DOI:
10.1039/D1RA05602F
(Paper)
RSC Adv., 2021,
11, 29741-29751
Design, synthesis and cytotoxic evaluation of a library of oxadiazole-containing hybrids†
Received
22nd July 2021
, Accepted 20th August 2021
First published on 6th September 2021
Abstract
The development of hybrid compounds led to the discovery of new pharmacologically active agents for some of the most critical diseases, including cancer. Herein, we describe a new series of oxadiazole-containing structures designed by a molecular hybridization approach. Penicillin derivatives and amino acids were linked to amino acid and aromatic moieties through the formation of a 1,2,4-oxadiazole ring. Alternatively, condensation between amino acid-derived hydrazides and an activated penicillanic acid led to a series of 1,3,4-oxadiazole penicillin-containing hybrids and non-cyclized diacylhydrazides. From the cytotoxicity assays it is highlighted that two 1,2,4-oxadiazoles and one 1,3,4-oxadiazole connecting a penicillin and aliphatic amino acids displayed a high degree of cytotoxic selectivity, ranging between being three and four times more potent against tumor cells than normal cells. The results give a very interesting perspective suggesting that these hybrid compounds can offer a novel antitumor scaffold with promising cytotoxicity profiles.
Introduction
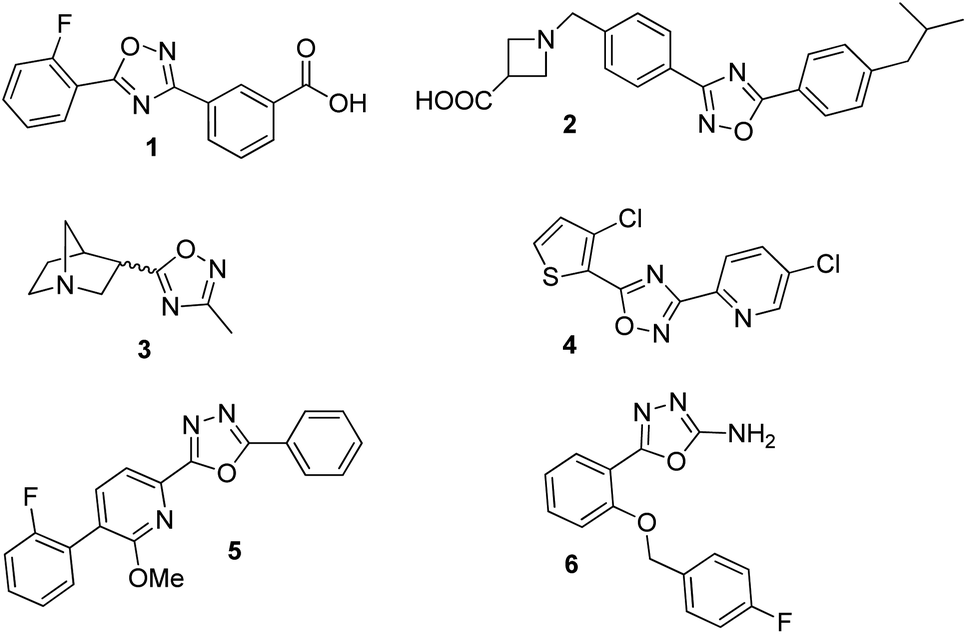
In the new millennium, organic synthesis has faced new challenges with respect to the generation of small molecules not only for finding lead compounds for drug discovery, but also to improve our understanding of different biological processes.1 The ability of small molecules to interact with biomacromolecules and disrupt their normal functioning, has made them crucial in chemical biology, nanomedicine and chemical genetics, and it has broadened the horizons of organic synthesis. Heterocycles are almost omnipresent in natural products as well as in many therapeutic agents, thus, being a key factor in the generation of small molecules.2 Among them, the 1,2,4-oxadiazole heterocycle is found in several drugs leads3 including ataluren (1) for the treatment of cystic fibrosis,4 a potent S1P1 agonist (2),5 a muscarinic receptor (3) for the treatment of alzheimer's disease6 and several potential anticancer agents7 (Fig. 1). Similarly, the 1,3,4-oxadiazole moiety can be found in many biologically active compounds.8 Particularly, 2,5-disubstituted 1,3,4-oxadiazoles have shown activity as antibacterial (5),9 anticancer,10 anticonvulsant (6),11 benzodiazepine receptor agonists,12 among others. In addition, these heterocycles are considered good bioisosteres of esters and amides, which increase bioactivity due to hydrogen-bond interactions with receptors.10b
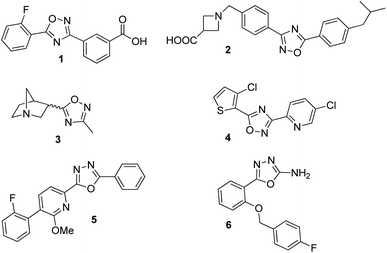 |
| | Fig. 1 Bioactive compounds containing 1,2,4 and 1,3,4-oxadiazole moiety: 1: ataluren; 2: CAY10734, agonist of sphingosine-1-phosphate receptor 1; 3: L 670548, muscarinic receptor agonist; 4: MX-74420, anticancer agent; 5: antibacterial agent; 6: anticonvulsant agent. | |
On the other hand, the concept of hybrid structures have emerged quite recently as an interesting twist in the search for new biologically promising small molecules.13 The development of hybrids led to the discovery of improved compounds for some of the most critical diseases, therefore, increasing the interest among the medicinal chemistry community.14 In previous studies, we have demonstrated that some triazolylaminoacyl (peptidyl) penicillins show a promising selectivity profile having up to 30-fold greater antiproliferative potency over HeLa and B16-F0 cells compared to normal cells.15 One of them inhibits in vitro proliferation of mouse melanoma cells by arresting the cell cycle and induces apoptosis, while it was also demonstrated the in vivo efficiency to reduce tumor growth and stimulate apoptotic response through the activation of both death receptor and mitochondria-dependent pathways.16 Taking into account the above mentioned analysis, we decided to apply a molecular hybridization strategy to conjugate structures of biological interest into a single entity. Therefore, we describe herein our results on the synthesis and biological evaluation of a small library of oxadiazole-containing hybrids.
Results and discussion
Chemistry
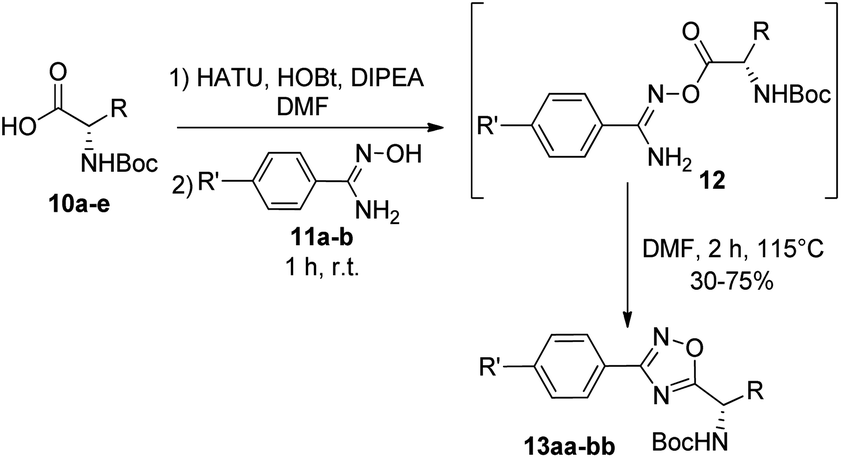
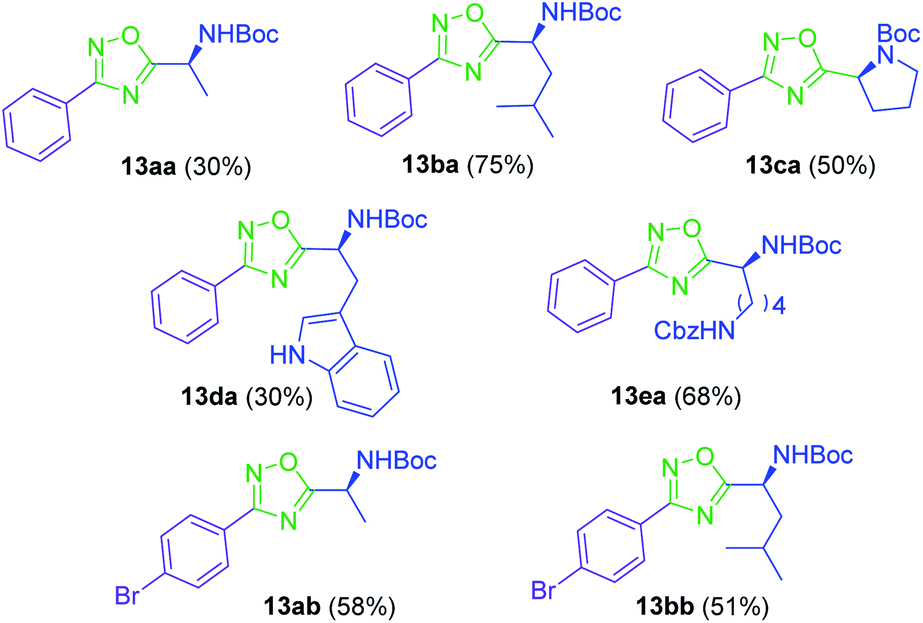
Synthesis of 1,2,4-oxadiazoles. Although the synthesis of 1,2,4-oxadiazoles by condensation of amidoximes and carboxylic acids is well established, their formation using amino acids is not always so trivial. In our hands, treatment of a variety of N-Boc amino acids (10a–e) with aryl amidoximes 11a–b, gave the corresponding 3,5-disubstituted 1,2,4-oxadiazoles (13aa–bb) in isolated yields from 30 to 75% (Scheme 1) (Fig. 2). Activation of the carboxylic acids (10a–e) for the O-acylation of the aryl amidoxime was carried out by the combination of HATU, HOBt and DIPEA, affording intermediates 12.17 Cyclization to the 1,2,4-oxadiazoles (13aa–bb) was finally achieved by heating the ring-opened intermediates in DMF at 115 °C.
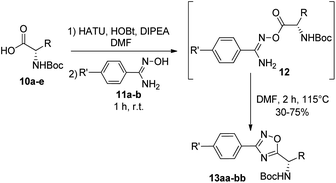 |
| | Scheme 1 Synthesis of 1,2,4-oxadiazoles from N-Boc amino acids 10a–e and amidoximes 11a–b. | |
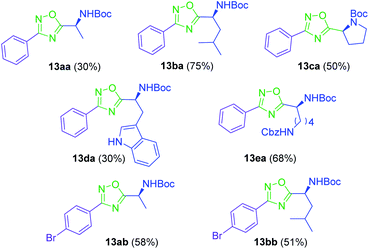 |
| | Fig. 2 3,5-Disubstituted 1,2,4-oxadiazoles 13 obtained from N-Boc amino acids and aryl amidoximes. | |
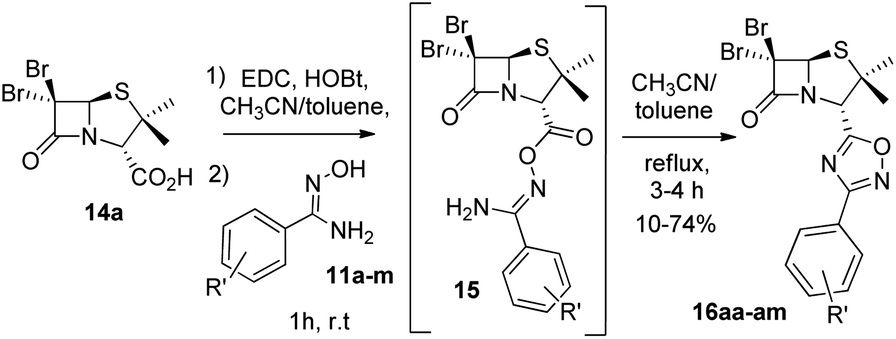
Next, we studied the conjugation of a penicillin derivative to an aromatic ring through an 1,2,4-oxadiazole heterocycle. Although a penicillin-based amidoxime could theoretically be obtained, the basic conditions required to synthesize the amidoxime from the corresponding nitrile, lead to the decomposition of the penicillin through the opening of the β-lactam ring. For this reason, we decided to carry out an inverse approach, in which different aryl amidoximes (11a–m)18 reacted with a penicillanic acid derivative (14a) (Scheme 2).
