
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring proteins in H2O with 2D-IR spectroscopy†
Samantha
Hume
a,
Gordon
Hithell
a,
Gregory M.
Greetham
b,
Paul M.
Donaldson
b,
Michael
Towrie
b,
Anthony W.
Parker
 b,
Matthew J.
Baker
c and
Neil T.
Hunt
*d
b,
Matthew J.
Baker
c and
Neil T.
Hunt
*d
aDepartment of Physics, University of Strathclyde, SUPA, 107 Rottenrow East, Glasgow, G4 0NG, UK
bSTFC Central Laser Facility, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
cWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Technology and Innovation Centre, 99 George Street, Glasgow, G1 1RD, UK
dDepartment of Chemistry, York Biomedical Research Institute, University of York, Heslington, York, YO10 5DD, UK. E-mail: neil.hunt@york.ac.uk
First published on 14th May 2019
Abstract
The amide I infrared band of proteins is highly sensitive to secondary structure, but studies under physiological conditions are prevented by strong, overlapping water absorptions, motivating the widespread use of deuterated solutions. H/D exchange raises fundamental questions regarding the impact of increased mass on protein dynamics, while deuteration is impractical for biomedical or commercial applications of protein IR spectroscopy. We show that 2D-IR spectroscopy can avoid this problem because the 2D-IR amide I signature of proteins dominates that of water even at sub-millimolar protein concentrations. Using equine blood serum as a test system, we investigate the significant implications of being able to measure the spectroscopy and dynamics of proteins in water, demonstrating relevance in areas ranging from fundamental science to the clinic. Measurements of vibrational relaxation dynamics of serum proteins reveals that deuteration slows down the rate of amide I vibrational relaxation by >10%, indicating a dynamic impact of isotopic exchange in some proteins. The unique link between protein secondary structure and 2D-IR amide I lineshape allows differentiation of signals due to albumin and globulin protein fractions in serum leading to measurements of the biomedically-important albumin to globulin ratio (AGR) with an accuracy of ±4% across a clinically-relevant range. Furthermore, we demonstrate that 2D-IR spectroscopy enables differentiation of the structurally similar globulin proteins IgG, IgA and IgM, opening up a straightforward spectroscopic approach to measuring levels of serum proteins that are currently only accessible via biomedical laboratory testing.
Infrared spectroscopy is sensitive to molecular structure, but its application to proteins is hindered by the broad nature of the amide I band, essentially the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the peptide link. 2D-IR spectroscopy overcomes this problem by measuring vibrational couplings between peptide units in macromolecular structures and the 2D-IR amide I signal is diagnostic of secondary structure content and ligand binding.1–6 Water is an obstacle common to both IR absorption and 2D-IR spectroscopy. The biological solvent absorbs widely across the mid-IR, but the H–O–H bending mode (δH–O–H, 1644 cm−1) directly obscures the protein amide I transition, motivating the widespread use of deuterated solvents (D2O) for protein IR spectroscopy (Fig. 1(a)). Deuteration is an imperfect solution however. D2O is non-physiological, raising fundamental questions regarding the impact of solvent isotope exchange on our ability to measure natural protein structural dynamics. The kinetic isotope effect is well-known while changes in vibrational coupling between solvent and protein in deuterated systems arising from the removal of spectral overlap between the amide I band and the δH–O–H mode may alter energy transfer dynamics and impact upon function in ways that are yet to be revealed.7 The use of D2O also leads to practical issues surrounding incomplete proton exchange and elevated cost. These severely limit applications of protein IR spectroscopy in the biomedical or commercial sector where H/D exchange is either impractical or viewed as economically unfeasible.
O stretching mode of the peptide link. 2D-IR spectroscopy overcomes this problem by measuring vibrational couplings between peptide units in macromolecular structures and the 2D-IR amide I signal is diagnostic of secondary structure content and ligand binding.1–6 Water is an obstacle common to both IR absorption and 2D-IR spectroscopy. The biological solvent absorbs widely across the mid-IR, but the H–O–H bending mode (δH–O–H, 1644 cm−1) directly obscures the protein amide I transition, motivating the widespread use of deuterated solvents (D2O) for protein IR spectroscopy (Fig. 1(a)). Deuteration is an imperfect solution however. D2O is non-physiological, raising fundamental questions regarding the impact of solvent isotope exchange on our ability to measure natural protein structural dynamics. The kinetic isotope effect is well-known while changes in vibrational coupling between solvent and protein in deuterated systems arising from the removal of spectral overlap between the amide I band and the δH–O–H mode may alter energy transfer dynamics and impact upon function in ways that are yet to be revealed.7 The use of D2O also leads to practical issues surrounding incomplete proton exchange and elevated cost. These severely limit applications of protein IR spectroscopy in the biomedical or commercial sector where H/D exchange is either impractical or viewed as economically unfeasible.
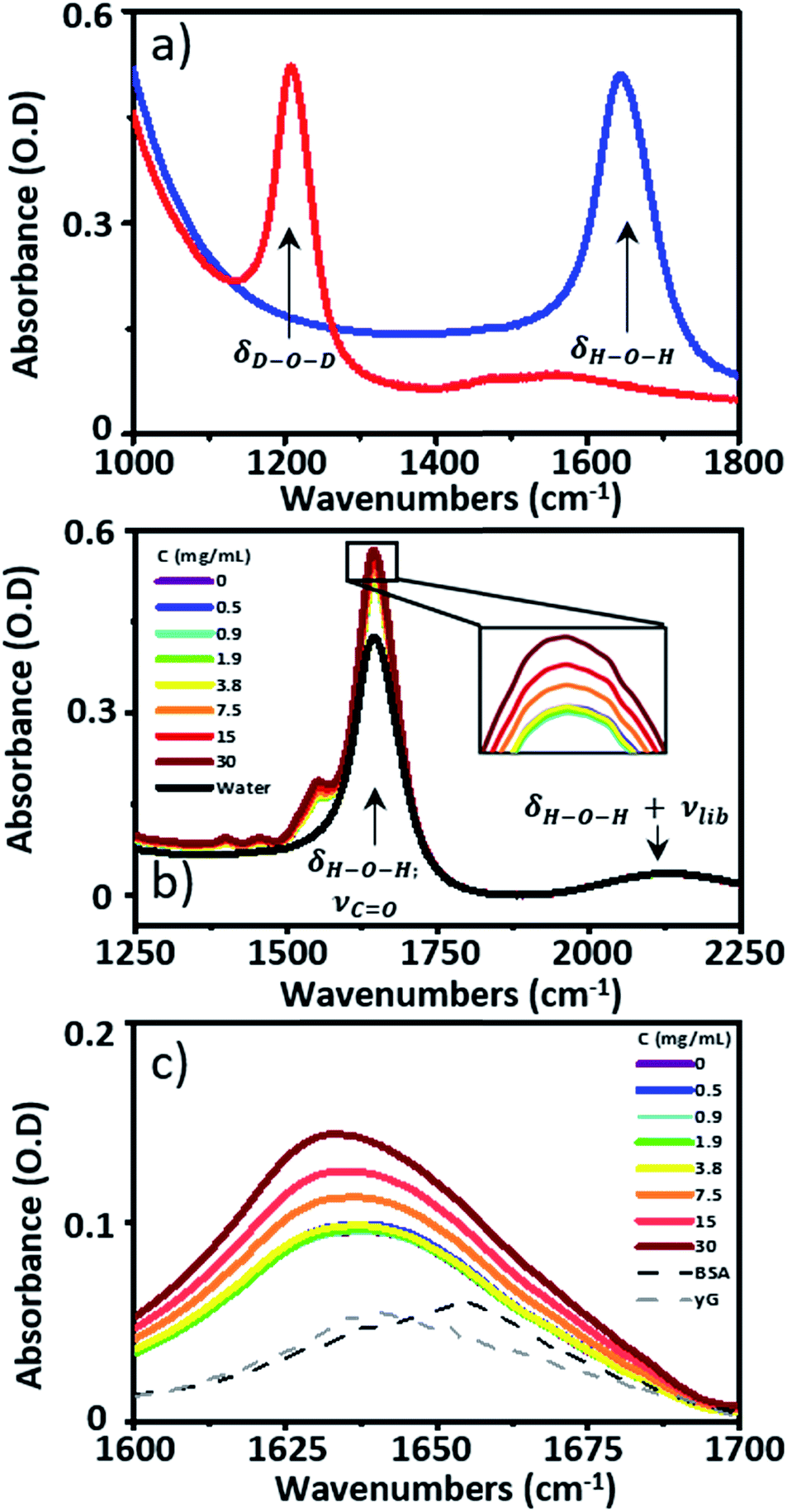 | ||
| Fig. 1 (a) The IR absorption spectra of D2O (red) and H2O (blue). (b) IR absorption spectra of serum samples spiked with γ-globulins, pure H2O is shown in black. The inset shows an expansion of the tip of the peak due to the H–O–H bending mode of water and the protein amide I band at 1650 cm−1. (c) IR absorption spectra of serum spiked with γ-globulins from (b) following subtraction of the H2O spectrum. | ||
In the biomedical arena, spectroscopic interrogation of biofluids holds attractions as a label-free, minimally-invasive screening technology.8 Blood serum is easily obtained, with minimal patient discomfort, and contains a range of potentially diagnostic chemical markers by virtue of contact with most of the major organs.9,10 Current technologies use antibody assays to enhance the signal associated with a target biomolecule, relying critically on the availability of specific antibodies for proteins of interest and requiring significant sample preparation. Moreover, the heterogeneous nature of disease means that single-metabolite detection may be inferior to a broad biomolecular fingerprint of metabolic function as an early warning of deteriorating patient health9,10 or, for example, to indicate the presence of cancers.10,11 The protein content of blood serum represents an ideal substrate for holistic analysis. Human serum contains ∼70 mg mL−1 of proteins composed of albumin (∼35–50 mg mL−1) and the globulins (∼25–35 mg mL−1). Diagnostically, measurement of the albumin to globulin ratio (AGR) is valuable. Changes in the AGR are linked to an inflammatory response and can correlate with the ability of the patient to survive cancer therapy, complementing progress towards personalized treatment and precision medicine.12–16 Currently, the globulin level is derived indirectly, from laboratory-based measurements of total protein and albumin content rather than by direct measurement. Moreover, the globulins encompass a huge number of proteins. The γ-globulins constitute the bulk of the serum globulin fraction. Of these, immunoglobulin-G (IgG) is the most abundant, accounting for ∼80% of the γ-globulins, while IgA (∼13%) and IgM (∼6%) are the next most abundant.16 As well as bulk changes in globulin concentration, changes in serum-levels of each of these individual globulin components are associated with health-related issues. IgG levels are found to increase in cases of liver disease or chronic infection. IgA is linked to cirrhosis while changes in IgM levels can warn of the presence of trypanosomiasis or antibody deficiency syndrome.16
