
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of anionic processes in Li1−xNi0.44Mn1.56O4 studied by resonant inelastic X-ray scattering†
Felix
Massel
a,
Burak
Aktekin
b,
Yi-Sheng
Liu
 c,
Jinghua
Guo
c,
Magnus Helgerud
Sørby
d,
Daniel
Brandell
b,
Reza
Younesi
b,
Maria
Hahlin
ab and
Laurent-Claudius
Duda
*a
c,
Jinghua
Guo
c,
Magnus Helgerud
Sørby
d,
Daniel
Brandell
b,
Reza
Younesi
b,
Maria
Hahlin
ab and
Laurent-Claudius
Duda
*a
aDepartment of Physics and Astronomy, Division of X-ray Photon Science, Uppsala University, Box 516, S-751 20 Uppsala, Sweden. E-mail: Laurent.Duda@physics.uu.se
bDepartment of Chemistry – Ångström Laboratory, Structural Chemistry, Uppsala University, Box 538, 751 21 Uppsala, Sweden
cAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
dDepartment for Hydrogen Technology, Institute for Energy Technology, P.O. Box 40, NO-2027 Kjeller, Norway
First published on 23rd January 2023
Abstract
We investigated the first lithiation cycle of the positive electrode material Li1−xNi0.44Mn1.56O4 (LNMO) using soft X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) at the transition metal L- and oxygen K-edges. Our XAS results show that charge compensation in LNMO takes place mostly within the Ni–O bonds, which is consistent with previous similar studies. O K- and Ni L-RIXS reveals how the holes that are created by removal of electrons during delithiation are distributed between the Ni- and O-ions. Non-trivial anionic activity is revealed by O K-RIXS features such as the appearance of low-energy intra-band excitations and re-hybridization with Ni 3d-states forming a new intense band close to the top of the oxygen valence band. At the same time, Ni L-RIXS compares more favorably with covalently than with ionically bonded Ni-oxide based compounds. Thus, a picture emerges where delithiation leads to a gradual transition of the ground state of LNMO from Ni 3d8 to one with non-negligible amounts of ligand holes, i.e. Ni 3d8−x![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) 2−x (0 < x < 2, where stands for a ligand hole) instead of a highly ionic state e.g. Ni 3d6. Our observations highlight the importance of studying the anionic character of redox processes in lithium ion batteries.
2−x (0 < x < 2, where stands for a ligand hole) instead of a highly ionic state e.g. Ni 3d6. Our observations highlight the importance of studying the anionic character of redox processes in lithium ion batteries.
I. Introduction
Modern battery technology is ubiquitous, which is evidenced by its wide-spread usage in areas such as consumer electronics, electric vehicles, and grid energy storage. The demand for high-energy–density batteries is ever-growing and requires rapid development of rechargeable battery technology. Novel materials must be found that overcome the limitation factors of current materials. This will rely on quickly achieving accurate and more detailed knowledge about the energy storage mechanisms in advanced materials. The energy density of lithium ion batteries (LIBs) depends strongly on the amount of charge that their cathode material can store. In fact, the cathode presents the limiting factor of energy storage capacity in commercial batteries.Rechargeable batteries are based on moving atoms that have loosely bound valence electrons and form small positive ions, e.g. Li and Na. Electron transfer in redox processes facilitate the reversible storage of chemical energy in the partially filled bands of transition metal oxides. Traditionally, one has considered the cationic part of this process to be of prime importance and only recently the full potential of anionic redox processes has moved into focus.1,2 Various mechanisms have been suggested to underlie anionic redox: Saubanère et al.3 suggested a metal-driven reductive coupling mechanism for oxo- and peroxo-like transition and cation migration was put forward by Gent et al.4 Major research efforts have addressed the anionic redox active class of Li-rich, layered materials such as e.g. Li1.2Ni0.2Mn0.6O2,5,6 where localized oxygen species have been shown to contribute extra capacity in addition to the cationic redox processes. Subsequently, these localized states have been shown to belong to trapped oxygen molecules that form in the crystal when the battery is charged.7 Seo et al. have theoretically described an anionic redox process that is predicated on extracting electrons from Li–O–Li configurations, so-called orphaned oxygen states8 but the occurrence of these has yet to be experimentally verified. On the other hand, some evidence has emerged for the reductive coupling mechanism in certain sodium ion battery cathode materials.9 Finally, oxygen holes have also been shown to play a major role for understanding redox processes in the classical cathode material LixCoO2.10 Thus there is an intriguing range of possible mechanisms for reversible and irreversible anionic processes that stimulates investigation of their role in other classes of cathode materials that appear to be driven by cationic processes.
The non-Li-rich, high-voltage spinel material LiNiyMn2−yO4 (LNMO) is a promising high-energy density cathode material due to its high-voltage plateau and low voltage hysteresis, while containing relatively cheap and abundant components.11,12 LNMO, as an intercalation material for positive electrodes in Li-ion batteries, is based on the extraction of Li+-ions during delithiation (charging process) and reinsertion of these ions during relithiation (discharge process). The conventional approach is to attribute charge compensation to the oxidation and reduction of cations during de- and relithiation, respectively. As Ni substitution in LiNiyMn2−yO4 approaches y = 0.5, the Mn3+-to-Mn4+ ratio of the pristine material decreases and the operating voltage shifts from 4.1 V (vs. Li/Li+) associated with a charge compensation via a Mn3+/4+ redox to a higher voltage plateau of 4.7 V13,14 that has attracted much attention. This high-voltage plateau has been attributed to cationic redox between Ni2+ and Ni4+ based upon X-ray absorption spectroscopy.15–18 At the same time however, O K-edge X-ray absorption spectroscopy (XAS) shows that oxygen anions participate appreciably in the charge compensation processes.17 This has been interpreted as the consequence of Ni–O-bond hybridization, consistent with a formal transition between Ni2+ and Ni4+ during delithiation of LNMO. However, Ni-based battery cathodes can also involve oxygen ions for charge compensation, and an example of unexpectedly large contributions has just recently been revealed.19 Additionally, Ni-oxide systems are known to have strong electron correlations that may also affect the ability of LNMO to undergo a purely cationic redox. For example, when the seeming simple stoichiometric nickel oxide NiO (a prototypical Mott insulator with a gap between Ni 3d and oxygen 2p states) is made nickel deficient it undergoes a profound change in its band structure, i.e. an electronic phase transition, and it has been shown to then become a negative-charge-transfer insulator with a gap that is of the oxygen p–p type.20 Thus electronic phase transitions during delithiation may occur and must be taken into account for achieving a proper understanding of these battery electrode materials.
We investigated the promising LNMO material with a Mn-rich composition Li1−xNi0.44Mn1.56O4 using Ni L- and O K-edge resonant inelastic X-ray scattering (RIXS) to elucidate the anionic character of the redox processes. We find that while the Ni L-XAS spectra are consistent with the oxidation of Ni–O bonds when LNMO is delithiated, the formation of holes in the O 2p-bands is significant and changes the character of the electronic structure. On one hand, O K-RIXS of lithiated LNMO exhibits localized transition metal dd-excitations of about 2.5 eV, on the other hand, delithiated LNMO has localized excitations that are of O 2p-character with low energy (≪1 eV). Additionally, a strong resonant excitation of hybridized Ni 3d-states at the top of the oxygen valence band is observed in O K-RIXS. Thus delithiated LNMO shows evidence of being a negative charge-transfer Ni oxide system with electronic states of predominantly O 2p-character across the Fermi level.