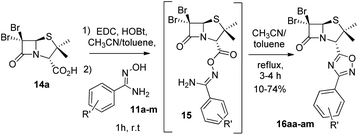 |
| | Scheme 2 Synthesis of 1,2,4-oxadiazoles 16 prepared from 6,6-dibromopenicillanic acid 14a and amidoximes 11a–m. | |
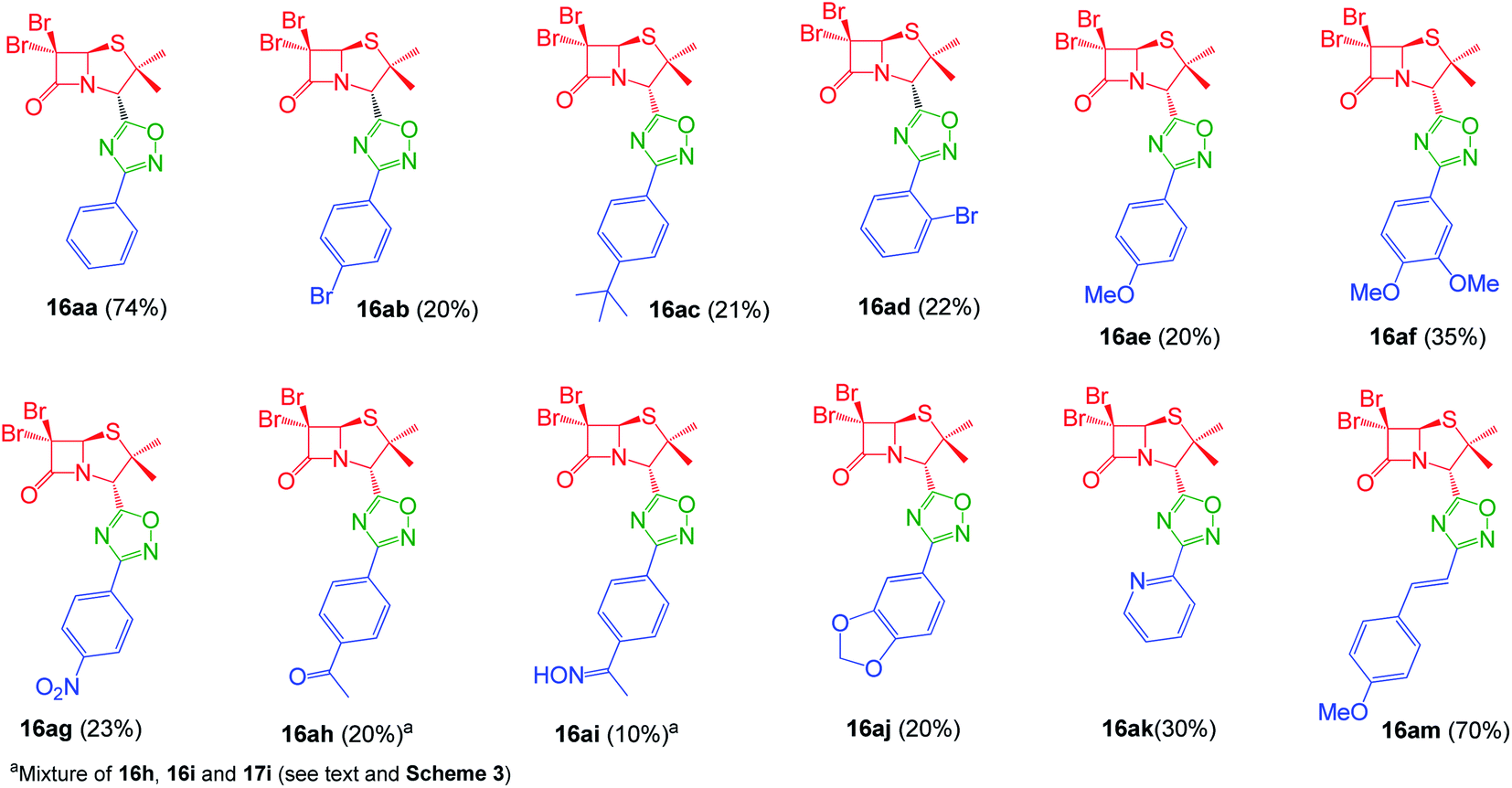
Interestingly, the previously mentioned combination HATU/HOBt for carboxylic acid activation failed to give the expected 1,2,4-oxadiazoles in most of the cases and a variant of this method was necessary. When the combination EDC/HOBt was used as carboxylic acids activator and diglyme as solvent,19 we obtained the corresponding penicillin-1,2,4-oxadiazoles in acceptable yields. Since diglyme is a very hygroscopic and high-boiling point solvent, we considered a mixture of anhydrous solvents![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile for dissolving the reaction mixture and toluene for reaching the temperature for the cyclization of the O-acylated amidoxime precursor. In this last stage, the reaction vessel was equipped with a Dean Stark trap to remove water. Using this methodology, we obtained a small library of 1,2,4-oxadiazolylaryl penicillins (16aa–am) (Fig. 3).
:acetonitrile for dissolving the reaction mixture and toluene for reaching the temperature for the cyclization of the O-acylated amidoxime precursor. In this last stage, the reaction vessel was equipped with a Dean Stark trap to remove water. Using this methodology, we obtained a small library of 1,2,4-oxadiazolylaryl penicillins (16aa–am) (Fig. 3).
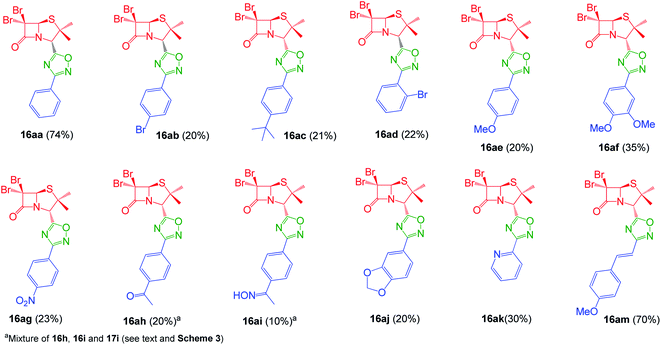 |
| | Fig. 3 1,2,4-Oxadiazolylaryl penicillins. | |
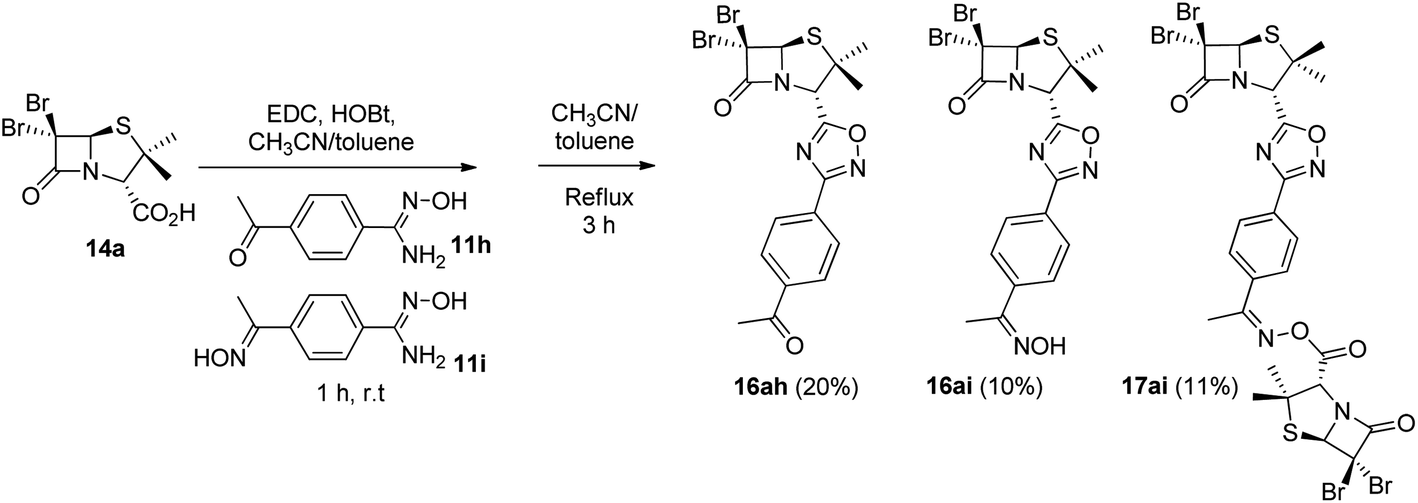
As expected, when 4-acetylbenzonitrile was used to prepare the corresponding the aryl amidoxime, we obtained a mixture of the desired 4-acetylbenzamidoxime (11h) and its oxime (11i). Consequently, during coupling and cyclization we obtained a mixture of three different 1,2,4-oxadiazoles: oxadiazoles from the acetyl and the oxime derivatives (16ah and 16ai, respectively), and a dimer O-acyl oxime (17ai) from the reaction of 16ai and a fraction of unreacted carboxylic acid 14a (Scheme 3). These three compounds were chromatographically isolated, characterized and incorporated into our library, in order to increase its diversity.
