
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible pH-responsive MRI contrast with paramagnetic polymer micelles†
Connor M.
Ellis
 a,
Daohe
Yuan
a,
Ferenc E.
Mózes
b,
Jack J.
Miller
bcd and
Jason J.
Davis
*a
a,
Daohe
Yuan
a,
Ferenc E.
Mózes
b,
Jack J.
Miller
bcd and
Jason J.
Davis
*a
aDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: jason.davis@chem.ox.ac.uk; Tel: +44 (0)1865 275 914
bOxford Centre for Clinical Magnetic Resonance Research, Radcliffe Department of Medicine, University of Oxford, Level 0, John Radcliffe Hospital, Oxford, OX3 9DU, UK
cThe MR Research Centre, Aarhus University, Aarhus 8200, Denmark
dDepartment of Physics, Clarendon Laboratory, Parks Road, Oxford, OX1 3PU, UK
First published on 11th January 2023
Abstract
Paramagnetically-doped polymer micelles, containing an ionizable poly(acrylic acid) (PAA) block, support high-contrast MR imaging at clinically relevant field strengths in a manner that is strongly pH responsive. A reversible switch in polymer strand charge specifically has a direct impact on local rigidity, and rotational correlation time characteristics, of the integrated Gd-chelate, driving a ∼50% amplitude switch in positive contrast.
The design of novel magnetic resonance imaging (MRI) contrast agents suitable for clinical applications that are translatable for use in vivo is paramount in improving the diagnostic potency of derived scans. The use of nanoparticulate agents offers such a possibility with notably improved relaxivities over their corresponding molecular analogues; this can translate into either higher contrast specificity or the requirement of a lower effective dose.1 They additionally possess enhanced blood circulation times, improved biocompatibilities, an ability to incorporate additional imaging or therapeutic modes and a potentially engineered responsive character to facilitate disease specific imaging.2,3 There exist a broad number of configurations of paramagnetically-doped nanoparticles, iron oxide nanoparticles and doped liposomes that can underpin such characteristics.4–6
pH-responsive T1 or T2 contrast is clinically valuable in conditions ranging from cancer to cardiac ischaemia, and has been demonstrated (although often irreversibly) with a range of paramagnetic, superparamagnetic, and metabolic imaging agents.2,7–10 Underpinning this, a structural or dynamic parameter intrinsic to the contrast agent must be pH-sensitive, with common approaches manipulating governing Solomon–Bloembergen–Morgan (SBM) predicted correlation times (e.g., chelate bound hydration or water exchange rates).3,8 For in vivo applications it is essential that the contrast agent resists degradation/fast metabolic clearance and, ideally, possesses a reversible response such that it reaches the desired site both intact and (still) responsive.11 Whilst reversible pH reporting has been described for inorganic nanoparticles such as those based on modified mesoporous silica,8 there are no published studies with organic nanoparticles, despite these having advantageous blood retention times and physicochemical tunability.12,13
Block copolymer micelles are an exciting novel class of MR contrast agent offering a highly biocompatible platform whose composition, size and shape are tuneable at the point of synthesis.12 Individual polymer blocks can, for example, be generated with a chosen monomer able to impart a desired stimulus-response, or support a further downstream modification e.g., with an active targeting moiety.14 High colloidal stability and biocompatibility are a necessity in preventing challenges associated with fast renal clearance and related biodistribution of the nanoparticulate agent,13 and polymers present much in this regard.12 A number of MRI active micellar systems have been shown to irreversibly fragment or aggregate with pH change.2,15–17 There are no prior reports of paramagnetic micelles able to support an engineered reversible pH-responsive contrast.
The integration of paramagnetic chelates within a nanoparticulate architecture can dramatically boost relaxivity,18–23 an observation most typically associated with a reduction in chelate rotational mobility.20 This is generally assumed to be fixed at the point of particle synthesis/paramagnet integration and has not been previously integrated into a responsive imaging modality. Herein, we report the generation of pH-sensitive poly(styrene)-block-poly(acrylic acid) micelles (PS-b-PAA), where triggered changes in polymer conformation directly impact the associated longitudinal relaxivity. These changes we assign to reversible modulation of the associated rotational correlation characteristics of the entrapped chelate (Fig. 1) as supported by an analytical treatment given by SBM theory (see ESI†). Specifically, differences in PAA chain conformational mobility, on which gadolinium(III)-1,4,7-tris(carbonylmethyl)-10-(aminoethyl-N′-acetyl)-1,4,7,10-tetraazacyclododecane (Gd-DO3A) units sit, modulate mechanical coupling to the, slowly rotating, micelle core (ESI 1†); see details below.24 It has been prior noted that PAA occupies a globular conformation below its associated pKa (= 4.7), swelling at higher pH as the polymer backbone is deprotonated.25,26 Under such conditions one would expect that the integrated Gd-chelates become confined within the polymer matrix such that relaxivity is much more dictated by the rotation of the core (F2 → 1) and relaxivity increases.