There is thus considerable benefit in a straightforward spectroscopic measurement that can not only deliver the AGR directly but also differentiate between the major globulin components. Current IR spectroscopic studies of blood serum employ dried samples to avoid the problem of water absorption, which can introduce artefacts from the drying process. Attenuated total reflection methods enable studies in aqueous liquids, but such linear spectroscopic methods cannot separate albumin and globulin signals or identify contributions from individual globulin proteins.8
Using blood serum as our exemplar system, we show that H/D exchange is not necessary for label-free protein IR studies in H2O-based media. Typical serum protein concentrations in humans are in the sub mM range (35–50 mg mL−1 albumin corresponds to 0.5–0.7 mM; 25–30 mg mL−1 γ-globulins ∼0.15–0.25 mM), which correspond closely to those used for 2D-IR spectroscopic studies of proteins in D2O. Thus, our approach extends beyond blood serum, allowing detailed amide I studies of proteins in physiological solvents for the first time. Our measurements show that deuteration slows down the vibrational relaxation dynamics of serum proteins, while the enhanced spectral resolution of 2D-IR relative to IR absorption enables accurate differentiation of protein signals in the complex aqueous serum environment, even when their secondary structure composition is similar. These results indicate that the ability to measure the 2D-IR spectrum of proteins in water has relevance in areas ranging from fundamental molecular science to healthcare. Applications of 2D-IR spectroscopy in the biomedical arena are particularly timely in light of recent advances enabling 2D-IR spectral-acquisition in a few seconds17–20 alongside demonstrations of high throughput screening methodologies.21
Materials and methods
Sample preparation
Pooled equine serum, serum albumin (bovine), γ-globulins (bovine), IgG, IgA and IgM (human) were obtained from Sigma Aldrich and used without further purification. Measurements of individual proteins were performed using aqueous Tris buffer (pH = 7.5) to mimic the pH of the serum samples. To study the spectroscopy of serum samples at a range of AGR values, γ-globulin was spiked into pooled horse serum at concentrations of 30, 15, 7.5, 3.8, 1.9, 0.9 and 0.5 mg mL−1. Yielding a total of 8 samples (7 spikes and pure serum).IR spectroscopy
IR absorption spectra were measured using a Thermo Scientific Nicolet iS10 Fourier Transform spectrometer. Spectra were the result of 20 co-added scans at a resolution of 1 cm−1 in the spectral region 400–4000 cm−1. To measure IR spectra in water using transmission mode, the sample thickness was carefully controlled to avoid saturation of the δH–O–H mode of water at 1650 cm−1. Samples were housed between two CaF2 windows. No spacer was used, but the tightness of the sample holder was adjusted to obtain approximately consistent absorbance values of ∼0.1 OD for the δOH + νlibr combination mode of water located at 2130 cm−1. Based upon the measured molar extinction coefficient of water, this corresponded to a sample thickness of ∼2.75 μm. This method limited the absorbance at 1650 cm−1 to <0.6 (Fig. 1(b)). A background spectrum was measured before each sample and subtracted following scaling to the amplitude of the δOH + νlibr mode. Each measurement was made in triplicate.2D-IR spectroscopy
2D-IR spectra were recorded using the Ultra laser spectrometer at the Central Laser Facility using the Fourier Transform 2D-IR method employing a sequence of three mid-IR laser pulses arranged in a pseudo pump–probe beam geometry, as described elsewhere.22,23 Comparable results were obtained irrespective of the use of a scanning interferometer or pulse shaper to deliver the sequence of two pump pulses. The pulses were generated by a Ti:sapphire laser (Coherent Legend Elite Duo, 20 W, 50 fs, 10 kHz pulse repetition rate) producing 5 W of 800 nm light pumping a home-built white-light seeded BBO optical parametric amplifier (OPA) equipped with difference frequency mixing of the signal and idler in AgGaS2. Mid-IR pulses with a temporal duration of <50 fs; a central frequency of 1650 cm−1 and a bandwidth of ∼400 cm−1 were obtained. All 2D-IR spectra were recorded at a waiting time (Tw) of 250 fs between pump and probe pulses using a parallel pump–probe polarization relationship. Each measurement was made in triplicate using identical sample conditions to those used for IR absorption measurements and spectra were vector normalised.Results
Blood serum spectroscopy
Infrared absorption spectra of equine serum samples spiked with 0–30 mg mL−1 γ-globulins are shown in Fig. 1(b). The dominant feature near 1650 cm−1 is assignable to overlapping contributions arising from the δH–O–H mode of water (Fig. 1(b), black) and the amide I mode of the serum protein component. The spectra were normalised to the amplitude of the combination band of the δH–O–H and librational modes of water (νlibr), located at 2130 cm−1 and the water contribution subtracted (Fig. 1(c)). The resulting amide I band of the serum proteins was largely featureless, but gained in amplitude as the added quantity of γ-globulins increased. The IR absorption spectra of the individual serum albumin and γ-globulin protein components are shown as dashed lines in Fig. 1(c). Strong overlap of their respective amide I bands prevented quantitative determination of the AGR from IR absorption data.By contrast to the IR absorption spectra (Fig. 1 and 2(a)), the 2D-IR spectrum of pure serum shows considerable structure (Fig. 2(b)). The negative feature (red) located on the 2D-IR diagonal near 1650 cm−1 is assigned to the v = 0–1 transitions of modes observed in the IR absorption spectrum and contains two distinct contributions with pump frequencies of 1639 and 1656 cm−1 (arrows). Positive (blue) peaks due to the accompanying v = 1–2 transitions are shifted to lower probe frequencies by vibrational anharmonicity.