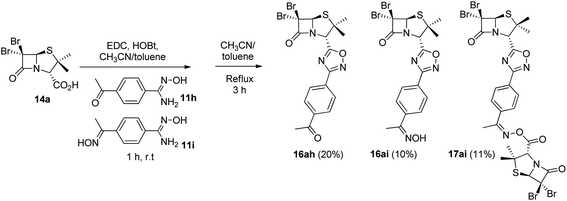 |
| | Scheme 3 1,2,4-Oxadiazoles from the acetyl (16ah) and the oxime (16ai) derivatives and a dimer O-acyl oxime (17ai) from 16ai and carboxylic acid 14a. | |
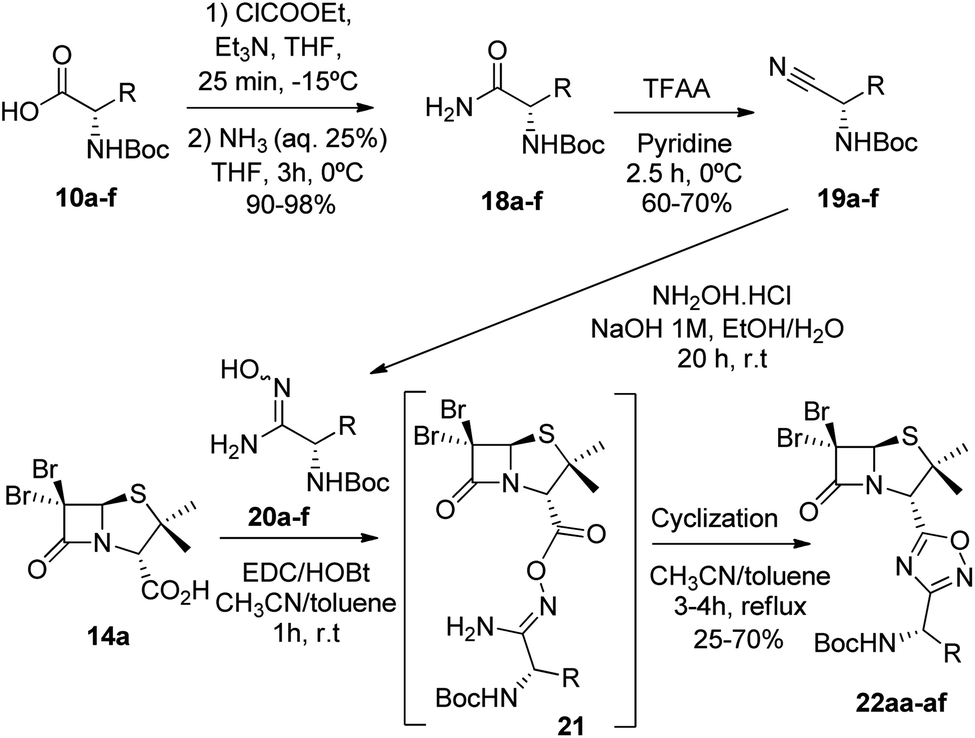
Having succeeded in the preparation of a small library of 1,2,4-oxadiazolylaryl penicillins, we next turned our attention to the development of penicillin–1,3,4-oxadiazole–amino acid hybrids. Then, we decided to generate a series of amino acid-based amidoximes.20
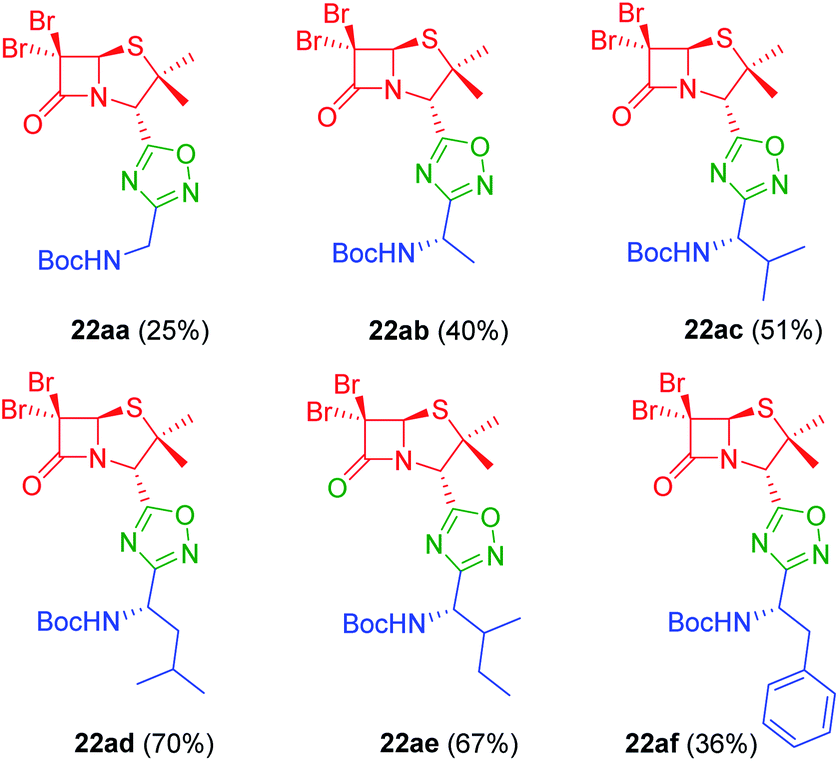
To achieve this goal, the N-Boc amino acids (10a–f) were first treated with ethyl chloroformate, triethylamine and then aqueous ammonia in THF to obtain the corresponding primary amides (18a–f) in excellent yields and pure enough to use it in the next step without further purification (Scheme 4). Subsequently, we converted the N-Boc amino amides 18 into the corresponding nitriles (19a–f) by dehydration using trifluoroacetic anhydride (TFAA) in anhydrous pyridine as solvent. We purified the N-Boc amino nitriles (19a–f) by column chromatography, and finally performed the synthesis of amidoximes (20a–f) by treating the nitriles with hydroxylamine hydrochloride and NaOH in ethanol at room temperature. This procedure was adapted from literature using a more volatile solvent than reported,21 and gave shorter reaction time and better yields than other alternative methods.22 The final step was the reaction of the amino amidoximes with the penicillanic acid derivative 14a to afford the corresponding 1,2,4-oxadiazolylaminoacyl penicillins (22aa–af), under similar conditions to those described above (EDC/HOBt and cyclization in CH3CN/toluene at reflux) (Fig. 4).
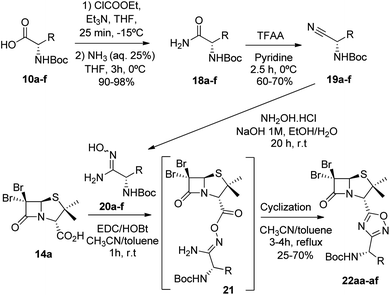 |
| | Scheme 4 Synthesis of 1,2,4-oxadiazoles from 6,6-dibromopenicilanic 14a and amidoximes 20a–f. | |
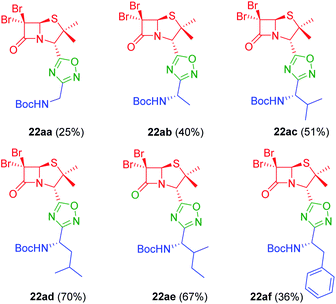 |
| | Fig. 4 1,2,4-Oxadiazolylaryl penicillins from amino amidoximes. | |
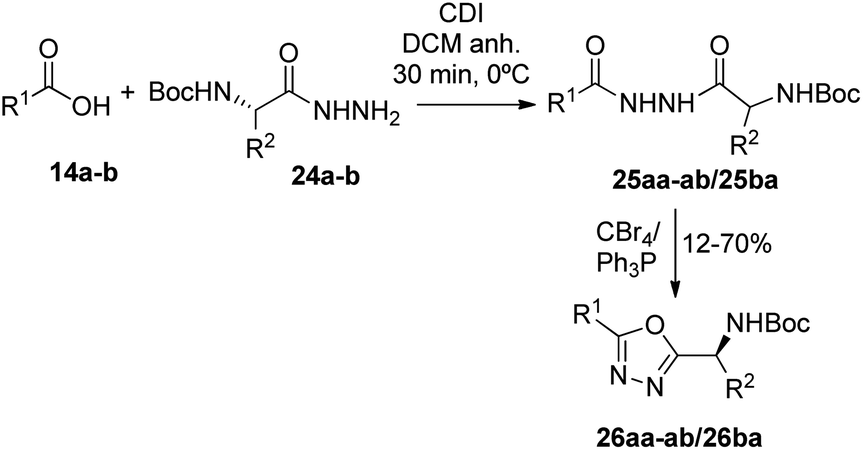
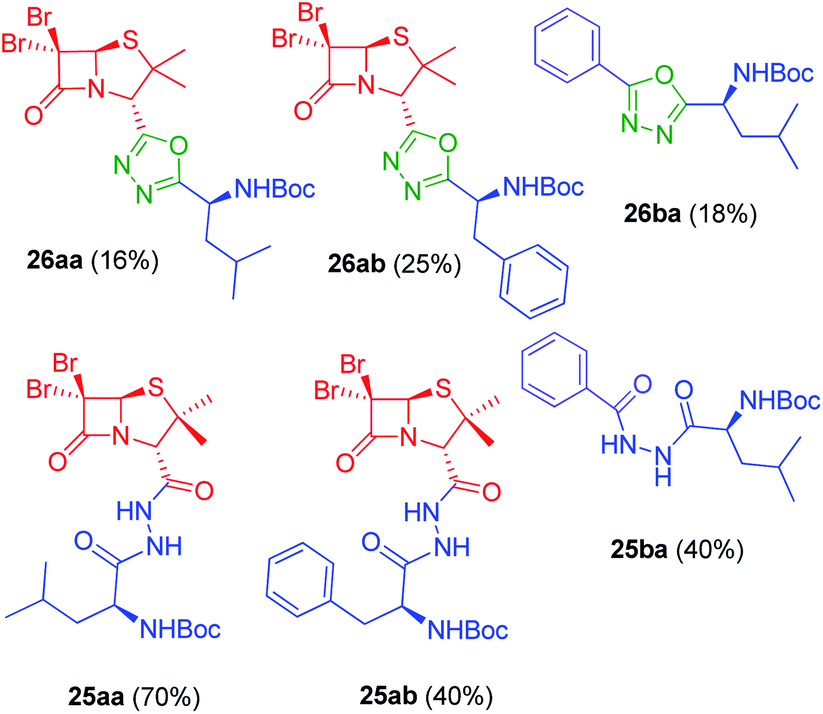
Synthesis of 1,3,4-oxadiazoles. Bearing in mind our interest in the development of penicillin-containing hybrids, we next decided to synthesize 1,3,4-oxadiazoles by condensation between hydrazides and activated carboxylic acids. Considering that penicillin hydrazides would be difficult to synthesize due to the basic conditions imposed by the hydrazine, we decided to use amino acid-derived hydrazide partners for the generation of these oxadiazoles. Thus, activation of N-Boc-protected amino acids with 1,1-carbonyldiimidazole (CDI) followed by the addition of hydrazine gave the corresponding hydrazides 24a–b.23 As a model, we carried out the reaction of CDI-activated benzoic acid (14b, R1 = Ph) with the leucine-derived hydrazide (24a, R2 = CH2CH(CH3)2), followed by an in situ dehydration of the disubstituted hydrazide to yield the corresponding 1,3,4-oxadiazole 26ba (Scheme 5) (Fig. 5). In order to obtain the penicillin hybrids, we used 6,6-dibromopenicillanic acid (14a) as carboxylic acid partner; thus, some novel and interesting 1,3,4-oxadiazolylaminoacyl penicillins were synthesized (26aa–ab). The dehydrative cyclization has proven to be difficult, we have tried a variety of procedures, such as, POCl3 under microwave heating24 and trifluoroacetic anhydride/pyridine,25 with little success. Finally, the corresponding 1,3,4-oxadiazoles were obtained by treatment of the disubstituted hydrazides with CBr4 and Ph3P, although conversion was incomplete. The corresponding disubstituted hydrazide intermediates (25ba, 25aa–ab) were also isolated to be tested as antiproliferative agents (Fig. 5).
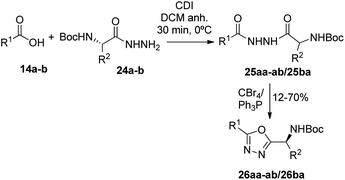 |
| | Scheme 5 Synthesis of 1,3,4-oxadiazoles 26 from carboxylic acids 1a–b and hydrazides 24a–c. | |
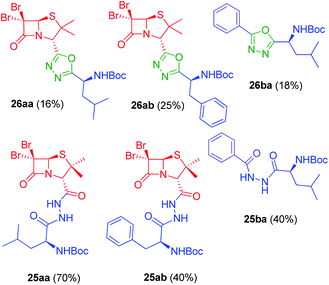 |
| | Fig. 5 1,3,4-Oxadiazoles and disubstituted hydrazides intermediates synthesized from acids 14a–b and corresponding hydrazides 24a–b. | |
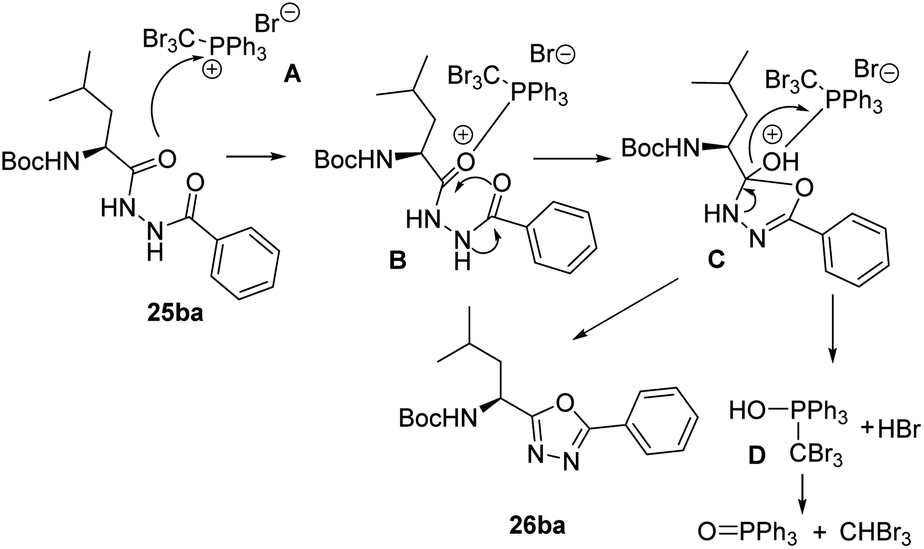
A reasonable mechanism for the cyclization of the disubstituted hydrazides with CBr4 and Ph3P, would involve an initial formation of salt A which undergoes nucleophilic attack by the carbonyl oxygen giving the intermediate B. Cyclization followed by release of adduct D26 and aromatization gives the corresponding 1,3,4-oxadiazole (Scheme 6).
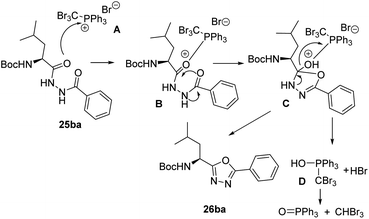 |
| | Scheme 6 Proposed reaction mechanism for the cyclodehydration of diacylhydrazines using CBr4 and PPh3. | |
Biological evaluation
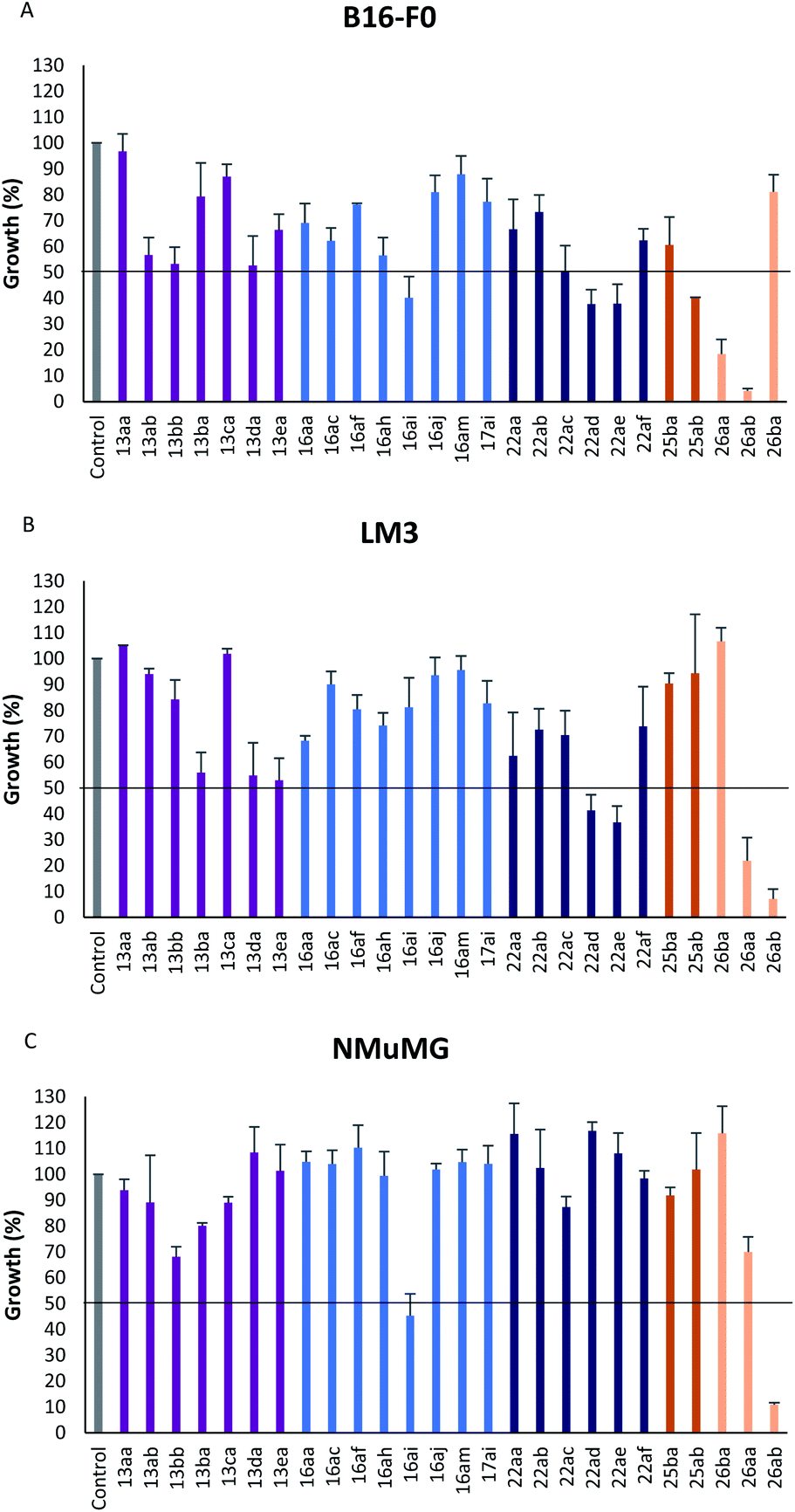
In vitro antiproliferative activity of twenty-six 1,2,4 and 1,3,4-oxadiazole hybrid compounds was determined at a concentration of 20 μM in two murine tumor lines: B16-F0 (melanoma) (Fig. 6A) and LM3 (mammary adenocarcinoma) (Fig. 6B). In order to evaluate the selectivity against tumor cells, cytotoxic effect against non-tumor NMuMG line (normal murine mammary epithelium) was also determined (Fig. 6C).
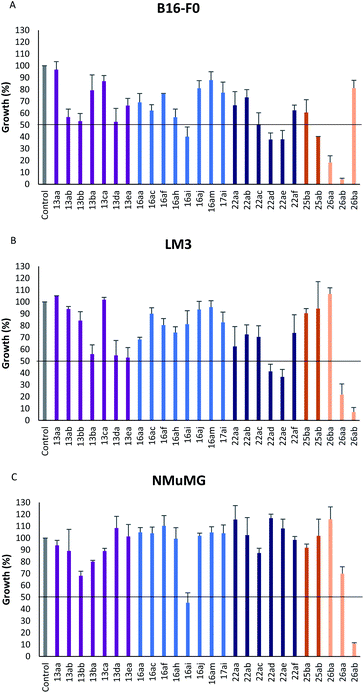 |
| | Fig. 6 Effect of hybrid compounds on the proliferation of non-neoplastic and tumor cell lines. 2 × 104 cells per well (NMuMG and LM3) (C and B, respectively) or 1 × 104 cells per well (B16-F0) (A) were incubated in the presence or absence of 20 μM of different compounds for 72 h at 37 °C. Cell proliferation was determined by colorimetric determination of hexosaminidase levels. Results are expressed as the percentage of growth obtained in the absence of compounds (control) and represented as mean ± S. E. M. of three different experiments. | |
As can be seen in Fig. 6C, only two compounds were found to be cytotoxic against NMuMG cells, namely, 16ai and 26ab. From the non-cytotoxic compounds against NMuMG cells, diacylhydrazide 25ab, and oxadiazoles 22ad, 22ae and 26aa (Fig. 7) were the most active against B16-F0 (Fig. 6A), while 22ad, 22ae and 26aa were the most active against LM3 (Fig. 6B). The presence of the penicillin component seems to be crucial since all the active compounds have that moiety. Interestingly, active compounds 22ad, 22ae and 26aa are closely related: they are all penicillins attached to a leucine or isoleucine by either an 1,2,4-oxadiazole or the 1,3,4 isomer.
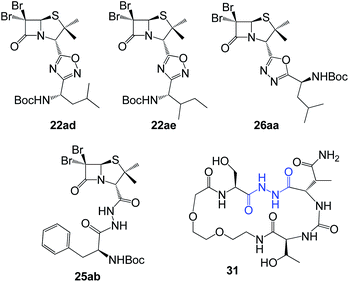 |
| | Fig. 7 Active compounds against B16-F0 (melanoma) and LM3 (breast adenocarcinoma) cells, not cytotoxic against NMuMG normal cells. | |
The activity of 25ab is very interesting since diacylhydrazides have been described as peptidomimetics and are of interest in immuno-oncology therapeutics.27 For instance, compound 31 has been reported as immune checkpoint inhibitor targeting the ligand of the protein PD-1 (Programmed Death-1, CD279).28
The interesting results obtained with the penicillin–oxadiazole–aminoacid hybrids 22ad, 22ae and 26aa, were reflected in their respective IC50 values (Table 1, entries 1–6). The selectivity index (SI) for 22ad, 22ae is ranging between 3.06 and 4.13, for both cell lines, which indicate that the cytotoxicity caused by them to the tumor cells is at least three times higher than that against normal cells. Similarly, IC50 values for 25ab against B16F0, showed also a SI around four (3.75). Many clinically used anticancer drugs are hindered by their highly toxic side-effects. Cis-platinum, widely used in clinical cancer therapy, shows a SI of less than one.29
Table 1 IC50 values of most active hybrids
| Entry |
Compound |
IC50 B16F0 (μM) |
IC50 LM3 (μM) |
IC50 NMuMG (μM) |
SI |
| 1 |
22ad |
15 ± 2 |
— |
62 ± 5 |
4.13 |
| 2 |
22ad |
— |
18 ± 4 |
62 ± 5 |
3.44 |
| 3 |
22ae |
19 ± 1 |
— |
62 ± 2 |
3.06 |
| 4 |
22ae |
— |
15 ± 1 |
62 ± 2 |
4.13 |
| 5 |
26aa |
13 ± 3 |
— |
34 ± 8 |
2.16 |
| 6 |
26aa |
— |
12 ± 1 |
34 ± 8 |
2.83 |
| 7 |
25ab |
24 ± 8 |
— |
90 ± 3 |
3.75 |
These promising results open the door for further development of penicillin-containing hybrids as new potential chemotherapeutic antitumor agents, particularly those bearing diacylhydrazide, 1,3,4- or 1,2,4-oxadiazole moieties.
Conclusion
In summary, we have developed a series of strategies for the synthesis of some novel hybrids to conjugate three biologically interesting structures in a single compound. Thus, penicillin derivatives were connected to amino acid and aromatic moieties through a 1,2,4-oxadiazole ring using the corresponding penicillanic acid derivative and an aryl or amino amidoxime. The sensitivity of the β-lactam ring to basic conditions required for the generation of the amidoxime precursor, prevented us from synthesizing the “inverse” variant of these hybrids. Likewise, other amino acid-containing 1,2,4-oxadiazoles have been obtained. On the other hand, 1,3,4-oxadiazole penicillin-containing hybrids were synthesized by the condensation between amino acid-derived hydrazides and activated penicillanic acids. An in vitro antiproliferative study of twenty-six 1,2,4 and 1,3,4-oxadiazole hybrids determined that the most actives were 25ab against B16-F0 line, and 22ad and 22ae, against both tumor lines, B16-F0 and LM3. Hydrazide 25ab was found to be 3.75 times more active against B16-F0 cells than NMuMG, while 1,3,4-oxadiazole derivatives 22ad and 22ae were ranging between three and four times more potent against tumor cells. Due to the interesting levels of selectivity, these compounds can be regarded as promising structures in order to find the best candidates for developing new chemotherapeutic antitumor agents. Further studies on the cascade of molecular events that lead to cell death will provide more insight on these encouraging results.
Experimental
Chemistry
Selected NMR spectra are reported in the ESI.†
General information
Chemical reagents were purchased from commercial suppliers and used without further purification, unless otherwise noted. Solvents were analytical grade or were purified by standard procedures prior to use. NMR spectra were recorded with Bruker spectrometer brand model Avance-300 DPX at 300 MHz for 1H NMR (using the Me4Si signal as internal reference standard, δ = 0.00 ppm) and at 75 MHz for 13C NMR (using the solvent signal as internal reference standard). 1H NMR spectra are informed indicating the chemical shifts of the signals (δ) and then, in parentheses, the multiplicity of the signal, the coupling constants (J) and integration. The 13C NMR spectra are reported indicating the chemical shifts of the signals. Abbreviations used to indicate the multiplicities of the signals were s: singlet; brs: broad singlet; d: doublet; dd: doublet of doublets; td: triplet of doublets; dt: doublet of triplets; t: triplet; sept: septet and m: multiplet. HRMS spectra were performed at the Unit of Microanalysis and Physical Methods Applied to Organic Chemistry (UMYMFOR-UBA) or Mass Spectrometry Service at Córdoba Food Science and Technology Institute (ICYTAC), using a Bruker mass spectrometer micrOTOF-Q II. The detection of ions was carried out by electrospray ionization (ESI), positive mode. Reactions were monitored by thin layer chromatography performed on silica gel 60 F254 pre-coated aluminum sheets, visualized by a 254 nm UV lamp and/or stained with an ethanolic solution of 4-anisaldehyde or a 0.2% ninhydrin solution. Preparative separations were carried out by liquid column chromatography using Merck 60H silica gel (230–400 mesh).
(A) General procedure for the synthesis of 1,2,4-oxadiazoles
A round bottom flask equipped with a magnetic stir bar was charged with the Boc-protected amino acid 10a–e (0.3 mmol). Dry DMF (5 mL for aliphatic amino acids, 10 mL for the rest of amino acids) was added, followed by HATU (0.3 mmol), HOBt (0.06 mmol) and DIPEA (1.5 mmol). The reaction mixture was stirred at room temperature for one minute. Then, amidoxime 11a–b (0.3 mmol) was added and the reaction mixture was stirred at room temperature for 1 h (absence of starting material was checked by TLC). Finally, the reaction was heated at 115 °C for 2 h. After this time, the solvent was removed by evaporation under reduced pressure. The crude mixture was purified by column chromatography (hexane/AcOEt (70:30)).
tert-Butyl(S)-(1-(3-phenyl-1,2,4-oxadiazol-5-yl)ethyl)carbamate (13aa)30. Obtained following general procedure A as a white solid, mp 82–82.5 °C; 30% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.43 (s, 9H), 1.60 (d, J = 7.05 Hz, 3H), 5.07–5.23 (m, 1H), 5.35 (br s, 1H), 7.40–7.50 (m, 3H), 8.03–8.06 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 19.86, 28.17, 44.16, 80.34, 126.48, 127.37, 128.72, 131.14, 154.74, 168.16, 180.04. HRMS (ESI) m/z found: 312.1394 (M + Na+), estimated for C15H19N3NaO3: 312.13186.
tert-Butyl(S)-(3-methyl-1-(3-phenyl-1,2,4-oxadiazol-5-yl)butyl)carbamate (13ba). Obtained following general procedure A as a white solid, mp 54–55 °C; 75% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.97 (d, J = 6.22 Hz, 3H), 0.99 (d, J = 6.22 Hz, 3H), 1.45 (s, 9H), 1.65–1.84 (m, 3H), 4.94–5.28 (m, 2H), 7.44–7.49 (m, 3H), 8.05–8.08 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 21.95, 22.53, 24.66, 28.25, 43.23, 46.79, 80.41, 126.63, 127.48, 128.79, 131.20, 154.96, 168.26, 180.01. HRMS (ESI) m/z found: 354.1778 (M + Na+), estimated for C18H25N3NaO3: 354.1788.
tert-Butyl(S)-2-(3-phenyl-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (13ca)31. Obtained following general procedure A as a white solid, mp 116–117 °C; 50% yield. 1H NMR (CDCl3, 300 MHz) δ 1.29 (s, 6H), 1.46 (s, 3H), 1.94–2.24 (m, 2H), 2.24–2.49 (m, 2H), 3.43–3.78 (m, 2H), 5.04–5.26 (m, 1H), 7.42–7.54 (m, 3H), 8.02–8.11 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 23.66, 24.30, 28.11, 31.46, 46.32, 53.78, 80.50, 126.68, 127.41, 128.70, 128.84, 131.02, 131.19, 153.56, 154.26, 168.32, 180.19, 180.62. HRMS (ESI) m/z found: 338.1467 (M + Na+), estimated for C17H21N3NaO3: 338.1475.
tert-Butyl(S)-(2-(1H-indol-3-yl)-1-(3-phenyl-1,2,4-oxadiazole-5-yl)ethyl)carbamate (13da). Obtained following general procedure A as a white solid, mp 140–141 °C; 25% yield. 1H NMR (CDCl3, 300 MHz) δ 1.45 (s, 9H), 3.49–3.50 (d, J = 3.98 Hz, 2H), 5.33 (d, J = 6.51 Hz, 1H), 5.41–5.55 (m, 1H), 6.87 (br s, 1H), 7.07–7.19 (m, 2H), 7.25–7.32 (m, 1H), 7.46–7.54 (m, 4H), 8.04–8.04 (d, J = 6.24, 2H), 8.27 (br s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 28.30, 49.47, 80.51, 109.20, 119.79, 122.29, 123.13, 126.60, 127.52, 128.86, 131.28, 136.14, 155.07, 168.28, 179.35. HRMS (ESI) m/z found: 427.1736 (M + Na+), estimated for C23H24N4NaO3: 427.1741.
(S)-Benzyl-tert-butyl(1-(3-phenyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl)dicarbamate (13ea). Obtained following general procedure A as a yellow solid, mp 119–112 °C; 68% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.44 (s, 9H), 1.50–1.60 (m, 4H), 1.86–2.03 (m, 2H), 3.18–3.20 (m, 2H), 4.90–4.94 (m, 1H), 5.08 (s, 2H), 5.38 (d, J = 7.08 Hz, 1H), 7.28–7.38 (m, 5H), 7.42–7.50 (m, 3H), 8.05–8.08 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 22.34, 28.29, 29.35, 33.64, 40.43, 48.20, 66.67, 80.50, 126.56, 127.51, 128.09, 128.51, 128.84, 131.28, 136.55, 155.18, 156.56, 168.27, 179.49. HRMS (ESI) m/z found: 503.2280 (M + Na+), estimated for C26H32N4NaO5: 503.2265.
tert-Butyl(S)-(1-(3-(4-bromophenyl)-1,2,4-oxadiazol-5-yl)ethyl)carbamate (13ab). Obtained following general procedure A as a white solid, mp 118–119 °C; 58% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.45 (s, 9H), 1.62 (d, J = 6.87 Hz, 3H), 5.08–5.24 (m, 2H), 7.61 (d, J = 8.60 Hz, 2H), 7.94 (d, J = 8.60 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 20.03, 28.28, 44.28, 80.61, 125.55, 125.85, 128.98, 132.15, 154.74, 167.60, 180.34. HRMS (ESI) m/z found: 390.0438 (M + Na+), estimated for C15H18BrN3NaO3: 390.0424.
tert-Butyl(S)-(1-(3-(4-bromophenyl)1,2,4-oxadiazol-5-yl)-3-methylbutyl)carbamate (13bb). Obtained following general procedure A as a white solid, mp 67–68 °C; 51% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.98 (d, J = 6.22 Hz, 3H), 1.00 (d, J = 6.22 Hz, 3H), 1.44 (s, 9H), 1.71–1.77 (m, 3H), 4.77–5.12 (m, 2H), 7.60 (d, J = 8.55 Hz, 2H), 7.94 (d, J = 8.55 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 20.91, 21.54, 21.96, 27.25, 42.16, 45.80, 79.51, 124.60, 124.64, 127.98, 131.10, 153.94, 166.57, 179.29. HRMS (ESI) m/z found: 432.0879 (M + Na+), estimated for C18H24BrN3NaO3: 432.0893.
(B) General procedure for the synthesis of N-protected amino acid-derived amides
A solution of the N-Boc-(L)-amino acid (0.5 mmol) and Et3N (0.55 mmol) in THF (4.5 mL) was cooled to −15 °C (ice-salt bath) under N2 atmosphere. A solution of ethyl chloroformate (0.55 mmol) in THF (1.5 mL) was added dropwise and the mixture stirred for 25 min at −15 °C. Then a 25% NH3 aqueous solution (2.5 mmol) was added in a single portion and continued stirring at 0 °C for 3 h. The solvent was evaporated under reduced pressure and the pH was adjusted to 2–3 with a KHSO4 solution (1 N). The aqueous phase was extracted twice with AcOEt. The combined organic phases were washed with NaHCO3 solution (1 N), water and brine, and finally dried over Na2SO4. After evaporation, the crude was used in the next step without purification.
(C) General procedure for the synthesis of N-protected amino acid-derived nitriles
Under a N2 atmosphere, a solution of the amide derived from the N-Boc-L-amino acid (0.4 mmol) in pyridine (3 mL) was cooled to 0 °C with an ice bath and trifluoroacetic anhydride (0.56 mmol) added dropwise. Then, the solution was stirred for 2.5 h at 0 °C. Finally, the solvent was evaporated, the residue was dissolved in AcOEt and washed with KHSO4 (1 N), water, brine and then dried over Na2SO4. The crude mixture was purified by column chromatography using hexane–AcOEt as eluent.
(D) General procedure for the synthesis of N-protected amino acid-derived amidoxime
NaOH (1 M solution, 1 mmol) was slowly and carefully added to a hydroxylamine hydrochloride solution (1 mmol) in ethanol–water (1:1) (5 mL) in an ice bath. The mixture was stirred for 1 h at room temperature. Subsequently, the N-Boc-(L)-aminonitrile (0.4 mmol) was added and the reaction was left stirring at room temperature overnight. The solvent was evaporated under reduced pressure and the extraction was carried out with AcOEt. The collected organic phases were dried over Na2SO4. Finally, the crude was purified by column chromatography using hexane/AcOEt as eluent.
(E) General procedure for the synthesis of penicillin-containing 1,2,4-oxadiazoles
A round bottom flask equipped with a magnetic stir bar was charged with the 6,6-dibromopenicillanic acid 14a (0.5 mmol) dry MeCN (4.5 mL) was added, followed by EDC (0.57 mmol) and HOBt (0.6 mmol). The reaction mixture was stirred at room temperature for 1 h. After this time, amidoxime 11a–m or 20a–f (0.5 mmol) and dry toluene (2:1 MeCN:toluene) was added. The reaction was refluxed using a Dean–Stark trap with activated molecular sieves, for 4 h. Finally, the solvent was removed by evaporation under reduced pressure. The crude mixture was purified by column chromatography (hexane/AcOEt).
(2S,5R)-6,6-Dibromo-3,3-dimethyl-2-(3-phenyl-1,2,4-oxadiazol-5-yl)-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16aa). Obtained following general procedure E as a white solid, mp 142.5–143 °C; 74% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.30 (s, 3H), 1.75 (s, 3H), 5.38 (s, 1H), 6.02 (s, 1H), 7.60–7.44 (m, 3H), 8.14–8.04 (m, 2H). 13C NMR (75 MHz, CDCl3) δ (ppm): 24.54, 32.23, 57.54, 63.65, 64.57, 79.76, 124.91, 126.58, 127.95, 130.66, 163.3, 167.46, 172.16. HRMS (ESI) m/z found: 457.91598 (M + H+), estimated for C15H14Br2N3O2S: 457.91680.
(2S,5R)-6,6-Dibromo-2-(3-(4-bromophenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[ 3.2.0]heptan-7-one (16ab). Obtained following general procedure E as a white solid, mp 194–194.5 °C; 20% yield. 1H NMR (CDCl3, 300 MHz) δ 1.30 (s, 3H), 1.75 (s, 3H), 5.36 (s, 1H), 6.00 (s, 1H), 7.64 (d, J = 8.48 Hz, 2H), 7.96 (d, J = 8.48 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.59, 33.23, 58.47, 64.66, 65.57, 80.78, 124.87, 126.36, 129.08, 132.30, 164.34, 167.83, 173.48. HRMS (ESI) m/z found: 557.8083 (M + Na+), estimated for C15H12Br3N3NaO2S: 557.8093.
(2S,5R)-6,6-Dibromo-2-(3-(4-(tert-butyl)phenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ac). Obtained following general procedure E as a gummy material; 20% yield. 1H NMR (CDCl3, 300 MHz) δ 1.29 (s, 3H), 1.36 (s, 9H), 1.74 (s, 3H), 5.37 (s, 1H), 6.01 (s, 1H), 7.52 (d, J = 8.60 Hz, 2H), 8.01 (d, J = 8.60 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.55, 31.15, 33.28, 35.04, 58.60, 64.68, 65.61, 80.80, 123.08, 125.95, 127.38, 155.25, 164.32, 168.41, 172.97. HRMS (ESI) m/z found: 535.95862 (M + Na+), estimated for C19H21Br2N3NaO2S: 535.96134.
(2S,5R)-6,6-Dibromo-2-(3-(2-bromophenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ad). Obtained following general procedure E as a white solid, mp 131–131.5 °C; 22% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.33 (s, 3H), 1.75 (s, 3H), 5.40 (s, 1H), 6.01 (s, 1H), 7.38 (td, J = 7.62, 1.62 Hz, 1H), 7.46 (td, J = 7.62, 1.62 Hz, 1H), 7.75 (dd, J = 7.76, 1.56 Hz, 1H), 7.83 (dd, J = 7.76, 1.56 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.63, 33.21, 58.47, 64.63, 65.72, 80.77, 122.29, 127.33, 127.53, 131.93, 132.24, 134.29, 164.36, 168.16, 172.86. HRMS (ESI) m/z found: 557.8093 (M + Na+), estimated for C15H12Br3N3NaO2S: 557.8093.
(2S,5R)-6,6-Dibromo-2-(3-(4-methoxyphenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ae). Obtained following general procedure E as a yellow solid, mp 183–183.5 °C; 20% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.30 (s, 3H), 1.74 (s, 3H), 3.87 (s, 3H), 5.35 (s, 1H), 6.00 (s, 1H), 6.99 (d, J = 8.88 Hz, 2H), 8.01 (d, J = 8.88 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.53, 33.23, 55.40, 58.56, 64.63, 65.57, 80.74, 114.33, 118.31, 129.23, 162.26, 164.28, 168.13, 172.82. HRMS (ESI) m/z found: 487.92594 (M + H+), estimated for C16H16Br2N3O3S: 487.92736.
(2S,5R)-6,6-Dibromo-2-(3-(3,4-dimethoxyphenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16af). Obtained following general procedure E as a yellow solid, mp 141–141.5 °C; 35% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.30 (s, 3H), 1.74 (s, 3H), 3.94 (s, 3H), 3.96 (s, 3H), 5.35 (s, 1H), 6.00 (s, 1H), 6.96 (d, J = 8.42 Hz, 1H), 7.55 (d, J = 1.95 Hz, 1H), 7.69 (dd, J = 8.42, 1.95 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.54, 33.19, 55.98, 56.04, 58.50, 64.65, 65.54, 80.72, 109.83, 111.04, 118.39, 121.25, 149.23, 151.91, 164.29, 168.29, 172.87. HRMS (ESI) m/z found: 539.918273 (M + Na+), estimated for C17H17Br2N3NaO4S: 539.919872.
(2S,5R)-6,6-Dibromo-3,3-dimethyl-2-(3-(4-nitrophenyl)-1,2,4-oxadiazol-5-yl)-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ag). Obtained following general procedure E as a yellow solid, mp 184–184.5 °C; 23% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.31 (s, 3H), 1.76 (s, 3H), 5.40 (s, 1H), 6.01 (s, 1H), 8.29 (d, J = 8.98 Hz, 2H), 8.36 (d, J = 8.98, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.59, 33.15, 58.33, 64.65, 65.53, 80.77, 124.18, 128.63, 131.71, 149.73, 164.34, 168.99, 174.16. HRMS (ESI) m/z found: 524.8826 (M + Na+), estimated for C15H12Br2N4NaO4S: 524.8838.
(2S,5R)-2-(3-(4-Acetylphenyl)-1,2,4-oxadiazol-5-yl)-6,6-dibromo-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ah). Obtained following general procedure E as a white solid, mp 166–166.5 °C; 20% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.30 (s, 3H), 1.75 (s, 3H), 2.67 (s, 3H), 5.39 (s, 1H), 6.01 (s, 1H), 8.07 (d, J = 8.65 Hz, 2H), 8.19 (d, J = 8.65 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.57, 26.76, 33.18, 58.42, 64.65, 65.54, 80.76, 127.83, 128.81, 129.95, 139.29, 164.33, 167.73, 173.66, 197.29. HRMS (ESI) m/z found: 521.9105 (M + Na+), estimated for C17H15Br2N3NaO3S: 521.9093.
(2S,5R)-6,6-Dibromo-2-(3-(4-(1-(hydroxyimino)ethyl)phenyl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16ai). Obtained from previous reaction as a yellow solid, mp 170–171 °C; 10% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.30 (s, 3H), 1.75 (s, 3H), 2.32 (s, 3H), 5.40 (s, 1H), 6.02 (s, 1H), 7.77 (d, J = 8.60 Hz, 2H), 8.10 (d, J = 8.60 Hz, 2H), 8.57 (br s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 11.98, 25.57, 33.21, 58.48, 64.65, 65.58, 80.76, 126.43, 126.52, 127.71, 139.57, 155.25, 164.36, 168.03, 173.32. HRMS (ESI) m/z found: 514.9385 (M + H+), estimated for C17H17Br2N4O3S: 514.9383.
(2S,5R)-6,6-dibromo-2-((((Z)-(1-(4-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethylidene)amino)oxy)carbonyl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (17ai). Obtained from previous reaction as a white solid, m.p 111–111.5 °C; 11% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm) 1.25 (s, 3H), 1.31 (s, 3H), 1.59 (s, 3H), 1.69 (s, 3H), 1.75 (s, 3H), 2.47 (s, 3H), 4.82 (s, 1H), 5.38 (s, 1H), 5.86 (s, 1H), 6.01 (s, 1H), 7.89 (d, J = 8.70, 2H), 8.15 (d, J = 8.64, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm) 14.82, 25.60, 25.99, 33.20, 33.51, 58.46, 64.67, 64.80, 65.55, 69.00, 80.77, 80.92, 127.70, 127.90, 128.36, 136.93, 163.64, 163.96, 164.34, 164.55, 167.76, 173.58. HRMS (ESI) m/z found: 875.772517 (M + Na+), estimated for C25H23Br4N5NaO5S2: 875.776630.
(2S,5R)-2-(3-(Benzo[d][1,3]dioxol-5-yl)-1,2,4-oxadiazol-5-yl)-6,6-dibromo-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16aj). Obtained following general procedure E as a white solid, m.p 174–175 °C; 20% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.29 (s, 3H), 1.73 (s, 3H), 5.34 (s, 1H), 5.99 (s, 1H), 6.04 (s, 2H), 6.90 (d, J = 8.15 Hz, 1H), 7.50 (d, J = 1.60 Hz, 1H), 7.64 (dd, J = 8.15, 1.60 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.51, 33.20, 58.52, 64.59, 65.54, 80.72, 101.68, 107.48, 108.69, 119.66, 122.65, 148.16, 150.48, 164.26, 168.06, 172.88. HRMS (ESI) m/z found: 523.88711 (M + Na+), estimated for C16H13Br2N3NaO4S: 523.88857.
(2S,5R)-6,6-Dibromo-3,3-dimethyl-2-(3-(pyridin-2-yl)-1,2,4-oxadiazol-5-yl)-4-thia-1-azabicyclo [3.2.0]heptan-7-one (16ak). Obtained following general procedure E as a white solid, m.p 132–132.5 °C; 30% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.31 (s, 3H), 1.75 (s, 3H), 5.43 (s, 1H), 5.98 (s, 1H), 7.44–7.49 (m, 1H), 7.85–7.91 (m, 1H), 8.13–8.16 (m, 1H), 8.80–8.83 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.69, 33.18, 58.43, 64.82, 65.59, 80.69, 123.498, 125.93, 137.18, 145.58, 150.58, 164.42, 168.23, 173.99. HRMS (ESI) m/z found: 458.9124 (M + H+), estimated for C14H13Br2N4O2S: 458.9120.
(2S,5R)-6,6-Dibromo-2-(3-((E)-4-methoxystyryl)-1,2,4-oxadiazol-5-yl)-3,3-dimethyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one (16am). Obtained following general procedure E as a yellow solid, m.p 159.5–160 °C; 70% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.29 (s, 3H), 1.73 (s, 3H), 3.84 (s, 3H), 5.32 (s, 1H), 5.99 (s, 1H), 6.91 (d, J = 16.12 Hz, 1H), 6.93 (d, J = 8.70 Hz, 2H), 7.51 (d, J = 8.70 Hz, 2H), 7.65 (d, J = 16.12 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.52, 33.20, 55.34, 58.52, 64.62, 65.51, 80.69, 109.46, 114.33, 127.74, 129.05, 139.81, 160.90, 164.29, 168.13, 172.23. HRMS (ESI) m/z found: 535.92426 (M + Na+), estimated for C18H17Br2N3NaO3S: 535.92496.
tert-Butyl((5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl) methyl) carbamate (22aa). The N-protected amino acid-derived amidoxime 20a was obtained following general procedure D.32 The oxadiazole 22aa was obtained following general procedure E as a white solid, mp 60–61 °C; 25% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.23 (s, 3H), 1.45 (s, 9H), 1.70 (s, 3H), 4.48 (d, J = 5.73 Hz, 2H), 5.10 (br s, 1H), 5.29 (s, 1H), 5.92 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.51, 28.30, 29.69, 33.15, 36.71, 58.39, 64.55, 65.50, 80.44, 80.66, 155.41, 164.31, 168.43, 173.69. HRMS (ESI) m/z found: 532.946137 (M + Na+), estimated for C15H20Br2N4NaO4S: 532.946421.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)ethyl)carbamate (22ab). The N-protected amino acid-derived amidoxime 20b was obtained following general procedure D. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.35 (d, J = 6.99 Hz, 3H), 1.45 (s, 9H), 4.26–4.29 (m, 1H), 4.89–4.92 (m, 3H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 17.53, 28.32, 46.99, 80.22, 155.37, 156.03. The oxadiazole 22ab was obtained following general procedure E as a white solid, mp 162–163 °C; 40% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.21 (s, 3H), 1.42 (s, 9H), 1.51 (d, J = 6.75 Hz, 3H), 1.69 (s, 3H), 4.92–5.12 (m, 2H), 5.28 (s, 1H), 5.91 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 20.29, 25.49, 28.30, 29.68, 33.15, 43.59, 58.43, 64.60, 65.51, 80.21, 80.66, 154.75, 164.32, 172.02, 173.46. HRMS (ESI) m/z found: 546.96042 (M + Na+), estimated for C16H22Br2N4NaO4S: 546.96207.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)-2-methylpropyl)carbamate (22ac). The N-protected amino acid-derived amidoxime 20c was obtained following general procedure D.33 The oxadiazole 22ac was obtained following general procedure E as a white solid, mp 165–165.5 °C; 51% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.90–0.95 (m, 6H), 1.20 (s, 3H), 1.43 (s, 9H), 1.70 (s, 3H), 2.12–2.19 (m, 1H), 4.78–4.83 (m, 1H), 5.05 (d, J = 8.73 Hz, 1H), 5.92 (s, 1H), 5.92 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 17.81, 18.81, 25.48, 28.27, 32.09, 33.16, 52.70, 58.48, 64.55, 65.54, 80.09, 80.68, 155.22, 164.31, 170.37, 173.19. HRMS (ESI) m/z found: 574.9940 (M + Na+), estimated for C18H26Br2N4NaO4S: 574.9934.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)-3-methylbutyl)carbamate (22ad). The N-protected amino acid-derived amidoxime 20d was obtained following general procedure D. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.89 (d, J = 6.15 Hz, 3H), 0.91 (d, J = 6.15, 3H), 1.42 (s, 9H), 1.54–1.70 (m, 3H), 4.05–4.18 (m, 1H), 4.96–4.99 (m, 2H), 5.25–5.28 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 21.88, 22.80, 24.58, 22.27, 40.84, 49.95, 80.02, 155.36, 156.09. The oxadiazole 22ad was obtained following general procedure E as a white solid, mp 128–129 °C; 70% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.95 (d, J = 6.15 Hz, 3H), 0.96 (d, J = 6.15 Hz, 3H), 1.21 (s, 3H), 1.42 (s, 9H), 1.62–1.67 (m, 3H), 1.70 (s, 3H), 4.80–5.05 (m, 2H), 5.28 (s, 1H), 5.92 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 22.11, 24.47, 24.64, 25.48, 28.26, 29.66, 33.14, 43.16, 45.89, 58.44, 64.61, 65.49, 80.10, 80.65, 154.87, 164.30, 171.44, 173.30. HRMS (ESI) m/z found: 567.02560 (M + H+), estimated for C19H29Br2N4O4S: 567.02708.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)-2-methylbutyl)carbamate (22ae). The N-protected amino acid-derived amidoxime 20e was obtained following general procedure D. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.84–0.94 (m, 6H), 1.04–1.16 (m, 2H), 1.42 (s, 9H), 1.77–1.92 (m, 1H), 3.74–3.89 (m, 1H), 4.70 (brs, 1H), 4.93 (brs, 1H), 5.42–5.55 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 10.92, 15.59, 24.94, 28.31, 36.83, 56.76, 79.82, 154.40, 156.02. The oxadiazole 22ae was obtained following general procedure E as a white solid, mp 148–149 °C; 67% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.86 (d, J = 6.75 Hz, 3H), 0.93 (t, J = 7.37 Hz, 3H), 1.12–1.28 (m, 2H), 1.20 (s, 3H), 1.43 (s, 9H), 1.70 (s, 3H), 1.84–1.97 (m, 1H), 4.86–4.91 (m, 1H), 5.06 (d, J = 9.03 Hz, 1H), 5.28 (s, 1H), 5.93 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 11.34, 15.25, 24.97, 25.48, 28.28, 33.17, 38.52, 51.64, 58.48, 64.56, 65.54, 80.09, 80.69, 155.11, 164.31, 170.18, 173.11. HRMS (ESI) m/z found: 589.00935 (M + Na+), estimated for C19H28Br2N4NaO4S: 589.00902.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,2,4-oxadiazol-3-yl)-2-phenylethyl)carbamate (22af). The N-protected amino acid-derived amidoxime 20f was obtained following general procedure D. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.36 (s, 9H), 2.93–3.18 (m, 2H), 4.26–441 (m, 1H), 4.72 (brs, 1H), 4.98 (brs, 1H), 5.27 (brs, 1H), 7.19–7.28 (m, 5H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 28.24, 29.69, 38.32, 52.92, 80.29, 126.75, 128.49, 129.32, 137.13, 154.74, 155.91. The oxadiazole 22af was obtained following general procedure E as a yellow solid, mp 72–73 °C; 36% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.16 (s, 3H), 1.40 (s, 9H), 1.69 (s, 3H), 3.18 (d, J = 6.61 Hz, 2H), 5.04 (d, J = 6.93, 1H), 5.17–5.23 (m, 1H), 5.26 (s, 1H), 5.93 (s, 1H), 7.08–7.11 (m, 2H), 7.22–7.25 (m, 3H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.44, 28.26, 33.19, 40.04, 48.63, 58.50, 64.55, 65.61, 80.35, 80.69, 127.12, 128.59, 129.41, 135.54, 154.72, 164.33, 170.35, 173.48. HRMS (ESI) m/z found: 622.9910 (M + Na+), estimated for C22H26Br2N4NaO4S: 622.9934.
(F) General procedure for the synthesis of 1,3,4-oxadiazoles
A round bottom flask equipped with a magnetic stir bar was charged with the carboxylic acid 14a–b (0.5 mmol) dry DCM (6 mL) was added, followed by CDI (0.5 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 30 minutes. After this time, hydrazide 24a–c (0.5 mmol) was incorporated. The coupling process was carried out for 45 minutes at 0 °C. Then CBr4 (1 mmol) and PPh3 (1 mmol) were added in one portion. The reaction mixture was stirred at 0 °C for 2 h. Finally, the residue is purified by column chromatography using silica gel as adsorbent and a mixture of hexane:AcOEt (70:30) as elution solvent.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,3,4-oxadiazol-2-yl)-3-methylbutyl)carbamate (26aa). Obtained following general procedure F as a white solid, mp 120–121 °C; 16% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.96 (d, J = 6.29 Hz, 3H), 0.97 (d, J = 6.29 Hz, 3H), 1.23 (s, 3H), 1.43 (s, 9H), 1.55–1.83 (m, 3H), 1.65 (s, 3H), 4.87–5.08 (m, 2H), 5.30 (s, 1H), 5.88 (s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 21.97, 22.48, 24.68, 25.58, 28.26, 33.10, 42.36, 45.66, 58.59, 63.87, 65.41, 80.61, 154.79, 161.27, 164.28, 168.14. HRMS (ESI) m/z found: 589.0060 (M + Na+), estimated for C19H28Br2N4NaO4S: 589.0090.
tert-Butyl((2S)-1-(2-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-carbonyl)hydrazinyl)-4-methyl-1-oxopentan-2-yl)carbamate (25aa). Obtained from previous reaction, as a yellow solid, mp 125–126 °C; in 70% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.95 (d, J = 6.72 Hz, 3H), 0.93 (d, J = 6.72 Hz, 3H), 1.43 (s, 9H), 1.54 (s, 3H), 1.60–1.77 (m, 6H), 4.19–4.33 (m, 1H), 4.62 (s, 1H), 5.12 (d, J = 6.09 Hz, 1H), 5.30 (s, 1H), 9.14 (br s, 1H), 9.32 (br s, 1H). 13C NMR (CDCl3, 75 MHz) δ 21.68, 22.92, 24.66, 26.02, 28.32, 31.73, 40.83, 51.57, 56.52, 64.87, 69.41, 79.86, 80.80, 156.03, 163.21, 165.96, 170.20. HRMS (ESI) m/z found: 607.0197 (M + Na+), estimated for C19H30Br2N4NaO5S: 607.0196.
tert-Butyl((1S)-1-(5-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-yl)-1,3,4-oxadiazol-2-yl)-2-phenylethyl)carbamate (26ab). Obtained following general procedure F as a white solid, mp 153–153.5 °C; 25% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.09 (s, 3H), 1.41 (s, 9H), 1.64 (s, 3H), 3.15–3.35 (m, 2H), 5.15–5.29 (m, 3H), 5.71 (s, 1H), 7.09–7.14 (m, 2H), 7.23–7.32 (m, 3H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 25.39, 28.19, 33.02, 39.91, 48.62, 58.63, 63.74, 65.18, 80.35, 127.47, 128.78, 129.14, 135.00, 154.66, 161.23, 164.24, 167.33. HRMS (ESI) m/z found: 601.01030 (M + H+), estimated for C22H27Br2N4O4S: 601.01143.
tert-Butyl((2S)-1-(2-((2S,5R)-6,6-dibromo-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptan-2-carbonyl)hydrazinyl)-1-oxo-3-phenylpropan-2-yl)carbamate (25ab). Obtained from previous reaction, as a white solid, mp 124–125 °C; in 40% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 1.36 (s, 9H), 1.51 (s, 3H), 1.62 (s, 3H), 2.95–3.08 (m, 1H), 3.09–3.27 (m, 1H), 4.57–4.72 (m, 1H), 4.78 (s, 1H), 5.51 (d, J = 6.45 Hz, 1H), 5.83 (s, 1H), 7.15–7.32 (m, 5H), 9.64 (br s, 1H), 9.95 (d, J = 4.71 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ 25.80, 28.29, 32.55, 38.55, 54.24, 57.52, 64.66, 68.83, 80.48, 127.14, 128.70, 128.77, 129.26, 135.95, 155.81, 162.78, 165.63, 168.87. HRMS (ESI) m/z found: 641.0046 (M + Na+), estimated for C22H28Br2N4NaO5S: 641.0039.
tert-Butyl((S)-3-methyl-1-(5-phenyl-1,3,4-oxadiazol-2-yl)butyl)carbamate (26ba). Obtained following general procedure F as a yellow solid, mp 91–91.5 °C; 18% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.99 (d, J = 6.25 Hz, 3H), 0,98 (d, J = 6.25 Hz, 3H), 1.45 (s, 9H), 1.68–1.86 (m, 3H), 4.99–5.22 (m, 2H), 7.45–7.54 (m, 3H), 8.01–8.06 (m, 2H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 22.00, 22.60, 24.68, 28.27, 43.01, 45.74, 80.38, 123.77, 126.91, 129.01, 131.74, 154.96, 164.85, 167.19. HRMS (ESI) m/z found: 354.17875 (M + Na+), estimated for C18H25N3NaO3: 354.17881.
tert-Butyl(1-(2-benzoylhydrazinyl)-4-methyl-1-oxopentan-2-yl)carbamate (25ba). Obtained from previous reaction, as a white solid, mp 128.5–129 °C; in 40% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 0.88 (d, J = 5.82 Hz, 3H), 0.89 (d, J = 5.82 Hz, 3H), 1.39 (s, 9H), 1.52–1.75 (m, 3H), 4.44–4.55 (m, 1H), 5.56 (d, J = 8.49 Hz, 1H), 7.31–7.36 (m, 2H), 7.42–7.47 (m, 1H), 7.81–7.84 (m, 2H), 9.81 (br s, 1H), 10.00 (br s, 1H). 13C NMR (CDCl3, 75 MHz) δ (ppm): 21.78, 22.88, 24.59, 28.25, 41.44, 51.53, 80.00, 127.49, 128.38, 131.53, 132.02, 155.85, 164.71, 170.74. HRMS (ESI) m/z found: 372.1891 (M + Na+), estimated for C18H27N3NaO4: 372.1894.