II. Results
A. Electrochemical cycling
Composite electrodes consisting of LiNi0.44Mn1.56O4 powders (90 wt%), carbon black (5 wt%), and a binder (5 wt%) were electrochemically cycled in half-cell setups with Li metal as counter electrodes. The initial cycle of the charge–discharge curve is reflected in Fig. 1(a). After a steep initial rise from below 3.4 V and a subsequent less steep, short ascent to a potential of about 4.75 V a plateau is reached that extends to nearly the end of the initial charge where there is another short rise in potential, upon which the charging is terminated when about 4.95 V are reached. The discharge branch nearly mirrors the charge branch with a plateau that is only about 0.05 V lower, i.e. at about 4.70 V, and a specific charge uptake shortened only by about 30 mA h g−1. LNMO electrode samples at different states of charge (SoC) were thus obtained by galvanostatic cycling and stopping the current at the points shown in Fig. 1(a) before disassembly (the crystal structure of the pristine material is depicted in Fig. 1(b)). The SoC were selected at salient points of intererest, namely at the beginning of the plateau (BoP), at the end of the plateau (EoP), at the end of charge (EoC), and at the end of discharge (EoD). Transport of the removed cathodes to the synchrotron facilities was done in a manner avoiding any air exposure prior to the spectroscopic measurements.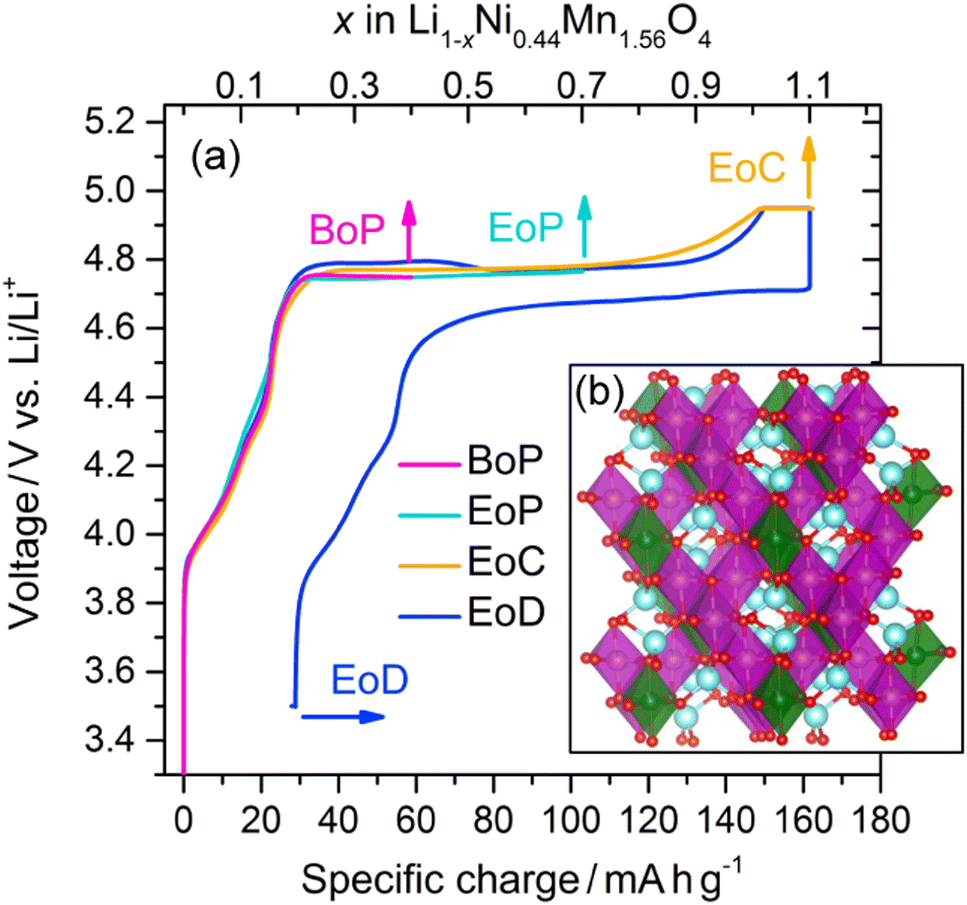 | ||
| Fig. 1 (a) Li1−xNi0.44Mn1.56O4 charge–discharge profile obtained from the partial curves of the individual cathodes from different states of charge (see text for the definitions): BoP, EoP, EoC, and EoD, respectively. The inset (b) shows the spinel crystal structure in the cation ordered P4332 space group of LiNi0.5Mn1.5O4; Li (blue), O (red), Ni (green), and Mn (purple).21 | ||
A more detailed description of the sample preparation is presented in the Methods section.
B. XAS: Mn L-edge
Fig. 2(a) and (b) show the Mn L3-edge XAS of Li1−xNi0.44Mn1.56O4 at different states of charge (SoC). The bulk-sensitive inverse partial fluorescence yield (IPFY) mode is shown in the left panel and in the surface-sensitive total electron yield (TEY) mode is shown in right panel. At this edge, Mn 2p32 core electrons are excited into empty states of the Mn 3d band and the resulting spectrum contains information about the oxidation states of the probed element. It has been shown that L-edge absorption profiles of Mn compounds in octahedral site symmetry share key-features with compounds of the same Mn valency despite having different crystal structures.22 For this reason, the reference spectra of compounds with known nominal Mn valency are shown at the bottom spectra of Fig. 2(a) and (b). This comparison gives good insight into the oxidation states that are present at each SoC.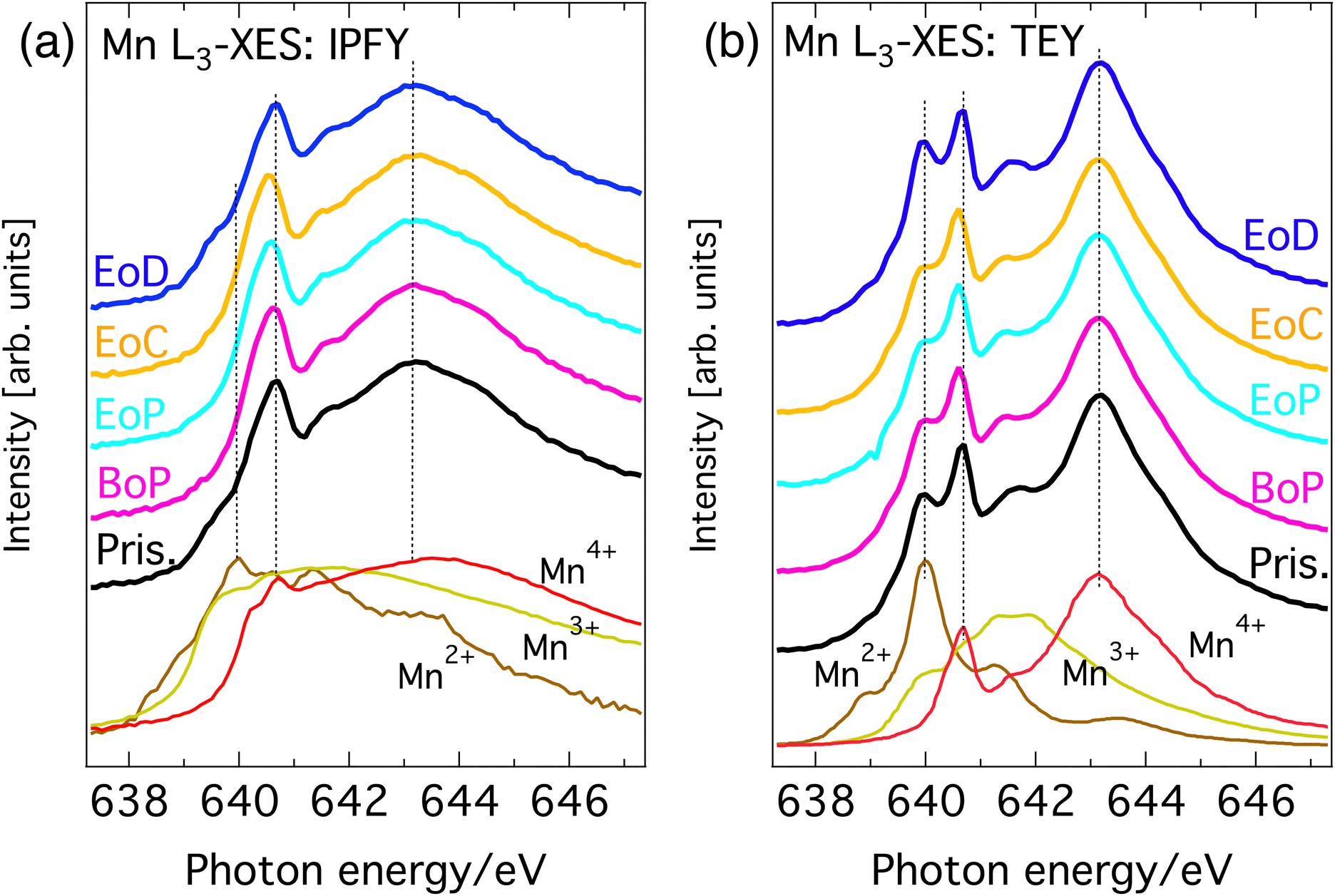 | ||
| Fig. 2 Mn L3-edge XAS of Li1−xNi0.44Mn1.56O4 at different SoC and reference spectra. (a) Bulk sensitive IPFY-signal. The bottom three traces are IPFY spectra from (Mn(NO)3·4H2O) (Mn2+), Mn2O3 (Mn3+), and MnO4 (Mn4+). (b) Surface sensitive TEY-signal. The bottom three traces are TEY reference spectra of MnO (Mn2+), Mn2O3 (Mn3+), and Li2MnO3 (Mn4+) adapted from Qiao et al.23 | ||
All (TEY) IPFY spectra share a strong spectral fingerprint with the tetravalent MnO2 (Li2Mn4+O3) reference compounds in the form of two main features at 639.3 and 641.8 eV (the two rightmost dashed vertical lines in both panels). This confirms Mn4+ as the expected dominant oxidation state for LiNi0.44 Mn1.56O4. Additionally, for the both the IPFY and the TEY spectra there is a shoulder at the low energy side of the edge and extra intensity between the two main features unaccounted for by the Mn4+ reference. Both can be attributed to the presence of small amounts of Mn3+ or Mn2+, which is also expected as per stoichiometry. Upon partial delithiation to the beginning of the voltage plateau of the charge–discharge curve (BoP), the contributions from lower oxidation states in the Mn L-spectra slightly decrease and then remain stable until complete delithiation (EoC) is reached. Re-lithiation (EoD) leads to a reversal of this process, reinstating the situation of the pristine material.
Summarizing, the overall rather small and reversible changes in the predominantly Mn4+-character L-edge implies that Mn plays a minor role in the charge compensating redox chemistry and is mostly limited to the charge–discharge region before the voltage-plateau, where a redox of a small amount Mn between its 3+ and 4+ oxidation states occurs. This is in line with expectations from the electrochemical measurements and application of formal oxidation rules to the stoichiometry.
C. XAS and RIXS: O K-edge