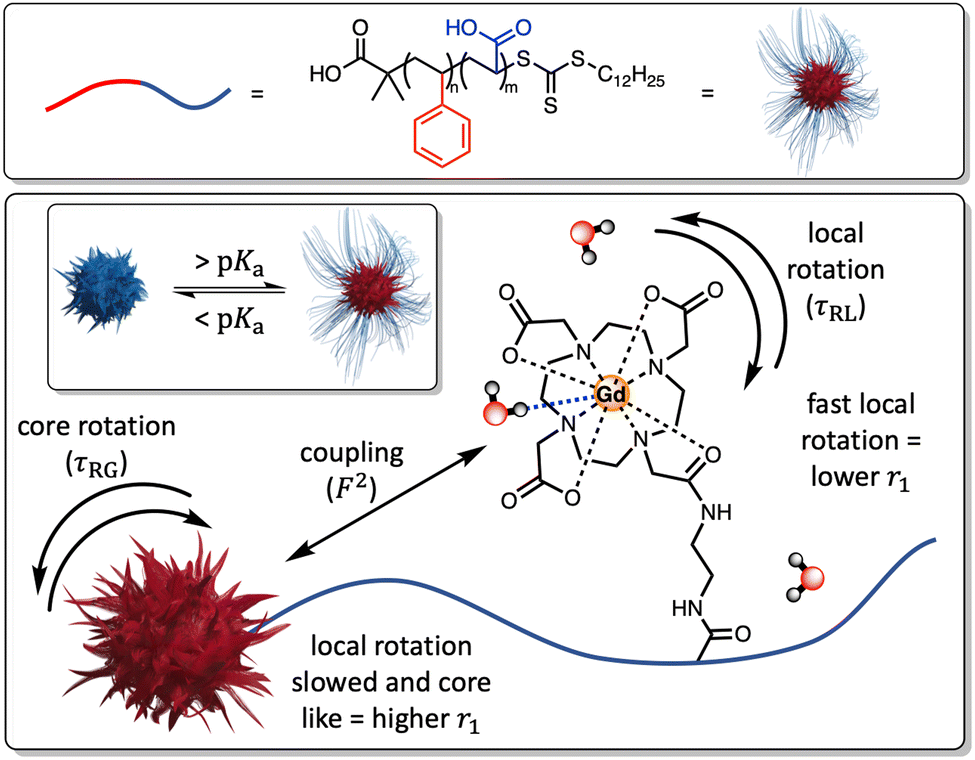 | ||
| Fig. 1 A schematic highlighting how differences in the mechanical flexibility of the PAA backbone can affect the coupling of the paramagnet local rotational correlation characteristics to the core of the micelle. This is defined by an order parameter (F2), where 0 < F2 < 1, with larger F2 values increasing r1 values in a manner that is reversed as the polymer chains charge and become more conformationally dynamic. | ||
The block copolymers were synthesised through a reversible addition-fragmentation chain transfer polymerisation (RAFT) method, with a polystyrene (2-(dodecylthiocarbonothioylthio)-2-methylproponic acid terminated) (PS (DDMAT terminated)) backbone macroinitiator (ESI 3†). The hydrophobic nature of PS is responsible for a natively collapsed micelle core in aqueous solution. The AA monomer forms the extended micellular shell, with an abundance of pH-responsive and chemically modifiable carboxylic acid groups. Polymerisation was monitored by NMR and can be seen by the assigned 1H NMR spectrum for PS-b-PAA (ESI 15†), with notable peaks at δ 6.55–7.08 ppm and δ 12.27 ppm arising from the benzene and carboxylic acid motifs respectively. The “living” character of this was confirmed by pseudo-first order kinetic plots, with a complete reaction time of 18 h (ESI 16†). Gel permeation chromatography measurements (GPC) resolve the number-average molecular weight (Mn), weighted-average molecular weight (Mw) and polydispersity (Mn/Mw) to be 10.5 kDa, 10.9 kDa and 1.03 respectively. Pre-metalated DO3A-R-NH2 was then subsequently tethered by standard active ester methods to generate Gd-PAA-b-PS; see ESI 2–14.†27
Micellar ATR-IR and dynamic light scattering fingerprints were as expected (ESI 17 and 18†). Measured ζ-potentials were negative, consistent with expectations for a PAA block (ESI 18(b)†). The micelles were shown to be stable (PDI < 0.3) across a full pH range (pH 4.5–8.0; for at least 5 weeks, ESI 19†) with, as expected, a substantial increase in hydrodynamic size (88%, 52.67 ± 5.43 nm at pH 3.5, 98.96 ± 8.69 nm at pH 7.6, ESI 18(a)†) as the pKa is traversed. These DLS resolved pH-triggered swelling/de-swelling and associated ζ-potential changes are consistent