Biological evaluation
Cell lines and proliferation assay. B16-F0 cell line (murine melanoma, ATCC CRL-6322) was grown in RPMI-1640 (Gibco BRL) supplemented with 10% FBS, 2 mmol L−1 L-glutamine, 50 U mL−1 penicillin and 50 μg mL−1 streptomycin. LM3 cell line34 (mammary adenocarcinoma) and NMuMG cells (normal murine mammary gland, ATCC CRL-1636), kindly provided by the Institute of Oncology ‘Angel H. Roffo’ (Buenos Aires, Argentina), were cultured in Dulbecco's modified Eagle's Medium-F12 containing 10% FBS, 2 mmol L−1 L-glutamine, 0.6% HEPES, 50 U ml−1 penicillin and 50 μg mL−1 streptomycin. For harvesting, cells were treated with 0.05% trypsin/EDTA using standard procedures.Cells were incubated in 96-well microplates at a density of 1 × 104 cells per well (B16-F0) or 2 × 104 cells per well (LM3 or NMuMG) for 72 h at 37 °C in the presence of a 20 μM concentration of the different hybrid compounds or vehicle (control) (dimethyl sulfoxide diluted 1/10 in ethanol; final concentration 20 μL vehicle per mL of medium), in a total volume of 0.2 mL of the corresponding culture medium. Cell number was evaluated by colorimetric determination of the levels of the ubiquitous lysosomal enzyme hexosaminidase.35
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Support from CONICET (PUE-2016), ANPCyT (PICT 2017-2694), Agencia Santafesina de Ciencia, Técnica e Innovación (ASACTEI) (AC – 2015-00005) and Universidad Nacional de Rosario (BIO 514). Is gratefully acknowledged. Likewise, we acknowledge support of CONICET (PIP 0154), ANPCyT (PICT 2017-1278) and Universidad de Buenos Aires (2018–2020, UBACYT 20020170100041BA). We also thank Dr Julia C. Mata for careful reading the manuscript. CMC and MGP thank CONICET for fellowship.
Notes and references
-
(a) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 1–13 CrossRef PubMed;
(b) C. J. O' Connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332–4345 RSC;
(c) H. Beckmann, C. J. O' Connor and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444–4456 RSC;
(d) N. Mateu, S. L. Kidd, T. J. Osberger, H. L. Stewart, S. Bartlett, W. R. Galloway, A. J. P. North, H. F. Sore and D. R. Spring, The Application of Diversity-oriented Synthesis in Chemical Biology. in Chemical and Biological Synthesis: Enabling Approaches for Understanding Biology, ed. N. J. Westwood and A. Nelson, The Royal Society of Chemistry, Cambridge, 2018, pp. 8–44 Search PubMed.
- T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co, Weinheim, 2nd edn, 2003 Search PubMed.
- J. Boström, A. Hogner, A. Llinàs, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817–1830 CrossRef PubMed.
- A. M. Jone and J. M. Helem, Drugs, 2009, 69, 1903–1910 CrossRef PubMed.
- Z. Li, W. Chen, J. J. Hale, C. L. Lynch, S. G. Mills, R. Hajdu, C. A. Keohane, M. J. Rosenbach, J. A. Milligam, G. J. Shein, G. Chrebet, S. A. Parent, J. Bergstrom, D. Card, M. Forrest, E. J. Quackenbush, L. A. Wickham, H. Vargas, R. M. Evans, H. Rosen and S. J. Mandala, J. Med. Chem., 2005, 48, 6169–6173 CrossRef CAS PubMed.
- L. J. Street, R. Baker, T. Book, C. O. Kneen, A. M. MacLead, K. J. Merchant, G. A. Showell, J. Sounders, R. H. Herbert, S. B. Fredman and E. A. Harley, J. Med. Chem., 1990, 33, 2690–2697 CrossRef CAS PubMed.
- H. Z. Zhang, S. Kasibhatla, J. Kuemmerle, W. Kemnitzer, K. OllisMason, L. Qiu, C. Crogan-Grundy, B. Tseng, J. Drewe and S. X. Cai, J. Med. Chem., 2005, 48, 5215–5223 CrossRef CAS PubMed.
- A. Vaidya, S. Jain, P. Jain, P. Jain, N. Tiwari, R. Jain, R. Jain, A. K. Jain and R. K. Agrawal, Mini-Rev. Med. Chem., 2016, 16, 825–845 CrossRef CAS PubMed.
-
(a) V. Namani, B. B. K. Goud, Y. B. Kumari and R. Kumbham, Lett. Org. Chem., 2016, 13, 249–254 CrossRef CAS;
(b) V. Namani, K. Goud, B. Bharath and Y. B. Kumari, Asian J. Chem., 2015, 27, 4579–4582 CrossRef CAS.
-
(a) V. Raj, A. Rai, A. K Singh, A. K. Keshari, P. Trivedi, B. Ghosh, U. Kamar, D. Kumar and S. Saha, Anti Canc. Agents Med. Chem., 2018, 18, 719–738 CrossRef CAS PubMed;
(b) F. Qiao, Y. Yin, Y. N. Shen, S. F. Wang, S. Sha, X. Wu, A.-M. Lu, X. Chen, W.-M. Zhang and H. L. Zhu, RSC Adv., 2015, 5, 19914–19923 RSC.
- A. Zarghi, S. A. Tabatabai, M. Faizi, A. Ahadian, P. Navabi, V. Zanganeh and A. Shafiee, Bioorg. Med. Chem. Lett., 2005, 15, 1863–1865 CrossRef CAS PubMed.
- A. Zarghi, M. Faizi, B. Shafaghi, A. Ahadian, H. R. Khojastehpoor, V. Zanganeh, S. A. Tabatabai and A. Shafiee, Bioorg. Med. Chem. Lett., 2005, 15, 3126–3129 CrossRef CAS PubMed.
-
(a) N. Kerru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 142, 179–212 CrossRef CAS PubMed;
(b) C. Viegas-Junior, A. Danuello, V. Da Silva Bolzani, E. J. Barreiro and C. A. Fraga, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- For some recent examples, see:
(a) S. Shah, C. Lee, H. Choi, J. Gautam, H. Jang, G. J. Kim, Y.-J. Lee, C. L. Chaudhary, S. W. Park, T.-G. Nam, J.-A. Kim and B.-S. Jeong, Org. Biomol. Chem., 2016, 14, 4829–4841 RSC;
(b) M. T. Gabr and M. S. Abdel-Raziq, Bioorg. Med. Chem. Lett., 2018, 28, 2910–2913 CrossRef CAS PubMed;
(c) I. D'Agostino, I. Giacchello, G. Nannetti, A. L. Fallacara, D. Deodato, F. Musumeci, G. Grossi, G. Palù, Y. Cau, M. I. Trist, A. Loregian, S. Schenone and M. Botta, Eur. J. Med. Chem., 2018, 157, 743–758 CrossRef PubMed;
(d) T. Fröhlich, C. Reiter, M. E. Saeed, C. Hutterer, F. Hahn, M. Leidenberger, O. Friedrich, B. Kappes, M. Marschall, T. Efferth and S. B. Tsogoeva, ACS Med. Chem. Lett., 2018, 9, 534–539 CrossRef PubMed;
(e) H. K. Mahmoud, T. A. Farghaly, H. G. Abdulwahab, N. T. Al-Qurashi and M. R. Shaaban, Eur. J. Med. Chem., 2020, 208, 112752 CrossRef CAS PubMed;
(f) L. Gummidi, N. Kerru, P. Awolade, A. Raza, A. K. Sharma and P. Singh, Bioorg. Med. Chem. Lett., 2020, 30, 127544 CrossRef CAS PubMed.
- P. G. Cornier, C. M. L. Delpiccolo, F. C. Mascali, D. B. Boggián, E. G. Mata, M. G. Cárdenas, V. C. Blank and L. P. Roguin, RSC Med. Chem., 2014, 5, 214–218 RSC.
- V. Blank, Y. Bellizzi, E. Zotta, P. G. Cornier, C. M. L. Delpiccolo, D. B. Boggián, E. G. Mata and L. P. Roguin, Anti-Cancer Drugs, 2018, 29, 416–428 CrossRef CAS PubMed.
- R. F. Poulain, A. L. Tartar and B. P. Déprez, Tetrahedron Lett., 2001, 42, 1495–1498 CrossRef CAS.
- Phenyl amidoxime (11a) was obtained from the corresponding commercially available benzonitrile. However, the other arylnitriles were obtained from the corresponding carboxylic acids by dehydration with H2SO4, see: K. Mlinarić-Majerski, R. Margeta and J. Veljković, Synlett, 2005, 13, 2089–2091 CrossRef CAS ; or from the aldehydes, by reaction with trifluoromethanesulfonic acid and sodium azide, see: B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364–5370 CrossRef PubMed.
- G. Liang and D. D. Feng, Tetrahedron Lett., 1996, 37, 6627–6630 CrossRef CAS.
- A. Boeijen and R. M. J. Liskamp, Eur. J. Org. Chem., 1999, 2127–2135 CrossRef.
- J. H. Ansede, M. Anbazhagan, R. Brun, J. D. Easterbrook, J. E. Hall and D. W. J. Boykin, J. Med. Chem., 2004, 47, 4335–4338 CrossRef CAS PubMed.
- P. Raboisson, A. Breitholtz-Emanuelsson, H. Dahllöf, L. Edwards, W. L. Heaton, M. Isaac, K. Jarvie, A. Kers, A. B. Minidis, A. Nordmark, S. M. Sheehan, A. Slassi, P. Ström, Y. Terelius, D. Wensbo, J. M. Wilson, T. Xin and D. A. McLeod, Bioorg. Med. Chem. Lett., 2012, 22, 6974–6979 CrossRef CAS PubMed.
- J. Bondebjerg, H. Fuglsang, K. R. Valeur, D. W. Kaznelson, J. A. Hansen, R. O. Pedersen, B. O. Krogh, B. S. Jensen, C. Lauritzen, G. Petersen, J. Pedersen and L. Naerum, Bioorg. Med. Chem., 2005, 13, 4408–4424 CrossRef CAS PubMed.
- L. Dornelles, O. E. Rodrigues, E. F. Heck, C. R. Bender, M. B. Cansian, R. S. Schwab and W. A. Severo Filho, Arkivoc, 2015, 7, 131–144 Search PubMed.
- C. S. de Oliveira, B. F. Lira, J. M. Barbosa-Filho, J. G. Lorenzo and P. F. de Athayde-Filho, Molecules, 2012, 17, 10192–10231 Search PubMed.
- P. Gao and Z. Bai, Chin. J. Chem., 2017, 35, 1673–1677 CrossRef CAS.
- H. Weinmann, ChemMedChem, 2016, 11, 450–466 CrossRef CAS PubMed.
- G. J. Freeman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10275–10276 CrossRef CAS PubMed.
- X. Q. Song, Z. Y. Ma, Y. G. Wu, M. L. Dai, D. B. Wang, J. Y. Xu and Y. Liu, Eur. J. Med. Chem., 2019, 167, 377–387 CrossRef CAS PubMed.
- V. M. L. Braga, S. J. de Melo, R. M. Srivastava and E. P. da S. Falcão, J. Braz. Chem. Soc., 2004, 15, 603–607 CrossRef CAS.
- S. Nordhoff, S. Bulat, S. Cerezo-Galvez, O. Hill, B. Hoffmann-Enger, M. Lopez-Canet, C. Rosenbaum, P. J. Edwards and A. Feurer, Bioorg. Med. Chem. Lett., 2009, 19, 6340–6345 CrossRef CAS PubMed.
- L. A. Clutterbuck, C. G. Posada, C. Visintin, D. R. Riddall, B. Lancaster, P. J. Gane, J. Garthwaite and D. L. Selwood, J. Med. Chem., 2009, 52, 2694–2707 CrossRef CAS PubMed.
- J. E. Mangette, M. R. Johnson, V.-D. Le, R. A. Shenoy, H. Roark, M. Stier, T. Belliotti, T. Capiris and P. R. Guzzo, Tetrahedron, 2009, 65, 9536–9541 CrossRef CAS.
- V. Ladeda, A. P. Adam, L. Puricelli and E. Bal de Kier Joffé, Breast Canc. Res. Treat., 2001, 69, 39–51 CrossRef CAS PubMed.
- U. J. Landegren, J. Immunol. Methods, 1984, 67, 379–388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05602f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yanina Bellizzib,
Elizabeth Barrionuevob,
Viviana C. Blank
*a,
Yanina Bellizzib,
Elizabeth Barrionuevob,
Viviana C. Blank