A complete data set giving an overview with color-coded O K-RIXS maps scaled by acquisition time for each spectrum is found in Fig. S1 in the ESI.†Fig. 3(a) are 1D-waterfall line plots based on the same raw data (pristine and fully charged) scaled to equal area for better shape comparison. Fig. 3(b) shows the valence and conduction band region with O 2p-character using O K-edge XAS/RIXS plotted on a common energy scale. The charging evolution is visualized in the bottom panel of Fig. 3(b) as progressing from the lithiated pristine material (bottom) via partially delithiated (BoP and EoP) to the completely delithiated (EoC) material and finally to the relithiated material (EoD). For the empty states, we find that the most striking evolution is the development of a pronounced shoulder at the low energy side of the leading pre-edge XAS peak (highlighted by the pink shading at the top). This indicates that delithiation creates additional holes at the oxygen sites. A similar observation has been reported previously17 for Li1−xNi0.5Mn1.5O4. For convenience, a suggestion of the resulting energy scheme is illustrated in Fig. 3(c). | ||
| Fig. 3 (a) Waterfall plots for O K-edge RIXS of pristine (left panel) and (right panel) fully delithiated LNMO. The spectra are normalized to unit height. The magenta vertical dashed lines mark mediated Mn dd-excitations at a fixed energy loss of approximately 2.5 eV and the blue arrow in right panel highlights the fluorescence-like dispersion of the O 2p–Ni 3d band feature. (b) Top panel: O K-edge RIXS and XAS spectra at different SoC. Light green (pink) shading indicates increment (decrement) of intensity compared to the previous SoC. The long vertical dashed line marks the incident photon energy (at 528.5 eV) of the RIXS spectra. Bottom panel: Overlaid O K-edge RIXS spectra showing their natural intensity variation relative to each other. The color-shaded rectangles indicate various energy regions of interest: intra band O 2p excitations (orange), Mn dd-excitations (green), O 2p–Ni 3d-hybridization band (turquoise). (c) Energy diagram derived from the RIXS and XAS spectra illustrating the effects of delithiation on the oxygen states by re-hybridization with nickel states. | ||
The incidence energy used for the RIXS spectra, situated on the left side of Fig. 3(a), corresponds to the position of the dashed vertical line, i.e. the XAS shoulder. The changes of the occupied O 2p-states associated with the states at the XAS-shoulder (528.5 eV) are thus reflected by the correspondingly excited RIXS spectra. We have highlighted the successive relative intensity (and thus occupation) changes in the XAS and the RIXS spectra by shaded areas. Light green (pink) shading shows the incremental increase (decrease) between the RIXS spectra at consecutive SoC. As mentioned above there are some smaller spectral contributions present in the spectra as well that are not to be interpreted as bandlike but instead are of excitonic nature. These spectral parts are highlighted in Fig. 3(b) with green shading (Mn dd-excitation) and yellow shading (tentatively, O pp-excitations bridging the gap as seen in the energy scheme depicted in Fig. 3(c)), respectively. Note that although some caution is necessary when interpreting O K-RIXS in terms of “the partial occupied density of states”, oxygen bands are often dominated by their itinerant electron state character24 thereby reflecting both bandlike occupied states as well as excitonic charge-neutral excitations such as (mediated) dd-excitations simultaneously in one spectrum.
Comparing the delithiated (EoC) to the lithiated (pristine) material in the bottom panel of Fig. 3(b), we observe not only an absolute intensity increase, which is natural due to the increase in unoccupied states at 528.5 eV of the XAS spectra, but more importantly we point out a relative accumulation of spectral weight compared to the broad low-energy tail in the two following distinct features. One is found in the RIXS spectra at about 526.1 eV (light blue) as well as just below the elastic peak about 527.9 eV (orange). At first, such a behavior in O K-RIXS seems to be strikingly reminiscent of that discovered for Li-rich materials5 where an enhancement of one peak in the elastic peak vicinity together with a higher energy feature was found. Later, it was shown that these features belonged to the spectrum from O2 trapped in the crystal structure. However, several differences between the previous and the present observations are noteworthy, namely, the enhancements in the present material appear at much lower incident energy and the high-energy feature occurs at a much lower energy separation from the first. Therefore, these are distinct types of anionic redox processes.
We now focus on the nature of the green- and light-blue-shaded regions in the bottom panel of Fig. 3(b) which are close in energy yet have different origins. This best visualized by comparing the O K-RIXS energy dependence of the material in the pristine state and at EoC on an energy loss scale as in Fig. 3(a). The pristine material possesses an energy loss feature around 2.5 eV. It is visible in the RIXS spectra for most incident energies as a shoulder at constant energy loss that is centered at the vertical dashed red line, meaning it behaves exciton-like and tracks the incident energy at a fixed energy loss. This feature can be attributed to mediated Mn dd-excitations (green shading) as has been observed and described previously in other oxide compounds.24 In contrast, in the delithiated cathode the contribution from the Mn dd-excitations following the dashed red line is weak, although discernible upon close inspection. On the other hand, now a different, more intense feature dominates and disperses to the left (along the blue arrow in the left panel of Fig. 3) together with the main part of the spectra. This fluorescence-like behavior is a sign for emission from bandlike occupied states (resonantly enhanced at 528.5 eV), in contrast to the excitonic-like Mn dd-excitations.
We now have established that the green- and light-blue-shaded regions in the bottom panel of Fig. 3(b), originate from completely different states. We therefore tentatively assign the light-blue-shaded feature to a narrow band of occupied O 2p-states that are hybridized with Ni 3d-states formed when the cathode is delithiated. This assignment is corroborated in our discussion of the Ni L-spectra below. The corresponding empty band is naturally the shoulder in the XAS spectra at 528.5 eV. Finally, the spectral weight of the region in the O K-RIXS at energies within some 0.6 eV to the elastic peak (orange shading) is simultaneously hugely enhanced from a practically zero intensity. This may be indicative of the forming of a small gap during delithiation and an accumulation of occupied O 2p states below the Fermi energy (giving rise to O pp-excitations). Though the origin of these states is less certain (vibrational or other lattice excitations may be involved), this is nevertheless a significant change and could be an indication that a kind of electronic phase transition due to delithiating the cathode is taking place.
In previous studies nearly pure cationic Ni2+/Ni4+ redox processes have been considered for the charge compensation process.17 In view of our observation that also the oxygen states undergo rather profound changes during LNMO delithiation, we want to reassess the behavior of the Ni 3d-states here. In the next section we therefore address the nature of the formally Ni3+- and Ni4+-states formed during delithiation considering that we must account for the observed additional holes appearing in the oxygen band.
D. Ni L-edge XAS
Fig. 4(a) shows PFY-mode Ni L3,2-XAS at different SoC (pristine, BoP, EoP, EoC and EoD). For comparison we have also plotted Ni L3,2-spectra in Fig. 4(b) from compounds25 in which the Ni ions are in relatively pure ionic oxidation states (as denoted).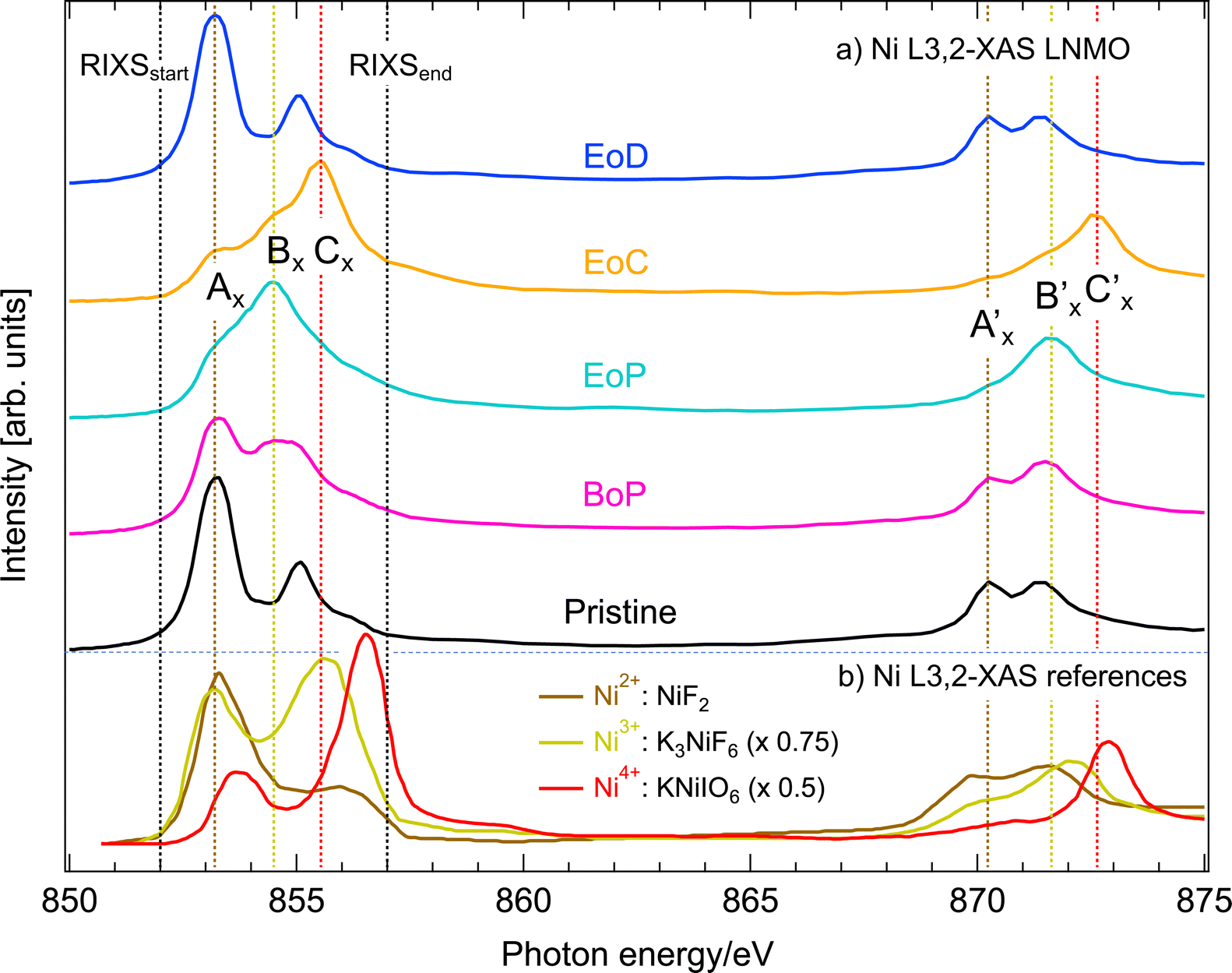 | ||
| Fig. 4 (a) Ni L3,2-edge XAS in PFY mode of Li1−xNi0.44Mn1.56O4 at different SoC (see Fig. 1). The black dashed, vertical lines mark the incident energy region straddling the L3-resonances covered by RIXS. (b) Ni L3,2-edge XAS reference spectra for Ni2+ (NiF2), Ni3+ (K3 NiF6), and Ni4+ (KNiIO6), respectively, as reported by Wang et al.25 with a respective intensity scaling factor of 0.75 and 0.5 for Ni3+ and Ni4+. The colored dashed, vertical lines are labelled to identify salient spectral features at the Ni L3,2-resonances. | ||
We observe that the pristine LNMO sample shows a strong Ni2+-signature with the characteristic low-energy peak and a small high-energy shoulder. The degree of separation between high- and low-energy peaks can be seen as a qualitative measure for ionic bonding character of Ni with its ligands, which has been shown by CT multiplet calculations on NiO.26 The slightly less separated multiplet structure of pristine LNMO compared to the NiF2 reference spectrum with very ionic bonding character, is a good indicator for GS admixture of a 3d9 ligand CT contribution to the otherwise 3d8 dominated configuration distribution. For LiNi0.5Mn1.5O4, Liu et al. present Ni L-XAS and a spectral simulation using a configuration interaction cluster model and quantify the configuration ratio of 3d8 to 3d9 at nine to one.27 This supports our interpretation where we expect slightly more CT between O and Ni in the present pristine material due to its lower Ni content of y = 0.44 and thus somewhat lower average oxidation state of Mn.
Delithiation of LNMO (BoP-EoC) leads to a reversal of the intensity ratio between the high energy and low energy features of Ni L3-edge XAS, as well as the shift of the L2-edge XAS centroid towards higher energies. After relithiation, the Ni ions return to the initial Ni2+ oxidation state. With the help of the Ni-edge XAS spectra of LNMO, the delithiation process has thus been described17 as successive oxidation between the Ni2+/3+ (BoP to EoP) and Ni3+/4+ (EoP to EoC) couples. Depending on the activity at the anionic site, the correct interpretation of the corresponding electron configurations for the ground states can be found between the two following extremes. Firstly, the “completely ionic” case, namely, Ni 3d8 → 3d7 → 3d6 or instead the “completely covalent” case, entailing the transitions Ni 3d8 → 3d8 → 3d82 where and 2 stand for one or two holes, respectively, in the oxygen valence band. A first indicator to this end is given by our comparison to the Ni L3-edge XAS with the spectra from the reference compounds with Ni3+ and Ni4+ oxidation states. The latter spectra differ in several ways from those of delithiated LNMO. An important distinction are the significantly larger separations of the multiplet features than delithiated LNMO. This observation is a good indicator for the presence of ligand CT in LNMO,26 and already reveals that the bonding between Ni and O is more covalent than the reference Ni–O compounds. Taking advantage of energy selective excitation of Ni L3-edge RIXS, we can find further evidence and further elucidate details about the nature the nature of the Ni oxidation states in LNMO. This is presented in the next section.
E. Ni L-edge RIXS
All raw Ni L3-edge RIXS data are shown in Fig. S2 (ESI†) as a waterfall plot (each spectrum had the same acquisition time at approximately constant photon flux but normalized to equal height). Fig. 5 highlights a selection of spectra in the immediate Ni L3 resonance region for different SoC. Incident energies span from 852 to 857 eV with 0.5 eV increments, which corresponds to the XAS region delimited by black vertical dashed lines in Fig. 4. As a guide, we indicated the most likely formal oxidation states of the XAS resonance energies Ni2+ (AX), Ni3+ (BX), and Ni4+ (CX), respectively, to the incident photon energy axis. The general observation is that substantial spectral changes occurs during cycling through different SoC whereas the changes are largely reversible as re-lithiation restores the pristine spectra again.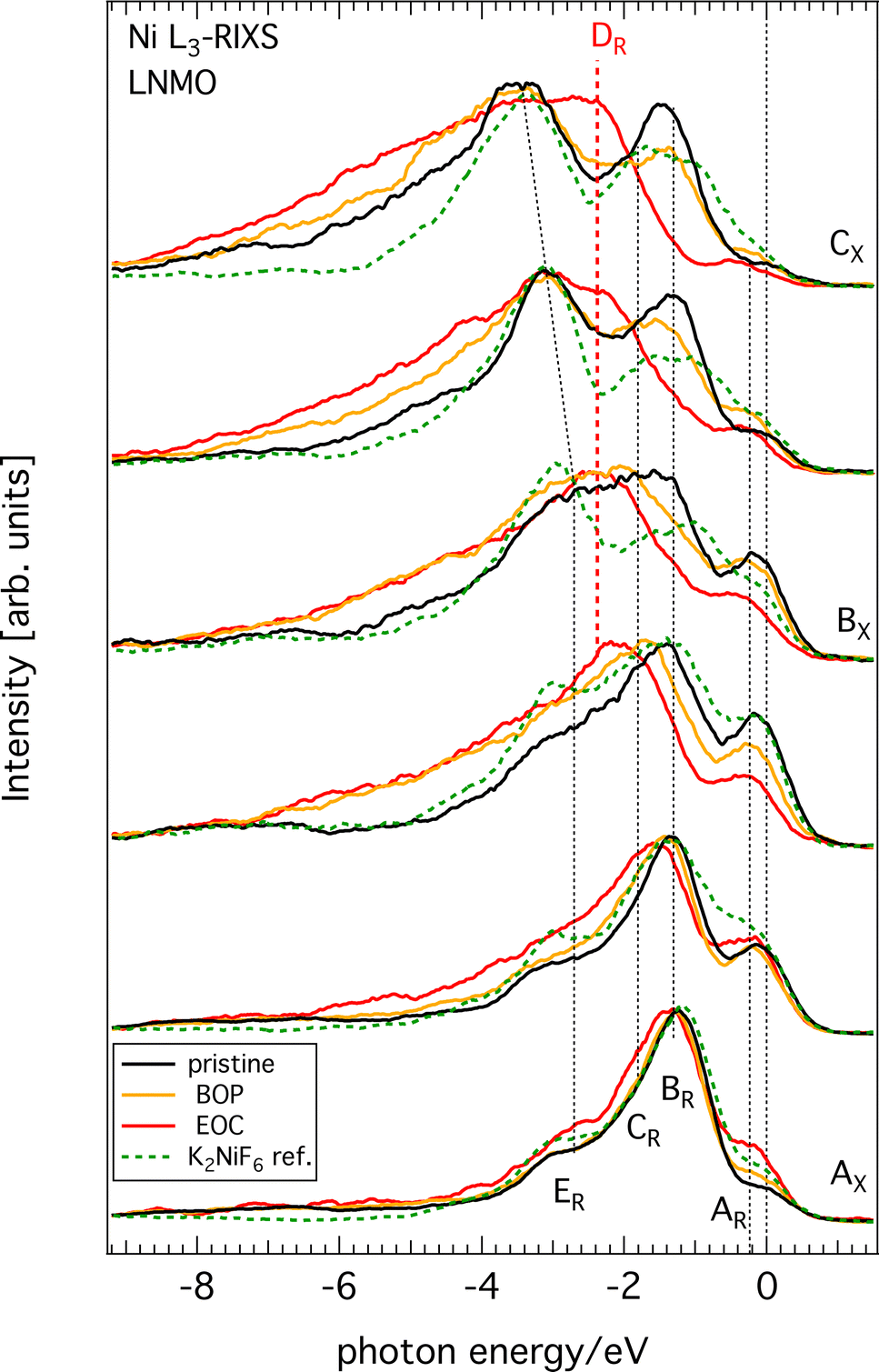 | ||
| Fig. 5 RIXS spectra across the Ni L3-resonances AX, BX, CX of Li1−xNi0.44Mn1.56O4 from three selected SoCs and from the Ni(IV) reference compound K2NiF6 (see color code legend). Each spectrum is normalized to its respective maximum to facilitate spectral shape comparison. | ||
We find that the behavior of the Ni L3-RIXS spectra of the pristine material (black traces) closely resembles that for a Ni2+ material such as NiO28 with salient features at the energy losses of approximately 0.2 eV (AR), 1.3 eV (BR), 1.8 eV (CR) and 3.1 eV (ER) at, which are indicated by the labelled, dashed black vertical lines in Fig. 5. Delithiation of LNMO leads to a gradual transformation and general broadening of the spectral profiles. More significantly however, is the emergence of a entirely new structure at about 2.4 eV (DR, dashed red vertical line) that resonates at the incident energy CX. Simultaneously, the peaks and BR and CR have lost most of their spectral weight at complete delithiation (EoC). It is not surprising to find that oxidation of the material also leads to spectral changes in Ni L3-RIXS, in particular at CX considering the changes observed in the Ni L-XAS. More unexpectedly however, there are substantial differences compared to e.g. the Ni L3-RIXS spectra of the ionically bonded Ni4+-model compound K2 NiF6 (see Fig. S3, ESI†). This suggests that the delithiated LNMO ground state deviates from a Ni 3d6 configuration. Particularly the Ni L3-RIXS spectra excited at incident energy CX in Fig. 5 suggest that key to the correct description of the delithiated LNMO ground state is the understanding of the emerging DR-feature.
III. Discussion
We observe that delithiation of LNMO leads to very small changes in the Mn L3-edge XAS during charging (Fig. 2). This suggests that Mn ions do not participate substantially to the charge compensation process. Instead, the high voltage plateau at 4.75 V of very similar materials has been attributed in earlier work to a possible double-electron-transfer in a Ni2+/4+ redox reaction29 whereas later Qiao et al.17 suggested two successive single-electron-transfers belonging to Ni2+/3+- and Ni3+/4+-redox reactions, respectively, that occur close in potential. The Ni L-edge XAS spectra in the present work (Fig. 4) corroborate the previous studies of similar compounds that observe a reversal in the intensity ratio between the high energy and low energy features of Ni L3-edge XAS upon delithiation. This is a clear sign that the Ni2+ ground state of the pristine cathode has oxidized in delithiated LNMO to form Ni3+ or Ni4+. On the other hand, we find that some important differences to ionic reference compound spectra from literature exist, namely, the peaks of the latter are more distinct, the ratio between the high- and low-energy peaks is larger, and there are differences in energy position. Although several factors can play a role for these differences, an important one is the greater contribution from ligand charge transfer states in LNMO than in the reference compounds. In other words, for the same amount of oxidation of the Ni–O bond in a given compound, the balance is tipped in favor of holes in the oxygen bands of delithiated LNMO. Even the calculations included in ref. 17 seem to indicate hole formation in itinerant bands of mixed oxygen/nickel orbital symmetry and achieves good agreement between the experimental and the calculated Ni L-XAS spectra. However, the calculations are still based on the formation of significant amounts of ligand oxygen holes. This is corroborated by our O K-RIXS observations (Fig. 3) revealing that significant restructuring takes place at the top of the oxygen valence band when charging LNMO across the high voltage plateau (delithiation).Turning to the Ni L-RIXS spectra (Fig. 5), we firstly note that they change gradually as a function of delithiation rather than in discrete steps that would be expected if successive transitions belonging to Ni2+/3+- and Ni3+/4+-redox reactions were involved. Secondly, the Ni L-RIXS spectra of delithiated LNMO compare poorly with a highly ionic Ni4+-compound suggesting that the ground state is not well-described ionically. Though some of the broadening at incident energy AX could be indicative of the admixture of a small degree of Ni 3d7 character (see e.g.30) it still cannot explain the overwhelming intensity from other spectral contributions. Additionally, we may compare to results of the isoelectronic Co2+ (3d7)31 and Fe2+ (3d6).32 RIXS spectra for incident energies around the main absorption feature of CoO31 show a strong dd-excitation at 1 eV and a number of peaks around 2.3 eV. While the former is also seen in our Ni RIXS spectra of LNMO, the latter dd-excitation pattern is clearly absent. RIXS spectra for incident photon energies at the main absorption feature of an Fe2+-compound32 with octahedral site symmetry reveal a rich RIXS spectrum with nine dd-excitations below 4 eV that have no counterpart in LNMO. Clearly, Ni RIXS spectra of delithiated LNMO show discrepancies to what one would expect from assuming a simple Ni 3d6 ground state.
Anionic activity of Ni-based cathode materials is not unprecedented and may be associated with various local crystal structures. While the binary, cation-disordered system LiNi2/3Nb1/3O2 has a linear Ni–O–Ni configuration that activates oxygen redox19 the local structure of LNMO is more complex33 and our experimental observations prompt future theoretical progress. On the other hand, we can compare the behavior of the O K-XAS/RIXS to that of NiO as a model system. For instance, when NiO is made Ni-deficient20 oxygen-derived states appear in the gap and the system becomes a negative charge transfer insulator with characteristics similar to our observations close to the Fermi level shown in Fig. 3(b) and (c). Note though that in the deficient-Ni case of ref. 20 the experimental Ni L-XAS is rather unchanged in contrast to delithiated LNMO in this work. However, the Ni L-XAS/RIXS of delithiated LNMO shows several intriguing similarities to the case of another charge transfer insulator as well, namely the low-temperature phase of NdNiO3.30 This system formally resides in a Ni3+-state but Bisogni et al. describe this as being a consequence of the formation of a Ni 3d8n-state (n = 1, 2) due to becoming a negative charge transfer insulator. The spectra of both NdNiO3 and delithiated LNMO excited at the Ni L-resonances AX and CX have well-matching energy loss structures together with a pronounced long tail of charge transfer excitations up to 9 eV energy loss. Furthermore, in NdNiO3 one expects an opening of a gap in the O 2p valence band that overlaps with a narrow Ni 3d-band (see Fig. 3(b) and (c)). Despite important differences in local bonding structure of LNMO compared to the mentioned Ni-oxide systems, Ni L- and O K-RIXS spectra reveal some commonalities in fundamental electronic structure. The anionic activity observed in LMNO during delithiation therefore appears to be (at least) supported by its non-ionic character Ni–O bonding characteristics.
In summary, a picture emerges where a gradual increase in oxygen hole density during delithiation of LNMO is leaving its fingerprints on both the O K-RIXS and the Ni L-RIXS spectra. This suggests that we instead consider here a ground state of delithiated LNMO where contributions from charge transfer states are included, i.e. Ni 3d8−x2−x (0 < x < 2). Judging from the Ni L-XAS, a value close to x ≤ 1 may be a reasonable estimate.
IV. Conclusions
We have studied the nature of the highly reversible redox processes during the first lithiation cycle of the intercalation compound Li1−xNi0.44Mn1.56O4 (LNMO) by using O K- and Ni L3-RIXS. Our O K- and Ni L-RIXS spectra shed a new light on the character of the formed holes and, generally, the description of the electronic structure of delithiated LNMO. Rehybridization during delithiation of LNMO is found to follow a non-trivial route including the formation of a narrow new O 2p band that is hybridized mostly with Ni 3d states while Mn 3d states experience very low activity during the entire first cycle. Low energy excitations (≤0.5 eV) observed in the O K-RIXS indicate a possible opening of a small gap in the O 2p states. The electronic structure of delithiated LNMO shows much more resemblance to negative charge transfer systems than with Ni oxides with highly ionic bonds. From this we conclude that delithiated LMNO is best described having a Ni 3d8−x2−x ground state with x ≲ 1. Finally, we point out that anionic contributions to the charge compensation processes in Li ion battery cathode materials can generally be highly non-trivial, thus inspiring future investigations with both high-resolution RIXS as well as with theoretical methods.
V. Methods
A. Synthesis
In order to obtain a cation-ordered (space group P4332) electrode material, powder material of micron-sized particles of LiNi0.44Mn1.56O4 (composition determined by ICP-OES) was heated up to 760 °C for 6 hours, and then cooled to 500 °C for 2 hours which was followed by re-heating to 760 °C for 2 hours and re-cooling to 710 °C for 2 hours. This heat treatment step was performed in order to compensate any oxygen deficiency that the starting powder material would have ref. 34. The powder was kept at 710 °C for 2 hours and then slowly cooled to 310 °C for 10 hours, where after it was furnace-cooled to room temperature. All the heat treatment was done under a pure oxygen atmosphere in an alumina crucible.B. SEM, PND, and Rietveld refinement
The morphology of powders were obtained using a Zeiss 1550 scanning electron microscope (SEM) and images are shown in Fig. S4 (ESI†). Structural characterization was made using the constant wavelength (1.5543 Å) powder neutron diffractometer PUS at the JEEP II reactor, Norway.35 Rietveld refinement was performed using the GSAS36 suite of programs with graphical user interface EXPGUI.37 Neutron diffraction and Rietveld refinement results are given in Fig. S5 (ESI†).C. Electrochemistry
Electrochemical cycling was performed on composite electrodes with an approximate capacity of 1.1 to 1.3 mA h cm−2. The composition of the electrodes was as follows: LiNi0.44Mn1.56O4 powders (90 wt%), carbon black (Imerys, C65, 5 wt%), and a poly(vinylidene difluoride)-based binder (5 wt%, PVdF-HFP, Kynar Flex 2801). The electrodes were dried under vacuum for 10 hours inside a glove box (Ar atmosphere, H2O < 5 ppm and O2 < 1 ppm). Li metal foil (125 m, Cyprus Foote Minerals) was used as a counter electrode. The cells were made as pouch cell using two layers of Celgard 2400 separator. The electrolyte was 120 μl of 1 M LiPF6 (Ferro Corp.) dissolved in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC (1:1 by volume, BASF). All cell assembly was performed in the same glovebox and cells were vacuum sealed.
:DEC (1:1 by volume, BASF). All cell assembly was performed in the same glovebox and cells were vacuum sealed.
Cycling of the cells was achieved by using a Novonix HPC (High Precision Charger) system at a temperature of 55 °C. Prior to applying a current the cells were kept at open circuit voltage (OCV) for a period of 10 hours. In order to prevent any artifacts due to kinetic limitations and to ensure a complete delithiation, charging was done relatively slowly at a C/5 current, followed by holding a constant voltage at 4.95 V (unless specified otherwise, all electrochemical potentials in this work are relative to Li/Li+) for 2 hours, and finally holding at OCV for 2 hours. The discharge rate was again C/5 followed by 2 hour constant voltage at 3.5 V. The uncycled electrode sample was obtained by inserting the pristine material electrode into a pouch cell and keeping it at OCV for 10 hours without running any current through it prior to disassembly. The other samples were obtained by cycling and stopping the current at points as shown in Fig. 1(a) before disassembly. The pouch cells were disassembled in an Ar-flow glovebox and the LNMO electrodes were removed. In order to remove any salt residues, the electrodes were washed with DMC three times (each time rinsing with 4 and 5 droplets). After drying, the electrodes were cut into a suitable size (∼2 mm × 5 mm) for spectroscopy and attached to strips of double-sided adhesive Cu tape. These were put into plastic boxes, vacuum sealed (twice) using Al laminated pouch cell case films in order to transport samples to the synchrotron facility and avoiding air exposure.
D. Soft X-ray spectroscopy
The LNMO electrode samples were transported in air-tight bags to the synchrotrons (iRIXS endstation at beamline 8.0.1 of the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory and BL27SU at SPring-8) where the strips were attached to the sample holders in an Ar-flow glovebox. The samples were then transferred in a sealed, air-tight suitcase to the vacuum chambers at the endstations BL 8.0.1 ALS38 and BL27SU SPring-8.39,40 Thus air exposure prior to the X-ray spectroscopy experiments was avoided. Both beamlines are equipped with high-resolution, high-brightness undulators and a spherical grating monochromator and variable line space grating spectrometers for performing RIXS measurements. BL27SU SPring-8 is equipped with an liquid nitrogen-cooled energy dispersive soft X-ray analyzer for recording partial fluorescence yield. XAS as well as RIXS measurements were performed. The monochromator bandwidth was set to 0.10–0.17 eV (5000 resolution) for XAS and 0.17–0.28 eV (3000 resolution) for RIXS. The combined instrumental (monochromator and spectrometer) resolution was 0.25 eV for O K-RIXS and 0.40 eV for Ni L-RIXS.XAS reflects the nature of the empty states above the Fermi level and is often used to determine the local oxidation states of the probed ion. XAS spectra were collected for Mn L-, Ni L-, and O K-edge by recording the bulk-sensitive partial fluorescence yield (PFY) and the surface-sensitive total electron yield (TEY), simultaneously. Due to the low-yield Mn L-emission, we extracted inverse partial fluorescence (IPFY) spectra from the O K-PFY. IPFY spectra are free from well-known distortions that total fluorescence yield or ordinary PFY spectra can suffer from. PFY (and IPFY) have a mean probing depth of about 100 nm while TEY reflects the surface-sensitive XAS signal probing only the outer layers to a depth of ∼10 nm. Following an established procedure,41 all XAS spectra have been normalized by the incident intensity I0, which was measured as drain current from an Au-coated mesh upstream of the sample. A linear pre-edge background was fitted and subtracted from each acquired raw spectrum. Thereafter the edge step between the pre-edge and a post-edge region beyond any absorption features was set to unity.
RIXS involves occupied and empty states around the Fermi level and the spectra can reflect both bandlike as well as excitonic states such as dd-excitations.24 The O K-RIXS spectra and the Ni L-RIXS spectra were acquired for equal amounts of time, respectively, yielding a normalization by incident photon intensity and reflecting the resonance variation of the XAS profile. In some Figures (as indicated), RIXS spectra have been normalized to peak area or peak intensity to facilitate shape comparison.
Data availability
The data that support the plots in this paper and other findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the Swedish Research Council (2014-6019, 2016-03545, and 2018-06465), STandUP for Energy, the Swedish Energy Agency (40495-1, 45518-1, 50745-1). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. B. Aktekin gratefully acknowledges the financial support from Volkswagen AG and Scania CV AB. The experiments at the BL27SU of SPring-8 were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2019A1459).References
- G. Assat and J.-M. Tarascon, Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- C. Zhao, Q. Wang and Y. Lu, et al., Review on anionic redox for high-capacity lithium- and sodium-ion batteries, J. Phys. D, 2017, 50(18), 183001 CrossRef.
- M. Saubanere, E. McCalla and J.-M. Tarascon, et al., The intriguing question of anionic redox in high-energy density cathodes for Li-ion batteries, Energy Environ. Sci., 2015, 9, 984–991 RSC.
- W. E. Gent, K. Lim and Y. Liang, et al., Coupling between oxygen redox and cation migration explains unusual electrochemistry in lithium-rich layered oxides, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- K. Luo, M. R. Roberts and N. Guerrini, et al., Anion Redox Chemistry in the Cobalt Free 3d Transition Metal Oxide Intercalation Electrode Li[Li0.2Ni0.2Mn0.6]O2, J. Am. Chem. Soc., 2016, 138(35), 11211–11218 CrossRef CAS PubMed.
- J. Xu, M. Sun and R. Qiao, et al., Elucidating anionic oxygen activity in lithium-rich layered oxides, Nat. Commun., 2018, 9, 947 CrossRef PubMed.
- R. A. House, U. Maitra and M. A. Pérez-Osorio, et al., Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- D.-H. Seo, J. Lee and A. Urban, et al., The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Liexcess cathode materials, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- E. J. Kim, L. Anh, M. Laurent and C. Duda, et al., Oxygen Redox Activity through a Reductive Coupling Mechanism in the P3-Type Nickel-Doped Sodium Manganese Oxide, ACS Appl. Energy Mater., 2020, 3(1), 184–191 CrossRef CAS.
- T. Mizokawa, Y. Wakisaka and T. Sudayama, et al., Role of Oxygen Holes in LixCoO2 Revealed by Soft X-Ray Spectroscopy, Phys. Rev. Lett., 2013, 111, 056404 CrossRef CAS PubMed.
- X.-L. Xu, S.-X. Deng and H. Wang, et al., Research Progress in Improving the Cycling Stability of High-Voltage LiNi0.5Mn1.5O4 Cathode in Lithium-Ion Battery, Nano-Micro Lett., 2017, 9(2), 22 CrossRef PubMed.
- T.-F. Yi, J. Mei and Y.-R. Zhu, Key strategies for enhancing the cycling stability and rate capacity of LiNi0.5Mn1.5O4 as high-voltage cathode materials for high power lithium-ion batteries, J. Power Sources, 2016, 316, 85–105 CrossRef CAS.
- Y. Wei, K.-B. Kim and G. Chen, Evolution of the local structure and electrochemical properties of spinel LiNixMn2−xO4 (0 < x < 0:5), Electrochim. Acta, 2006, 51(16), 3365–3373 CrossRef CAS.
- H. Duncan, B. Hai and M. Leskes, et al., Relationships between Mn3+ Content, Structural Ordering, Phase Transformation, and Kinetic Properties in LiNixMn2−xO4 Cathode Materials, Chem. Mater., 2014, 26(18), 5374–5382 CrossRef CAS.
- T. Okumura, M. Shikano and H. Kobayashi, Contribution of oxygen partial density of state on lithium intercalation/de-intercalation process in LixNi0.5Mn1.5O4 spinel oxides, J. Power Sources, 2013, 244(1–2), 544–547 CrossRef CAS.
- J. Zhou, D. Hong and J. Wang, et al., Electronic structure variation of the surface and bulk of a LiNi0.5Mn1.5O4 cathode as a function of state of charge: X-ray absorption spectroscopic study, Phys. Chem. Chem. Phys., 2014, 16(27), 13838–13842 RSC.
- R. Qiao, L. A. Wray and J.-H. Kim, et al., Direct Experimental Probe of the Ni(II)/Ni(III)/Ni(IV) Redox Evolution in LiNi0.5Mn1.5O4 Electrodes, J. Phys. Chem. C, 2015, 119(49), 27228–27233 CrossRef CAS.
- R. Qiao, Y. Wang and P. Olalde-Velasco, et al., Direct evidence of gradient Mn(II) evolution at charged states in LiNi0.5Mn1.5O4 electrodes with capacity fading, J. Power Sources, 2015, 273, 1120–1126 CrossRef CAS.
- R. Fukuma, M. Harada and W. Zhao, et al., Unexpectedly Large Contribution of Oxygen to Charge Compensation Triggered by Structural Disordering: Detailed Experimental and Theoretical Study on a Li3NbO4-NiO Binary System, ACS Cent. Sci., 2022, 8, 775–794 CrossRef CAS PubMed.
- H. Peng, Y. Li and W. Lin, et al., Deterministic conversion between memory and threshold resistive switching via tuning the strong electron correlation, Sci. Rep., 2012, 2, 442 CrossRef PubMed.
- J. Cabana, M. Casas-Cabanas and F. O. Omenya, et al., Composition-Structure Relationships in the Li-Ion Battery Electrode Material LiNixMn2−xO4, Chem. Mater., 2012, 24(15), 2952–2964 CrossRef CAS PubMed.
- R. Qiao, T. Chin and S. J. Harris, et al., Spectroscopic fingerprints of valence and spin states in manganese oxides and fluorides, Curr. Appl. Phys., 2013, 13(3), 544–548 CrossRef.
- R. Qiao, K. Dai and J. Mao, et al., Revealing and suppressing surface Mn(II) formation of Na0.44MnO2 electrodes for Na-ion batteries, Nano Energy, 2015, 16(3), 186–195 CrossRef CAS.
- L.-C. Duda, J. Downes and C. McGuinness, et al., Bandlike and excitonic states of oxygen in CuGeO3: Observation using polarized resonant soft-X-ray emission spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61(6), 4186 CrossRef CAS.
- H. Wang, P. Ge and C. G. Riordan, et al., Integrated X-ray L Absorption Spectra. Counting Holes in Ni Complexes, J. Phys. Chem. B, 1998, 102(42), 8343–8346 CrossRef CAS.
- F. M. F. de Groot, Multiplet effects in X-ray spectroscopy, Coord. Chem. Rev., 2005, 249(1), 31–63 CrossRef CAS.
- H. Liu, J. Zhou and L. Zhang, et al., Insight into the Role of Metal–Oxygen Bond and O 2p Hole in High-Voltage Cathode LiNixMn2−xO4, J. Phys. Chem. C, 2017, 121(30), 16079–16087 CrossRef CAS.
- G. Ghiringhelli, M. Matsubara and C. Dallera, et al., NiO as a test case for high resolution resonant inelastic soft X-ray scattering, J. Phys.: Condens. Matter, 2005, 17(35), 5397–5412 CrossRef CAS.
- Z. Moorhead-Rosenberg, A. Huq and J. B. Goodenough, et al., Electronic and Electrochemical Properties of Li1−xMn1.5Ni0.5O4 Spinel Cathodes As a Function of Lithium Content and Cation Ordering, Chem. Mater., 2015, 27(20), 6934–6945 CrossRef CAS.
- V. Bisogni, S. Catalano and R. J. Green, et al., Groundstate oxygen holes and the metal-insulator transition in the negative charge–transfer rare-earth nickelates, Nat. Commun., 2016, 7, 13017 CrossRef CAS PubMed.
- M. M. van Schooneveld, R. Kurian and A. Juhin, et al., Electronic Structure of CoO Nanocrystals and a Single Crystal Probed by Resonant X-ray Emission Spectroscopy, J. Phys. Chem. C, 2012, 116(29), 15218–15230 CrossRef CAS.
- A. W. Hahn, B. E. Van Kuiken and M. Al Samarai, et al., Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p 3d RIXS, Inorg. Chem., 2017, 56(14), 8203–8211 CrossRef CAS PubMed.
- J.-H. Kim, S.-T. Myung and C. S. Yoon, et al., Comparative Study of LiNi0.5Mn1.5O4-delta and LiNi0.5Mn1.5O4 Cathodes Having Two Crystallographic Structures: Fd3m and P4332, Chem. Mater., 2004, 16, 906–914 CrossRef CAS.
- B. Aktekin, M. Valvo and R. I. Smith, et al., Cation Ordering and Oxygen Release in LiNi0.5−xMn1.5+xO4−y (LNMO): In Situ Neutron Diffraction and Performance in Li Ion Full Cells, ACS Appl. Energy Mater., 2019, 2(5), 3323–3335, DOI:10.1021/acsaem.8b02217.
- B. C. Hauback, H. Fjellvag and O. Steinsvoll, The High Resolution Powder Neutron Diffractometer PUS at the JEEP II Reactor at Kjeller in Norway, J. Neutron Res., 2000, 8, 215–232 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR, 2004, pp. 86–748 Search PubMed.
- B. H. Toby, EXPGUI, a Graphical User Interface for GSAS, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- R. Qiao, Q. Li and Z. Zhuo, et al., High-efficiency in situ resonant inelastic X-ray scattering (iRIXS) endstation at the Advanced Light Source, Rev. Sci. Instrum., 2017, 88(3), 033106 CrossRef PubMed 3350–3355.
- H. Ohashi, E. Ishiguro and Y. Tamenori, et al., Outline of soft X-ray photochemistry beamline BL27SU of SPring-8, Nucl. Instrum. Methods Phys. Res., 2001, 467–468, 529–532 CrossRef CAS.
- RIKEN/JASRI, BL27SU C-branch, 2019, URL: https://www.spring8.or.jp/wkg/BL27SU/instrument/lang-en/INS-0000001446/instrument_view#c3 (visited on 08/04/2019).
- J. Stöhr, NEXAFS Spectroscopy, Springer-Verlag, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00321j |
| This journal is © The Royal Society of Chemistry 2023 |