with expectations, and reversible (ESI 20†). TEM images of the paramagnetically-doped PAA-b-PS micelles indicated the formation of spherical particles, with an associated size of 20.78 ± 3 nm (expectedly lower than the hydrated radius, ESI 21†). The ICP-MS estimated number of chelated Gd3+ ions tethered to the PAA micellular backbone is 6450 ± 640 Gd3+/micelle. The number of lanthanide-bound H2O molecules (q) is constant above and below the pKa of PAA, as resolved by the time-resolved luminescence lifetime measurements of the corresponding Eu-doped micelles in both H2O and D2O, with q = 1.0 ± 0.1 at both pH 4.0 and pH 7.0 (ESI 23 and 24†).
To investigate the ability of the paramagnetically-doped micelles to act as effective MRI contrast agents, longitudinal relaxivity values (calculated per mM of Gd3+) were recorded at a clinically relevant 1.4 T (Fig. 2). Relaxivities recorded at pH 4.0 (i.e., < pKa of PAA) are some 10.5 times larger than the corresponding commercially available and clinically used molecular complex agent (Gd-DOTA).28 Notably, at physiological pH the micelles possess an associated r1 = 20.79 ± 0.02 mM−1 s−1 (pH = 7.5), which was shown to increase significantly as pH drops below the associated PAA pKa (r1 = 30.49 ± 0.12 mM−1 s−1 at pH 4.0, Δr1 = 9.70 mM−1 s−1). As pH increases, the micelles charge and swell, with strands extending into solution and chelate rotational flexibility increasing (being less mechanically coupled to the core). These r1 switches are highly reversible (Fig. 3), persisting over multiple pH cycles. The transverse relaxivity values for the Dy-doped micellular analogue (Dy-PAA-b-PS) were additionally recorded at 11.7 T, highlighting the potential for high field T2 weighted imaging; see ESI.†
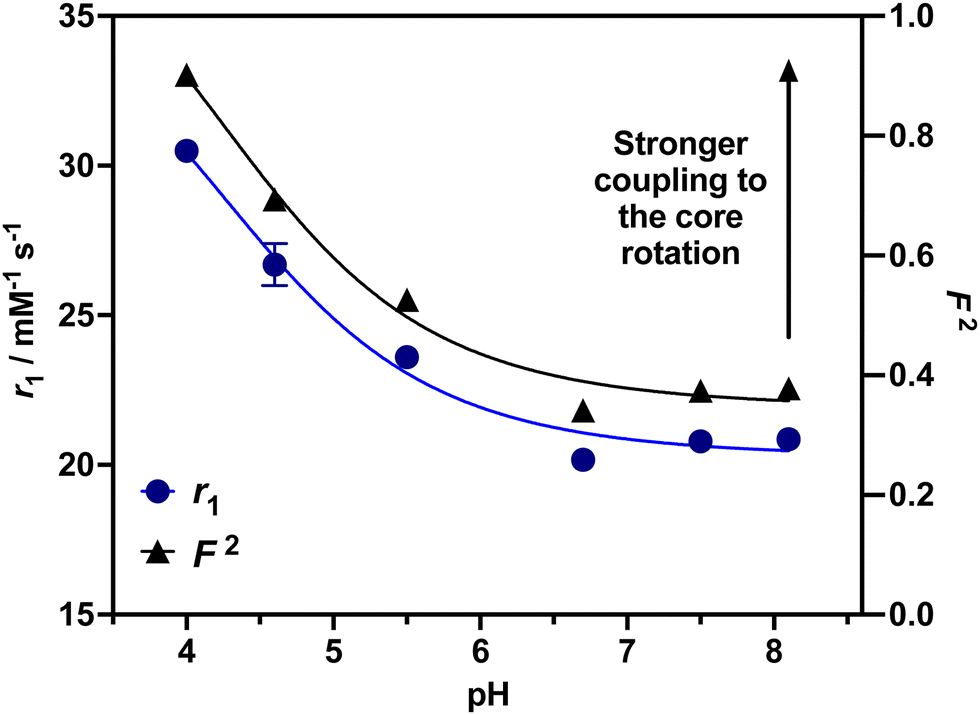 | ||
| Fig. 2 Relaxivity values for Gd-PAA-b-PS micelles over a range of pH values, exhibiting a marked ∼50% switch as pH decreases. The calculated F2 values, based on the Lipari–Szabo approach, for each recorded relaxivity value are included. The aligned experimental and theoretical observations highlight the dominance of the chelate rotational characteristics in the observed relaxivity and contrast responses. | ||
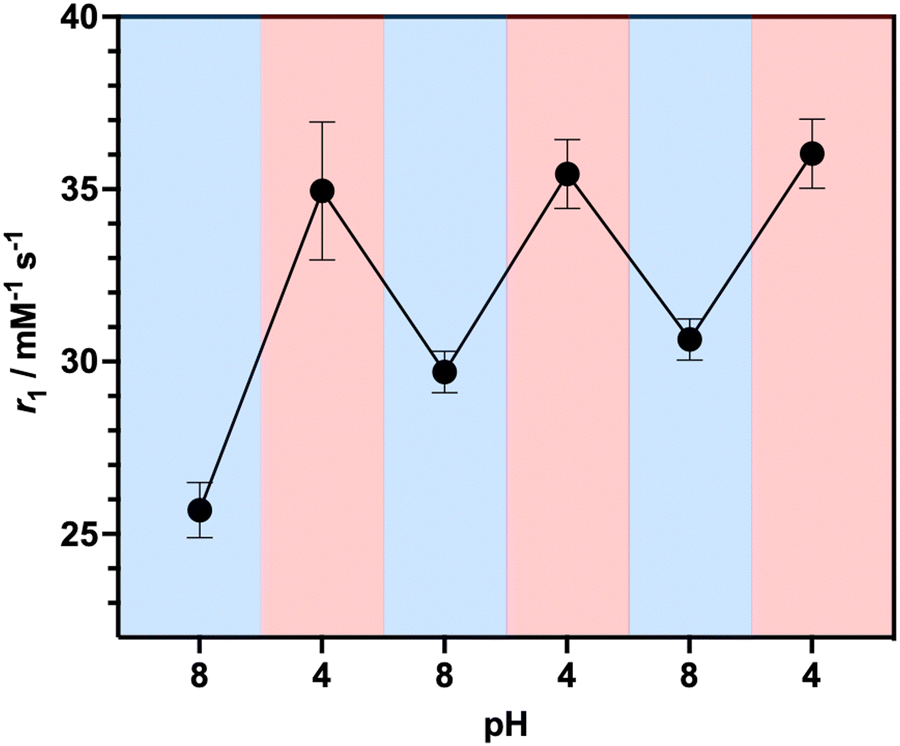 | ||
| Fig. 3 Reversible relaxivity measurements highlighting a reversible switch in r1 as pH decreases from pH 8.0 to pH 4.0 for three repeated cycles. | ||
To understand how differences in the flexibility of the PAA tether affect chelate rotation and hence relaxivity, a Lipari–Szabo approach was considered, and is detailed in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24,29 For this, the value of τRG was estimated from the TEM derived micellular radius and the Stokes–Einstein equation (τRG = 4πa3η/3kBT; with η = the viscosity of water, kB = Boltzmann's constant, T = temperature and a = particle radius).30 All other parameters were estimated and are included in the ESI.†31 As can be observed in ESI 25(a),† increasing F2 significantly enhances r1 as the rotational characteristics of the Gd-doped chelate become core-like (and thus at a frequency much closer to the proton Larmor precession). It should be noted that swelling-induced changes in global rotation are not significant and cannot account for the r1 changes observed (ESI 25(b)†). Responsive local correlation times are dominant here (ESI 25(c)†); specifically, in solving the Lipari–Szabo modified equations for the experimentally derived relaxivities (Fig. 2), an estimate for the change in the order parameter (F) can be obtained where F2 = 1 is indicative of isotropic motion and chelate rotation strongly coupled to the micelle core. Conversely, a F2 = 0 value correlates fast local motion of the chelate, with rotational characteristics independent to the core of the micelle.32 At pH = 4.0 where F2 approaches 0.9, the paramagnetic centre is much more tightly coupled to the slow global rotation of the core and r1 grows markedly. Conversely, at pH 7.5, F2 is estimated to be 0.4, and the paramagnetic chelate has a much higher level of independent rotational freedom and a lower associated relaxivity. It should be noted that this is the observed lower limit reflecting some permanent non-zero coupling of chelate to core motion; associated relaxivities accordingly always exceed those of the molecular analogue. The differences in F2 across a full clinical pH range are included in Fig. 2, with relaxivity changes fully accounted for by these.
24,29 For this, the value of τRG was estimated from the TEM derived micellular radius and the Stokes–Einstein equation (τRG = 4πa3η/3kBT; with η = the viscosity of water, kB = Boltzmann's constant, T = temperature and a = particle radius).30 All other parameters were estimated and are included in the ESI.†31 As can be observed in ESI 25(a),† increasing F2 significantly enhances r1 as the rotational characteristics of the Gd-doped chelate become core-like (and thus at a frequency much closer to the proton Larmor precession). It should be noted that swelling-induced changes in global rotation are not significant and cannot account for the r1 changes observed (ESI 25(b)†). Responsive local correlation times are dominant here (ESI 25(c)†); specifically, in solving the Lipari–Szabo modified equations for the experimentally derived relaxivities (Fig. 2), an estimate for the change in the order parameter (F) can be obtained where F2 = 1 is indicative of isotropic motion and chelate rotation strongly coupled to the micelle core. Conversely, a F2 = 0 value correlates fast local motion of the chelate, with rotational characteristics independent to the core of the micelle.32 At pH = 4.0 where F2 approaches 0.9, the paramagnetic centre is much more tightly coupled to the slow global rotation of the core and r1 grows markedly. Conversely, at pH 7.5, F2 is estimated to be 0.4, and the paramagnetic chelate has a much higher level of independent rotational freedom and a lower associated relaxivity. It should be noted that this is the observed lower limit reflecting some permanent non-zero coupling of chelate to core motion; associated relaxivities accordingly always exceed those of the molecular analogue. The differences in F2 across a full clinical pH range are included in Fig. 2, with relaxivity changes fully accounted for by these.
To demonstrate the translational relevance of these pH-responsive micelles, we performed MR imaging on clinical imaging scanners at both 1.5 T and 3 T with a widely used clinical T1-mapping sequence commonly applied to body imaging.33,34 As shown in Fig. 4(a) the micelle generated contrast is clear and uniform at both field strengths and notably responds strongly to pH in a manner that mirrors the relaxivity measurement observations (MR resolved relaxivities are within 6% of those determined by NMR, and are across independent syntheses). The switching effects are, predictably, greatest at 1.5 T where rotational correlation mediated relaxivity is highest.35,36 We note that ShMOLLI (shortened modified look-locker inversion recovery), like other clinically used T1 mapping sequences, is a highly optimised technique capable of consecutive inversion-recovery measurements during the length of time a sick patient can hold their breath.33 For this reason, it is reassuring to note that the magnitude of the change in T1 created by our micellular contrast agents at a plausible intravascular concentration is readily resolvable (ESI 26†), even at 3 T.
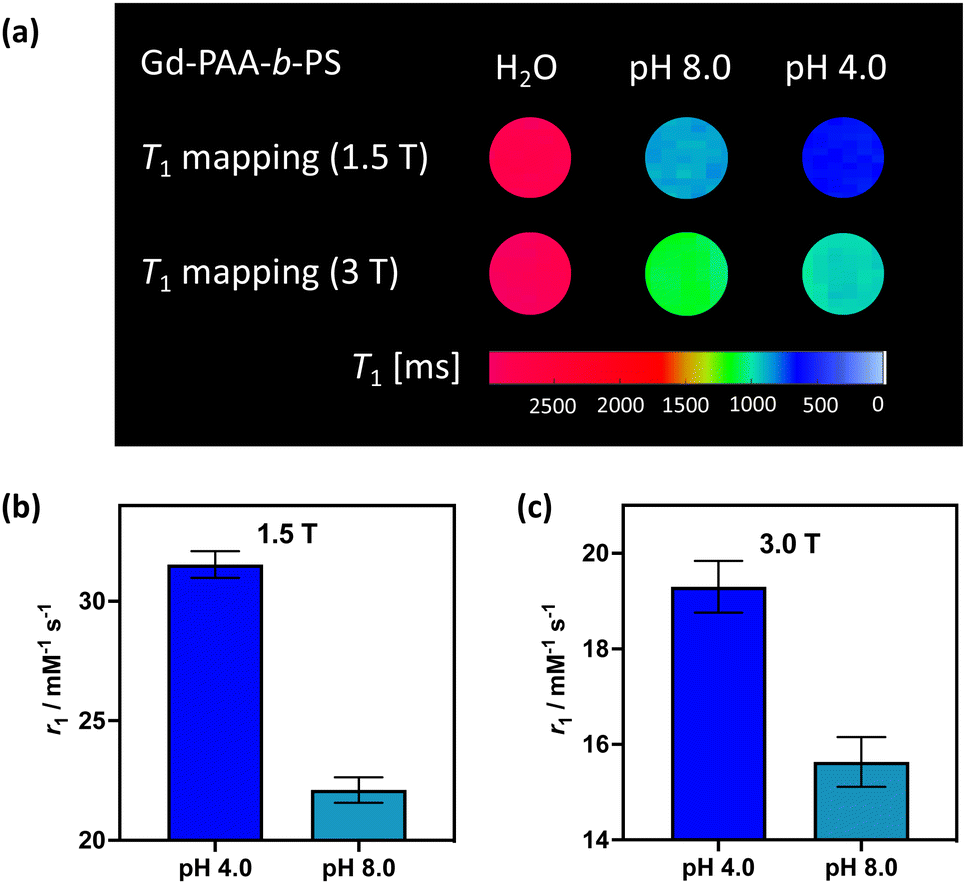 | ||
| Fig. 4 (a) T1 weighted MRI maps, recorded at both 1.5 T and 3 T, for the paramagnetically-doped micelles. The associated T1 values are 2809 ± 45 ms (1.5 T) and 2873 ± 71 ms (3.0 T) for water, 871 ± 21 ms (1.5 T) and 1101 ± 36 ms (3.0 T) for the micelles at pH 8.0, and 673 ± 12 ms (1.5 T) and 962 ± 27 ms (3.0 T) for the micelles at pH 4.0. The MR derived relaxivities for the micelles at both pH extremes, for 1.5 T (b) and 3 T (c). | ||
The paramagnetically-doped micellular platform presented here can facilitate effective, and reversibly responsive, imaging of local pH. This occurs through triggered changes in mechanical coupling of polymer strands to the micelle core and is mechanistically novel. The responsive contrast, where ∼50% relaxivity enhancements are observed at low pH, is both significant and clinically valuable.2
C. M. E. synthesised the micelles, conducted the ligand synthesis, and acquired the experimental data. D. Y. conducted the ligand synthesis. F. E. M. acquired the MRI data. J. J. M. assisted with the MRI analysis and relaxation theory modelling. J. J. D. conceptualised and designed the project. The manuscript was written by contributions from all authors. JJM would like to acknowledge a Novo Fonden (Foundation) Faculty Grant, ref. NNF21OC0068683.
Conflicts of interest
There are no conflicts to declare.References
- H. Bin Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef.
- C. M. Ellis, J. Pellico and J. J. Davis, Materials, 2019, 12, 1–13 CrossRef PubMed.
- G. L. Davies, I. Kramberger and J. J. Davis, Chem. Commun., 2013, 49, 9704–9721 RSC.
- J. Pellico, C. M. Ellis and J. J. Davis, Contrast Media Mol. Imaging, 2019, 2019, 1–13 CrossRef PubMed.
- Z. Shen, A. Wu and X. Chen, Mol. Pharmacol., 2017, 14, 1352–1364 CrossRef CAS PubMed.
- D. Ni, W. Bu, E. B. Ehlerding, W. Cai and J. Shi, Chem. Soc. Rev., 2017, 46, 7438–7468 RSC.
- R. J. Gillies, N. Raghunand, M. L. Garcia-Martin and R. A. Gatenby, IEEE Eng. Med. Biol. Mag., 2004, 23, 57–64 CrossRef PubMed.
- J. Pellico, C. M. Ellis, J. Miller and J. J. Davis, Chem. Commun., 2019, 55, 8540–8543 RSC.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D. E. Hu, J. H. Ardenkjær-Larsen, R. In’t Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–943 CrossRef CAS PubMed.
- A. Z. Lau, J. J. Miller and D. J. Tyler, Magn. Reson. Med., 2017, 77, 1810–1817 CrossRef CAS PubMed.
- C. Gao, Y. Wang, Z. Ye, Z. Lin, X. Ma and Q. He, Adv. Mater., 2021, 33, 2000512 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- M. Elsabahy, G. S. Heo, S. M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- Y. Liu, L. Feng, T. Liu, L. Zhang, Y. Yao, D. Yu, L. Wang and N. Zhang, Nanoscale, 2014, 6, 3231–3242 RSC.
- K. S. Kim, W. Park, J. Hu, Y. H. Bae and K. Na, Biomaterials, 2014, 35, 337–343 CrossRef CAS PubMed.
- T. Wang, D. Wang, H. Yu, M. Wang, J. Liu, B. Feng, F. Zhou, Q. Yin, Z. Zhang, Y. Huang and Y. Li, ACS Nano, 2016, 10, 3496–3508 CrossRef CAS PubMed.
- K. Shiraishi, K. Kawano, Y. Maitani and M. Yokoyama, J. Controlled Release, 2010, 148, 160–167 CrossRef CAS PubMed.
- D. Ni, W. Bu, E. B. Ehlerding, W. Cai and J. Shi, Chem. Soc. Rev., 2017, 46, 7438–7468 RSC.
- J. J. Davis, W. Y. Huang and G. L. Davies, J. Mater. Chem., 2012, 22, 22848–22850 RSC.
- J. S. Ananta, B. Godin, R. Sethi, L. Moriggi, X. Liu, R. E. Serda, R. Krishnamurthy, R. Muthupillai, R. D. Bolskar, L. Helm, M. Ferrari, L. J. Wilson and P. Decuzzi, Nat. Nanotechnol., 2010, 5, 815–821 CrossRef CAS PubMed.
- K. Ni, Z. Zhao, Z. Zhang, Z. Zhou, L. Yang, L. Wang, H. Ai and J. Gao, Nanoscale, 2016, 8, 3768–3774 RSC.
- J. B. Livramento, É. Tóth, A. Sour, A. Borel, A. E. Merbach and R. Ruloff, Angew. Chem., Int. Ed., 2005, 44, 1480–1484 CrossRef CAS PubMed.
- L. Wang, H. Lin, L. Ma, C. Sun, J. Huang, A. Li, T. Zhao, Z. Chen and J. Gao, J. Mater. Chem. B, 2017, 5, 8004–8012 RSC.
- P. Caravan, C. T. Farrar, L. Frullano and R. Uppal, Contrast Media Mol. Imaging, 2009, 4, 89–100 CrossRef CAS PubMed.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- P. B. Vilela, C. A. Matias, A. Dalalibera, V. A. Becegato and A. T. Paulino, J. Environ. Chem. Eng., 2019, 7, 103327 CrossRef.
- W. S. Perry, S. J. A. Pope, C. Allain, B. J. Coe, A. M. Kenwright and S. Faulkner, Dalton Trans., 2010, 39, 10974–10983 RSC.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H.-J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef PubMed.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- G. M. Nicolle, É. Tóth, H. Schmitt-Willich, B. Radüchel and A. E. Merbach, Chem. – Eur. J., 2002, 8, 1040–1048 CrossRef CAS PubMed.
- S. K. Piechnik, V. M. Ferreira, E. Dall’Armellina, L. E. Cochlin, A. Greiser, S. Neubauer and M. D. Robson, J. Cardiovasc. Magn. Reson., 2010, 12, 1–11 CrossRef PubMed.
- A. J. Taylor, M. Salerno, R. Dharmakumar and M. Jerosch-Herold, JACC: Cardiovasc. Imaging, 2016, 9, 67–81 Search PubMed.
- D. J. Mastarone, V. S. R. Harrison, A. L. Eckermann, G. Parigi, C. Luchinat and T. J. Meade, J. Am. Chem. Soc., 2011, 133, 5329–5337 CrossRef CAS PubMed.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019–3042 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06255k |
| This journal is © The Royal Society of Chemistry 2023 |