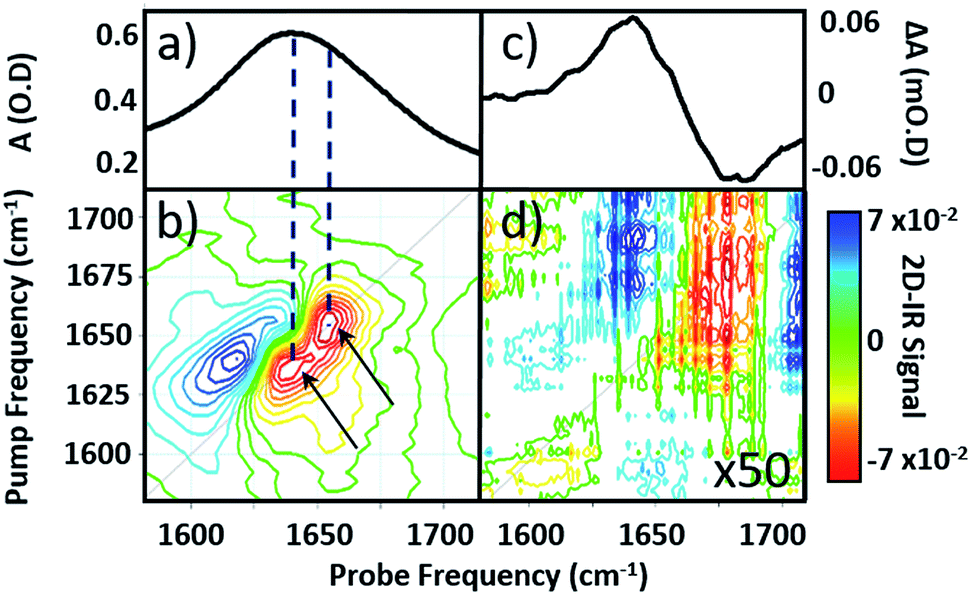 | ||
| Fig. 2 (a) IR absorption and (b) 2D-IR spectrum of pure serum in the amide I region. Arrows identify two components of the v = 0–1 transition discussed in the text. (c) IR pump–probe spectrum of H2O at a pump–probe time delay of 300 fs. (d) 2D-IR spectrum of H2O, magnified 50×. The spectra in (b) and (d) were both obtained with a waiting time (Tw) of 250 fs and are plotted on the same scale, see colour bar. | ||
Comparison of 2D-IR spectra of serum and pure water (Fig. 2(d)) shows that the 2D-IR signal of water is significantly weaker than that of the serum under the same sample conditions. Though weak, the measured 2D-IR response of water was found to be in good agreement with previous observations.24–26 The corresponding IR pump–probe spectrum is also shown for comparison (Fig. 2(c)).27
At 1650 cm−1, the molar extinction coefficient of the serum proteins is at least two orders of magnitude larger than that of water.28 As 2D-IR signals are dependent upon the 4th power of the vibrational transition dipole moment, this leads to enhancement of the strong amide I mode of the biological macromolecules relative to the more plentiful, but weakly-absorbing, water molecules.1,5 As a result, the 2D-IR response of proteins is the dominant feature in the serum spectrum despite the large absorbance of the δH–O–H mode of water.
Comparing the 2D-IR spectrum of pure serum to the spectra of serum albumin and γ-globulins obtained individually in water (Fig. 3) allows assignment of the peaks at 1656 and 1639 cm−1 in the serum spectrum to the albumin and the globulin components respectively. The difference in frequency of the two protein signals arises from the fact that serum albumin has a largely α-helical secondary structure while the globulins have a higher proportion of β-sheet, which shifts the center of mass of the amide I band to lower frequency.3 The higher-order dependence of the 2D-IR signal upon the transition dipole moment means that the lineshapes appearing on the diagonal of a 2D-IR spectrum are narrower than those found in the IR absorption spectrum. This leads to the appearance of two well-resolved peaks along the diagonal of the 2D-IR plot, where only one broad signal was observed in the IR absorption spectrum (Fig. 1(c) and 2(a)).1,5
 | ||
| Fig. 3 IR absorption spectra of (a) serum (b) serum albumin (c) γ-globulins. 2D-IR spectra of (d) serum (e) serum albumin (f) γ-globulins. Dashed grey horizontal lines show peak positions of albumin and γ-globulins. The color scale is as shown in Fig. 2. | ||
Protein dynamics
To establish the impact of solvent isotopic exchange on the vibrational dynamics of the serum proteins, IR pump–probe data (Fig. 4) were obtained for bovine serum albumin, the γ-globulins and the individual γ-globulin protein IgG in both H2O and D2O (Fig. 4(d)–(f)). A measurement of the vibrational lifetime of the amide I band of the albumin component of the spectrum of neat serum was also carried out for comparison (Fig. 4(c)). The IR pump–probe spectrum is equivalent to the 2D-IR response projected onto the probe frequency axis.1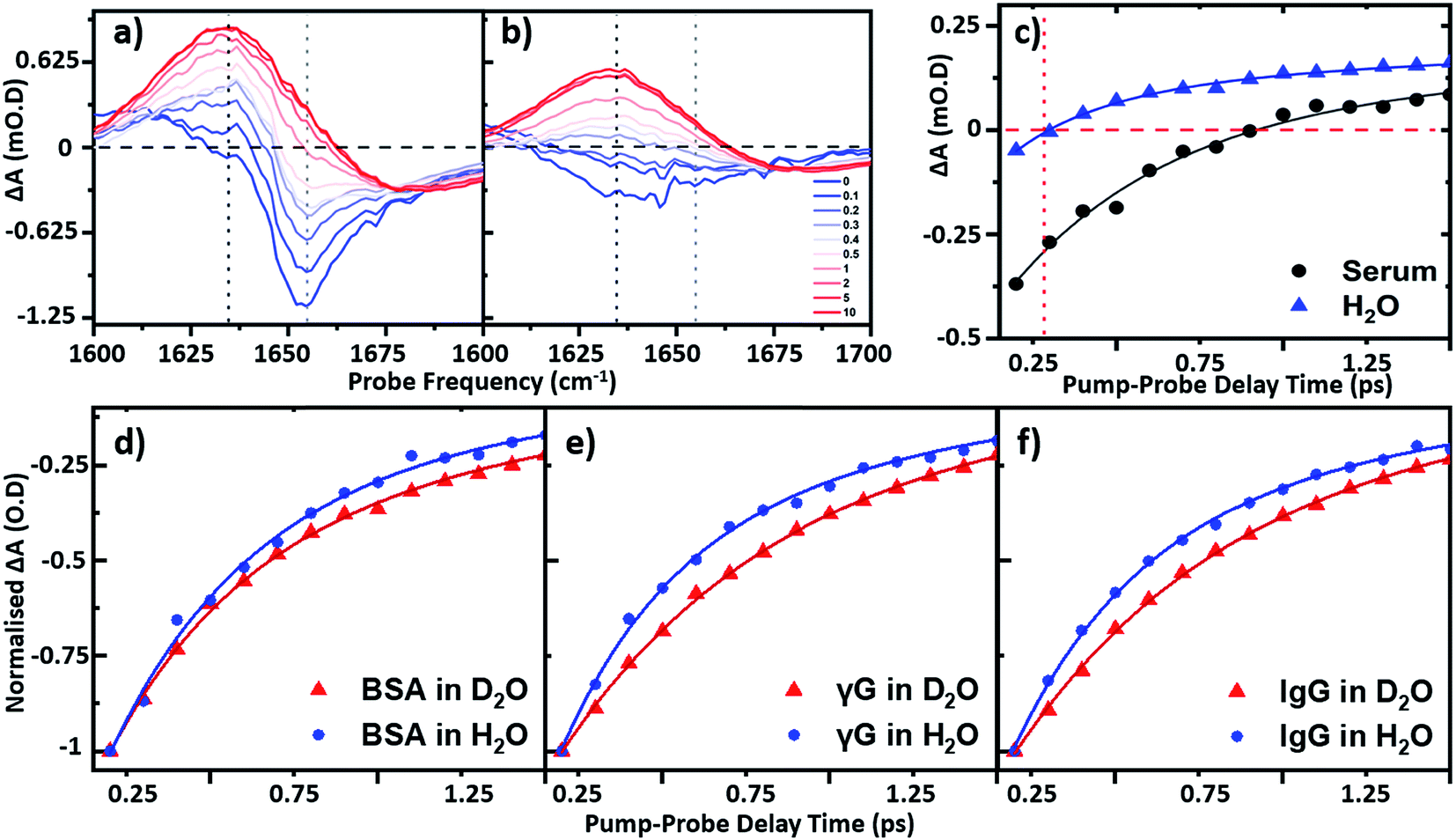 | ||
| Fig. 4 IR pump–probe spectra of (a) serum albumin and (b) H2O. Waiting times range from 0 ps (blue) to 10 ps (red). (c) Temporal variation of the bleach signal observed in the IR pump–probe spectrum of H2O (blue) and serum (black) at a frequency of 1650 cm−1. Solid lines show fits to the data using a single exponential function with a decay time constant of 220 fs (H2O) and 0.83 ps (serum) respectively. Temporal variation of the amide I v = 0–1 bleach signal for (d) serum albumin (e) γ-globulins and (f) IgG in both D2O (red) and H2O (blue) solvents. All pump probe data were obtained with magic angle polarization of pump and probe pulses. | ||
In the case of serum albumin, the γ-globulins and IgG in D2O, the dynamics of the peak of the amide I v = 0–1 transition were well-represented by single exponential decays with lifetimes of 0.89, 0.90 and 0.93 ps respectively (Fig. 4(d)–(f), red). In the case of samples made in H2O, the amide I v = 0–1 peak (Fig. 4(a)) is overlapped by the bleach of the v = 0–1 transition of the H2O δH–O–H mode (Fig. 4(b)). The latter was significantly smaller in amplitude than the protein response (<20% of the total amplitude at Tw = 0 ps) and was well-represented by a bi-exponential function featuring a ∼220 ± 40 fs decay and a ∼1.2 ± 0.2 ps rise-time due to the effects of residual sample heating, which persisted to Tw values longer than 5 ps (Fig. 4(b) and (c)). This behaviour of the water band is in agreement with previous work.27 In the data shown in Fig. 4(d)–(f), the water response has been subtracted by scaling the signals of water and the protein/serum to the Tw signal at 5 ps where no protein contribution is observed. Using this method, single-exponential vibrational relaxation times of 0.78, and 0.74 and 0.78 ps were observed for the amide I v = 0–1 transition for albumin, the γ-globulins and IgG in water. These values were found to be robust using other data analysis approaches, including fitting to tri-exponential functions to account for the bi-exponential relaxation of the water signal and the protein relaxation behaviour.
It is important to note that the vibrational relaxation time of the amide I band of a large protein is, by definition, a weighted average over a large number of coupled amide I oscillators. In the case of the γ-globulins, this is a mixture of proteins. It has been shown previously using fibrillar aggregates of short chain peptides that the lifetime of the amide I band is sensitive to secondary structure and to the level of solvation of a given residue.29 However, the consistent observation of a reduction in vibrational lifetime by around 10% upon moving from D2O to H2O indicates that the average lifetime of the amide I mode of these proteins is being reduced. It is perhaps to be expected that this may be arising from a combination of responses from solvent-exposed and buried residues, which are perturbed differently by H/D exchange, but this is a topic for further study. These findings are however consistent with previous studies of solvent isotope-dependent vibrational dynamics, suggesting that isotopic exchange of the solvent may be responsible for altering the observed protein dynamics.30 In addition to pump–probe data, the spectral diffusion of the 2D-IR amide I lineshape of the proteins in water was compared to that in D2O. Indications of altered linewidths and spectral diffusion processes in H2O were present in the data (ESI Fig. S1†) but as these observations relate to the entire amide I lineshape, which is underpinned by considerable structure arising from different secondary structural contributions a numerical analysis of this as a whole is not physically-relevant.
In relation to analytical studies of complex protein mixtures such as serum, the relative dynamics of the signals due to water and proteins can be exploited to optimise the contrast between the 2D-IR protein response and that of water. The faster relaxation time of the δH–O–H mode than the protein amide I response of serum (∼0.83 ± 0.1 ps at 1650 cm−1) means that, at Tw = 250 fs, the water signal is at a minimum prior to the onset of the small rising signal due to water heating (Fig. 4(c)). The result is that the spectrum shows only the protein signature at this waiting time and on this basis, the following spectral analysis of protein samples was carried out using a Tw of 250 fs, though it is stressed that the water signal at other values of Tw is not sufficiently large as to prevent measurement of protein relaxation dynamics.
2D-IR biofluid analysis – using 2D-IR to measure the AGR
Using 2D-IR spectra of a range of serum samples spiked with differing quantities of γ-globulins (Fig. S2†), an attempt was made to capitalize on the spectral differentiation of the peaks assigned to the albumin and globulin protein components (Fig. 3) to quantify the AGR directly from 2D-IR spectra. Three approaches were employed to determine the AGR values of the serum samples:(i) Using the relative amplitudes of the peaks assigned to albumin and globulins on the 2D-IR spectrum diagonal (Fig. 5(a) and (b)). In this approach, the ratio of the absolute values of the amplitudes of two distinct peaks at 1656 cm−1 and 1639 cm−1 on the diagonal of each 2D spectrum, assigned to the albumin and globulin fractions respectively, was used to determine the AGR. Scaling of the globulin amplitude by a factor of 1.8 was performed to account for the measured differences in signal amplitude between albumin and the γ-globulins per unit concentration (see Fig. S3†).
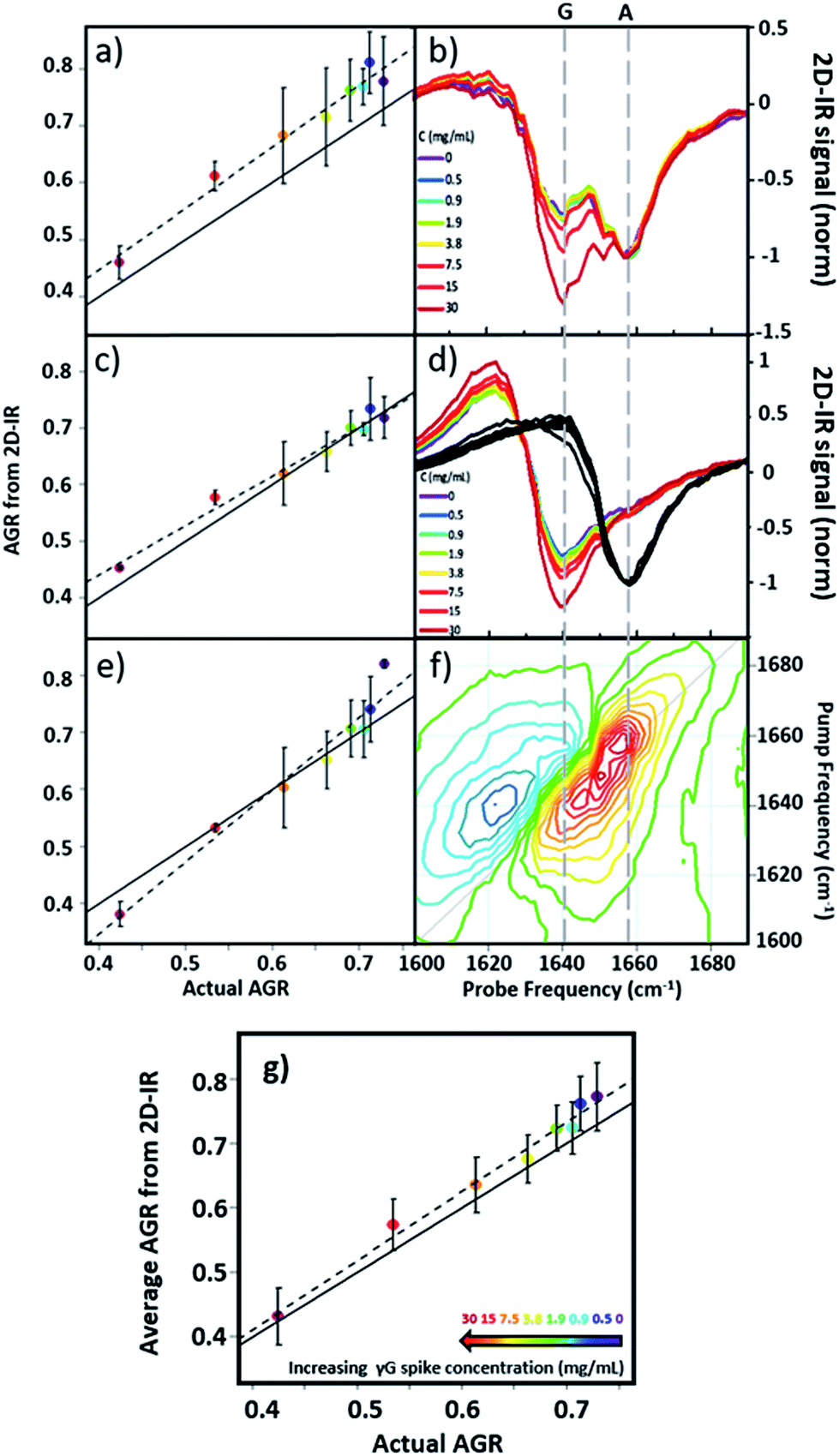 | ||
| Fig. 5 AGR of serum samples from 2D-IR spectroscopy. (a and b) AGR obtained using the 2D-IR diagonal method. (c and d) AGR obtained using the pump slice method. (e and f) AGR values obtained using the 2D-IR linear combination method. The spectral basis of the method is shown in (b), (d) and (f) respectively. (g) AGR obtained from averaging the results in (a), (c) and (e). In panels (a), (c), (e) and (g) the solid black line indicates the actual AGR of the samples. Error bars show 2σ variation. Dashed lines show linear fits to the experimental AGR values. | ||
(ii) Using the amplitudes of the v = 0–1 peaks due to albumin and globulins taken from pump-frequency slices through the 2D spectra (Fig. 5(c) and (d)). This method utilised slices through the 2D-IR spectrum at pump frequencies of 1656 cm−1 (albumin) and 1639 cm−1 (globulin). The ratio of the absolute values of the amplitude of the globulin pump slice and that of the albumin slice was used to determine the AGR following application of the scaling factor (1.8) to the globulin signal.
(iii) Analysis using a linear combination of the 2D-IR spectra of albumin and γ-globulins (Fig. 5(e) and (f)). All 2D-IR spectra were normalised to the albumin peak at 1656 cm−1. A linear combination analysis fitted the serum 2D-IR spectrum to the linear sum of the independent 2D-IR spectra of albumin and the globulins. The coefficients of the relative contributions of the two protein spectra were then used to evaluate the AGR, following scaling of the globulin fraction by 1.8. The methods are described in detail in the ESI† alongside all 2D-IR spectra (Fig. S2†).
All three methods for determining the AGR spectroscopically produced a linear relationship when the measured value (points and dashed lines, Fig. 5(a), (c) and (e)) was plotted against the actual AGR. The latter was obtained via sending a sample of the as-received equine blood serum for standard laboratory testing at the Glasgow School of Veterinary Medicine and adding the quantity of the known γ-globulin spike to the globulin component. Ideal agreement between the known and 2D-IR-measured AGR values is represented by the solid black line in Fig. 5(a), (c) and (e). Of the three 2D-IR methods used, the pump slice approach (Fig. 5(c) and (d)) was most accurate at the higher values of the AGR, which correspond most closely to the expected human clinical range of 1–2 (horse serum AGR values are slightly lower than human levels). At lower AGR values, the agreement obtained with the pump-slice method was less effective, possibly owing to the very large γ-globulin spike distorting the albumin response. The results obtained from the 2D-IR diagonals were good across the full range of the samples studied (Fig. 5(a) and (b)), with most 2D-IR-derived values being within the measurement error of the actual AGR value, though a constant offset from the actual AGR value was noted. This is attributed to differences in the anharmonicities of the proteins. Finally, the linear combination yielded excellent agreement over the mid-range of the spiked samples (AGR = 0.5–0.7), but was less effective at the extremities. Taking an average of the three analysis approaches (Fig. 5(g)) produced agreement with actual AGR values across the full range of samples, within the experimental uncertainty and is the best approach. Leave one out-type tests of the analysis protocol also showed accuracy to within the expected error of the measurement (Fig. S4†). Overall, the 2D-IR measurements tested here show accuracy over a clinically-relevant range.
Based on sample-to-sample variation, the accuracy of the 2D-IR-derived AGR measurement was ±0.03 (∼4%). Direct comparisons with the current wet assay technique are not possible because these tests derive the AGR value from the difference in total protein and albumin concentrations and so do not directly measure globulin content, however typical quoted accuracies are ∼1%. Although the spectroscopic approach is less accurate than current technologies, this is a first demonstration and there is considerable scope for improvement of the accuracy through engineering approaches to sample path length repeatability and improved data collection protocols.
Spectrum diagnostics – beyond the AGR
In principle, 2D-IR offers the scope to go beyond the AGR measurement by virtue of the information-rich 2D lineshape of the proteins, which gives the opportunity to resolve more than albumin and the γ-globulins.4 The major protein constituent of the γ-globulins is IgG (80%). IgA (13%) and IgM (6%) are the next most abundant globulin proteins and so represent realistic test targets. The aim was to differentiate increases in signal from IgA and IgM from that of the albumin and γ-globulin contributions to a spectrum of a serum sample. The challenge for 2D-IR lies in the fact that IgA and IgM both have similar β-sheet-rich structures to IgG and so it is necessary to identify regions of the 2D-IR spectrum where the response due to IgA and IgM proteins can be differentiated from IgG and the generic γ-globulin response.2D-IR spectra of IgG, IgA and IgM are shown in Fig. 6(a)–(c). As expected, the spectral features are all very similar due to their comparable secondary protein structures and compositions. In order to highlight the subtle discrepancies between the signals, difference 2D-IR spectra (Fig. 6(d)–(f)) were constructed by subtracting the average of triplicate measurements of IgG, IgA and IgM at a known concentration (Fig. 6(a)–(c)) from the spectrum of the equivalent concentration of γ-globulins to account for different maximum solubility levels of the immunoglobulin proteins studied. The results of this process shows that there is little difference between the spectrum of IgG and that of the γ-globulins (Fig. 6(d)). Although somewhat trivial, this expected result acts as an effective control for the process. By contrast, the difference spectra obtained for IgA and IgM do show regions of spectral differences with the γ-globulins. IgA in particular (Fig. 6(e)) clearly shows regions of decreased negative (red) spectral density in the diagonal region near ∼1640 cm−1 and increases in the diagonal part of the amide I band (blue) near 1657 cm−1. IgM also shows spectral differences to the γ-globulins in the diagonal region of the spectrum (Fig. 6(f)), though the effect is less marked than for IgA and the amplitude of the difference signal is reduced.
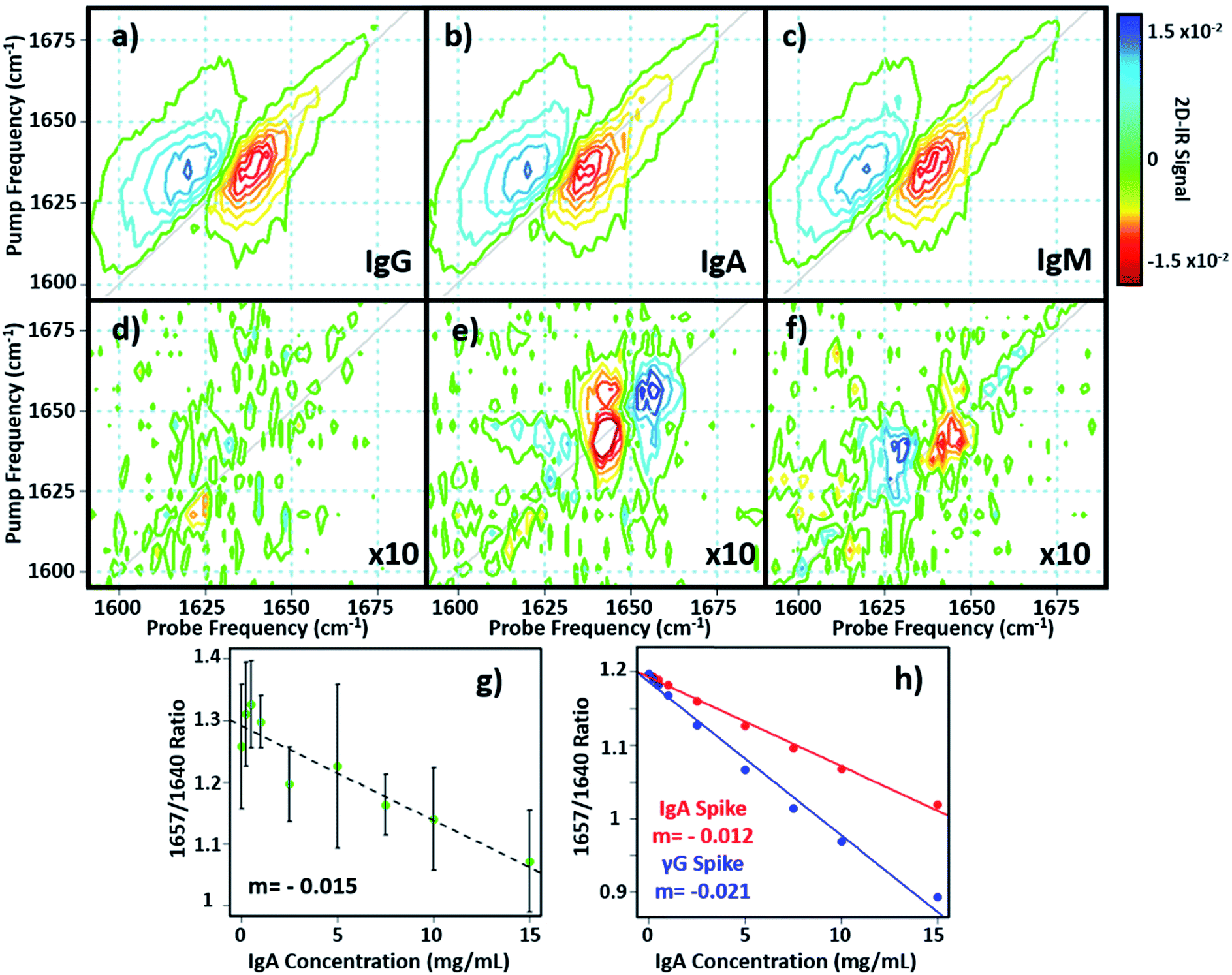 | ||
| Fig. 6 (a–c) 2D-IR spectra of IgG, IgA and IgM in H2O. (d–f) Difference spectra obtained by subtracting the 2D-IR response of the spectra in (a–c) from that of the γ-globulins. The spectral features have been magnified by a factor of 10 for clarity and are shown on the same scale as (a–c). (g and h) show the variation in 2D-IR signal at the peak of the difference spectral response for IgA as a function of added protein for the experimental data (g) and calculations (h). The linear increase in the negative signal shows a correlation with protein content. | ||
In order to determine whether the signals observed in the difference spectra are sufficient to quantify changes in serum levels of IgA and IgM, measurements were carried out on a range of serum samples spiked with additional quantities of IgA and IgM. The positions of the peaks in the (γ-globulins–IgA) difference spectrum (Fig. 6(e), 1640 and 1657 cm−1) are not the same as those used to measure the γ-globulin fraction for the above AGR analysis (Fig. 3(f)). Furthermore, the opposing sign of the two components gives two points of reference that can be used to separate the contributions from IgA and γ-globulins.
The 2D-IR spectra of serum samples spiked with concentrations of IgA from 0–15 mg mL−1 are shown in Fig. S5.† To determine the ability of the 2D-IR spectrum to determine the IgA content, the ratio of the amplitudes on the spectrum diagonal at 1657![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1640 cm−1 (the positive and negative peaks in the (γ-globulin–IgA) difference spectrum) was plotted as a function of IgA concentration (Fig. 6(g)). The measurement is difficult because the IgA and γ-globulin signals overlap strongly and there is no portion of the spectrum that is unique to either the γ-globulins or to IgA. However, calculation of the spectra expected from this experiment using reconstructions from the individually-measured albumin, γ-globulin, and IgA spectra (Fig. 6(h)) show that the 1657:1640 cm−1 amplitude ratio should decrease as the IgA level was increased if IgA is influencing the signal. Importantly, the gradient of the decrease in this ratio would be significantly shallower than that observed if the generic γ-globulin response increased (Fig. 6(g)). It can be seen from a comparison of the calculated (Fig. 6(h)) and measured (Fig. 6(g)) data that the 2D-IR response recovered matches well with that expected for an IgA-specific signal increase. The gradient of −0.0015 from the experimental data matches well with the value of −0.0012 derived from calculated data. Furthermore, the gradient of the measured amplitude ratio is much closer to that predicted for a change in IgA levels than for a change in γ-globulin fraction. The fact that the correlation with IgA levels persists down to ∼1 mg mL−1 compares well with expected serum levels of IgA, which are in the range of 13% of ∼30 mg mL−1 (4 mg mL−1).16
:1640 cm−1 (the positive and negative peaks in the (γ-globulin–IgA) difference spectrum) was plotted as a function of IgA concentration (Fig. 6(g)). The measurement is difficult because the IgA and γ-globulin signals overlap strongly and there is no portion of the spectrum that is unique to either the γ-globulins or to IgA. However, calculation of the spectra expected from this experiment using reconstructions from the individually-measured albumin, γ-globulin, and IgA spectra (Fig. 6(h)) show that the 1657:1640 cm−1 amplitude ratio should decrease as the IgA level was increased if IgA is influencing the signal. Importantly, the gradient of the decrease in this ratio would be significantly shallower than that observed if the generic γ-globulin response increased (Fig. 6(g)). It can be seen from a comparison of the calculated (Fig. 6(h)) and measured (Fig. 6(g)) data that the 2D-IR response recovered matches well with that expected for an IgA-specific signal increase. The gradient of −0.0015 from the experimental data matches well with the value of −0.0012 derived from calculated data. Furthermore, the gradient of the measured amplitude ratio is much closer to that predicted for a change in IgA levels than for a change in γ-globulin fraction. The fact that the correlation with IgA levels persists down to ∼1 mg mL−1 compares well with expected serum levels of IgA, which are in the range of 13% of ∼30 mg mL−1 (4 mg mL−1).16
Repeating the exercise for IgM also showed a linear relationship between the measured 2D-IR signal at the peaks of the (IgM–γ-globulin) difference spectrum and the IgM concentration (Fig. S6†). In the case of IgM, the smaller magnitude of the spectral differences between IgM and γ-globulins led to a significantly more noisy correlation, while the calculated differences show that a less clear separation of the IgM and γ-globulin responses would be anticipated.
The reported proof of concept experiments illustrate the promising potential for development of 2D-IR as a tool for differentiating IgA and IgM protein contributions from albumin and γ-globulins based on their 2D-IR responses. Our approach utilizes two characteristic points of spectral difference that separate IgA/M from albumin and the γ-globulins. Whilst acknowledging that applying this approach to samples of unknown protein levels would be challenging, we believe that this first study gives a clear indicator that the potential exists to sub-divide the γ-globulin spectral component into its major constituents. This result encourages further developments in sample handling protocols and data analysis strategies, including absolute calibration methods needed to extend the capabilities of this approach.
Conclusions
These experiments show that 2D-IR measures the amide I band of protein samples at sub millimolar concentrations in water, without the need for H/D exchange of the solvent. The results demonstrate the capability to measure the molecular dynamics of proteins in water, rather than heavy water solutions, increasing the physiological relevance of 2D-IR studies. We further demonstrate that modern 2D-IR instrumentation can perform accurate quantitative measurements of proteins in H2O. The ability to use 2D-IR to suppress the water background signal enables 2D-IR data collection in simple transmission mode and the unique lineshapes allow clear separation of albumin and globulin signals. This confers a simple, robust approach to determining the AGR of serum using a single spectroscopic measurement without the need for time consuming sample preparation or complex data analysis. Indicative experiments showing that scope for more subtle protein differentiation exists. To our knowledge, this is the first label-free optical measurement of the AGR in as-received blood serum.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Funding is gratefully acknowledged from STFC for programme access to Central Laser Facility systems. The assistance of Mr James Harvie (University of Glasgow School of Veterinary Medicine) in obtaining the AGR value for horse serum samples is also gratefully acknowledged.References
-
P. Hamm and M. T. Zanni, Concepts and Method of 2D Infrared Spectroscopy, Cambridge University Press, Cambridge, 2011 Search PubMed
.
- N. T. Hunt, Ultrafast 2D-IR Spectroscopy – Applications to Biomolecules, Chem. Soc. Rev., 2009, 38, 1837–1848 RSC
- C. R. Baiz, C. S. Peng, M. E. Reppert, K. C. Jones and A. Tokmakoff, Coherent two-dimensional infrared spectroscopy: Quantitative analysis of protein secondary structure in solution, Analyst, 2012, 137(8), 1793–1799 RSC
- L. Minnes, D. J. Shaw, B. Cossins, P. M. Donaldson, G. M. Greetham, M. Towrie, A. W. Parker, M. J. Baker, A. Henry, R. Taylor and N. T. Hunt, Quantifying Secondary Structure Changes in Calmodulin using 2D-IR Spectroscopy, Anal. Chem., 2017, 89, 10898–10906 CrossRef CAS PubMed
- E. B. Dunkelberger, M. Grechko and M. T. Zanni, Transition Dipoles from 1D and 2D Infrared Spectroscopy Help Reveal the Secondary Structures of Proteins: Application to Amyloids, J. Phys. Chem. B, 2015, 119(44), 14065–14075 CrossRef CAS PubMed
- D. J. Shaw, R. E. Hill, N. Simpson, F. S. Husseini, K. Robb, G. M. Greetham, M. Towrie, A. W. Parker, D. Robinson, J. D. Hirst, P. A. Hoskisson and N. T. Hunt, Examining the role of protein structural dynamics in drug resistance in Mycobacterium tuberculosis, Chem. Sci., 2017, 8, 8384–8399 RSC
- D. Czurlok, J. Gleim, J. Lindner and P. Voehringer, Vibrational Energy Relaxation of Thiocyanate Ions in Liquid-to-Supercritical Light and Heavy Water. A Fermi's Golden Rule Analysis, J. Phys. Chem. Lett., 2014, 5(19), 3373–3379 CrossRef CAS PubMed
- M. J. Baker, S. R. Hussain, L. Lovergne, V. Untereiner, C. Hughes, R. A. Lukaszewski, G. Thiefin and G. D. Sockalingum, Developing and understanding biofluid vibrational spectroscopy: a critical review, Chem. Soc. Rev., 2016, 45(7), 1803–1818 RSC
- S. Hu, J. A. Loo and D. T. Wong, Human body fluid proteome analysis, Proteomics, 2006, 6(23), 6326–6353 CrossRef CAS PubMed
- E. F. Petricoin, C. Belluco, R. P. Araujo and L. A. Liotta, The blood peptidome: a higher dimension of information content for cancer biomarker discovery, Nat. Rev. Cancer, 2006, 6(12), 961–967 CrossRef CAS PubMed
- F. Grosserueschkamp, T. Bracht, H. C. Diehl, C. Kuepper, M. Ahrens, A. Kallenbach-Thieltges, A. Mosig, M. Eisenacher, K. Marcus, T. Behrens, T. Bruning, D. Theegarten, B. Sitek and K. Gerwert, Spatial and molecular resolution of diffuse malignant mesothelioma heterogeneity by integrating label-free FTIR imaging, laser capture microdissection and proteomics, Sci. Rep., 2017, 7, 44829 CrossRef CAS PubMed
- T. Koyama, N. Kuriyama, E. Ozaki, D. Matsui, I. Watanabe, F. Miyatani, M. Kondo, A. Tamura, T. Kasai, Y. Ohshima, T. Yoshida, T. Tokuda, I. Mizuta, S. Mizuno, K. Yamada, K. Takeda, S. Matsumoto, M. Nakagawa, T. Mizuno and Y. Watanabe, Serum albumin to globulin ratio is related to cognitive decline via reflection of homeostasis: a nested case-control study, BMC Neurol., 2016, 16, 253 CrossRef PubMed
- A. S. Peters, J. Backhaus, A. Pfutzner, M. Raster, G. Burgard, S. Demirel, D. Bockler and M. Hakimi, Serum-infrared spectroscopy is suitable for diagnosis of atherosclerosis and its clinical manifestations, Vib. Spectrosc., 2017, 92, 20–26 CrossRef CAS
- J. J. Liu, S. X. Chen, Q. R. Geng, X. C. Liu, P. F. Kong, Y. Q. Zhan and D. Z. Xu, Prognostic value of pretreatment albumin–globulin ratio in predicting long-term mortality in gastric cancer patients who underwent D2 resection, OncoTargets Ther., 2017, 10, 2155–2162 CrossRef CAS PubMed
- N. Beamer, B. M. Coull, G. Sexton, P. Degarmo, R. Knox and G. Seaman, Fibrinogen and the albumin–globulin ratio in recurrent stroke, Stroke, 1993, 24(8), 1133–1139 CrossRef CAS PubMed
-
F. W. Putnam, The plasma proteins, AP London, 2nd edn, 1975, vol. 1 Search PubMed
- S. H. Shim, D. B. Strasfeld, Y. L. Ling and M. T. Zanni, Automated 2D IR Spectroscopy Using a Mid-IR Pulse Shaper and Application of this Technology to the Human Islet Amyloid Polypeptide, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(36), 14197–14202 CrossRef CAS PubMed
- P. M. Donaldson, G. M. Greetham, D. J. Shaw, A. W. Parker and M. Towrie, A 100 kHz Pulse Shaping 2D-IR Spectrometer Based on Dual Yb:KGW Amplifiers, J. Phys. Chem. A, 2018, 122(3), 780–787 CrossRef CAS PubMed
- M. Srisa-Art, S. D. Noblitt, A. T. Krummel and C. S. Henry, IR-Compatible PDMS microfluidic devices for monitoring of enzyme kinetics, Anal. Chim. Acta, 2018, 1021, 95–102 CrossRef CAS PubMed
- K. M. Tracy, M. V. Barich, C. L. Carver, B. M. Luther and A. T. Krummel, High-Throughput Two-Dimensional Infrared (2D IR) Spectroscopy Achieved by Interfacing. Microfluidic Technology with a High Repetition Rate 2D IR Spectrometer, J. Phys. Chem. Lett., 2016, 7(23), 4865–4870 CrossRef CAS PubMed
- R. Fritzsch, P. M. Donaldson, G. M. Greetham, M. Towrie, A. W. Parker, M. J. Baker and N. T. Hunt, Rapid screening of DNA-ligand complexes via 2D-IR spectroscopy and ANOVA-PCA, Anal. Chem., 2018, 90, 2732–2740 CrossRef CAS PubMed
- K. Adamczyk, M. Candelaresi, R. Kania, K. Robb, C. Bellota-Antón, G. M. Greetham, M. R. Pollard, M. Towrie, A. W. Parker, P. A. Hoskisson, N. P. Tucker and N. T. Hunt, The Effect of Point Mutation on the Protein–Ligand Interactions in Equilibrium Structural Fluctuations of Myoglobin, Phys. Chem. Chem. Phys., 2012, 14, 7411–7419 RSC
- G. M. Greetham, P. Burgos, Q. Cao, I. P. Clark, P. S. Codd, R. C. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. R. Pollard, D. A. Robinson, Z.-J. Xin and M. Towrie, ULTRA: A Unique Instrument for Time-Resolved Spectroscopy, Appl. Spectrosc., 2010, 64, 1311–1319 CrossRef CAS PubMed
- D. Kraemer, M. L. Cowan, A. Paarmann, N. Huse, E. T. J. Nibbering, T. Elsaesser and R. J. D. Miller, Temperature dependence of the two-dimensional infrared spectrum of liquid H2O, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(2), 437–442 CrossRef CAS PubMed
- W. B. Carpenter, J. A. Fournier, R. Biswas, G. A. Voth and A. Tokmakoff, Delocalization and stretch-bend mixing of the HOH bend in liquid water, J. Chem. Phys., 2017, 147(8), 084503 CrossRef PubMed
- L. Chuntonov, R. Kumar and D. G. Kuroda, Non-linear infrared spectroscopy of the water bending mode: direct experimental evidence of hydration shell reorganization?, Phys. Chem. Chem. Phys., 2014, 16(26), 13172–13181 RSC
- N. Huse, S. Ashihara, E. T. J. Nibbering and T. Elsaesser, Vibrational couplings and ultrafast relaxation of the O–H bending mode in liquid H2O, Chem. Phys. Lett., 2005, 404, 389 CrossRef CAS
- S. Y. Venyaminov and F. G. Prendergast, Water (H2O and D2O) molar absorptivity in the 1000–4000 cm−1 range and quantitative infrared spectroscopy of aqueous solutions, Anal. Biochem., 1997, 248(2), 234–245 CrossRef CAS PubMed
- C. T. Middleton, L. E. Buchanan, E. B. Dunkelberger and M. T. Zanni, Utilizing Lifetimes to Suppress Random Coil Features in 2D IR Spectra of Peptides, J. Phys. Chem. Lett., 2011, 2(18), 2357–2361 CrossRef CAS PubMed
- K. Adamczyk, N. Simpson, G. M. Greetham, A. Gumiero, M. A. Walsh, M. Towrie, A. W. Parker and N. T. Hunt, Ultrafast Infrared Spectroscopy Reveals Water-mediated Coherent Dynamics in an Enzyme Active Site, Chem. Sci., 2015, 6, 505–516 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01590f |
| This journal is © The Royal Society of Chemistry 2019 |