.4.13 XRD characterization
X-ray powder diffraction experiments were conducted on a Rigaku Ultima IV using Cu Kα radiation. The operation voltage and current were 40 kV and 30 mA, respectively. The scanning speed was set to be 15 ° min−1.4.14 HAADF-STEM characterization
High-angle annular-dark-field (HAADF) images were acquired using a JEOL 200F transmission electron microscope operated at 200 keV. The attainable spatial resolution of the microscope was 78 pm with a probe spherical-aberration corrector. Images were acquired with the illumination semi-angle of 25 mrad and probe current of 100 pA. The dwell time for image acquisition was set at 10 microseconds per pixel to ensure a desirable signal-to-noise ratio.4.15 Computational details
The spin-polarized density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP5.4.4).64–67 The core electrons were replaced by the projector augmented wave (PAW) pseudopotentials,65,68 and the valence electrons were described by a plane wave basis set with a cut-off energy of 400 eV. The electron exchange and correlation were treated by Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA).69 The k-point sampling was generated by following the Monkhorst–Pack procedure with a 3 × 3 × 1 mesh.70 The transition states (TSs) were determined using the nudged elastic band (NEB) approach. The TS structures were optimized using a quasi-Newton algorithm. The steady states and TSs were converged to a residual force smaller than 0.03 eV Å−1 and 0.05 eV Å−1, respectively. All the minima and TSs were confirmed by the vibrational frequency calculations. The reaction barrier was defined as ΔEa = ETS − ER, where ER and ETS were the energies of the reactant on the slab and the corresponding transition states, respectively.Data availability
All the data supporting this article have been included in ESI.†Author contributions
N. F. Z. conceived and supervised the research project. Q. Y. W. synthesized and characterized the samples, as well as investigated the catalytic performances and mechanism. R. X. Q. performed DFT calculations. M. S. Z., Y. Y. Z and S. S. Y. contributed to the NMR and TPD measurements. G. F. provided some suggestions. N. F. Z., and Q. Y. W. wrote and revised the manuscript. X. D. Y and H. S. revised the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (grant no. 92261207, 22202164, and NSFC Center for Single-Atom Catalysis under grant no. 22388102) and the New Cornerstone Science Foundation. Q. Y. W. thanks the China Postdoctoral Science Foundation Project (2023M732946). R. X. Q. acknowledges support from National Key R&D Program of China (2022YFA1504500), the Natural Science Foundation of Fujian Province (2023J05006) and the Fundamental Research Funds for the Central Universities (20720230002). We also thank Prof. Lin Gu and Dr Qinghua Zhang at the Institute of Physics of the Chinese Academy of Sciences for the TEM measurements.References
- G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC.
- G. J. Kubas, Acc. Chem. Res., 1988, 21, 120 CrossRef.
- D. M. Heinekey, A. Lledos and J. M. Lluch, Chem. Soc. Rev., 2004, 33, 175 RSC.
- P. G. Jessop and R. H. Morris, Coord. Chem. Rev., 1992, 121, 155 CrossRef CAS.
- A. I. Krasna and D. Rittenberg, J. Am. Chem. Soc., 1954, 76, 3015 CrossRef CAS.
- D. Schilter, J. M. Camara, M. T. Huynh, S. S. Hammes and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305 CrossRef CAS PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Frohlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 119, 8196 CrossRef.
- G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407 RSC.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS.
- Y. Ma, S. Zhang, C. Chang, Z. Huang, J. Ho and Y. Qu, Chem. Soc. Rev., 2018, 47, 5541 RSC.
- G. Lu, P. Zhang, D. Sun, L. Wang, K. Zhou, Z. Wang and G. Guo, Chem. Sci., 2014, 5, 1082 RSC.
- K. C. Szeto, W. Sahyoun, N. Merle, J. L. Castelbou, N. Popoff, F. Lefebvre, J. Raynaud, C. Godard, C. Claver, L. Delevoye, R. M. Gauvin and M. Taoufik, Catal. Sci. Technol., 2016, 6, 882 RSC.
- J. Y. Xing, J. C. Buffet, N. H. Rees, P. Norby and D. O'Hare, Chem. Commun., 2016, 52, 10478 RSC.
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Phys. Chem. Chem. Phys., 2015, 17, 14623 RSC.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206 CrossRef CAS PubMed.
- L. He, T. E. Wood, B. Wu, Y. Dong, L. B. Hoch, L. M. Reyes, D. Wang, C. Kübel, C. Qian, J. Jia, K. Liao, P. G. O'Brien, A. Sandhel, J. Y. Y. Loh, P. Szymanski, N. P. Kherani, T. C. Sum, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 5578 CrossRef CAS PubMed.
- L. B. Hoch, L. He, Q. Qiao, K. Liao, L. M. Reyes, Y. Zhu and G. A. Ozin, Chem. Mater., 2016, 28, 4160 CrossRef CAS.
- A. Primo, F. Neatu, M. Florea, V. Parvulescu and H. Garcia, Nat. Commun., 2014, 5291 CrossRef CAS.
- J. Zhao, X. Liu and Z. Chen, ACS Catal., 2017, 7, 766 CrossRef CAS.
- X. Zhao, J. Wang, M. Yang, N. Lei, L. Li, B. Hou, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2017, 10, 819 CrossRef CAS PubMed.
- H. Lee, Y. N. Choi, D. W. Lim, M. M. Rahman, Y. I. Kim, I. H. Cho, H. W. Kang, J. H. Seo, C. Jeon and K. B. Yoon, Angew. Chem., Int. Ed., 2015, 54, 13080 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511 CrossRef CAS PubMed.
- M. Trunk, J. F. Teichert and A. Thomas, J. Am. Chem. Soc., 2017, 139, 3615 CrossRef CAS PubMed.
- S. Zhang, Z. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS PubMed.
- S. G. Hindin and S. W. Weller, J. Phys. Chem., 1956, 60, 1501 CrossRef CAS.
- Y. Amenomiya, J. H. B. Chenier and R. J. Cvetanovic, J. Catal., 1967, 9, 28 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C. W. Yi, D. H. Kim, C. H. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670 CrossRef CAS.
- N. Tang, Y. Cong, Q. Shang, C. Wu, G. Xu and X. Wang, ACS Catal., 2017, 7, 5987 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- P. Liu, R. Qin, G. Fu and N. F. Zheng, J. Am. Chem. Soc., 2017, 139, 2122 CrossRef CAS PubMed.
- Q. Wu, R. Qin, D. Zang, W. Zhang, B. Wu and N. F. Zheng, Acta Chim. Sin., 2018, 76, 617 CrossRef CAS.
- R. Qin, K. Liu, Q. Wu and N. F. Zheng, Chem. Rev., 2020, 120, 11810 CrossRef CAS PubMed.
- Q. Wu, W. Zhou, H. Shen, R. Qin, Q. Hong, X. Yi and N. Zheng, CCS Chem., 2023, 5, 1215 CrossRef CAS.
- L. Shi, G. M. Deng, W. C. Li, S. Miao, Q. N. Wang, W. P. Zhang and A. H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994 CrossRef CAS.
- S. h. Xin, Q. Wang, J. Xu, N. D. Feng, W. Z. Li and F. Deng, Solid State Nucl. Magn. Reson., 2017, 84, 103 CrossRef CAS.
- L. Wang, G. Kehr, C. G. Daniliuc, M. Brinkkötter, T. Wiegand, A.-L. Wübker, H. Eckert, L. Liu, J. G. Brandenburg, S. Grimme and G. Erker, Chem. Sci., 2018, 9, 4859 RSC.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. El Labban, G. O. Ngongang Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608 CrossRef CAS PubMed.
- B. Werghi, A. Bendjeriou-Sedjerari, A. Jedidi, E. Abou-Hamad, L. Cavallo and J. M. Basset, Organometallics, 2016, 35, 3288 CrossRef CAS.
- M. Veith, J. Frères, V. Huch and M. Zimmer, Organometallics, 2006, 25, 1875 CrossRef CAS.
- R. Wischert, C. Coperet, F. Delbecq and P. Sautet, Angew. Chem., Int. Ed., 2011, 123, 3260 CrossRef.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944 CrossRef CAS PubMed.
- H. Shen, Q. Wu, M. S. Asre Hazer, X. Tang, Y.-Z. Han, R. Qin, C. Ma, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Chem, 2022, 8, 2380 CAS.
- H. Shen, Q. Wu, S. Malola, Y.-Z. Han, Z. Xu, R. Qin, X. Tang, Y.-B. Chen, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2022, 144, 10844 CrossRef CAS PubMed.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Hakkinen, B. K. Teo and N. Zheng, ACS Nano, 2019, 13, 5975 CrossRef CAS PubMed.
- T. Mizuno, T. Nemoto, M. Tansho, T. Shimizu, H. Ishii and K. Takegoshi, J. Am. Chem. Soc., 2006, 128, 9683 CrossRef CAS PubMed.
- W. Zhang, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2023, 145, 10178 CrossRef CAS PubMed.
- K. Li, R. Qin, K. Liu, W. Zhou, N. Liu, Y. Zhang, S. Liu, J. Chen, G. Fu and N. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 52193 CrossRef CAS PubMed.
- W. Zhang, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2021, 143, 15882 CrossRef CAS PubMed.
- R. Qin, L. Zhou, P. Liu, Y. Gong, K. Liu, C. Xu, Y. Zhao, L. Gu, G. Fu and N. F. Zheng, Nat. Catal., 2020, 3, 703 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. F. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- K. Liu, R. Qin, L. Zhou, P. Liu, Q. Zhang, W. Jing, P. Ruan, L. Gu, G. Fu and N. Zheng, CCS Chem., 2019, 1, 207 CAS.
- C. Xu, G. Chen, Y. Zhao, P. Liu, X. Duan, L. Gu, G. Fu, Y. Yuan and N. Zheng, Nat. Commun., 2018, 9, 3367 CrossRef.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- A. Dedieu, S. Humbel, C. Elsevier and C. Grauffel, Theor. Chem. Acc., 2004, 112, 305 Search PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1994, 49, 14251 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 48, 13115 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Reagents, FTIR, TPD-MS, BET, XRD, HAADF-STEM, NMR, Fig. S1–S14 and Table S1. See DOI: https://doi.org/10.1039/d3sc06425e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |