
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in alkynylation with hypervalent iodine reagents
Eliott
Le Du
 and
Jérôme
Waser
*
and
Jérôme
Waser
*
Laboratory of Catalysis and Organic Synthesis, Institute of Chemical Sciences and Engineering École Polytechnique Fédérale de Lausanne EPFL, SB ISIC, LCSO, BCH 4306, 1015, Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 19th January 2023
Abstract
Although alkynes are one of the smallest functional groups, they are among the most versatile building blocks for organic chemistry, with applications ranging from biochemistry to material sciences. Alkynylation reactions have traditionally relied on the use of acetylenes as nucleophiles. The discovery and development of ethynyl hypervalent iodine reagents have allowed to greatly expand the transfer of alkynes as electrophilic synthons. In this feature article the progress in the field since 2018 will be presented. After a short introduction on alkynylation reactions and hypervalent iodine reagents, the developments in the synthesis of alkynyl hypervalent iodine reagents will be discussed. Their recent use in base-mediated and transition-metal catalyzed alkynylations will be described. Progress in radical-based alkynylations and atom-economical transformations will then be presented.
 Eliott Le Du | Eliott Le Du studied Chemistry at École Normale Supérieure de Lyon in France and at École Polytechnique Fédérale de Lausanne (EPFL) in Switzerland. He conducted his Master's Thesis in the group of Prof. Kay Severin in collaboration with the group of Prof. Jérôme Waser at EPFL, working on triazene-activated donor–acceptor cyclopropanes. Since 2018, he has been pursuing his doctoral studies in the Waser group. His research interests focus on the development of new alkynyl hypervalent iodine reagents and the functionalization of biomolecules. |
 Jérôme Waser | Jerome Waser was born in Sierre, Valais, Switzerland. He studied chemistry at ETH Zurich, where he obtained his PhD in 2006 with Prof. Erick M. Carreira. In 2006, he joined Prof. Barry M. Trost at Stanford University as a SNF postdoctoral fellow. Since October 2007 he has been professor of organic chemistry at the Ecole Polytechnique Fédérale de Lausanne (EPFL), where he was promoted to full professor in 2019. Since 2020, he has also been co-director of the NCCR Catalysis of the Swiss National Science Foundation. |
1. Introduction and context
Alkynes are highly versatile functional groups in organic chemistry, which also have found applications in applied fields such as biochemistry and material sciences.1,2 Among the main transformations that alkynes can undergo, the 1,3-dipolar cycloaddition with organic azides, also known as Huisgen cycloaddition or “Click Chemistry”, is of the utmost importance and was recognized with the Nobel prize in 2022.3–5 The high interest of the scientific community in alkynes has led to constant efforts to develop new flexible and efficient strategies to access them.Traditionally, most methods to access alkynes by transfer of a triple bond relied on the deprotonation of terminal alkynes generating nucleophilic acetylide intermediates that could then react with electrophiles (Scheme 1A). For instance, it is a method of choice to access propargylic alcohols or amines.6,7 Alternatively, acetylides can be involved in cross-coupling reactions such as the Sonogashira coupling,8 the Glaser dimerization or the Cadiot–Chodkiewicz reaction.9 While terminal alkynes are intrinsically nucleophilic, their reactivity can be reversed by installing an electron-withdrawing leaving group at an extremity (Scheme 1B).10 Initially, haloalkynes have been investigated in transition-metal catalyzed carbon–carbon and heteroatom–carbon couplings.11 Later, alkynyl sulfones have emerged as valuable partners for the alkynylation of nucleophilic radicals.12 The in situ oxidation of terminal alkynes has been also investigated but initially relied on toxic and highly reactive oxidants, which limited the application of these strategies.10 Since the discovery of the exceptional reactivity of the hypervalent bond, hypervalent iodine reagents have attracted the interest of synthetic chemists.13–21 In particular, alkynyl iodonium salts and ethynylbenziodoxolone (EBX) reagents have been particularly prolific as electrophilic alkyne synthons.22–25
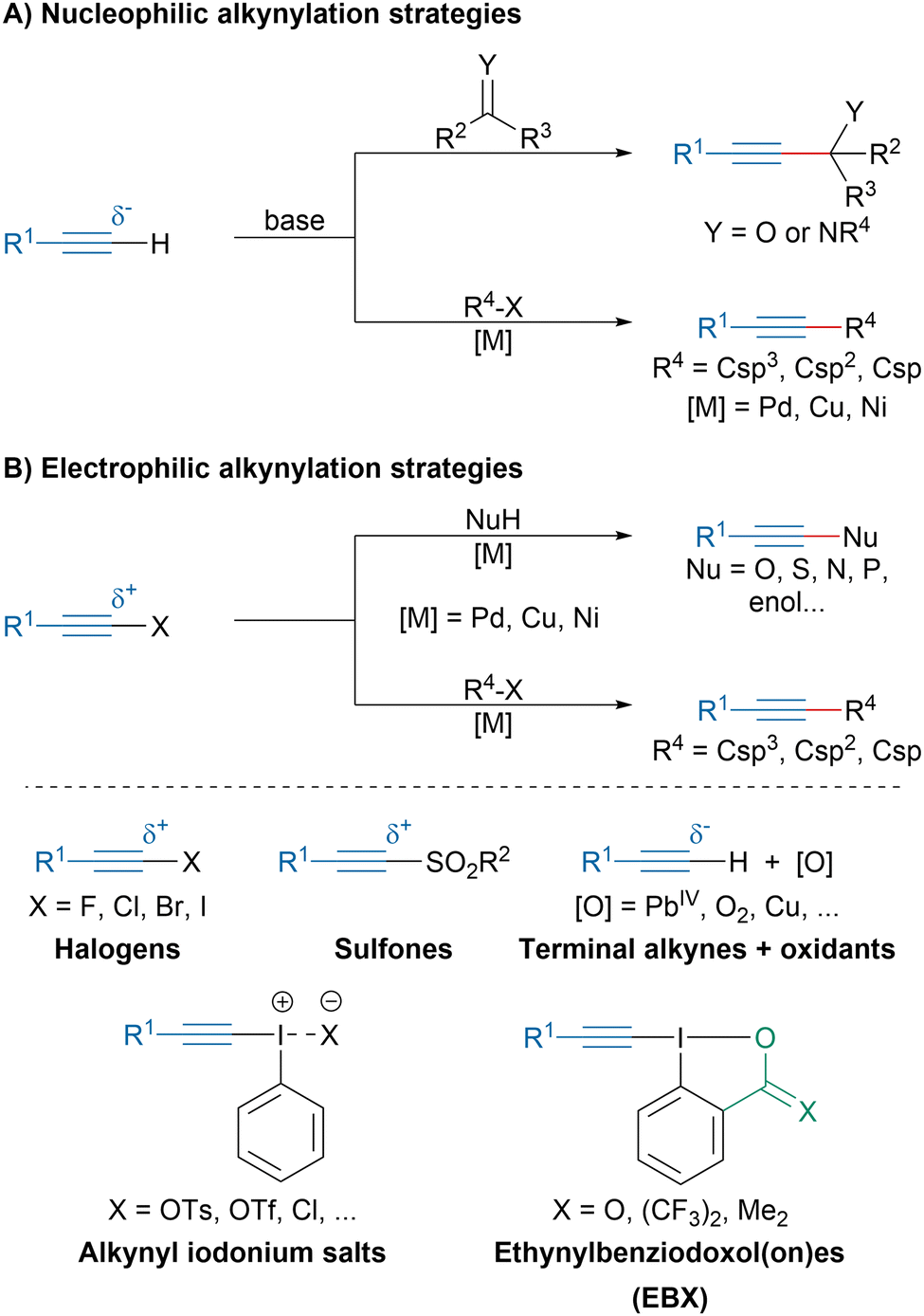 | ||
| Scheme 1 Nucleophilic and electrophilic alkynylation strategies. | ||
The peculiar reactivity of hypervalent iodine reagents arises from the 3-center-4-electron bond or hypervalent bond, which is longer, more polarized and weaker than a standard covalent bond leading to a higher electrophilic reactivity (Scheme 2A).26,27 Although the concept of hypervalency is still debated,28–30 it has been largely accepted to describe the unusual properties of hypercoordinated main-group elements. In recent years, most efforts have focused on cyclic hypervalent iodine reagents due to their higher stability (Scheme 2B).31 The additional stabilization has been proposed to arise from locking the iodine atom in an iodoheterocycle leading to an enhanced orbital overlapping.32 Moreover, the confinement of the oxygen lone pairs out of the 3c–4e plane disfavors the reductive elimination between the axial ligands.33
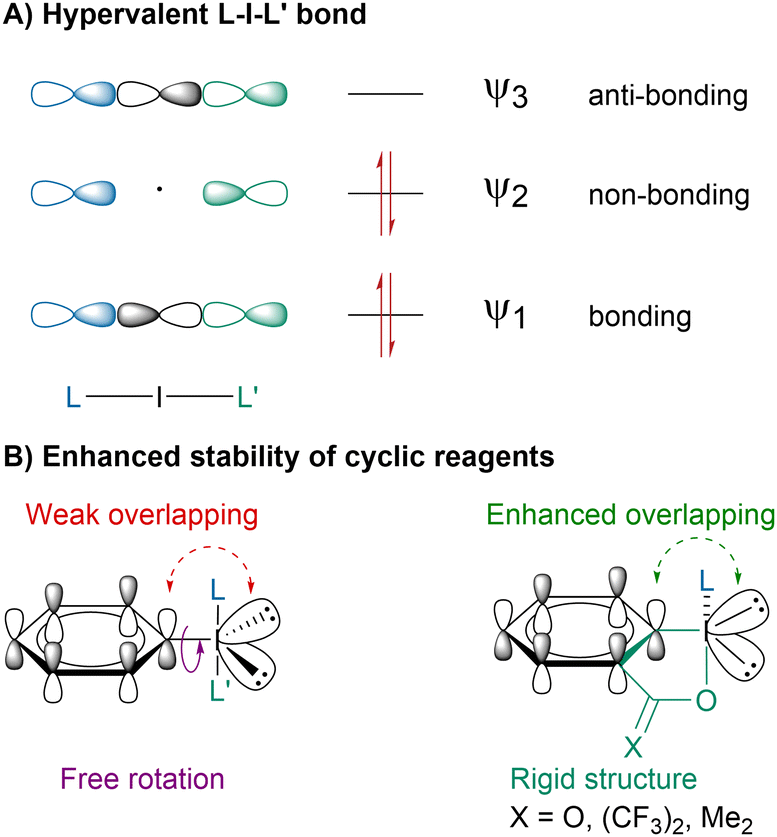 | ||
| Scheme 2 Hypervalent iodine bond and enhanced stability of cyclic reagents. | ||
Although iodonium salts were initially investigated for the alkynylation of nucleophiles, their instability limited their wider application.34,35 Since 2009, bench stable EBX reagents have emerged as powerful electrophilic alkyne synthons in metal-free and metal-catalyzed alkynylation of transient radicals, heteroatom and carbon-centered nucleophiles.31,36 The purpose of this feature article is to present the progress in the field since our last reviews in 201824,25 up to October 2022. The development of alkynyl hypervalent iodine reagents will first be described (Chapter 2). Then base-mediated (Chapter 3) and transition-metal mediated (Chapter 4) alkynylation reactions will be presented. Finally radical-based transfer of acetylenes (Chapter 5) and atom-economical transformations (Chapter 6) will be discussed.
2. Development of alkynyl hypervalent iodine reagents
Since the discovery of EBX reagents by the Ochiai group, efforts have focused on improving their synthesis.37 Zhdankin and coworkers reported a first general two-step procedure to access alkyl-, aryl- or silyl-substituted EBX reagents via hydroxybenziodoxole (2) (Scheme 3A).38 Following a renewed interest for these reagents, the Olofsson group developed a one-pot two-step procedure converting 2-iodobenzoic acid (1) into EBX reagents using pinacol alkynylboronates such as 5 (Scheme 3B).39 Interestingly this strategy could also be employed to access alkynyl iodonium salts. Our group later reported a similar protocol but using commercially available terminal alkyne 6 for the synthesis of TIPS-EBX (4a) on multigram scale (Scheme 3C).40 However, lower yields were obtained with alkyl or aryl substituted alkynes with this protocol and the Olofsson or Zhdankin protocols are usually preferred to synthesize them.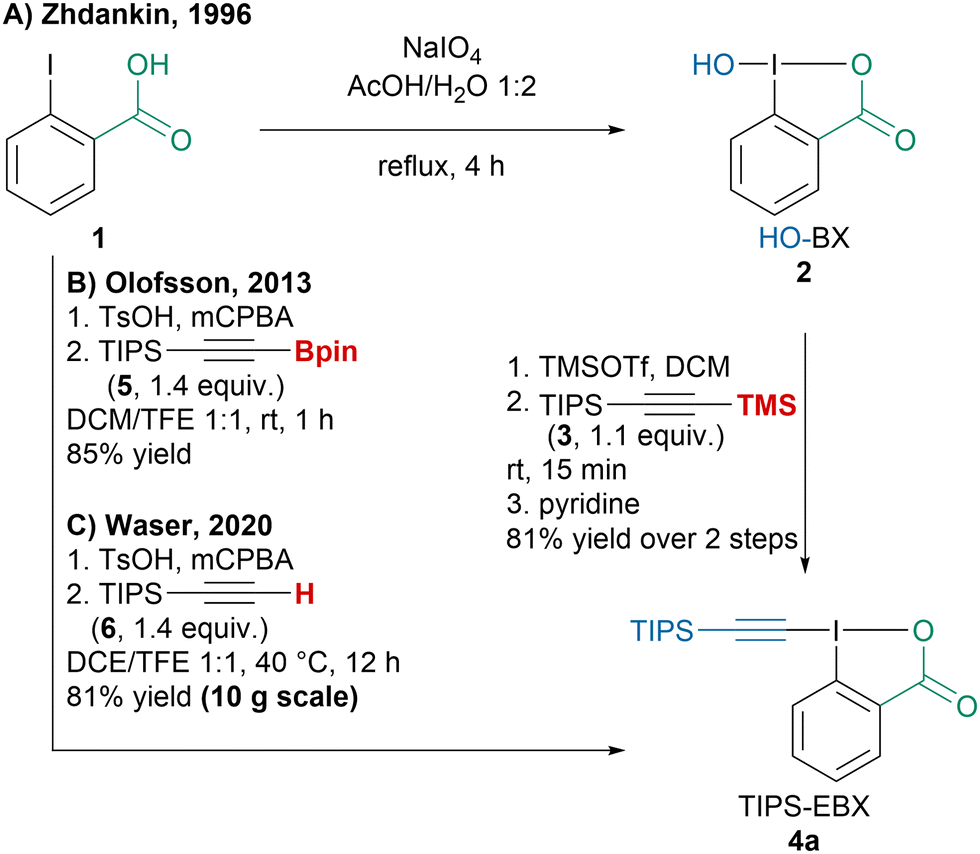 | ||
| Scheme 3 Improvements in the synthesis of EBX reagents (illustrated with TIPS-EBX (4a)). | ||
In 2022, our group disclosed a new procedure to access a broad variety of alkyl-, aryl- or silyl-substituted EBX reagents starting from TsO-BX (7) and more stable alkynyltrifluoroborates 8 (Scheme 4).41 The transformation tolerated a variety of solvents, did not require any additive and produced EBX reagents in high purity without purification, which allowed to directly apply them for the functionalization of different nucleophiles.
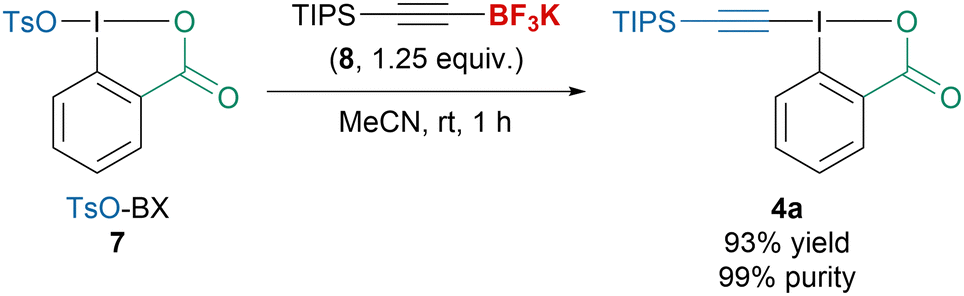 | ||
| Scheme 4 Synthesis of TIPS-EBX (4a) from TsO-Bx (7). | ||
Interestingly, Itoh, Tada and coworkers reported the synthesis of ethynylbenziodoxolone (4c) as a self-assembled double-layered honeycomb complex with MeCN, which allowed to isolate the otherwise highly unstable reagent (Scheme 5).42 Reagent 4c was then successfully used for the N-ethynylation of various sulfonamides and amino-acids. However, to avoid degradation of the reagent, all the reactions were run in the dark.
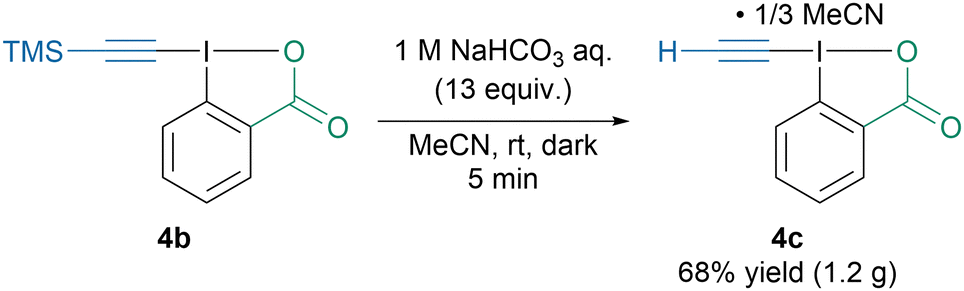 | ||
| Scheme 5 Synthesis of EBX-MeCN complex 4c. | ||
In 2021, Kang, Chen and coworkers reported the first synthesis of spirocyclic alkynyl hypervalent iodine reagents (Scheme 6).43 Instead of reacting with the alkyne to form ynamides or vinylbenziodoxolone reagents,44 under basic conditions, N-(oxy)-2-bromo-2-methylpropanamides reacted with the carbonyl group leading to spirocyclic reagents in good to excellent yields. These reagents could then be applied to the synthesis of benzoxazepine derivatives and diynes. Later, the spirocyclic reagents were used to access aryl ethers and thioethers.45,46
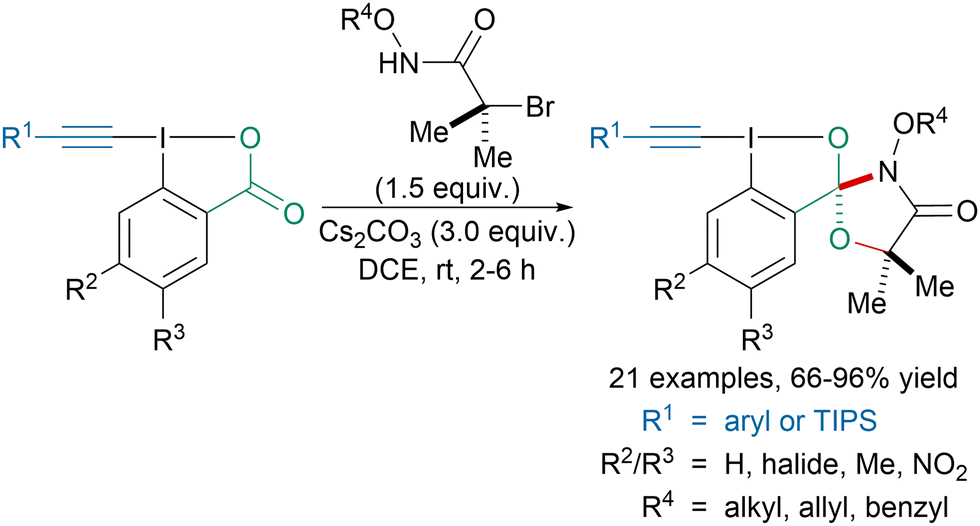 | ||
| Scheme 6 Synthesis of spirocyclic alkynyl hypervalent iodine reagents. | ||
With the aim of synthesizing α-alkynyl amino acid derivatives, our group developed a new class of reagents: ethynylbenziodazolone (EBZ) bearing an amide instead of a carboxylic acid in the iodoheterocycle (Scheme 7).47 The reagents were readily accessed via a one-pot two-step procedure from the corresponding iodobenzamide and either terminal or TMS-substituted alkynes. The developed regents exhibited similar reactivity as standard EBX reagents in alkynylation of β-ketoesters, thiols or indoles.
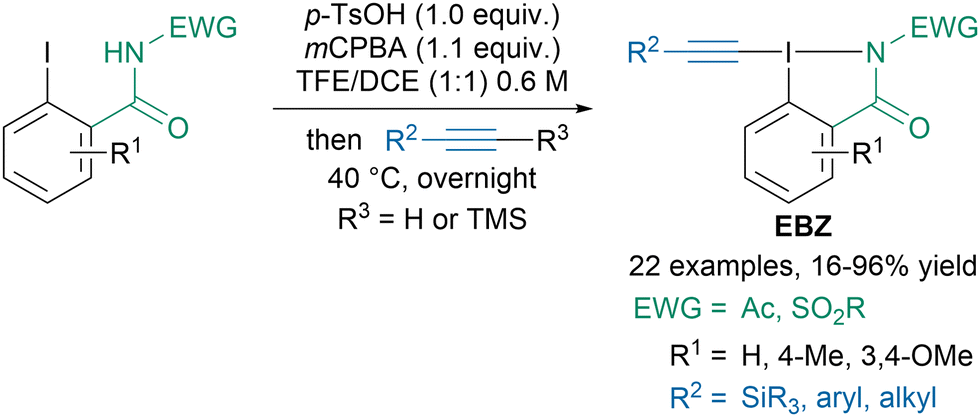 | ||
| Scheme 7 Synthesis of ethynylbenziodazolone (EBZ) reagents. | ||
As EBX reagents offer only limited possibilities for reactivity fine-tuning via structure modification, our group developed new N-heterocyclic reagents with increased structural flexibility (Scheme 8).48 For instance, we could synthesize mono- (10) and bis-protected (11) amidine based reagents as well as ethynylbenziodazole reagent (12) from 2-iodobenzonitrile. Through a collaboration with the Magnier group we could also access a racemic and an enantiopure sulfoximine based reagent (13). Unfortunately, they exhibited lower reactivity than the benchmark TIPS-EBX reagent (4a) in the reactions tested to date. More recently, the Nachtsheim group developed N-heteroaromatic alkynyl reagents.49
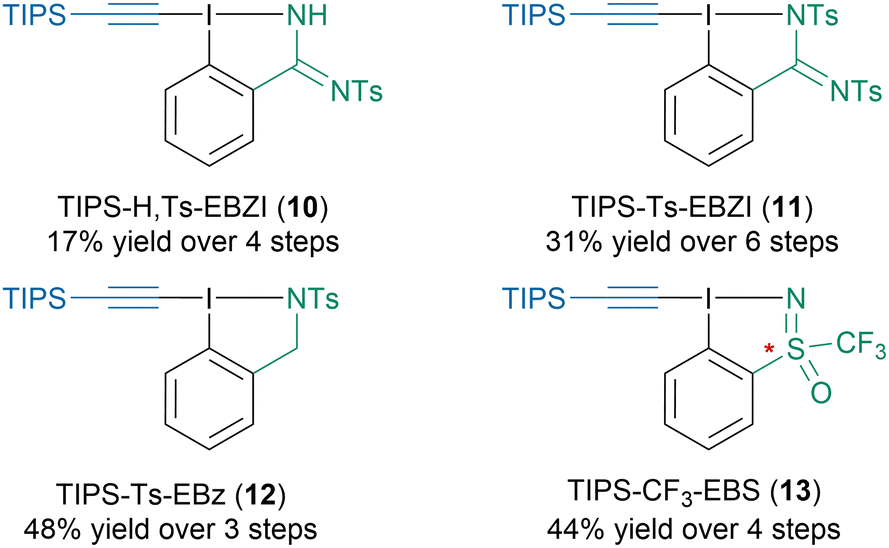 | ||
| Scheme 8 N-Heterocyclic alkynyl hypervalent iodine reagents developed in the Waser group. | ||
3. Base mediated alkynylation reactions
3.1. Alkynylation of C-nucleophiles
Since their first applications in electrophilic alkynylation of enolates,50,51 hypervalent iodine reagents have been widely used to install alkynes on the α-position of carbonyls, especially in total synthesis.52–58 In addition, the Bisai group recently reported a transition-metal free alkynylation of 2-oxindoles using EBX reagents.59 Using thiourea phosphonium salt catalysts, Wu and coworkers could develop an asymmetric alkynylation of azlactones and thiazolones leading to precursors of quaternary α-amino acids (Scheme 9).60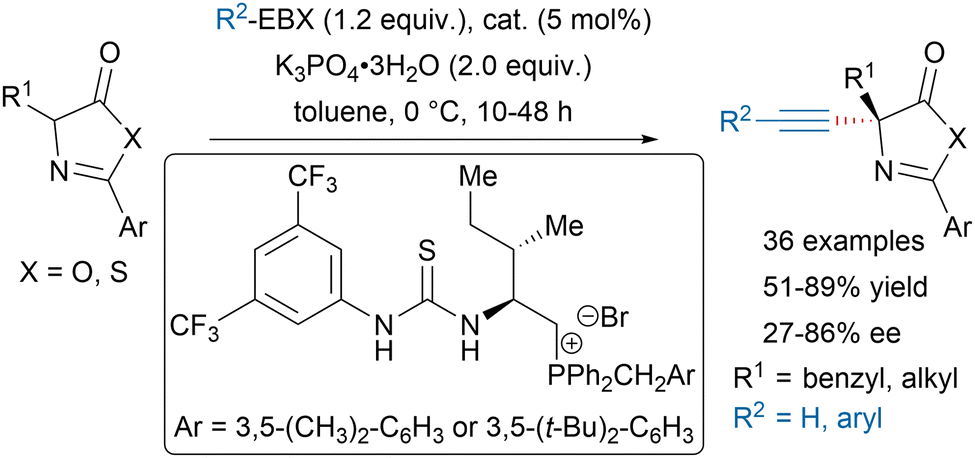 | ||
| Scheme 9 Asymmetric alkynylation of azlactones and thiazolones. | ||
In addition, the Teodoro and Silva reported a protocol to access α-alkynyl β-substituted ketones by trapping an enolate, generated via Michael addition, with EBX reagents.61
In 2020, the Kalek group disclosed a N-heterocyclic carbene (NHC) catalyzed alkynylation of aromatic aldehydes with iodonium salts leading to ynones in moderate to excellent yields (Scheme 10).6213C-labelling experiments and computational mechanistic studies revealed that the reaction might proceed via direct substitution at the α-acetylenic carbon. This report represented the first example in which this mechanism was significantly favored for a C-centered nucleophile compared to the typically prevalent pathway involving initial attack at the β-position. Such addition would lead to a vinylidene carbene, which upon 1,2-shift would provide the product as originally proposed by the Ochiai group.63
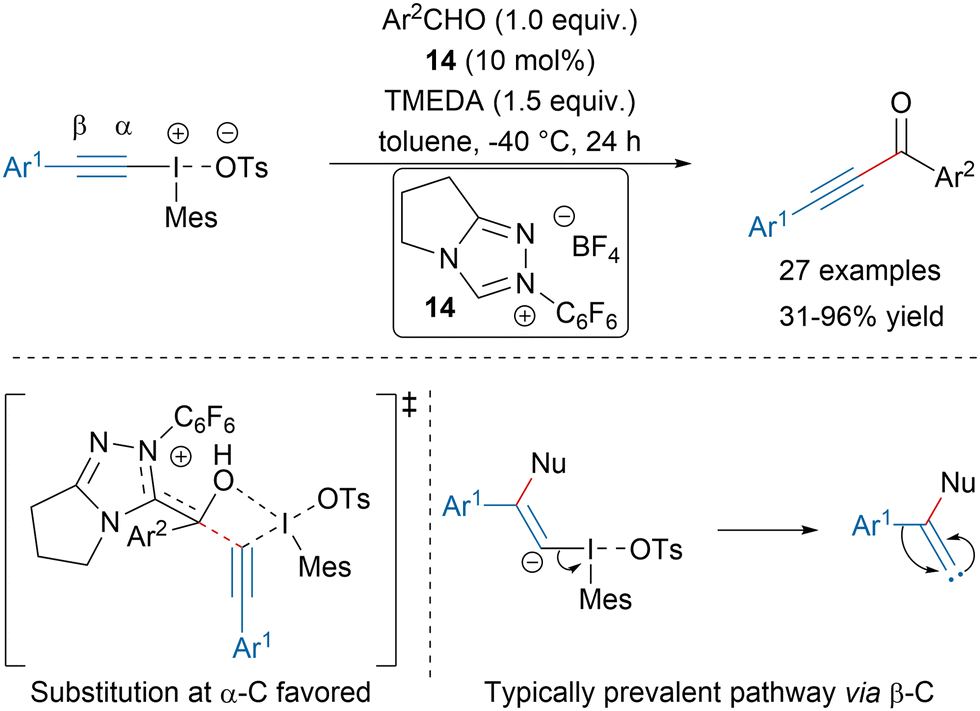 | ||
| Scheme 10 NHC-catalyzed synthesis of ynones. | ||
M. Waser and coworkers developed a transition-metal free Cadiot–Chodkiewicz coupling of terminal alkynes and EBX reagents leading to unsymmetrical 1,3-diynes.64 Gold-catalysis had previously been shown to be able to promote this transformation as well.65–67
3.2. Alkynylation of N-nucleophiles
The formation of N–Csp bonds has attracted a lot of attention in the last decades due to the high versatility of ynamides.68–70 Hypervalent iodine reagents have emerged as valuable tools especially for nitrogen nucleophiles reacting poorly in copper-catalyzed alkynylation transformations. Li, Zhang and coworkers recently reported a metal-free synthesis of ynamides with in situ generated alkynyl iodonium salts from PIDA (15) and dibenzylsulfonimide (Scheme 11).71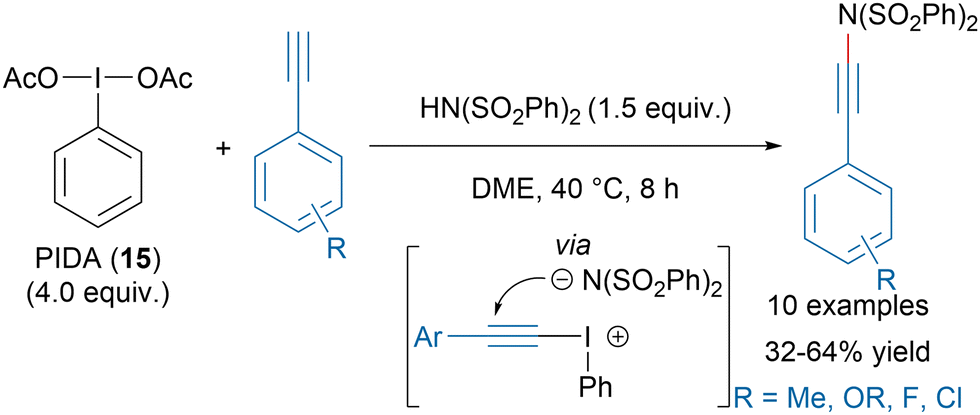 | ||
| Scheme 11 Ynamide synthesis with in situ generated alkynyl iodonium salts. | ||
Building upon their previous works on the formation of heterocycles with EBX reagents,72,73 Wen and coworkers developed a TBAF-mediated synthesis of benzofuran derivatives from N-phenoxyamides and TIPS-EBX (4a) (Scheme 12).74 When R2 is an alkyl, in situ formed H-EBX (4c) would be trapped by the N-phenoxyamide to form ynamide II after 1,2-hydride shift from intermediate I. [3,3]-Rearrangement followed by cyclization would lead to benzofurans. In contrast, when R2 is an aryl and in presence of a Na2CO3, vinylidene carbene I would undergo carbene insertion on the aryl ring followed by [3,3]-rearrangement, Michael addition and cyclization.
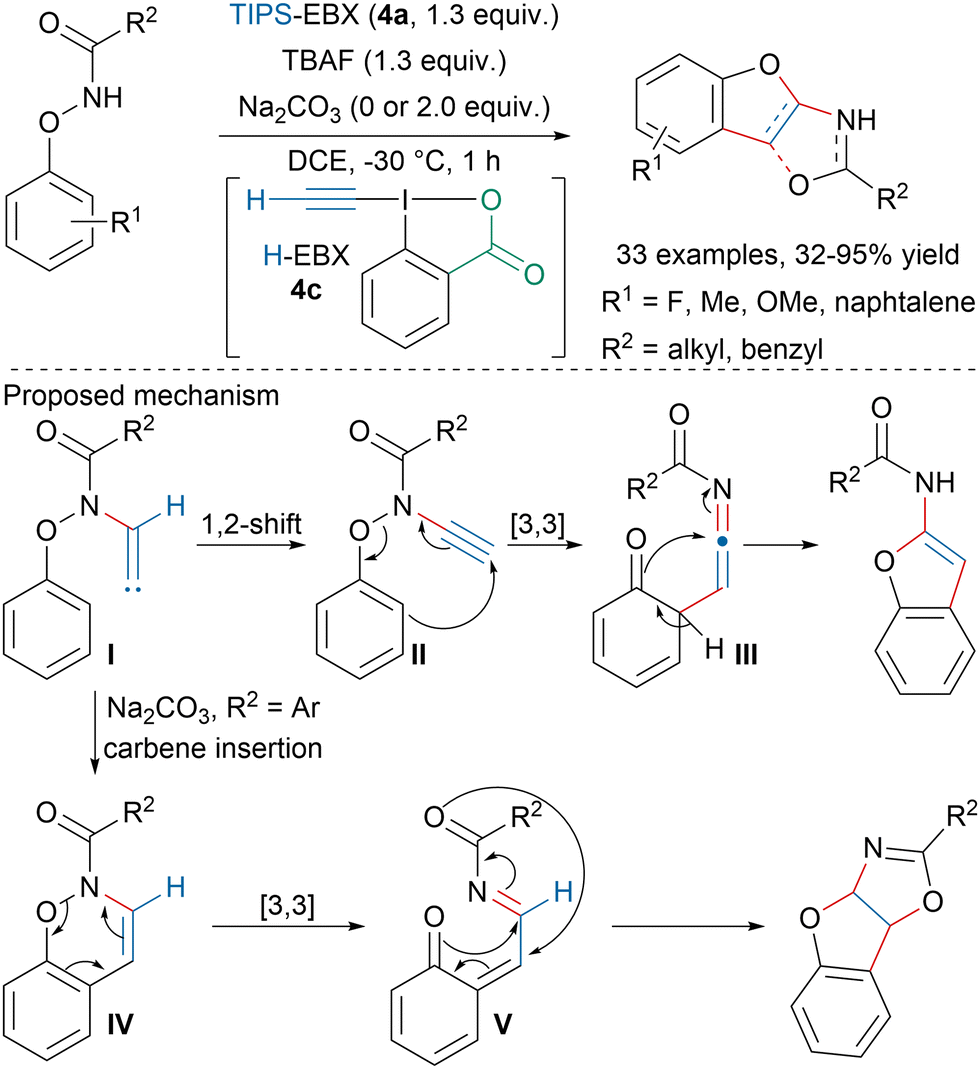 | ||
| Scheme 12 Synthesis of benzofuran derivatives from EBX and N-phenoxyamides. | ||
3.3. Alkynylation of S-nucleophiles and application to the functionalization of biomolecules
Alkynyl sulfoxides are valuable building blocks in organic chemistry with unique reactivities. However the lack of robust procedures to synthesize them has limited their wider applications. In 2019, our group disclosed a new method to access alkynyl sulfoxides from EBX reagents and sulfenate anions formed in situ via a retro-Michael reaction (Scheme 13).75 This protocol allowed to avoid the use of strong oxidants often leading to overoxidation, as well as nucleophilic alkyne derivatives, which could react with the products through 1,4-additions.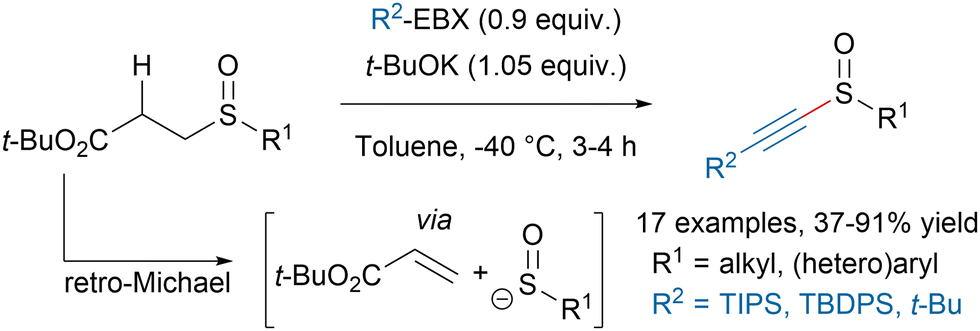 | ||
| Scheme 13 Metal-free synthesis of alkynyl sulfoxides. | ||
Owing to their high reactivity and biocompatibility, hypervalent iodine reagents have recently emerged as powerful tools for the functionalization of biomolecules.76 In the last decade, our group has extensively studied the alkynylation of thiols and especially of cysteines with the goal to functionalize biomolecules.77–79 Interestingly, our group showed that depending on the reaction conditions vinylbenziodoxolone (VBX) formation could also occur, which led to the development of a “doubly orthogonal” labeling of peptides with EBX reagents.80 Moreover, in 2020, in collaboration with the Adibekian and Chaubet groups, we reported a method to ethynylate cysteine residues on peptides and proteins in vitro and in living cells (Scheme 14).81 It was found that under slightly basic buffer conditions TMS-EBX reagents JW-RT-01 (4d) and JW-RT-03 (4e) would be desilylated in situ generating the corresponding highly reactive H-EBX reagents that could be trapped by cysteine residues. JW-RT-03 could efficiently alkynylate cysteines in both HeLa lysates in vitro and in living cells, showing that this reagent could be used for cysteine proteomic profiling. TMS-EBX (4b) was also evaluated for the bioconjugation of the bioactive antibody trastuzumab and showed promising reactivity.
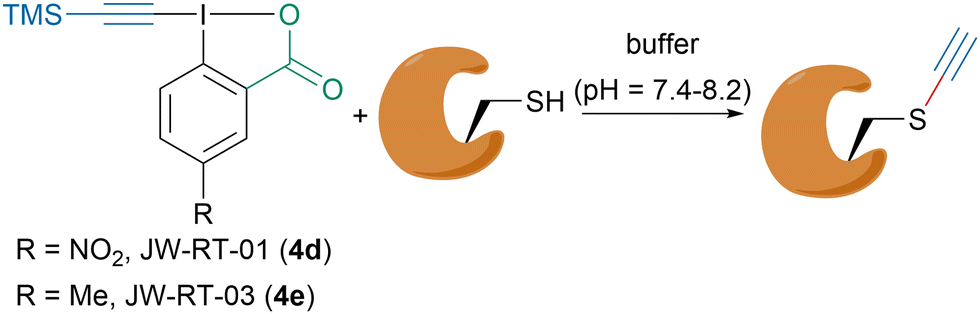 | ||
| Scheme 14 Ethynylation of cysteine residues with EBX 4d and 4e. | ||
Later, our group developed amphiphilic EBX-reagents 4f and 4g for the lipidation of cysteine residues (Scheme 15).82 The introduction of a sulfonate group on the aromatic ring favored water solubility. Peptides up to 18 amino acids as well as His6-Cys-Ubiquitin could be alkynylated in buffer and the modified peptides showed increased lipophilicity. Using TFA, the thioalkynes could be converted into thioesters, which could be cleaved in the presence of hydroxylamine regenerating the initial peptides.
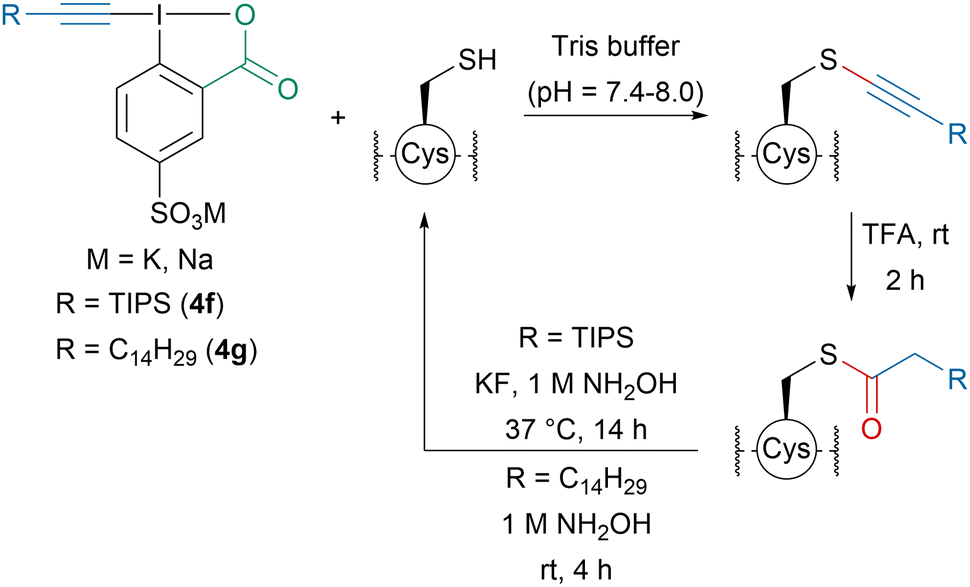 | ||
| Scheme 15 Lipophilization of peptides in water using amphiphilic reagents 4f and 4g. | ||
Leveraging the exquisite selectivity for cysteine alkynylation with EBX reagents, our group developed bifunctional reagents 4h–l, which could be used for i,i + 4 and i,i + 7 cysteine–cysteine and cysteine–lysine stapling of peptides (Scheme 16).83 Depending on the linker, changes in helicity were observed. A stapled peptide derived from the p53 protein showed increased helicity and binding affinity to MDM2 protein, a known cancer target and native binder to the p53 protein.
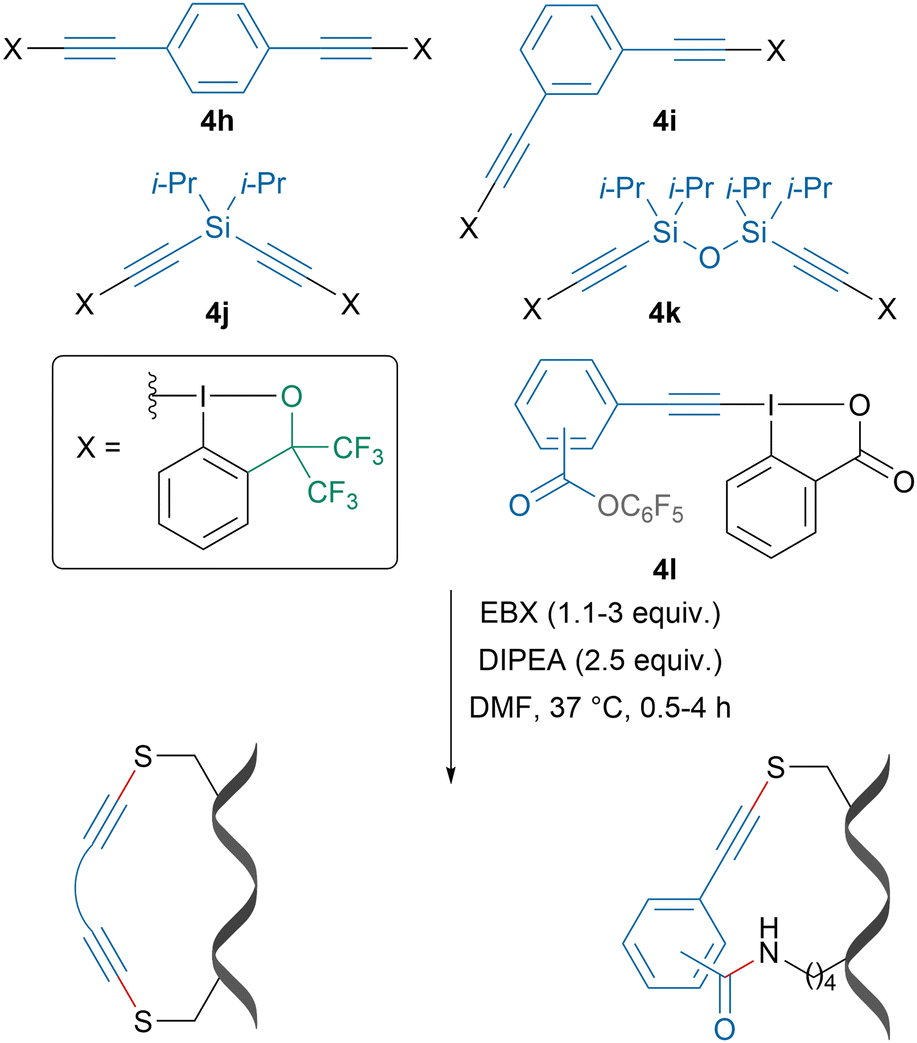 | ||
| Scheme 16 Cys–Cys and Cys–Lys stapling using bifunctional EBX reagents. | ||
Building upon their work on the fluorination of thioalkynes,84 O’Hagan, Bühl and coworkers synthesized fluorovinyl thioether acetyl coenzyme A analogue 16 (Scheme 17).85 The fluorovinyl thioether moiety was obtained by alkynylation of a thiol using TMS-EBX (4b) followed by treatment of the obtained thioalkyne with AgF/I2/triethylamine. The compound was shown to be a potent inhibitor of citrate synthase (Ki = 4.3 μM).
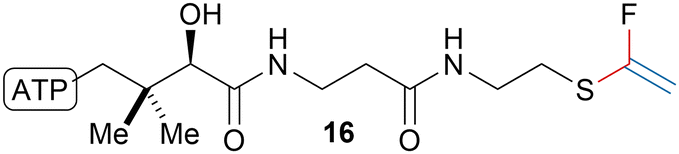 | ||
| Scheme 17 Fluorovinyl thioether acetyl coenzyme A analogue 16. | ||
In collaboration with the Matile group, several cyclic hypervalent iodine reagents were investigated as irreversible covalent inhibitors of thiol-mediated uptake. Although some showed promising activity, they usually exhibited early onset of toxicity.86
4. Transition-metal mediated alkynylation reactions
4.1. Alkynylation of metal-complexes
In 2019, the Hashmi group investigated the role of the trans-influence of ligands on the oxidative addition of ethynylbenziodoxole (EBx) reagents to gold(I) complexes (Scheme 18).87 The oxidative addition was initiated by the formation of a π-interaction complex I between the alkyne and the gold catalyst. Mechanistic studies showed that the lower the σ-donating ability of the ligands was, the higher the rate of oxidative addition was. The increase in oxidative addition rate was attributed to an enhanced accessibility of gold(I) intermediate for the oxidizing reagent with less σ-donating ligands.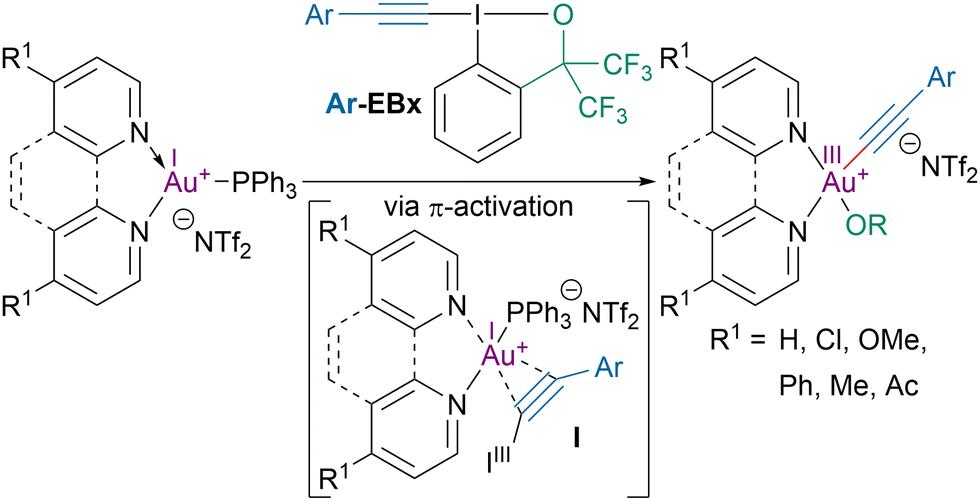 | ||
| Scheme 18 Oxidative addition of EBx reagents to gold(I) complexes. | ||
4.2. N–Csp bond formation
In 2021, Itoh, Tada and coworkers reported a copper-catalyzed N-alkynylation of sulfonamides with EBX reagents at room temperature (Scheme 19).88 Interestingly, the transformation was amenable to the alkynylation of the N-terminus of amino-acids and dipeptides. Moreover, mechanistic studies suggested that an electron-rich ligand, such as 17 or 18, and a protic solvent were required to reach high efficiency and promote the formation of oxidative addition intermediate I, allowing alkynes with bulky substituents to be used and preventing homodimerization.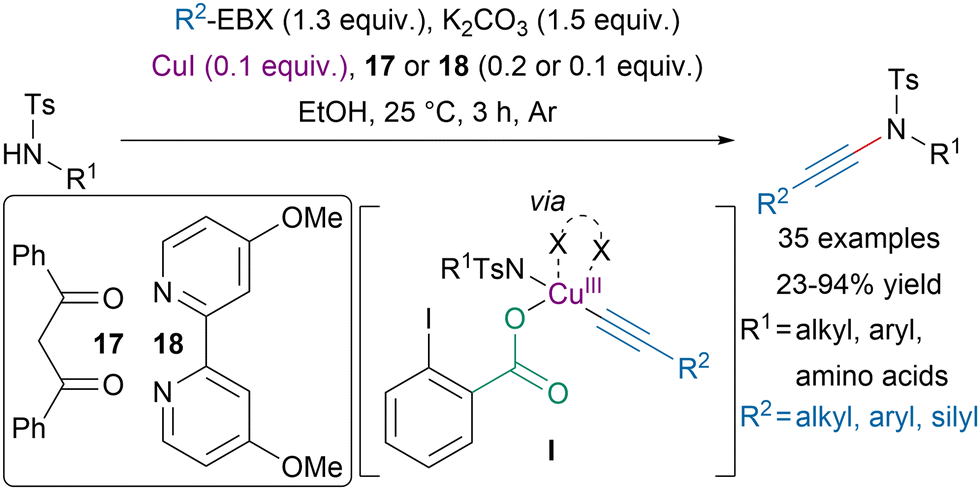 | ||
| Scheme 19 Copper-catalyzed ynamide synthesis with EBX reagents. | ||
While amides have been largely studied in alkynylation reactions, examples with hydrazides are scarce. The most popular method relied on the addition of acetylides on symmetrical diazodicarboxylates under strongly basic conditions.89 In 2022, our group reported a milder procedure for the direct alkynylation of hydrazides using copper-catalysis (Scheme 20).90 This method allowed to access functionalized azapeptide derivatives in moderate to excellent yields, tolerating a broad range of functional groups.
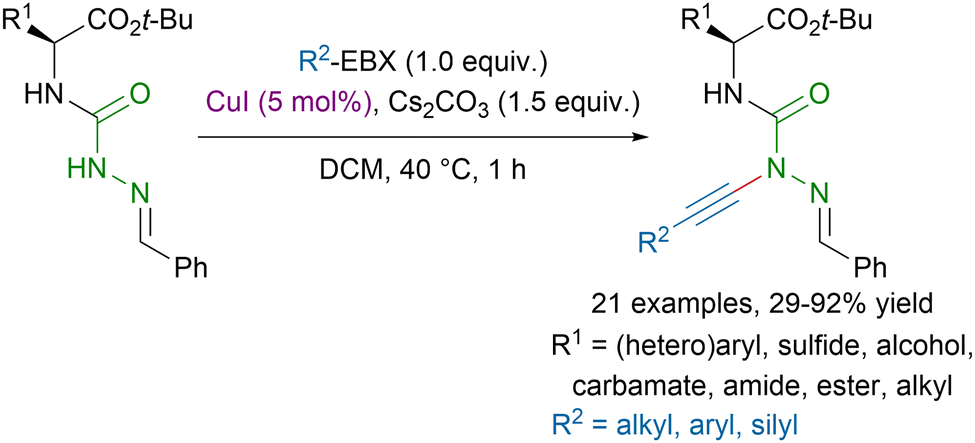 | ||
| Scheme 20 Copper-catalyzed alkynylation of azadipeptide derivatives. | ||
4.3. Aryl–Csp bond formation
Over the last two decades, the combination of hypervalent iodine reagents with gold,91 and other transition-metals has allowed the functionalization of a broad range of substrates.21 For example, the Hashmi group reported a bimetallic gold–silver catalytic system for the synthesis of 3-alkynyl benzofurans from phenols and EBx reagents (Scheme 21).92 Mechanistic studies, suggested that a bimetallic Au–Ag catalyst promoted a tandem ortho C(sp2)–H alkynylation/oxyalkynylation reaction by leveraging the exceptional redox property and carbophilic π-acidity of gold. Based on a similar approach, Hashmi and coworkers also developed tandem C(sp3)–H alkynylation/oxyalkynylation reactions to access tetra-substituted furans from acceptor-substituted carbonyl compounds and indolizines from ortho-substituted pyridine derivatives.93,94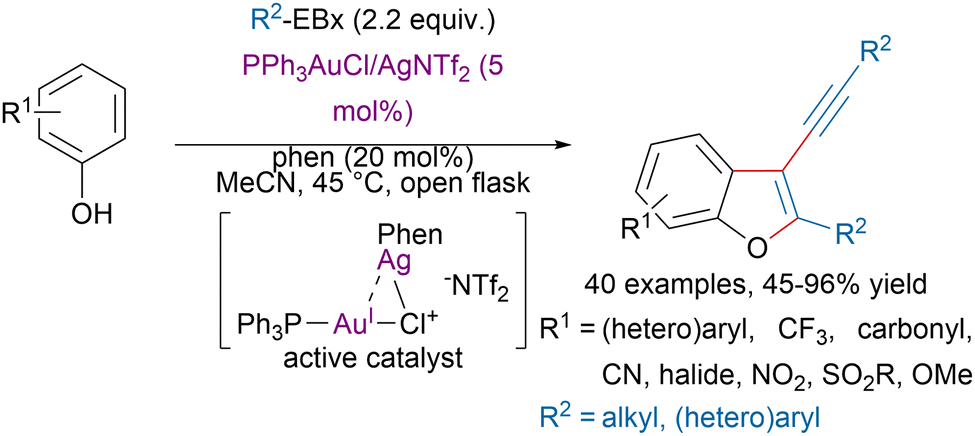 | ||
| Scheme 21 Synthesis of 3-alkynyl benzofurans with a bimetallic Au–Ag catalyst. | ||
In order to ensure high regioselectivity in C–H bond functionalizations the use of directing groups (DG) has become very popular, including in alkynylation transformations.95 Rohokale and coworkers developed an iridium-catalyzed ortho alkynylation of (hetero)aryls using TIPS-EBX (4a) and arylquinazolin-4-ones as directing groups (Scheme 22).96 Interestingly, switching the solvent for DCE and increasing the temperature to 70 °C allowed to access dialkynylated compounds in moderate to excellent yields.
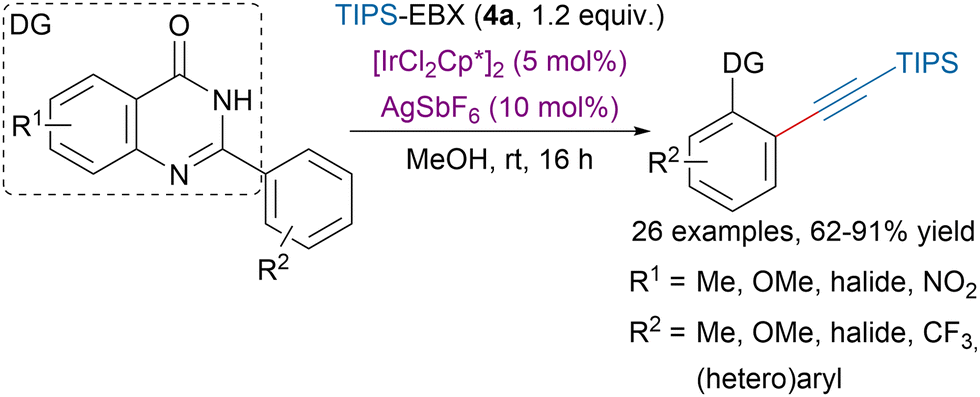 | ||
| Scheme 22 Arylquinazolin-4-ones directed alkynylation of (hetero)aryls. | ||
Alternatively, Xia, Zhang and coworkers developed a formal regiodivergent alkynylation of 1-arylpyrazolones (Scheme 23).97 With NH-free pyrazolone (A), a rhodium catalyst promoted a directed ortho C–H alkynylation via the proposed rhodacycle intermediate I. On the other hand, pyrazolones with alkylated nitrogen underwent C4-alkynylation under gold-catalysis via the proposed intermediate II (B). The authors suggested that the regioselectivity was determined by the nature of the substrate (alkylated pyrazolone or not) and the choice of metal catalyst.
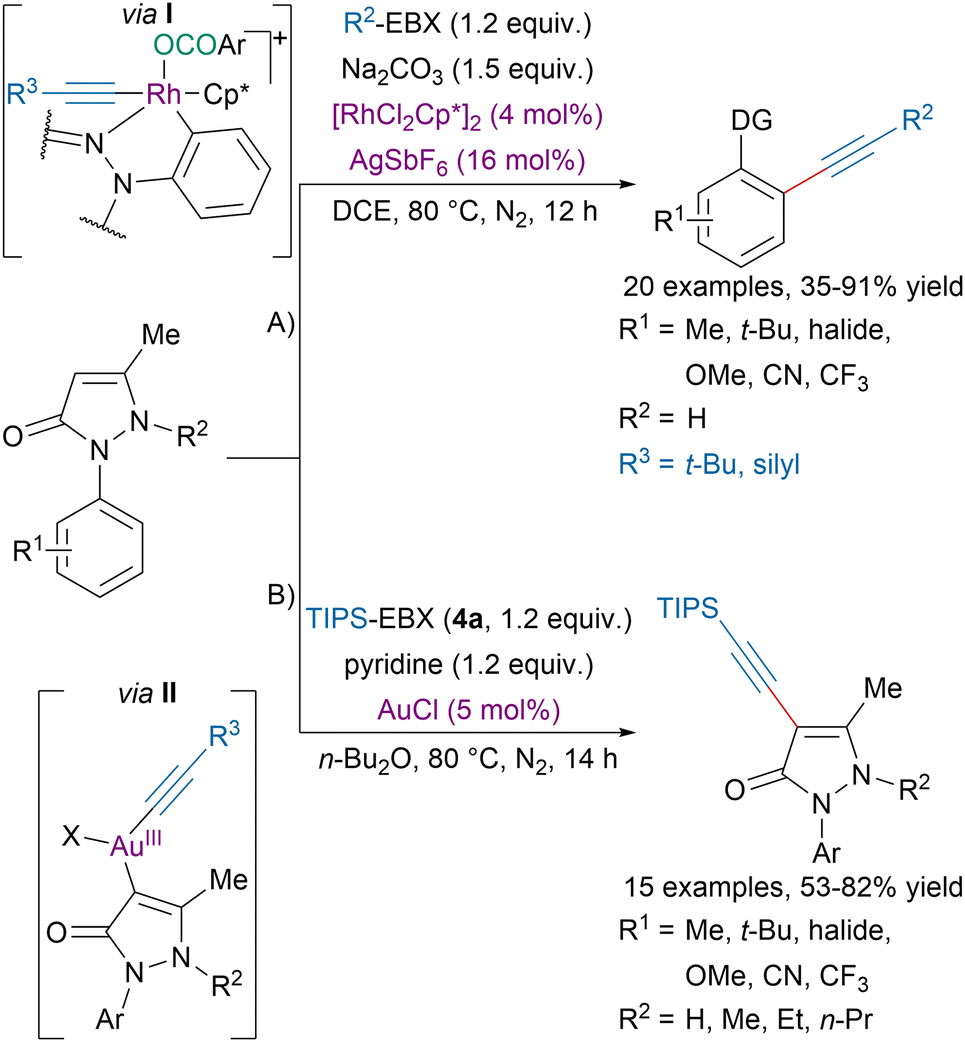 | ||
| Scheme 23 Regiodivergent alkynylation of 1-arylpyrazolones. | ||
A similar reactivity had been previously observed for the C–H alkynylation of N-methylisoquinolones.98 Computations showed that the difference of mechanism could also be explained by the dual reactivity of TIPS-EBX (4a) (Scheme 24).99 With the rhodium catalyst it behaved as a BrØnsted base, viaI, favoring a base-assisted concerted metalation-deprotonation (CMD) mechanism for the C8–H bond activation. In contrast, under gold-catalysis the iodine(III) center acted as a Lewis acid, viaII, to activate the alkyne. The computations suggested that in this case steric hindrance, rather than electronic effects, directed the regiochemistry of the reaction.
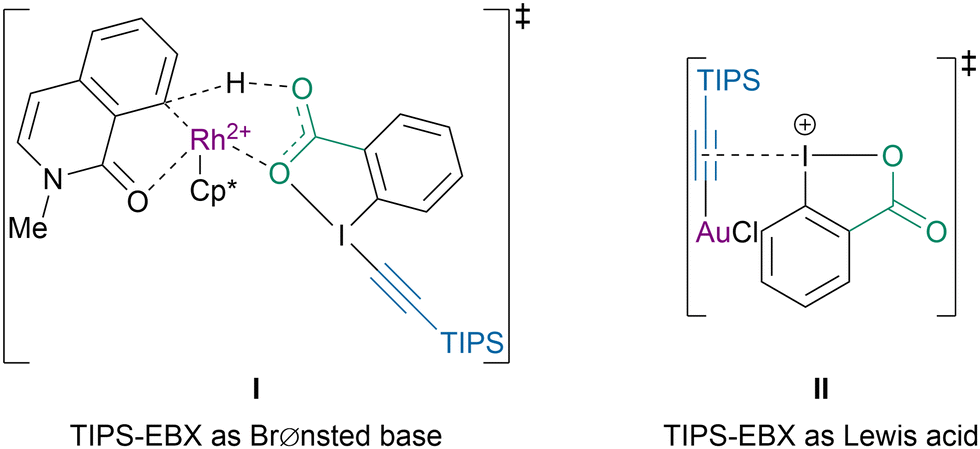 | ||
| Scheme 24 Dual reactivity of TIPS-EBX (4a) with Rh and Au catalysts. | ||
Hypervalent iodine reagents have been also widely used for the functionalization of heterocycles.100 Since our initial report on the alkynylation of indoles and pyrroles with EBX reagents using gold-catalysis,101 several researchers investigated this transformation. For instance, the Liu group reported in 2018 a Ru(II)-catalyzed C2-alkynylation of indoles using pyrimidine as a directing group.102 Using ball milling, Bolm and coworkers developed solventless alkynylations of indoles with either Rh(III)-(C2-selective) or Au(I)-catalysis(C2 or C3 selective).103 The mechanochemical conditions allowed to reduce the reaction time and the catalyst loading without requiring additional heating, still maintaining excellent functional group tolerance. Dai, Bai, Ma and coworkers developed a directed C2-alkynylation of indoles and used the triple bond as a handle for further functionalization.104 Using AuCl for the C2 or C3-alkynylation of pyrroles, Furuta, Ishida and coworkers could access functionalized BODIPY dyes with distinctive spectroscopic properties.105
4.4. Alkene–Csp bond formation
Building upon their previous works on synergistic gold-amine catalysis for the α-functionalization of carbonyls,106 the Huang group developed a procedure to access isolable diyneamines using gold catalysis (Scheme 25).107 The high conjugation of the π-system might explain why the corresponding ynones were not isolated. The alkynylation of enamines was also studied by the Hashmi group to access tetra-substituted 1,3-enynes from acceptor-substituted enamines.108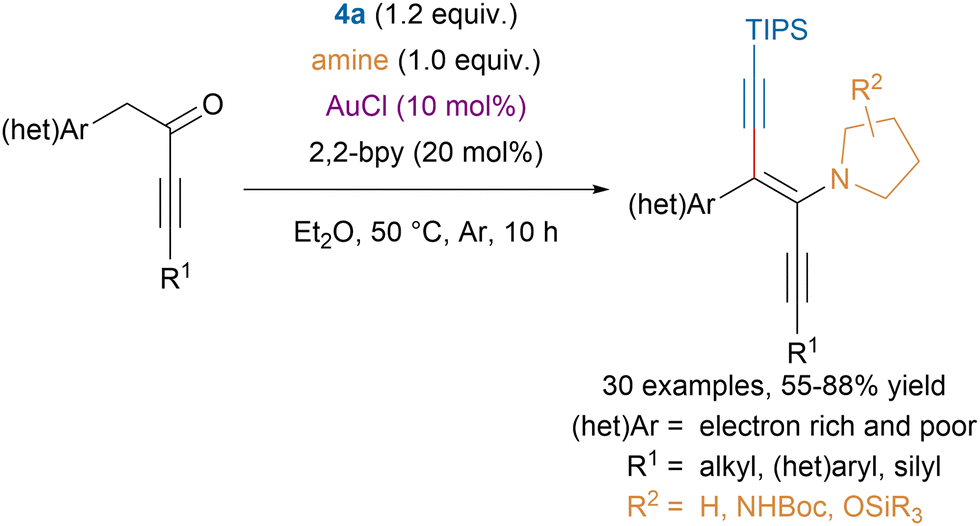 | ||
| Scheme 25 Gold-catalyzed diyneamines synthesis. | ||
In 2019, Hashmi and coworkers disclosed a dual gold/silver-catalyzed direct alkynylation of cyclopropenes with ethynylbenziodoxole reagents (Scheme 26).109
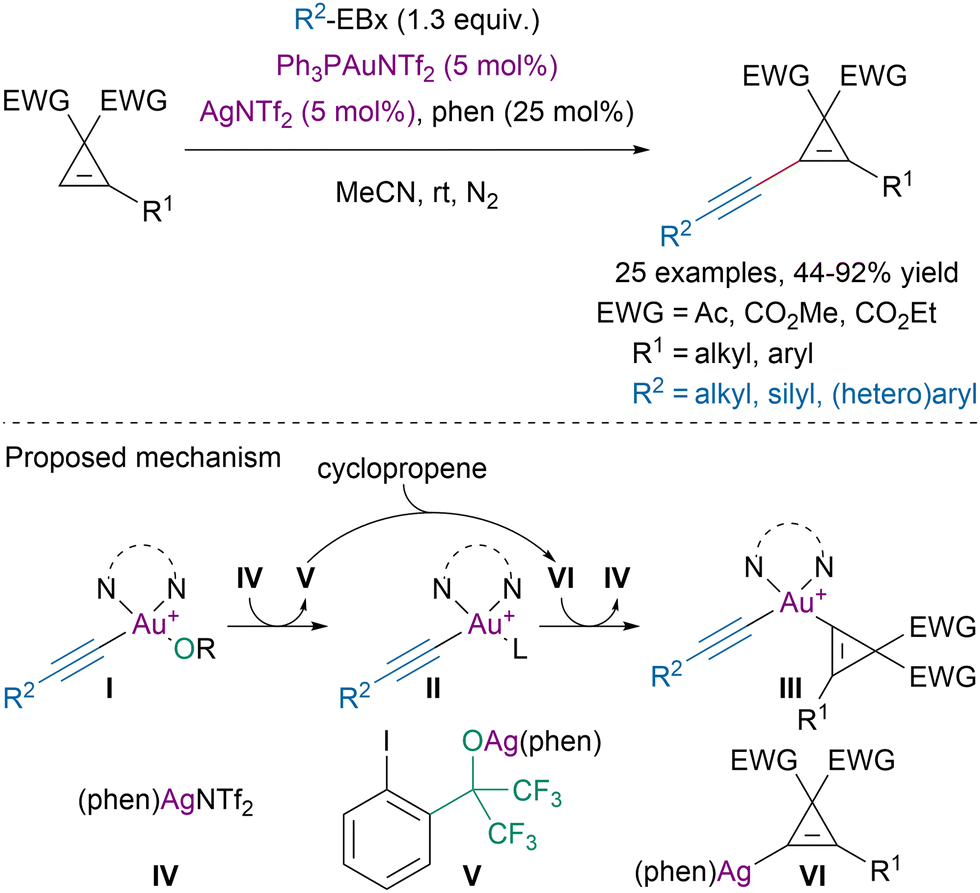 | ||
| Scheme 26 Dual Au/Ag-catalyzed alkynylation of cyclopropenes. | ||
Extensive mechanistic studies suggested that gold was involved in oxidative addition on EBx reagents leading to intermediate I. Ligand exchange with silver salt IV would lead to intermediates II and V. The later would be responsible for the C–H activation step affording intermediate VI. Transmetalation with intermediate II would generate III and reform the active silver catalyst IV. Reductive elimination from III would then provide the targeted alkynylated cyclopropene and close the gold-catalytic cycle.
The same group reported a stereoselective gold-catalyzed oxyalkynylation of N-propargylcarboxamides with EBx reagents leading to alkynyloxazolines (Scheme 27).110 The developed one-step procedure allowed to tolerate functional groups that would be prohibited for a Sonogashira coupling, which was traditionally used to access these scaffolds. Moreover, computations suggested that the observed stereoselectivity could be explained by kinetic control.
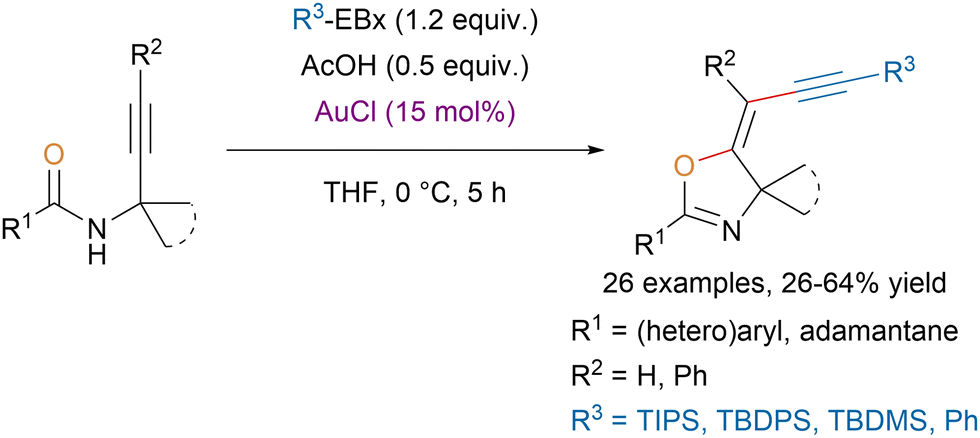 | ||
| Scheme 27 Au-catalyzed stereoselective synthesis of alkynyloxazolines. | ||
Alternatively, from propargyl alcohols, Patil, Senthilkumar and coworkers successfully developed a gold-catalyzed alkynylative Meyer-Schuster rearrangement affording enynones in moderate to excellent yields.111 The carbophilic π-acidity of gold was key for the success of the reaction as previous studies with palladium failed to promote an alkynylative Meyer-Schuster transformation.112
4.5. C(sp3)–Csp bond formation
From simple unactivated alkenes, Liu and coworkers could access β-alkynylcarboxylic esters via a palladium catalyzed intermolecular alkynylcarbonylation (Scheme 28).113 The mild reaction conditions allowed a broad functional group tolerance and moderate to excellent regioselectivity were observed.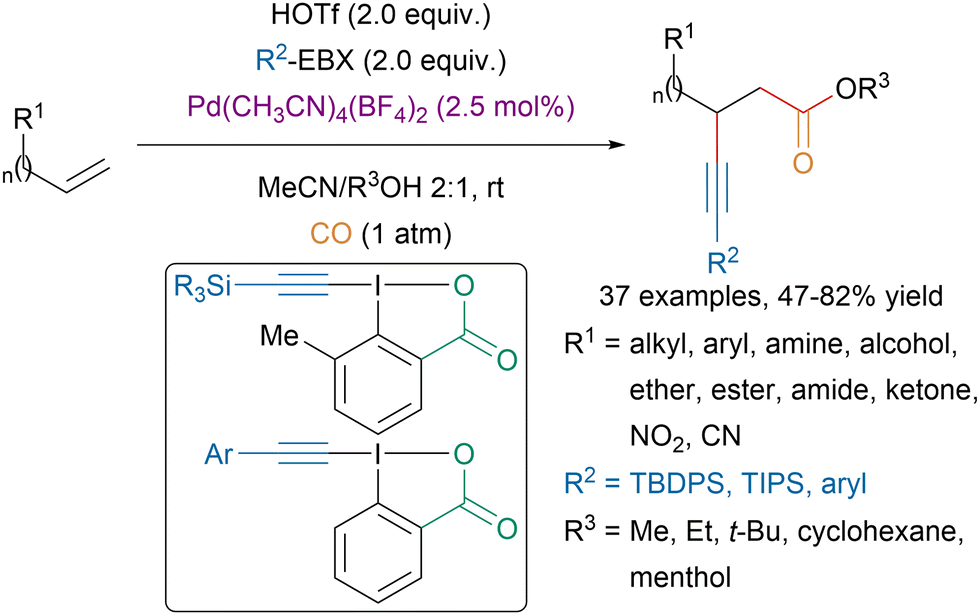 | ||
| Scheme 28 Pd-catalyzed alkynylcarbonylation of unactivated alkenes. | ||
Moreover, mechanistic studies suggested that the reaction involved cis-addition of the alkynyl and the carbonyl moiety. Interestingly, for the transfer of silyl-substituted alkynes, reagents bearing a methyl group ortho to the iodine on the aromatic core led to better yield. This enhancement of reactivity, also called hypervalent twist, had previously been observed and first reported in oxidation reactions and in the gold catalyzed alkynylation of indoles.114–116
Recently, the Chen group reported a palladium-catalyzed three-component cross-coupling of 1,4-dienes with indoles and EBZ reagents (Scheme 29).117 Interestingly, they showed that this class of reagents outperformed EBX reagents, which produced mixture of 1,2- and 1,4-functionalization products.
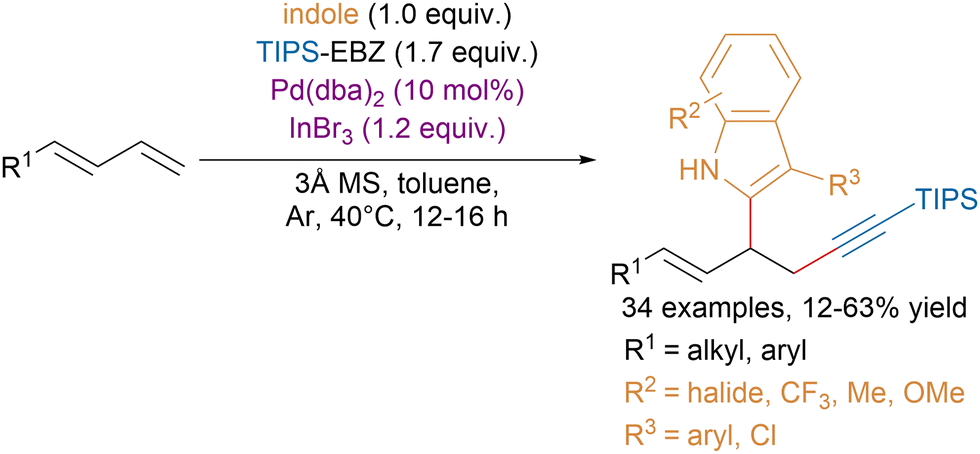 | ||
| Scheme 29 1,2-Functionalization of conjugated dienes with indoles and EBZ. | ||
In the last decade, the functionalization of diazo compounds with hypervalent iodine reagents has allowed to access numerous highly functionalized products.118 Our group took advantage of the ambiphilic reactivity of metal carbenes obtained from diazo compounds to develop atom-economical oxyalkynylation reactions.119,120 While studying product modifications, we discovered an unusual low-temperature [4 + 2]-cycloaddition of allenes and arenes, also known as Himbert reaction.121 Building upon this observation, our group reported a one-pot oxyalkynylation/Himbert reaction leading to bicyclo[2.2.2]octadiene products that could be used as diene ligands for rhodium catalysis (Scheme 30).122 The reaction is believed to involve first an oxyalkynylation of the diazo compounds. Then in the presence of fluoride or a base, an allene would be formed, which undergoes a Himbert cycloaddition. Computations suggested that the low activation energy for the cycloaddition arose from favorable dispersive interactions in the transition state I.
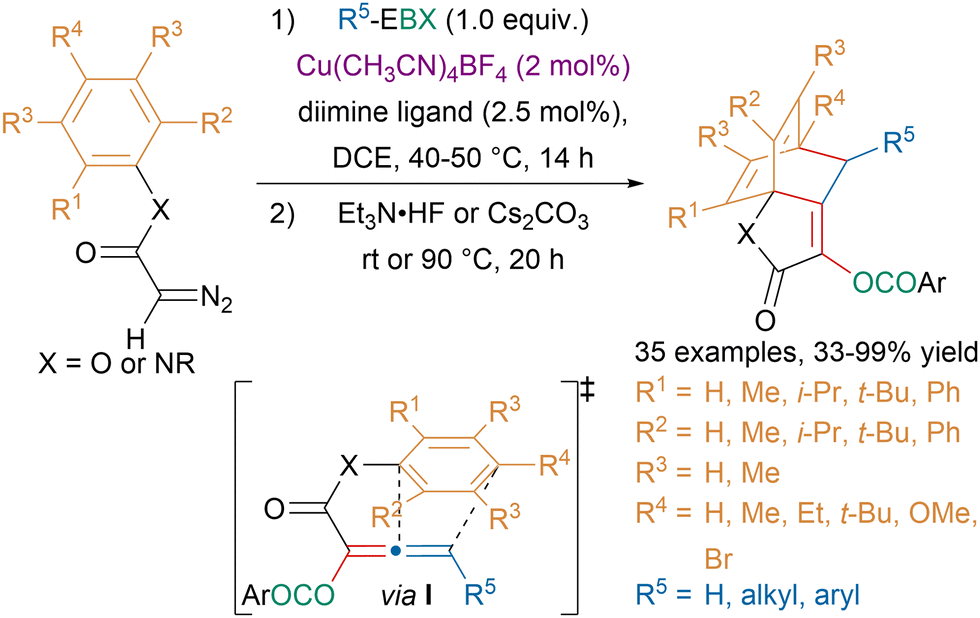 | ||
| Scheme 30 One-pot sequential oxyalkynylation/Himbert reaction. | ||
In order to increase structural diversity, our group later developed three-component reactions of diazo compounds, EBx reagents and alcohols or anilines (Scheme 31).123,124 The use of the benziodoxole core was critical for the success of the transformation as the corresponding fluorinated benzyl alcohol did not compete with external nucleophiles for the insertion in the metal carbene leading to ylide intermediate I. For the reaction with alcohols, a high structural diversity was achieved (A). On the other hand, the 3CR with amines has been limited so far to anilines and fluorinated diazo compounds (B).
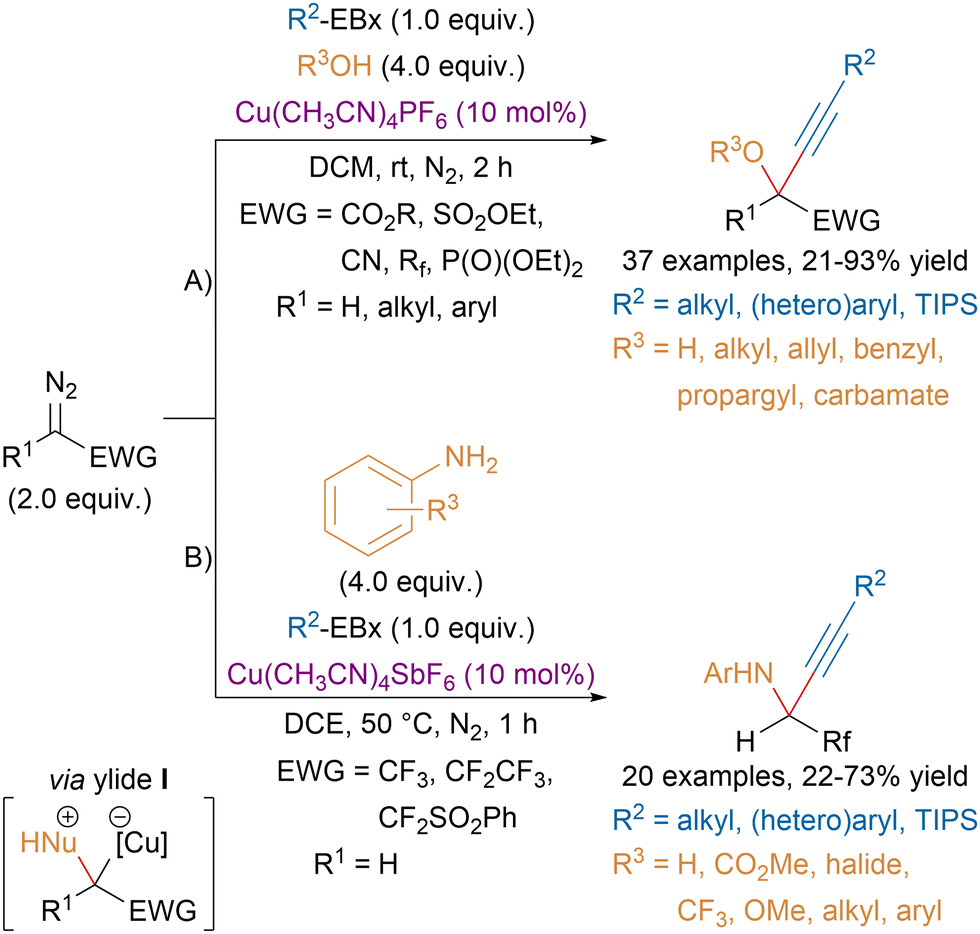 | ||
| Scheme 31 Cu-catalyzed gem-oxy- and aminoalkynylation of diazo compounds. | ||
5. Radical-based alkynylation reactions
Since the seminal work from Li, Cheng and coworkers,125 EBX reagents have emerged as powerful radical traps for alkynylation reactions. In the following chapter, progress in radical alkynylation transformations using hypervalent iodine reagents reported after our last review in the field will be presented.365.1. Non-photoredox-induced radical alkynylations
In 2019, Zhu and coworkers reported a visible light-induced alkynylation of acyl radicals leading to valuable alkyl and aryl ynones in moderate to good yields (Scheme 32A).126 The authors proposed that visible light irradiation of C2-acyl substituted benzothiazolines would promote a C–C bond homolytic cleavage leading to an acyl radical that would be trapped by EBX reagents. Similar products were obtained by the Maruoka group by heating aldehydes in presence of aryl ethynylbenziodoxole reagents, albeit in lower yields.127 Alternatively, the Wang group developed an electro-induced homolysis of 4-acyl-1,4-dihydropyridines generating acyl radicals that could react with EBX reagents (Scheme 32B).128 The mild reaction conditions allowed to access a broad range of ynones bearing functional groups and propiolamide derivatives. The late-stage functionalization of pharmaceutical molecules was also demonstrated.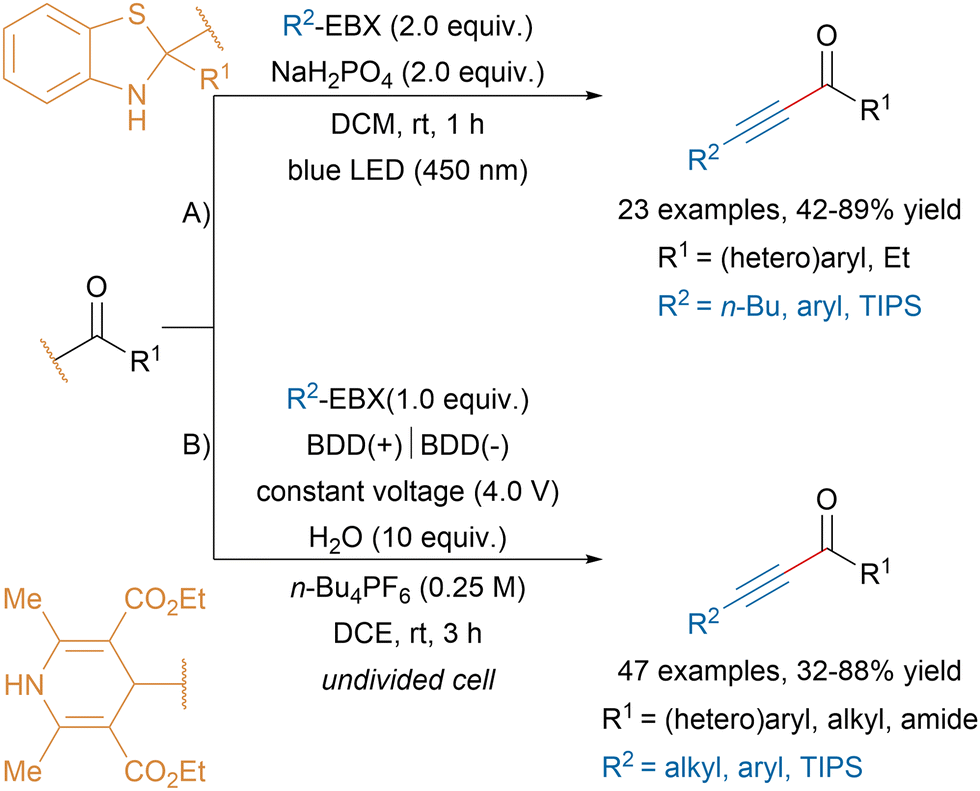 | ||
| Scheme 32 Ynone synthesis via radical alkynylation. | ||
In 2019, the Tsui group disclosed a silver-catalyzed trifluoromethylalkynylation of unactivated olefins tolerating a broad range of functional groups (Scheme 33).129 Mechanistic studies suggested that the reaction was proceeding through a radical mechanism with first addition of the trifluoromethyl radical and then trapping of the resulting nucleophilic radical by EBX reagents. NaOAc was required to initiate the formation of AgCF3 (I) which is responsible for the formation of CF3 radicals. MeO-BX (19) is believed to act as a source of iodanyl radical II, which is also generated after the radical alkyne transfer from EBX reagents. The authors proposed that radical II would oxidize AgCF3 to generate trifluoromethyl radicals.
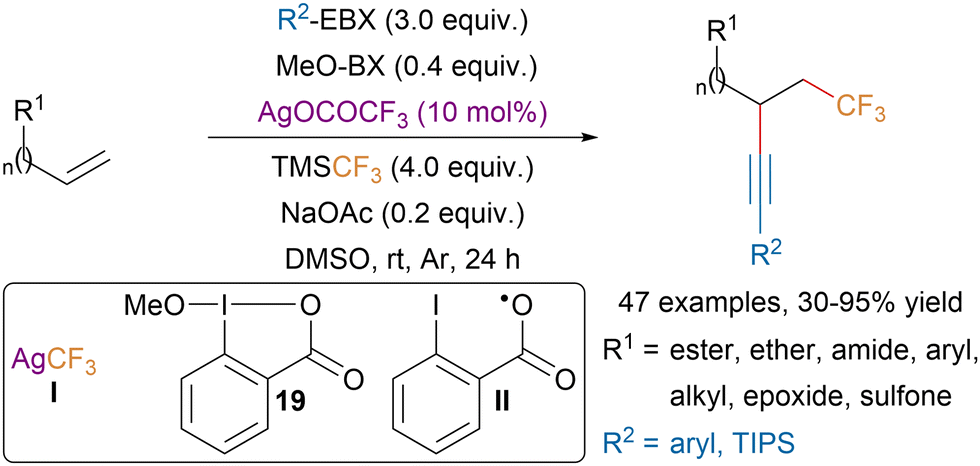 | ||
| Scheme 33 Ag-catalyzed trifluoromethylalkynylation of unactivated olefins. | ||
Starting from malonic acid derivatives, the Chen group developed a tandem monodecarboxylative alkynylation–lactonization affording 2(3H)-furanones in moderate to good yields (Scheme 34).130 Key for the success of the transformation was the dual role of the silver catalyst, first promoting the decarboxylative alkynylation in presence of an oxidant and a base, then acting as a Lewis acid to activate the triple bond to catalyze the lactonization step via intermediate I.
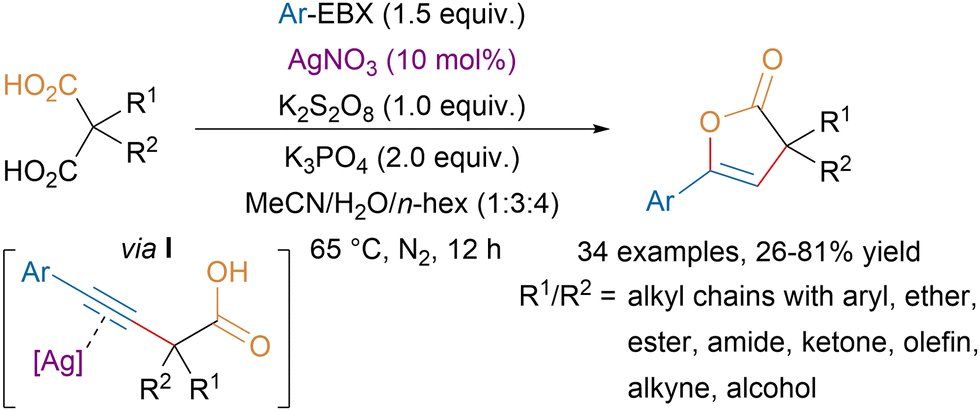 | ||
| Scheme 34 Ag-catalyzed monodecarboxylative alkynylation–lactonization of malonic acid derivatives. | ||
Alternatively, using an ammonium persulfate oxidant, Xu, Huang and coworkers could access arylthiodifluoromethylated alkynes via the decarboxylative alkynylation of arylthiodifluoroacetic acids.131
5.2. Photoredox-catalyzed C–H functionalization
In 2020, Nemoto, Nakajima and Matsumuto reported a benzophenone (20) promoted C(sp3)–H alkynylation of ethers and amides (Scheme 35).132 The use of violet LEDs (400 nm) allowed to selectively excite the photosensitizer and promote So → Tn transitions. The formed excited state I could then engage in CH abstraction on the substrate, which could then react with EBX reagents and the resulting iodanyl radical would then close the catalytic cycle.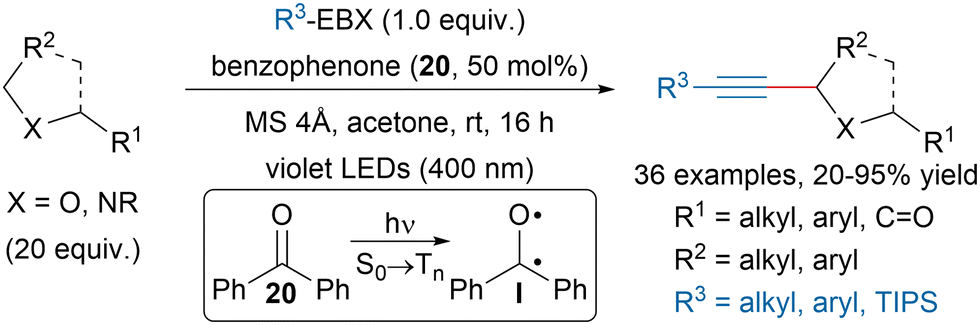 | ||
| Scheme 35 Benzophenone promoted C(sp3)–H alkynylation of ethers and amides. | ||
In collaboration with our group, Kokotos and coworkers developed a similar transformation but using phenylglyoxylic acid and white CFL lamps.133 Interestingly, under the reaction conditions thioethers underwent deconstructive alkynylation affording thioalkynes.
In 2022, the Chen group reported a photoredox-catalyzed remote C(sp3)–H alkynylation via the fragmentation of iminophenylacetic acids (Scheme 36).134 Based on literature precedence and mechanistic investigations, the authors suggested that first the substrate would engage in ligand exchange with 21 to afford intermediate I, which would be oxidized by a Ru(III) species leading to II. Ru(III) would be obtained after oxidative quenching of the excited-state Ru(II)* by a hypervalent iodine intermediate. Fragmentation of II would lead to alkoxyl radical intermediate III, which upon 1,5-HAT would generate intermediate IV that could react with EBX reagents and generate the targeted compounds.
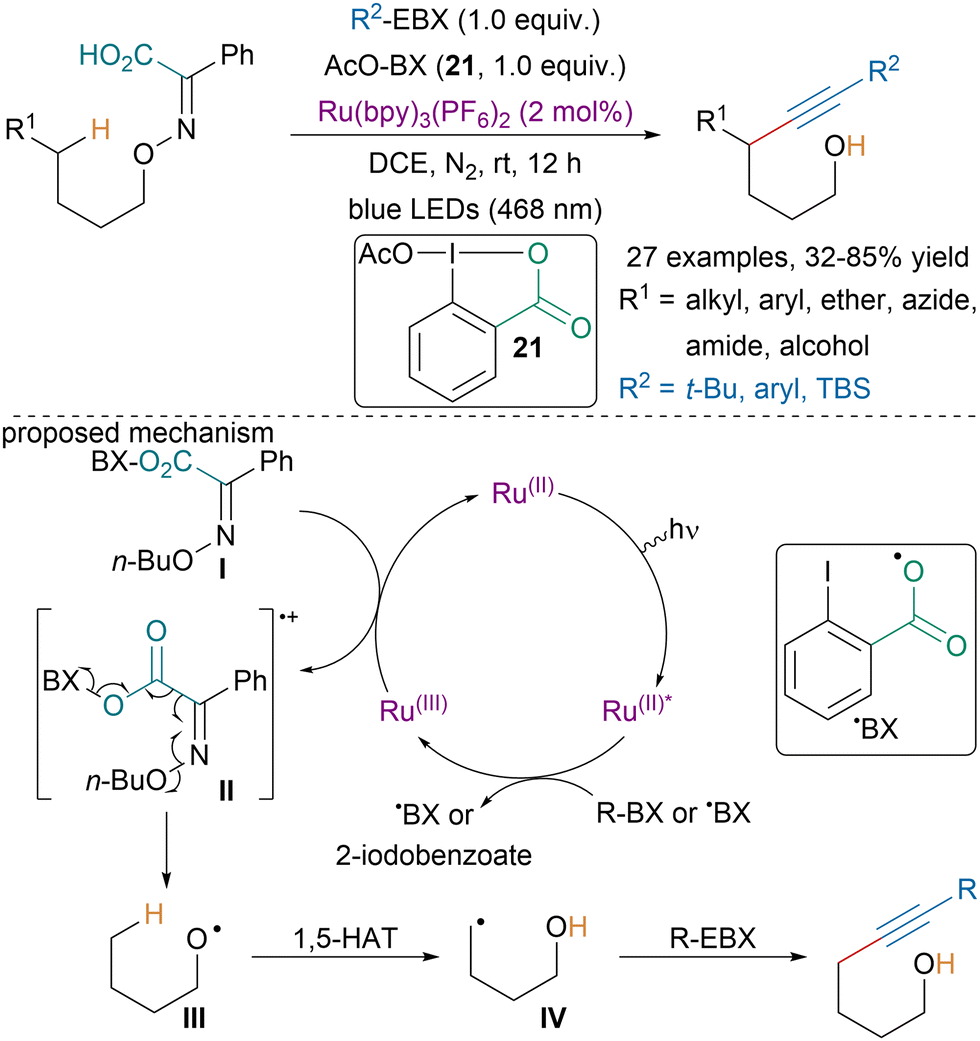 | ||
| Scheme 36 Photoredox-catalyzed remote C(sp3)–H alkynylation. | ||
Furthermore, Hammond and coworkers showed that aryl-EBX reagents were efficient traps for alkyl radicals generated from 1,4-dihydropyridines under iridium photocatalysis.135
5.3. Photoredox-catalyzed π-system functionalization
In 2022, the Han group described sequential catalytic annulations for the synthesis of N-heterocycles via radical [1,4]-oxygen atom transfer from alkyne tethered ketoximes (Scheme 37).136 Mechanistic studies suggested that the first annulation would be catalyzed by copper and involve the generation of an oxygen-centered radical on the ketoxime. Radical addition on the alkyne followed by trapping of the resulting vinyl radical by EBX reagents would provide isolable intermediate I and an iodanyl radical that would close the catalytic cycle by oxidizing copper(I) back to copper(II). Then triplet-state excitation of intermediate I by an iridium photocatalyst would generate diradical II, which would rearrange to afford the product via a [1,4]-oxygen atom transfer. Under Lewis acid catalysis a third annulation process was observed between the carbonyl group and the introduced alkyne.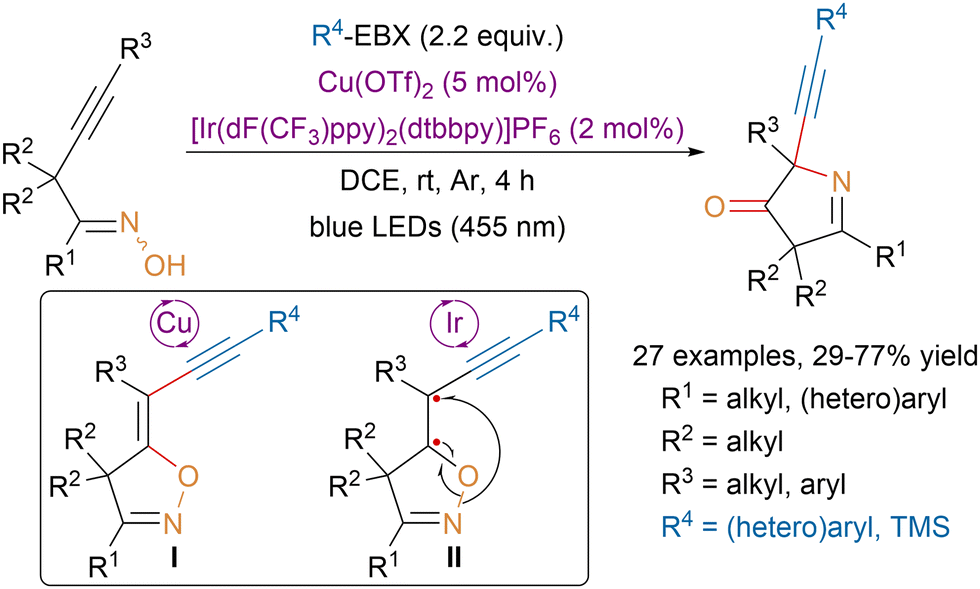 | ||
| Scheme 37 Synthesis of N-heterocycles via sequential Cu-catalyzed annulative alkynylation followed by Ir-catalyzed radical [1,4]-oxygen atom transfer. | ||
5.4. Photoredox-catalyzed alkynylation via C–C bond cleavage
In 2015, Xiao and our groups concurrently demonstrated that EBX reagents were valuable partners to develop decarboxylative alkynylation transformations.137,138 Zhang, Wang and coworkers showed that the transformation could also be catalyzed using recyclable graphitic carbon nitride polymers.139 Later our group could extend the concept to the photoredox catalyzed decarboxylative alkynylation of the C-terminus of peptides.140 Alternatively, in 2021, Zhang and coworkers reported a photoredox-catalyzed decarboxylative alkynylation of glycosides at the anomeric position (Scheme 38).141 A variety of alkynyl C-glycosides could be obtained in moderate to excellent yields with high diastereoselectivity.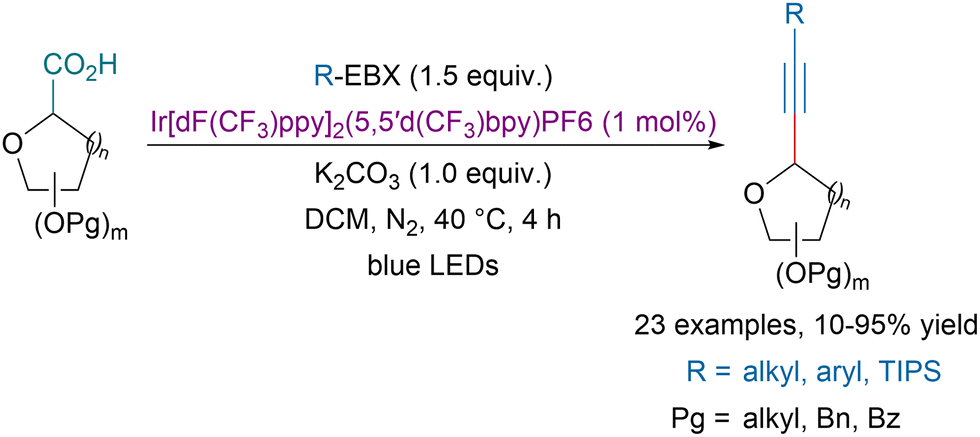 | ||
| Scheme 38 Photoredox-catalyzed decarboxylative alkynylation of glycosides. | ||
Chen and coworkers developed a photoredox-catalyzed ring opening alkynylation of cycloalkylamides using an acridinium photocatalyst (22) and hypervalent iodine reagents (Scheme 39).142 The authors proposed that the catalytic amount of AcO-(3,4-OMe)-BX reagent would facilitate the single-electron oxidation of cycloalkylamides leading to intermediate I. Then ring opening would generate an alkyl radical that could be trapped by EBX reagents and an imine that could react with nucleophiles.
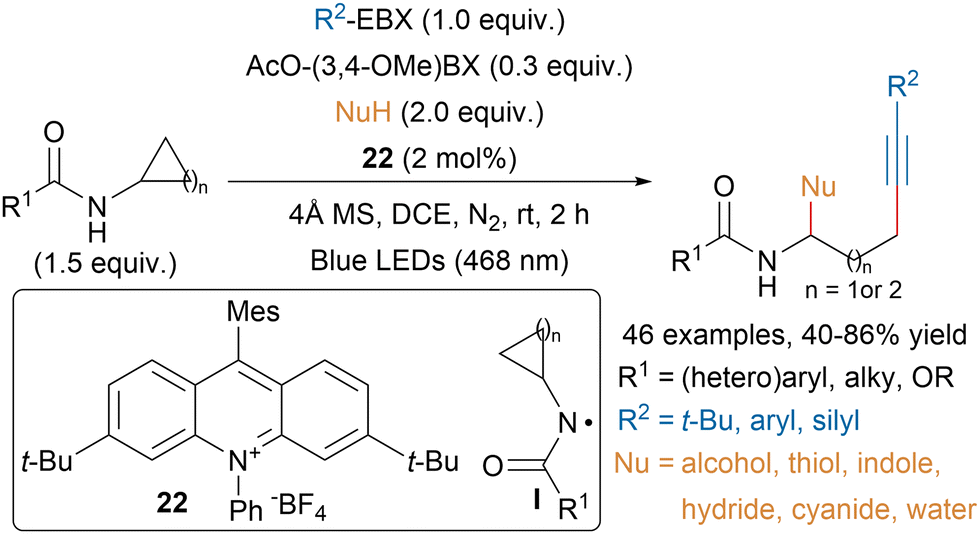 | ||
| Scheme 39 Photoredox-catalyzed alkynylative ring-opening of cycloalkylamides. | ||
5.5. Direct photo-excitation of Ar-EBX reagents
In 2021, our group discovered that the excited state of Ph-EBX (4i*), generated via direct light excitation using Kessil lamps, was a strong oxidant capable of oxidizing a broad range of substrate without the need for photocatalysts (Scheme 40A).143 For instance, the direct excitation of 4i promoted the deoxygenative alkynylation of cesium-oxalate salts (B). Slightly better yields were obtained with a photocatalyst after extensive optimization and was concurrently reported by the Xie group.144 The deboronative alkynylation of trifluoroborate salts developed by Chen and coworkers could also be initiated by direct excitation (C).145,146 In addition, 4i* was able to induce the alkynylation of THF (D) or the decarboxylative alkynylation of glyoxylic and aliphatic acids (E). The decarboxylative oxime fragmentation–alkynylation, previously described by our group,147 could be performed without the addition of organic dyes (F). Likewise, the atom-economical oxyalkynylation of enamides developed by our group could be promoted by the direct photoexcitation of Ph-EBX (4i) (G).148 Finally, an unprecedented deaminative-alkynylation of imines was also discovered using this approach (H).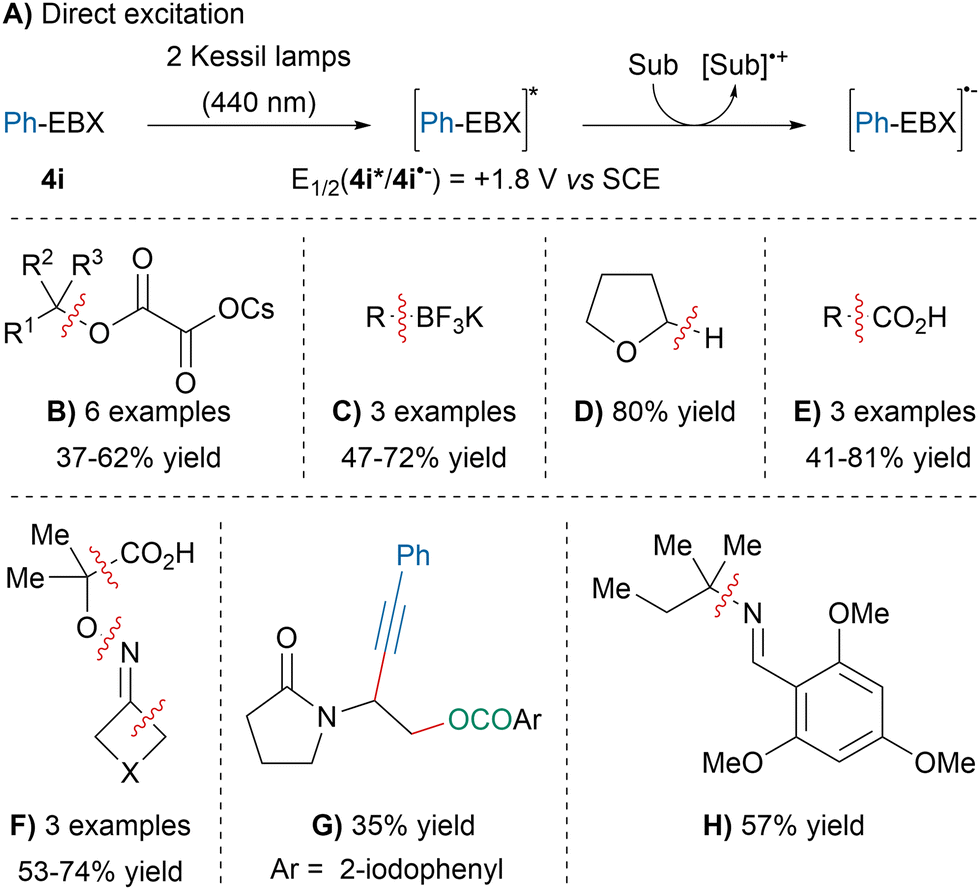 | ||
| Scheme 40 Direct photoexcitation of Ph-EBX with visible light and applications. | ||
Building upon this work, our group recently developed a substrate-controlled C–H alkynylation or C–C oxyalkynylation of aryl cyclopropanes via the direct photoexcitation of Ar-EBX reagents (Scheme 41).149
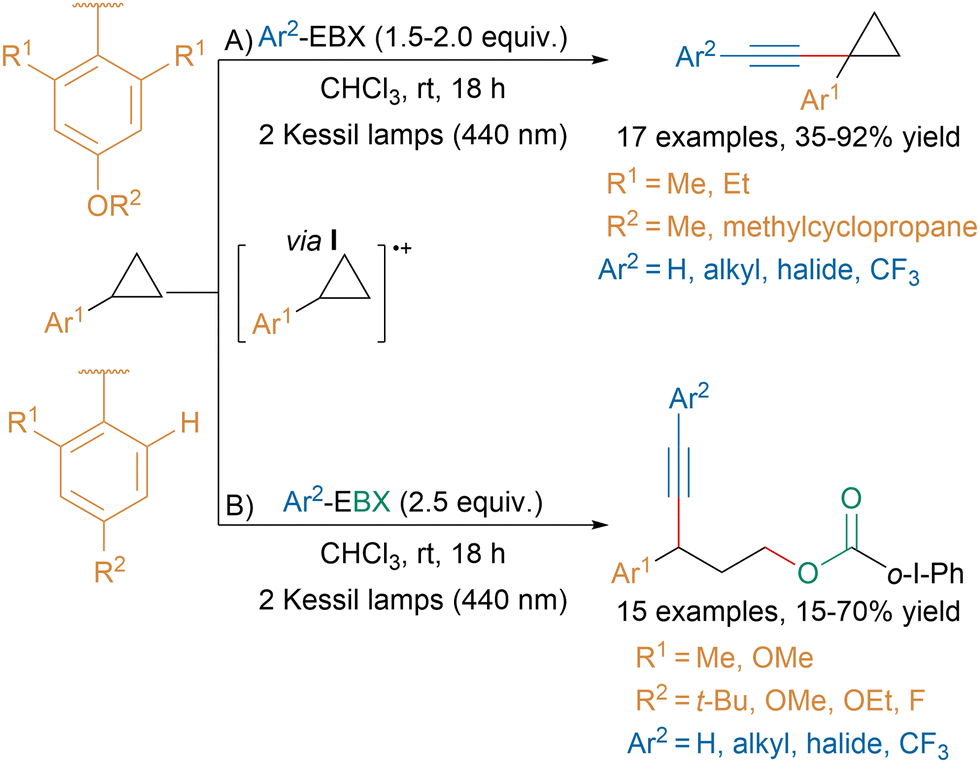 | ||
| Scheme 41 Photoexcitation of Ar-EBXs for the alkynylation of aryl cyclopropanes. | ||
When the aryl on the cyclopropane was bearing two ortho substituents, an unusual CH-alkynylation was taking place presumably via radical cation I (A). From this intermediate, computations suggested that the conformational constrains induced by the aryl ring favored H-abstraction followed by alkynylation over ring opening. In contrast, for aryl rings bearing only one ortho substituent, intermediate I underwent ring opening and then 1,3-oxyalkynylation (B). The same transformation had been previously reported by Studer and coworkers using an organic dye.150 Furthermore, our group showed that this reaction could be extended to the 1,3-oxyalkynylation of aminocyclopropanes and the 1,2-oxyalkynylation of styrenes.
6. Atom-economical transformations
The pursuit of greater efficiency in organic chemistry resulted in the development of atom economical reactions for which not only the alkyne of EBX reagents would be transferred but also the carboxylate moiety.151,152 For instance, our group had developed atom-economical reactions between EBX reagents and diazo compounds.119,120 In 2019, we tested the EBZ reagents in this transformation.47 A highly enantioselective copper-catalyzed oxyalkynylation of diazo compounds was developed with BOX ligand 23 leading to imidates in moderate to excellent yields via an ambiphilic copper-carbene intermediate I (Scheme 42).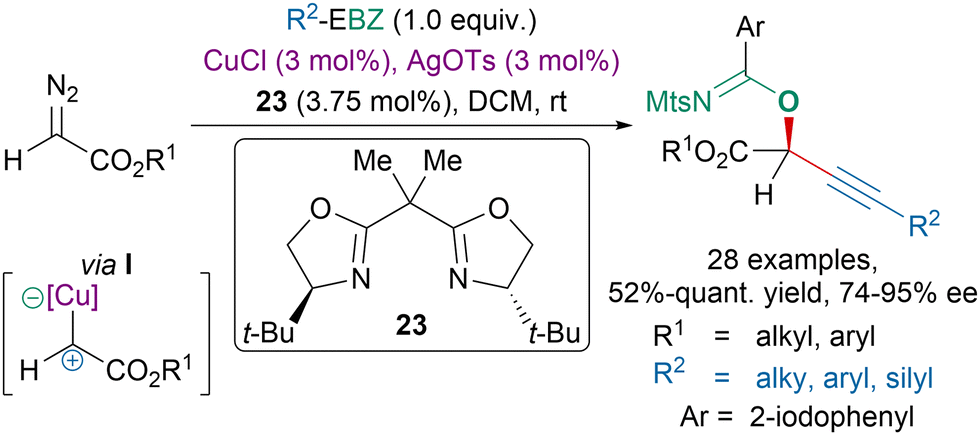 | ||
| Scheme 42 Copper-catalyzed oxyalkynylation of diazo compounds with EBZs. | ||
In 2020, our group disclosed a copper-catalyzed 1,3-oxyalkynylation of thiiranes and 1,4-oxyalkynylation of thiethanes with EBX reagents (Scheme 43).153 Literature precedence and mechanistic studies suggested that the reaction proceeds first via the activation of EBX reagents by a copper catalyst followed by addition of a sulfide leading to an episulfonium intermediate I. Ring opening by the nucleophilic attack of an aryl carboxylate species would generate the targeted compound.
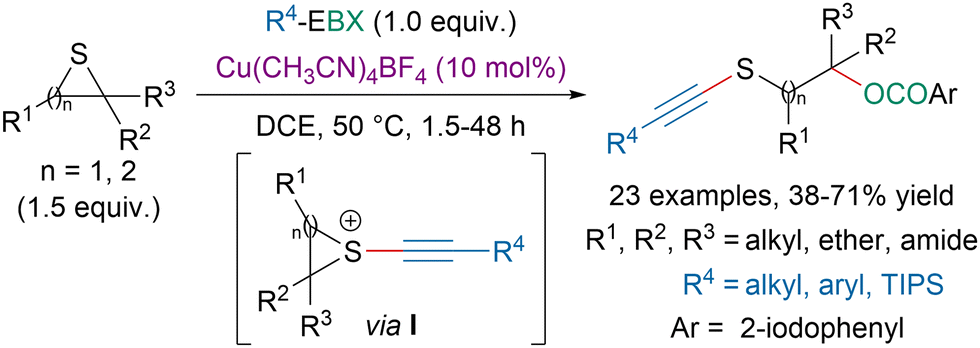 | ||
| Scheme 43 Copper-catalyzed oxyalkynylation of thiiranes and thiethanes. | ||
Chen, Liang and coworkers developed an enantioselective 1,3-oxyalknylation of Morita–Baylis–Hillman isatin carbonates using a chiral tertiary amine organocatalyst 24 (Scheme 44).154 The authors proposed that under the slightly basic conditions TES-EBX would be converted into H-EBX prior to the transfer of the alkyne. With the same strategy they could also develop an enantioselective 1,3-aminosulfenylation from N-(arylthio)succinimides.
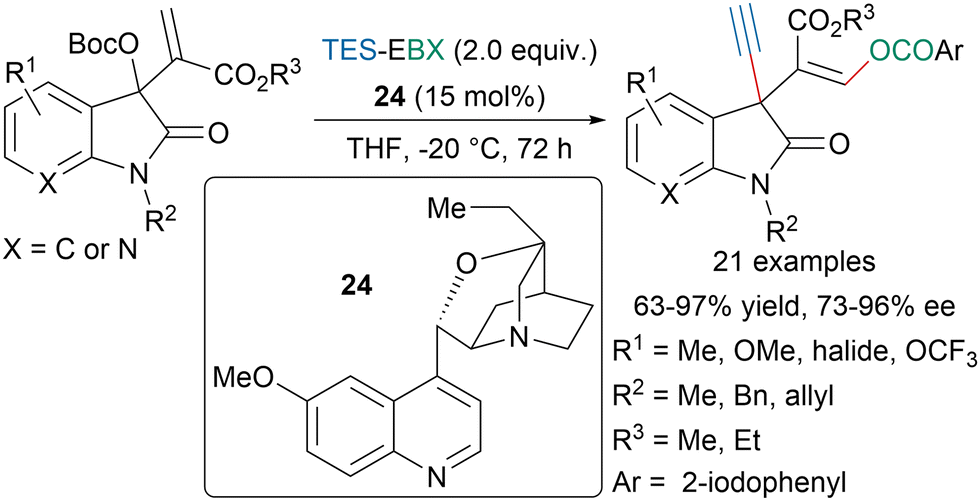 | ||
| Scheme 44 Enantioselective 1,3-oxyalkynylation of MBH isatin carbonates. | ||
In 2018, the Patil group reported a gold-catalyzed 1,2-oxyalkynylation of N-allenamides leading to 1,3-enynes (Scheme 45).155 The authors suggested that the reaction would proceed via intermediate I obtained after oxidative addition of a gold(I) species to the EBX reagent followed by coordination of the N-allenamide. Reductive elimination followed by nucleophilic addition of the aryl carboxylate from the EBX reagent would provide the targeted product. More recently, the same strategy was applied by the Hashmi group for the 1,2-oxyalkynylation of propargylamines leading to highly functionalized alkenes.156
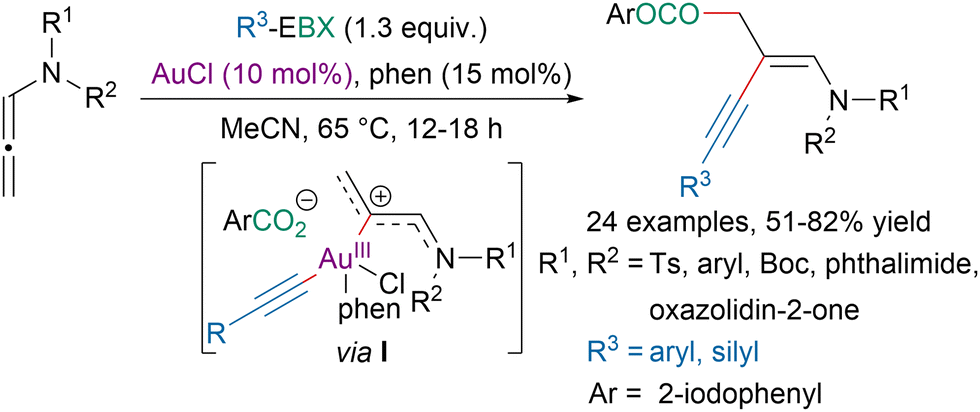 | ||
| Scheme 45 Gold-catalyzed 1,2-oxyalkynylation of N-allenamides. | ||
In 2020, our group reported a photoredox-catalyzed oxyalkynylation of enols and enamides (Scheme 46).148 It should be noted that better yields were obtained with an organic dye than via direct photoexcitation of EBX reagents (vide supra). The reaction is believed to involve the generation of radical cation intermediate I and 21 is suspected to initiate the reaction via the formation of iodanyl radicals.
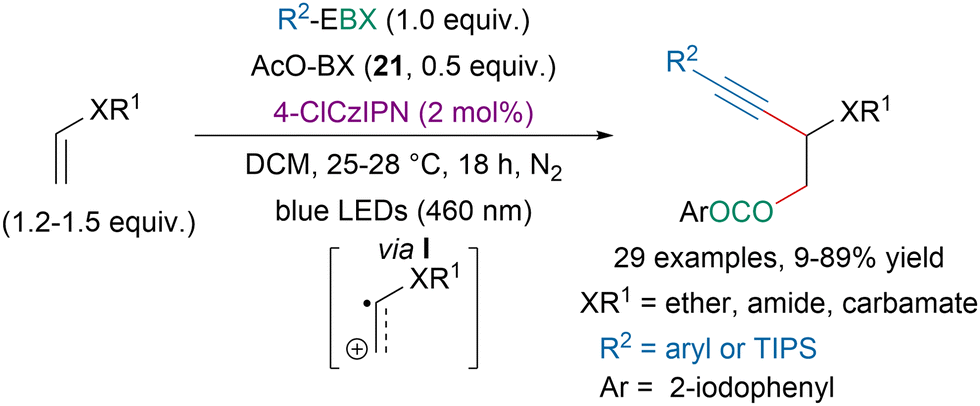 | ||
| Scheme 46 Photoredox-catalyzed oxyalkynylation of enols and enamides. | ||
7. Conclusions
The significance of alkynes in organic chemistry and other applied fields has been unquestionably established in the last decades. While the introduction of alkynes as nucleophiles into molecules has been the focus of intensive research, the development of electrophilic sources of alkynes has continued to attract more and more attention. Owing to their high reactivity and environmental friendliness, hypervalent iodine reagents have emerged as excellent alkyne electrophilic synthons. The limited stability of alkynyl iodonium salts has initially hampered the broad utilization of these reagents in alkynylation reactions. In contrast, since 2009, the more stable cyclic ethynylbenziodoxolone (EBX) reagents have found widespread applications. In this feature article, we presented the progress since 2018 in the development of new hypervalent iodine reagents, in base- or transition-metal-mediated as well as radical alkynylation transformations and in atom-economical reactions involving hypervalent iodine reagents.The application of EBX reagents for the functionalization of biomolecules is still in its infancy but could hold great promises for the future. The direct photoexcitation of aryl-EBX reagents allowed to discover and develop new radical alkynylation reactions. The simplicity of this method would make it well suitable for high-throughput experimentations to facilitate the discovery of new exciting transformations. Examples of enantioselective alkynylation reactions with hypervalent iodine reagents are still scarce in the literature and more effort to address this limitation would be needed in the future. Likewise, although EBX reagents have been widely studied in photoredox-catalyzed transformations, their use in electrochemistry is rare and would be worth more thorough investigations. Finally, one intrinsic limitation of these reagents, when used in alkynyl transfer reactions, is the stoichiometric generation of an aryl iodide side product. The development of alkynylation reactions catalytic in an organic iodine would be of high interest.
Author contributions
E. L. D. wrote and corrected the manuscript. J. W. proofread and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swiss National Science Foundation (Grant No. 200020_182798) and EPFL for financial support.Notes and references
- F. Diederich, P. J. Stang and R. R. Tykwinski, ed., Acetylene chemistry: chemistry, biology, and material science, Wiley-VCH; Weinheim, 2005 Search PubMed.
- B. M. Trost and C.-J. Li, ed., Modern alkyne chemistry: catalytic and atom-economic transformations, Wiley-VCH; Weinheim, 2015 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- Y. Sempere and E. M. Carreira, ed., The Catalytic, Enantioselective Favorskii Reaction: In Situ Formation of Metal Alkynylides and Their Additions to Aldehydes, in Organic Reactions, John Wiley & Sons, Inc., Wiley, 1st edn, 2019, pp. 207–254 Search PubMed.
- I. Jesin and G. C. Nandi, Eur. J. Org. Chem., 2019, 2704–2720 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763–2772 CrossRef CAS.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483–2504 CrossRef CAS PubMed.
- D. Ge, X. Wang and X.-Q. Chu, Org. Chem. Front., 2021, 8, 5145–5164 RSC.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523–2584 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- T. Dohi and Y. Kita, Chem. Commun., 2009, 2073–2085 RSC.
- V. V. Zhdankin, Hypervalent iodine chemistry: preparation, structure, and synthetic applications of polyvalent iodine compounds, John Wiley & Sons, Inc; Chichester, West Sussex, 2014 Search PubMed.
- T. Wirth, Hypervalent iodine chemistry, Springer Berlin Heidelberg; New York, NY, 2016 Search PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- M. Ghosh, A. Rajkiewicz and M. Kalek, Synthesis, 2019, 359–370 CAS.
- F. V. Singh and T. Wirth, ARKIVOC, 2021, 2021, 12–47 Search PubMed.
- N. Rani, R. Soni, M. Sihag, M. Kinger and D. K. Aneja, Adv. Synth. Catal., 2022, 364, 1798–1848 CrossRef CAS.
- J. Waser, Alkynylation with Hypervalent Iodine Reagents, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing; Cham, 2015, vol. 373, pp. 187–222 Search PubMed.
- J. Waser, Synlett, 2016, 2761–2773 CrossRef CAS.
- D. P. Hari, S. Nicolai and J. Waser, Alkynylations and Vinylations, in PATAI’S Chemistry of Functional Groups, ed. Z. Rappoport, John Wiley & Sons, Ltd; Chichester, UK, 2018, pp. 1–58 Search PubMed.
- D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS PubMed.
- J. I. Musher, Angew. Chem., Int. Ed. Engl., 1969, 8, 54–68 CrossRef CAS.
- J. C. Martin, Science, 1983, 221, 509–514 CrossRef CAS PubMed.
- R. Gillespie, Coord. Chem. Rev., 2002, 233–234, 53–62 CrossRef CAS.
- A. Stirling, Chem. – Eur. J., 2018, 24, 1709–1713 CrossRef CAS PubMed.
- S. S. Karandikar, A. Bhattacharjee, B. E. Metze, N. Javaly, E. J. Valente, T. M. McCormick and D. R. Stuart, Chem. Sci., 2022, 13, 6532–6540 RSC.
- Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed.
- R. L. Amey and J. C. Martin, J. Org. Chem., 1979, 44, 1779–1784 CrossRef CAS.
- T.-Y. Sun, X. Wang, H. Geng, Y. Xie, Y.-D. Wu, X. Zhang and H. F. Schaefer III, Chem. Commun., 2016, 52, 5371–5374 RSC.
- V. V. Zhdankin and P. J. Stang, Tetrahedron, 1998, 54, 10927–10966 CrossRef CAS.
- M. S. Yusubov, A. V. Maskaev and V. V. Zhdankin, ARKIVOC, 2011, 2011, 370–409 Search PubMed.
- F. Le Vaillant and J. Waser, Chem. Sci., 2019, 10, 8909–8923 RSC.
- M. Ochiai, Y. Masaki and M. Shiro, J. Org. Chem., 1991, 56, 5511–5513 CrossRef CAS.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547–6551 CrossRef CAS PubMed.
- M. J. Bouma and B. Olofsson, Chem. – Eur. J., 2012, 18, 14242–14245 CrossRef CAS PubMed.
- D. P. Hari, P. Caramenti, L. Schouwey, M. Chang, S. Nicolai, D. Bachert, T. Wright, C. Orella and J. Waser, Org. Process Res. Dev., 2020, 24, 106–110 CrossRef CAS.
- J. Borrel and J. Waser, Org. Lett., 2022, 24, 142–146 CrossRef CAS PubMed.
- M. Yudasaka, D. Shimbo, T. Maruyama, N. Tada and A. Itoh, Org. Lett., 2019, 21, 1098–1102 CrossRef CAS PubMed.
- X. Sun, X. Guo, L. Chen and T. Kang, Chem. – Eur. J., 2021, 27, 4312–4316 CrossRef CAS PubMed.
- N. Declas, G. Pisella and J. Waser, Helv. Chim. Acta, 2020, 103, e2000191 CrossRef CAS.
- S. He, X. Guo, J. Li, Y. Zhang, L. Chen and T. Kang, Eur. J. Org. Chem., 2022, e202200516 CAS.
- J. Li, C. Zhou, H. Liang, X. Guo, L. Chen and T. Kang, Eur. J. Org. Chem., 2022, e202200613 CAS.
- D. P. Hari, L. Schouwey, V. Barber, R. Scopelliti, F. Fadaei-Tirani and J. Waser, Chem. – Eur. J., 2019, 25, 9522–9528 CrossRef CAS PubMed.
- E. Le, Du, T. Duhail, M. D. Wodrich, R. Scopelliti, F. Fadaei-Tirani, E. Anselmi, E. Magnier and J. Waser, Chem. – Eur. J., 2021, 27, 10979–10986 CrossRef PubMed.
- T. J. Kuczmera, A. Boelke and B. J. Nachtsheim, Eur. J. Org. Chem., 2022, e202200276 CAS.
- F. M. Beringer and S. A. Galton, J. Org. Chem., 1965, 30, 1930–1934 CrossRef CAS.
- D. Fernández-González, J. P. Brand and J. Waser, Chem. – Eur. J., 2010, 16, 9457–9461 CrossRef PubMed.
- W. Shao, J. Huang, K. Guo, J. Gong and Z. Yang, Org. Lett., 2018, 20, 1857–1860 CrossRef CAS PubMed.
- S. Xie, G. Chen, H. Yan, J. Hou, Y. He, T. Zhao and J. Xu, J. Am. Chem. Soc., 2019, 141, 3435–3439 CrossRef CAS PubMed.
- R. Long and Z. Yang, Tetrahedron, 2019, 75, 1746–1750 CrossRef CAS.
- M. A. Baker, R. M. Demoret, M. Ohtawa and R. A. Shenvi, Nature, 2019, 575, 643–646 CrossRef CAS PubMed.
- R. M. Demoret, M. A. Baker, M. Ohtawa, S. Chen, C. C. Lam, S. Khom, M. Roberto, S. Forli, K. N. Houk and R. A. Shenvi, J. Am. Chem. Soc., 2020, 142, 18599–18618 CrossRef CAS PubMed.
- B. Hong, D. Hu, Y. Kadonaga, X. Lei, R. Tang and J. Wang, J. Am. Chem. Soc., 2020, 142, 2238–2243 CrossRef PubMed.
- H. Chen, Z. Li, P. Shao, H. Yuan, S.-C. Chen and T. Luo, J. Am. Chem. Soc., 2022, 144, 15462–15467 CrossRef CAS PubMed.
- A. Roy, M. K. Das, S. Chaudhuri and A. Bisai, J. Org. Chem., 2018, 83, 403–421 CrossRef CAS PubMed.
- B. Meng, Q. Shi, Y. Meng, J. Chen, W. Cao and X. Wu, Org. Biomol. Chem., 2021, 19, 5087–5092 RSC.
- B. V. M. Teodoro and L. F. Silva, J. Org. Chem., 2018, 83, 13604–13611 CrossRef CAS PubMed.
- A. A. Rajkiewicz, N. Wojciechowska and M. Kalek, ACS Catal., 2020, 10, 831–841 CrossRef CAS.
- M. Ochiai, M. Kunishima, Y. Nagao, K. Fuji, M. Shiro and E. Fujita, J. Am. Chem. Soc., 1986, 108, 8281–8283 CrossRef CAS.
- J. Schörgenhumer and M. Waser, Org. Biomol. Chem., 2018, 16, 7561–7563 RSC.
- X. Li, X. Xie, N. Sun and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 6994–6998 CrossRef CAS PubMed.
- S. Banerjee and N. T. Patil, Chem. Commun., 2017, 53, 7937–7940 RSC.
- Y. Liu, Y. Yang, R. Zhu, C. Liu and D. Zhang, Catal. Sci. Technol., 2019, 9, 4091–4099 RSC.
- G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed.
- K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed.
- A. M. Cook and C. Wolf, Tetrahedron Lett., 2015, 56, 2377–2392 CrossRef CAS PubMed.
- D. Xiang, H. Li, L. Zhang, Y. Zhang, Q. Zhang and D. Li, Asian J. Org. Chem., 2019, 8, 537–541 CrossRef CAS.
- M. Li, J.-H. Wang, W. Li and L.-R. Wen, Org. Lett., 2018, 20, 7694–7698 CrossRef CAS PubMed.
- M. Li, W. Li, C.-D. Lin, J.-H. Wang and L.-R. Wen, J. Org. Chem., 2019, 84, 6904–6915 CrossRef CAS PubMed.
- M. Li, J.-H. Wang, W. Li, C.-D. Lin, L.-B. Zhang and L.-R. Wen, J. Org. Chem., 2019, 84, 8523–8530 CrossRef CAS PubMed.
- S. G. E. Amos, S. Nicolai, A. Gagnebin, F. Le Vaillant and J. Waser, J. Org. Chem., 2019, 84, 3687–3701 CrossRef CAS PubMed.
- E. M. D. Allouche, E. Grinhagena and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202112287 CrossRef CAS PubMed.
- R. Frei and J. Waser, J. Am. Chem. Soc., 2013, 135, 9620–9623 CrossRef CAS PubMed.
- R. Frei, M. D. Wodrich, D. P. Hari, P.-A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563–16573 CrossRef CAS PubMed.
- D. Abegg, R. Frei, L. Cerato, D. P. Hari, C. Wang, J. Waser and A. Adibekian, Angew. Chem., Int. Ed., 2015, 54, 10852–10857 CrossRef CAS PubMed.
- R. Tessier, J. Ceballos, N. Guidotti, R. Simonet-Davin, B. Fierz and J. Waser, Chemistry, 2019, 5, 2243–2263 CrossRef CAS.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- A. K. Mishra, R. Tessier, D. P. Hari and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 17963–17968 CrossRef CAS PubMed.
- J. Ceballos, E. Grinhagena, G. Sangouard, C. Heinis and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 9022–9031 CrossRef CAS PubMed.
- R. Tomita, N. Al-Maharik, A. Rodil, M. Bühl and D. O’Hagan, Org. Biomol. Chem., 2018, 16, 1113–1117 RSC.
- D. Bello, M. G. Rubanu, N. Bandaranayaka, J. P. Götze, M. Bühl and D. O’Hagan, ChemBioChem, 2019, 20, 1174–1182 CrossRef CAS PubMed.
- B. Lim, Y. Cheng, T. Kato, A. Pham, E. Le Du, A. K. Mishra, E. Grinhagena, D. Moreau, N. Sakai, J. Waser and S. Matile, Helv. Chim. Acta, 2021, 104, e2100085 CrossRef CAS.
- Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414–17420 CrossRef CAS PubMed.
- R. Takai, D. Shimbo, N. Tada and A. Itoh, J. Org. Chem., 2021, 86, 4699–4713 CrossRef CAS PubMed.
- R. E. Beveridge and R. A. Batey, Org. Lett., 2012, 14, 540–543 CrossRef CAS PubMed.
- E. Le Du, J. Borrel and J. Waser, Org. Lett., 2022, 24, 6614–6618 CrossRef CAS PubMed.
- S. Banerjee, V. W. Bhoyare and N. T. Patil, Chem. Commun., 2020, 56, 2677–2690 RSC.
- L. Hu, M. C. Dietl, C. Han, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2021, 60, 10637–10642 CrossRef CAS PubMed.
- C. Han, X. Tian, L. Song, Y. Liu and A. S. K. Hashmi, Org. Chem. Front., 2021, 8, 6546–6552 RSC.
- C. Han, Y. Liu, X. Tian, F. Rominger and A. S. K. Hashmi, Org. Lett., 2021, 23, 9480–9484 CrossRef CAS PubMed.
- L. D. Caspers and B. J. Nachtsheim, Chem. – Asian J., 2018, 13, 1231–1247 CrossRef CAS PubMed.
- R. S. Rohokale, R. G. Kalshetti and C. V. Ramana, J. Org. Chem., 2019, 84, 2951–2961 CrossRef CAS PubMed.
- X. Wang, X. Li, Y. Zhang and L. Xia, Org. Biomol. Chem., 2018, 16, 2860–2864 RSC.
- A. C. Shaikh, D. R. Shinde and N. T. Patil, Org. Lett., 2016, 18, 1056–1059 CrossRef CAS PubMed.
- F. Zhao, B. Xu, D. Ren, L. Han, Z. Yu and T. Liu, Organometallics, 2018, 37, 1026–1033 CrossRef CAS.
- R. Budhwan, S. Yadav and S. Murarka, Org. Biomol. Chem., 2019, 17, 6326–6341 RSC.
- J. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346–9349 CrossRef CAS PubMed.
- Y. Liu, F. Chang, Q. Jiang, Z. Ma and C. Liu, Synlett, 2018, 658–662 CrossRef.
- G. N. Hermann, M. T. Unruh, S. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723–10727 CrossRef CAS PubMed.
- J. Zhang, M. Wang, H. Wang, H. Xu, J. Chen, Z. Guo, B. Ma, S.-R. Ban and H.-X. Dai, Chem. Commun., 2021, 57, 8656–8659 RSC.
- T. Shimada, S. Mori, M. Ishida and H. Furuta, Beilstein J. Org. Chem., 2020, 16, 587–595 CrossRef CAS PubMed.
- Z. Wang, X. Li and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 14219–14223 CrossRef CAS PubMed.
- S. Peng, Z. Wang, L. Zhang, X. Zhang and Y. Huang, Nat. Commun., 2018, 9, 375 CrossRef PubMed.
- C. Han, X. Tian, H. Zhang, F. Rominger and A. S. K. Hashmi, Org. Lett., 2021, 23, 4764–4768 CrossRef CAS PubMed.
- Y. Yang, P. Antoni, M. Zimmer, K. Sekine, F. F. Mulks, L. Hu, L. Zhang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 5129–5133 CrossRef CAS PubMed.
- X. Zhao, B. Tian, Y. Yang, X. Si, F. F. Mulks, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2019, 361, 3155–3162 CrossRef CAS.
- S. Banerjee, S. B. Ambegave, R. D. Mule, B. Senthilkumar and N. T. Patil, Org. Lett., 2020, 22, 4792–4796 CrossRef CAS PubMed.
- M. H. Babu, V. Dwivedi, R. Kant and M. S. Reddy, Angew. Chem., Int. Ed., 2015, 54, 3783–3786 CrossRef PubMed.
- X. Li, P. Chen and G. Liu, Chem. Commun., 2022, 58, 2544–2547 RSC.
- J. T. Su and W. A. Goddard, J. Am. Chem. Soc., 2005, 127, 14146–14147 CrossRef CAS PubMed.
- A.-A. Guilbault and C. Y. Legault, ACS Catal., 2012, 2, 219–222 CrossRef CAS.
- J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. – Eur. J., 2012, 18, 5655–5666 CrossRef CAS PubMed.
- J. Huang, L.-L. Chen and Z.-M. Chen, Org. Lett., 2022, 24, 5777–5781 CrossRef CAS PubMed.
- R. Zhao and L. Shi, Angew. Chem., Int. Ed., 2020, 59, 12282–12292 CrossRef CAS PubMed.
- D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190–2193 CrossRef CAS PubMed.
- D. P. Hari and J. Waser, J. Am. Chem. Soc., 2017, 139, 8420–8423 CrossRef CAS PubMed.
- G. Himbert and L. Henn, Angew. Chem., Int. Ed. Engl., 1982, 21, 620 CrossRef.
- D. P. Hari, G. Pisella, M. D. Wodrich, A. V. Tsymbal, F. Fadei-Tirani, R. Scopelliti and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 5475–5481 CrossRef CAS PubMed.
- G. Pisella, A. Gagnebin and J. Waser, Chem. – Eur. J., 2020, 26, 10199–10204 CrossRef CAS PubMed.
- N. P. Ramirez, G. Pisella and J. Waser, J. Org. Chem., 2021, 86, 10928–10938 CrossRef CAS PubMed.
- X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 CrossRef CAS PubMed.
- L. Li, S. Guo, Q. Wang and J. Zhu, Org. Lett., 2019, 21, 5462–5466 CrossRef CAS PubMed.
- S. Tsuzuki, R. Sakamoto and K. Maruoka, Chem. Lett., 2020, 49, 633–636 CrossRef CAS.
- X. Luo and P. Wang, Org. Lett., 2021, 23, 4960–4965 CrossRef CAS PubMed.
- X. Yang and G. C. Tsui, Org. Lett., 2019, 21, 8625–8629 CrossRef CAS PubMed.
- H.-L. Cheng, X.-H. Xie, J.-Z. Chen, Z. Wang and J.-P. Chen, Chem. Sci., 2021, 12, 11786–11792 RSC.
- Y.-L. Liu, X.-L. Zhu, Y. Huang, F.-L. Qing and X.-H. Xu, J. Fluorine Chem., 2021, 242, 109715 CrossRef CAS.
- K. Matsumoto, M. Nakajima and T. Nemoto, J. Org. Chem., 2020, 85, 11802–11811 CrossRef CAS PubMed.
- E. Voutyritsa, M. Garreau, M. G. Kokotou, I. Triandafillidi, J. Waser and C. G. Kokotos, Chem. – Eur. J., 2020, 26, 14453–14460 CrossRef CAS PubMed.
- Z. Liu, Y. Pan, P. Zou, H. Huang, Y. Chen and Y. Chen, Org. Lett., 2022, 24, 5951–5956 CrossRef CAS PubMed.
- S. Liang, R. A. Angnes, C. S. Potnis and G. B. Hammond, Tetrahedron Lett., 2019, 60, 151230 CrossRef.
- W.-J. Han, J.-W. Zhang, C.-X. Yan, J.-W. Wang, P.-P. Zhou and B. Han, Org. Lett., 2022, 24, 542–547 CrossRef CAS PubMed.
- Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 CrossRef CAS PubMed.
- F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 CrossRef CAS PubMed.
- J. Guo, Y. Wang, Y. Li, K. Lu, S. Liu, W. Wang and Y. Zhang, Adv. Synth. Catal., 2020, 362, 3898–3904 CrossRef CAS.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS PubMed.
- K. Lu, Y. Ma, S. Liu, S. Guo and Y. Zhang, Chin. J. Chem., 2022, 40, 681–686 CrossRef CAS.
- Z. Liu, S. Wu and Y. Chen, ACS Catal., 2021, 11, 10565–10573 CrossRef CAS.
- S. G. E. Amos, D. Cavalli, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 23827–23834 CrossRef CAS PubMed.
- M. Li, T. Liu, J. Li, H. He, H. Dai and J. Xie, J. Org. Chem., 2021, 86, 12386–12393 CrossRef CAS PubMed.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed.
- Y. Pan, K. Jia, Y. Chen and Y. Chen, Beilstein J. Org. Chem., 2018, 14, 1215–1221 CrossRef CAS PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn’ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 RSC.
- S. G. E. Amos, S. Nicolai and J. Waser, Chem. Sci., 2020, 11, 11274–11279 RSC.
- T. V. T. Nguyen, M. D. Wodrich and J. Waser, Chem. Sci., 2022, 13, 12831–12839 RSC.
- Z. Zuo and A. Studer, Org. Lett., 2022, 24, 949–954 CrossRef CAS PubMed.
- A. Boelke, P. Finkbeiner and B. J. Nachtsheim, Beilstein J. Org. Chem., 2018, 14, 1263–1280 CrossRef CAS PubMed.
- G. Grelier, B. Darses and P. Dauban, Beilstein J. Org. Chem., 2018, 14, 1508–1528 CrossRef CAS PubMed.
- J. Borrel, G. Pisella and J. Waser, Org. Lett., 2020, 22, 422–427 CrossRef CAS PubMed.
- Z.-C. Chen, P. Chen, Z. Chen, Q. Ouyang, H.-P. Liang, W. Du and Y.-C. Chen, Org. Lett., 2018, 20, 6279–6283 CrossRef CAS PubMed.
- S. Banerjee, B. Senthilkumar and N. T. Patil, Org. Lett., 2019, 21, 180–184 CrossRef CAS PubMed.
- Y. Liu, M. C. Dietl, C. Han, M. Rudolph, F. Rominger, P. Krämer and A. S. K. Hashmi, Org. Lett., 2022, 24, 7101–7106 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |