DOI:
10.1039/C7SC03404K
(Minireview)
Chem. Sci., 2018,
9, 546-559
Enantioselective synthesis of gem-diarylalkanes by transition metal-catalyzed asymmetric arylations (TMCAAr)
Received
4th August 2017
, Accepted 7th November 2017
First published on 8th November 2017
 Tao Jia | Tao Jia was born in Chengdu. He completed his undergraduate degree at Sichuan University in 2009. From 2011, he started to pursue his PhD degree at Chengdu Institute of Biology, Chinese Academy of Sciences, under the supervision of Prof. Jian Liao. His PhD project focused on the copper-catalyzed asymmetric difunctionalization of olefins. After completing his PhD he joined the Procter group at the University of Manchester as a postdoctoral researcher. |
 Peng Cao | Peng Cao completed his undergraduate degree at Sichuan Normal University in 2004 and received a PhD in chemistry at SIOC (Shanghai, China) with Prof. Yong Tang. He moved to a postdoctoral position at CaRLa (Heidelberg, Germany), a joint research laboratory of Heidelberg University and BASF. He then returned to China with a position at CIB (Chengdu, China). From 2017, he started his research career at Sichuan Normal University. |
 Jian Liao | Jian Liao completed his undergraduate degree at Shanghai Jiaotong University in 1994 and received his PhD in chemistry at the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences (CIOC, CAS) with Prof. Jingen Deng in 2002. He moved to a postdoctoral position at Penn State University (PA, USA) with Prof. Qing Wang (MatSE) and Prof. Xumu Zhang (department of Chemistry). He returned to China in early 2006 with a position at CIOC and started his research career. In 2010, he moved to CIB, CAS. His research interests are focused on chiral sulfoxide ligands and asymmetric catalysis. |
1 Introduction
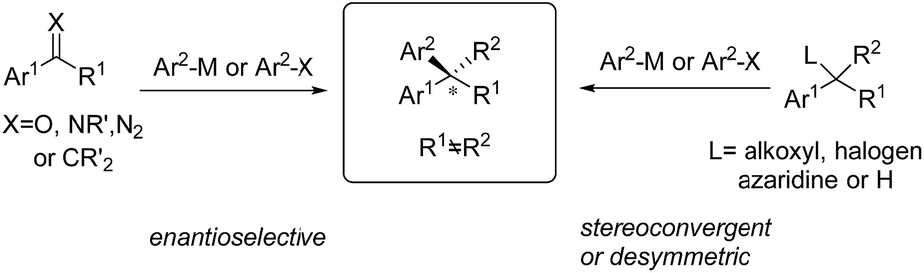
Chiral gem(1,1)-diaryl containing tertiary or quaternary stereogenic centers are present in many natural products and important pharmacophores that possess distinct bioactivities, such as anticancer, antidepressant and antifungal properties and so on.1 In most cases, a single enantiomer (R or S) of gem-diarylalkanes is therapeutically effective and most medicinal molecules are approved in the optically pure form. Thus, the development of effective methods to access enantiomerically enriched diaryl structural motifs will play a significant role in both academic and industrial settings. Enantiomerically pure drugs or their precursors are usually produced by the chiral kinetic resolution technique. However, access to 1,1-diarylalkanes with a high level of optical purity using this technique is challenging because little differentiates the two aryl groups installed on the stereogenic center electronically and sterically. This issue can be solved by asymmetric synthetic methods through either stereospecific or enantioselective transformations. In the last few decades, an array of catalytic enantioselective approaches towards the construction of nonracemic gem-diaryl compounds have been developed, including asymmetric Friedel–Crafts reactions, asymmetric aryl transfer reactions (arylations), asymmetric hydrogenation of 1,1-diarylalkenes, asymmetric C–H functionalization of enantiotropic diarylalkanes and so on. Among them, transition metal-catalyzed asymmetric arylations (TMCAArs), which install an aryl group onto the benzylic position of substrates in an enantioselective or stereoconvergent manner, represent the most powerful method. In this field, development of new reactions, chiral ligand families and metal complexes has enabled the precise construction of various chiral diaryl motifs, including dibenzyl alkanes and alkenes, 1,1-diarylmethanols, 1,1-diarylmethylamines and so on. To the best of our knowledge, TMCAAr for the synthesis of gem-diaryl compounds includes nucleophilic 1,2- or 1,4-additions of arylmetallic reagents across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and CC bonds; aryl cross-couplings to olefins, benzylic (pseudo)halides and aziridines; asymmetric aryl substitution reactions of allylic substrates; isotopic benzylic C–H arylation and so on (Scheme 1). These transformations feature a wide range of substrate scope, good functional group tolerance and the use of easily accessible feedstock chemicals. In contrast to conventional asymmetric methods, TMCAAr distinctively enable the assembly of both enantiomers through modulation of the reactants, instead of switching the absolute configuration of the chiral ligands.
O, CN and CC bonds; aryl cross-couplings to olefins, benzylic (pseudo)halides and aziridines; asymmetric aryl substitution reactions of allylic substrates; isotopic benzylic C–H arylation and so on (Scheme 1). These transformations feature a wide range of substrate scope, good functional group tolerance and the use of easily accessible feedstock chemicals. In contrast to conventional asymmetric methods, TMCAAr distinctively enable the assembly of both enantiomers through modulation of the reactants, instead of switching the absolute configuration of the chiral ligands.
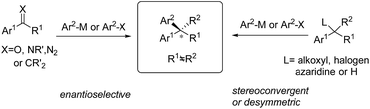 |
| | Scheme 1 Conceptual strategies of TMCAAr. | |
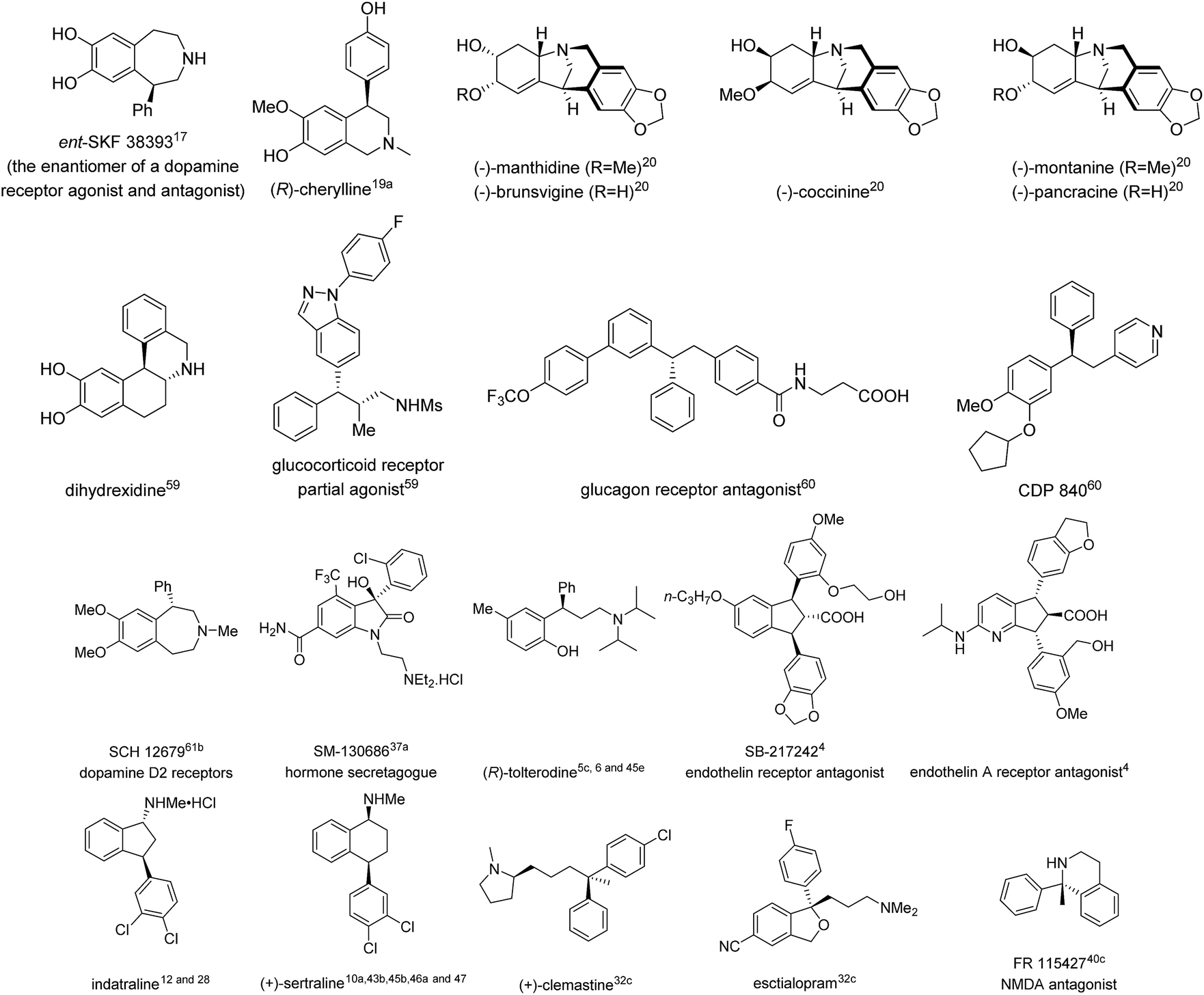
To date, there have been many excellent reviews summarizing various asymmetric arylation strategies,2 some of which consist of most of the approaches towards 1,1-diarylmethanols and 1,1-diarylmethylamines. Hence, this review will focus on TMCAAr for the synthesis of chiral gem-diarylalkanes whose alkyl moieties contain at least two carbons. Additionally, transition metal-catalyzed intramolecular arylation reactions for the construction of gem-diaryl containing fused rings are not included herein. In this review, the literature is organized according to the reaction type as well as the category of prochiral substrates. Furthermore, the natural products as well as bioactive compounds prepared in this review are also listed in Fig. 1.
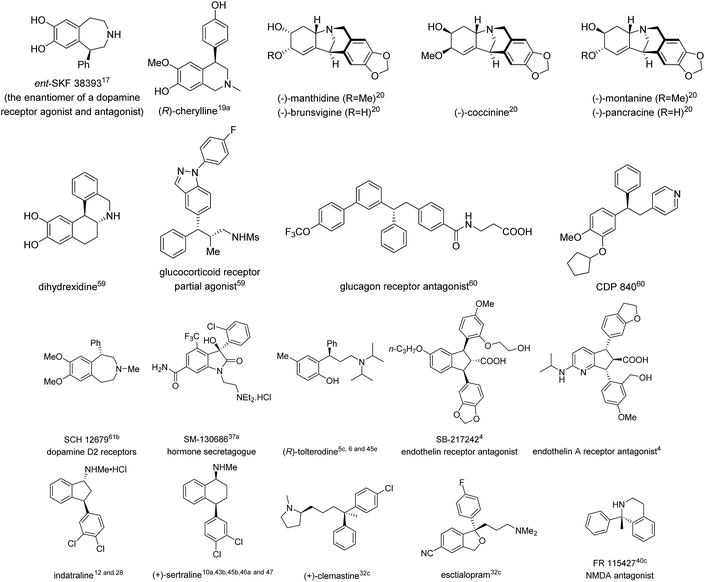 |
| | Fig. 1 Representative natural products and bioactive compounds synthesised using TMCAAr. | |
2 Asymmetric aryl addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C, CO and CN bonds
C, CO and CN bonds
Transition metal-catalyzed asymmetric aryl addition reactions to CC, CO and CN bonds represent a highly efficient method to construct tertiary or quaternary stereogenic centers, concomitant with the formation of Csp3–Csp2 bonds. These transformations are frequently used to prepare important chiral gem-diaryl containing compounds from activated styrene and aryl-substituted carbonyl substrates. Gem-diaryl stereogenic centers are generated in the key step of aryl migratory insertion across the unsaturated CC(O or N) bonds, followed by the hydrolysis or β-H elimination of the metal-binding intermediate (Scheme 2).
 |
| | Scheme 2 Conceptual strategies for conjugate or 1,2-arylation reactions. | |
2.1 Conjugate additions to unsaturated carbonyl compounds
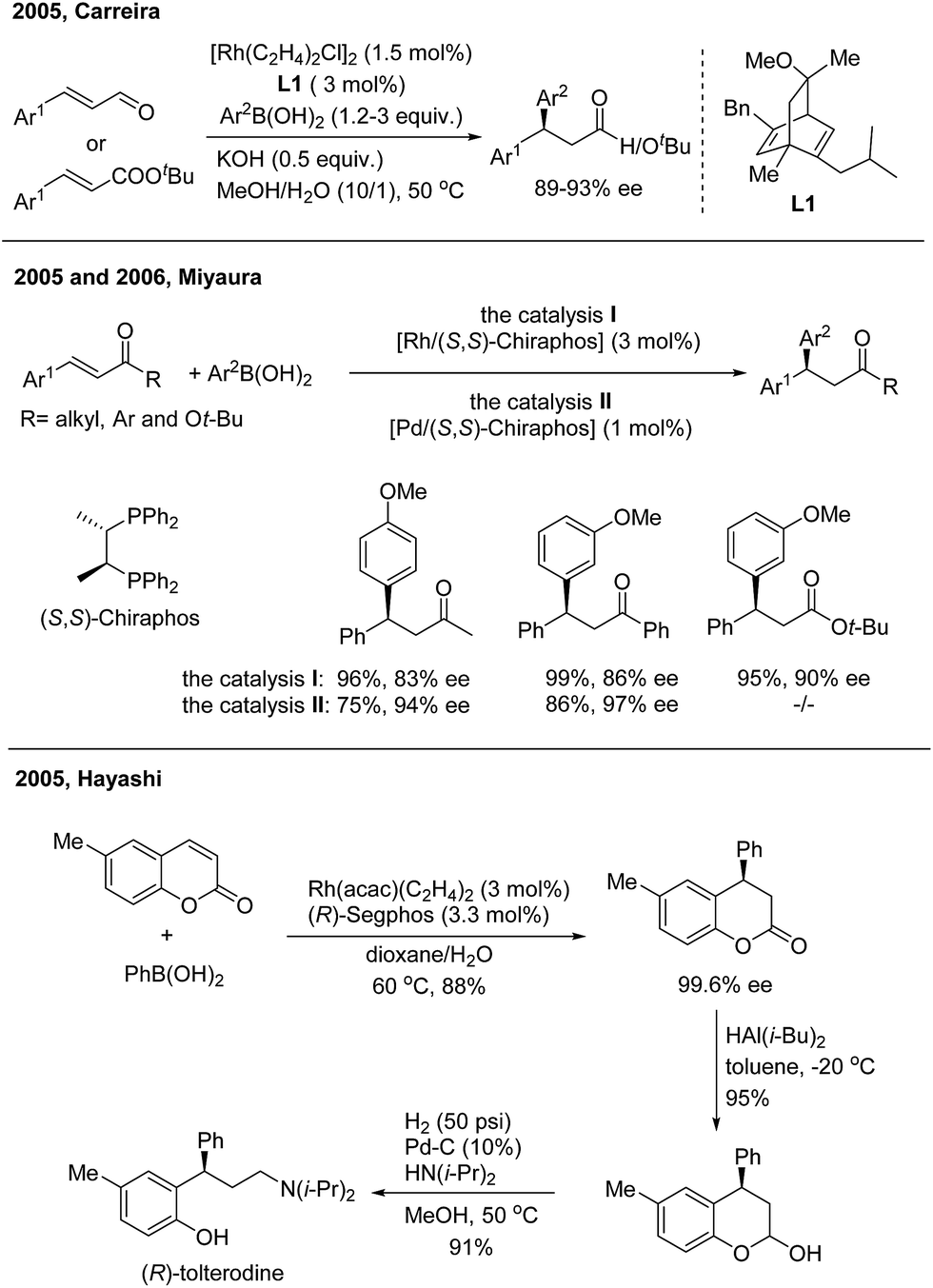
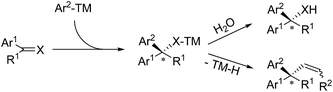
In 2005, Carreira successfully realized Rh(I)-catalyzed highly enantioselective 1,4-addition of arylboronic acids to β-aryl substituted unsaturated carbonyl derivatives using a carvone-derived chiral diene ligand (L1)3 (Scheme 3, top). Enantioenriched 3,3-diarylpropanals and tert-butyl 3,3-diarylpropanoates were afforded with 89–93% ee. Miyaura found that both Rh(I)4 and Pd(II)5 complexes with a (S,S)-chiraphos ligand are competent catalysts for TMCAAr of β-aryl-α,β-unsaturated ketones and esters (Scheme 3, middle). However, attempts to use indenone as the Michael acceptor gave only 20% yield of a nearly racemic product, the reason for which is yet unsolved. Hayashi found that coumarins undergo 1,4-arylation using a Rh/(R)-Segphos catalyst to provide enantiomerically pure 4-arylchroman-2-ones. The product, (R)-6-methyl-4-phenylchroman-2-one, was readily converted in two steps into (R)-tolterodine, an important urological drug6 (Scheme 3, bottom). For the 1,4-arylation of chalcones, Liao and coworkers demonstrated that a Rh(I) complex of sulfoxide–phosphine was an appropriate catalyst to afford chiral 1,3,3-triarylpropan-1-ones with up to 98% ee.7
 |
| | Scheme 3 The enantioselective 1,4-addition of arylboronic acids to β-aryl substituted unsaturated carbonyl derivatives. | |
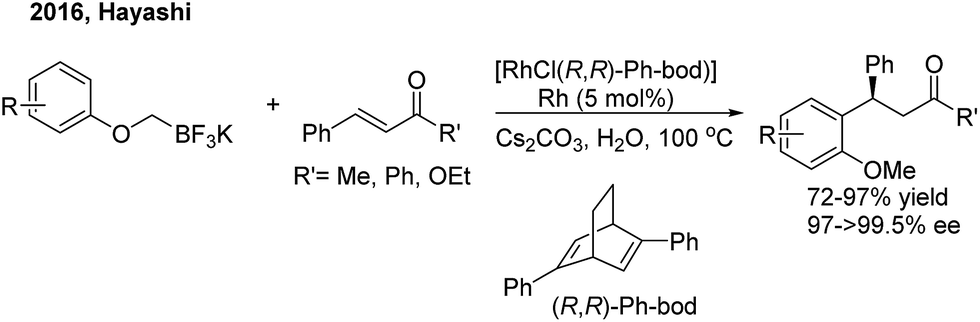
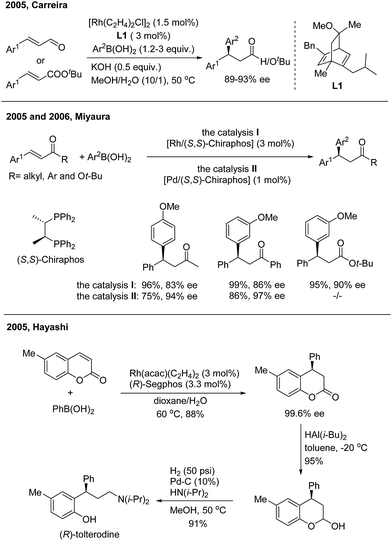
In 2016, Hayashi employed the 1,4-Rh migration/arylation strategy to realize conjugate addition of potassium aryloxymethyltrifluoroborates to α,β-unsaturated carbonyl compounds in the presence of a chiral diene–rhodium catalyst8 (Scheme 4). The desired β,β-diaryl ketones or esters were afforded in high yields with excellent enantioselectivities.
 |
| | Scheme 4 Rh-catalyzed migration/arylation of α,β-unsaturated carbonyl compounds. | |
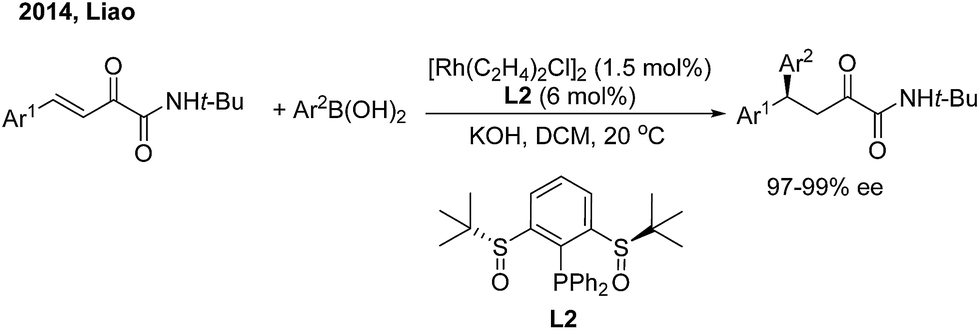
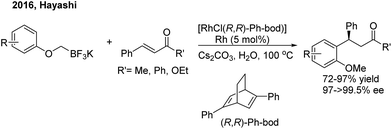
While chiral olefins or phosphines enable control of the 1,4-regioselectivity of α,β-unsaturated aldehyde/ketone/ester substrates, the conjugate arylation of β,γ-unsaturated α-keto carbonyl compounds is difficult to realize using these ligands, which only promote 1,2-addition.9 In 2014, Liao and co-workers10 demonstrated a highly regio- and enantioselective Rh-catalyzed 1,4-addition of arylboronic acids to β,γ-unsaturated α-keto carbonyl derivatives using a novel chiral sulfoxide–phosphine ligand (L2) (Scheme 5). Nonracemic γ,γ-diaryl, α-keto amides and esters were produced. The method was applied in the concise syntheses of sertraline and tetrahydroquinoline-2-carboxylamide.
 |
| | Scheme 5 Rh-catalyzed enantioselective 1,4-additon of β,γ-unsaturated-α-ketoamides. | |
In 2015, Kim and co-workers11 reported an elegant Rh-catalyzed asymmetric 1,4-addition of arylboronic acids to α,β-unsaturated N,N-dimethylsulfamoyl imino esters with a new bicyclic bridgehead phosphoramidite ligand (L3). Chiral (Z)-γ,γ-diaryl-α,β-dehydroamino esters were afforded with excellent yields and enantioselectivities (75–96% ee) (Scheme 6).
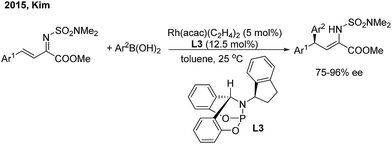 |
| | Scheme 6 Rh-catalyzed asymmetric 1,4-addition of arylboronic acids to α,β-unsaturated N,N-dimethylsulfamoyl imino esters. | |
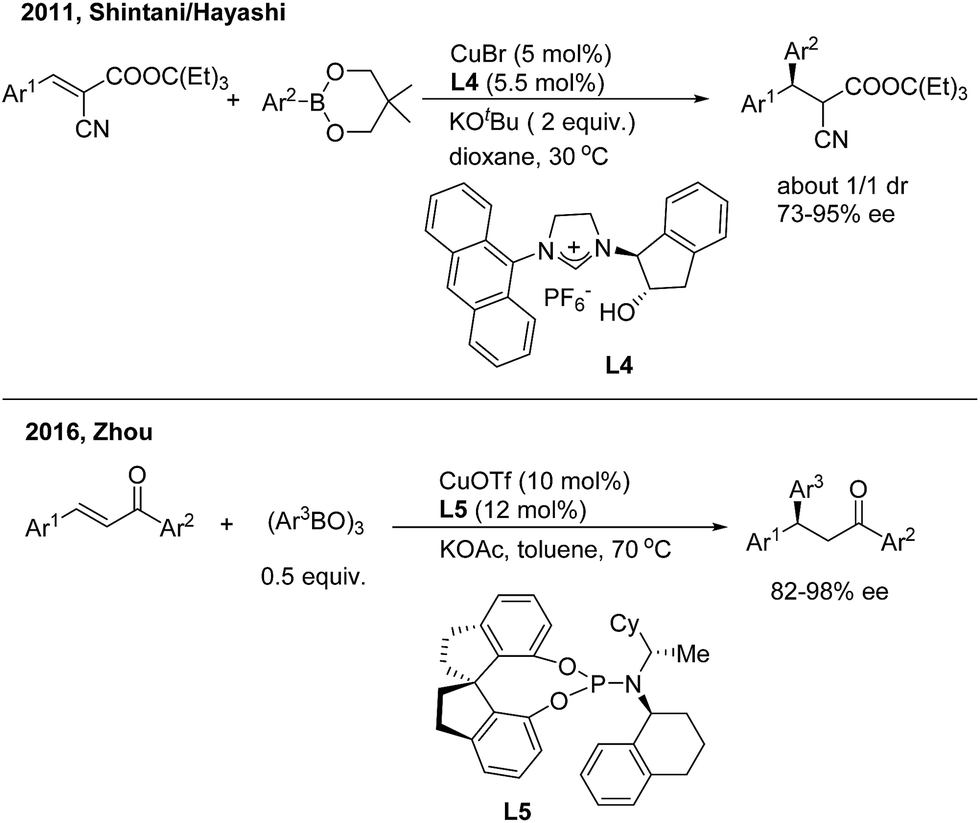
Asymmetric 1,4-addition of organoboronates to alkylidene cyanoacetates by copper catalysis was first demonstrated by Shintani/Hayashi12 using a chiral N-heterocyclic carbene ligand (L4) (Scheme 7, top). The transformation releases optically active 2-cyano-3,3-diaryl propanoates as a mixture of diastereomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The author conducted a series of stoichiometric reactions and indicated that only copper(I) mediated the catalytic cycle that consists of transmetalation/insertion/ligand exchange. Zhou and coworkers13 recently found that the chiral copper complex of phosphoramidite (L5) efficiently promoted the enantioselective 1,4-addition of chalcones with arylboroxines and a direct 1,4-insertion mechanism was proposed and supported by DFT calculations and natural-abundance 13C KIE experiments (Scheme 7, bottom).
:1). The author conducted a series of stoichiometric reactions and indicated that only copper(I) mediated the catalytic cycle that consists of transmetalation/insertion/ligand exchange. Zhou and coworkers13 recently found that the chiral copper complex of phosphoramidite (L5) efficiently promoted the enantioselective 1,4-addition of chalcones with arylboroxines and a direct 1,4-insertion mechanism was proposed and supported by DFT calculations and natural-abundance 13C KIE experiments (Scheme 7, bottom).
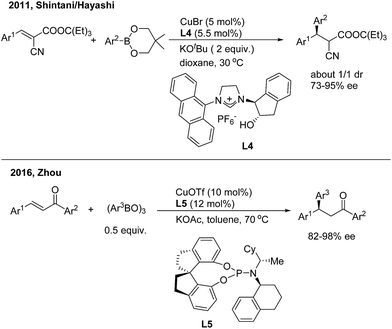 |
| | Scheme 7 Cu-catalyzed enantioselective 1,4-addition of α,β-unsaturated carbonyl derivatives. | |
2.2 Conjugate additions to nitro or sulfonyl olefins
In 2003, Minnaard/Feringa14 reported the first rhodium-catalyzed asymmetric addition of triphenyl boroxine to β-aryl nitroethylenes using chiral phosphoramidite ligands. Chiral 2,2-diaryl nitroethanes were afforded in excellent conversion and modest enantioselectivities. In this reaction, L6 could give 69% conversion but a low ee value (−7%), while the more sterically hindered L7 gave 28% ee but a low conversion (4%) (Scheme 8, top). Interestingly, the combination of L6 and L7 in a 1:1 ratio could improve the conversion (92%) as well as the enantioselectivity (31%). In 2013, Iuliano15 demonstrated that the deoxycholic acid-derived mono-Phos (L8) significantly improved the enantioselectivities (94–99%) as well as the yields (82–98%) of the desired products (Scheme 8, bottom).
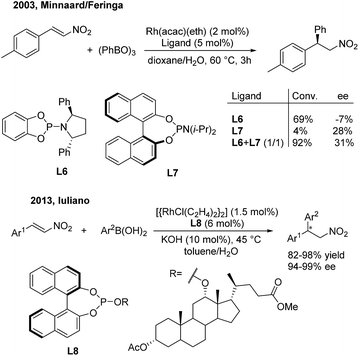 |
| | Scheme 8 Rh(I)-catalyzed conjugate addition of arylboron reagents to β-aryl nitroethylenes using chiral phosphoramidite and phosphite ligands. | |
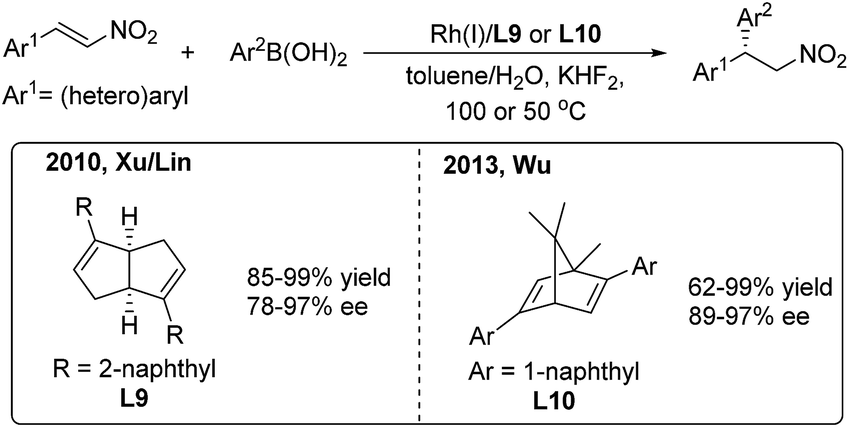
In 2010, Xu/Lin16 reported a highly enantioselective addition of organoboronic acids to nitroalkenes using a rhodium/chiral diene catalyst (Scheme 9, left). Enantioenriched 2,2-diaryl nitroalkanes were obtained with moderate to good enantioselectivities (78–97%) induced by the chiral [3.3.0]-diene ligand (L9). In 2013, Wu and coworkers17 used a chiral [2.2.1]-diene ligand (L10) in the arylation of nitroalkenes with high enantioselectivities (89–97%) (Scheme 9, right). The catalyst loading of the model reaction can be reduced to 0.1 mol%. Recently, Wu found that the amide-containing C1-symmetric [2.2.2]-diene ligand can promote the enantioselective reaction at room temperature.18
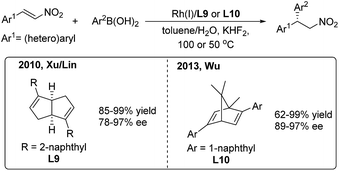 |
| | Scheme 9 Rh(I)-catalyzed conjugate addition of arylboronic acids to β-aryl nitroethylenes using chiral diene ligands. | |
In 2011, the highly efficient rhodium-catalyzed enantioselective addition of arylboronic acids to β-aryl and β-indolyl nitroalkenes was developed by the Liao group using the chiral sulfoxide–phosphine (SOP) ligand L1119 (Scheme 10, left). Moreover, the utility of this method was documented by Fan in the synthesis of montanine-type amaryllidaceae alkaloids.20 In 2012, Wan and co-workers21 reported a rhodium-catalyzed asymmetric addition of arylboronic acids to nitroalkenes using the chiral sulfoxide–olefin ligand L12 (Scheme 10, middle). They successfully enlarged the scope of the reaction to aryl, alkyl and heteroaryl nitroalkenes in one catalytic system. Recently, a P-chiral phosphine–olefin hybrid ligand L13 has been demonstrated by Sieber to efficiently promote this reaction22 (Scheme 10, right).
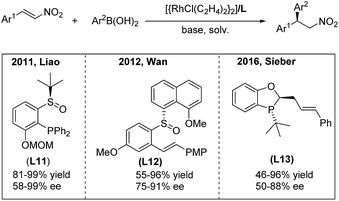 |
| | Scheme 10 Rh(I)-catalyzed conjugate addition of arylboronic acids to β-aryl nitroethylenes using chiral hybrid ligands. | |
Recently, Zhang and coworkers23 devoted themselves to developing a cheap and robust palladium catalysis system for conjugate aryl addition to nitroethylenes. When using iPr-IsoQuinox (L14) as a chiral ligand, enantioenriched 2,2-diaryl nitroalkanes can be produced in high yields and good enantioselectivities in air (Scheme 11).
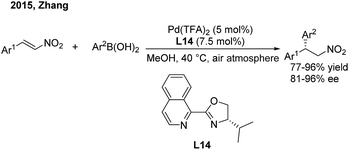 |
| | Scheme 11 Pd(II)-catalyzed conjugate addition of arylboronic acids to β-aryl nitroethylenes using chiral IsoQuinox ligands. | |
In contrast to the extensive studies on conjugate arylations of nitroalkene substrates, successful conjugate additions of sulfonyl olefins have rarely been reported.24 In 2012, Nishimura and Hayashi disclosed an elegant enantioselective addition of arylboronic acids to α,β-unsaturated sulfonyl compounds with a high enantioselectivity (97 to >99.5% ee)25 (Scheme 12, top). They demonstrated that the use of a diene ligand (L15) induces the protonation of the alkylrhodium intermediate faster than the β-H elimination process, thus selectively forming the addition product instead of the substitution product. Later on, Xu employed the chiral phosphine–olefin ligand (L16) in the same asymmetric reaction to achieve generally high yields and ee values26 (Scheme 12, bottom).
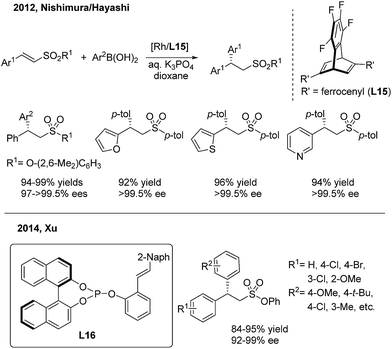 |
| | Scheme 12 Rh(I)-catalyzed conjugate aryl addition to α,β-unsaturated sulfonyl compounds. | |
2.3 Asymmetric Heck-type addition to unactivated styrenes
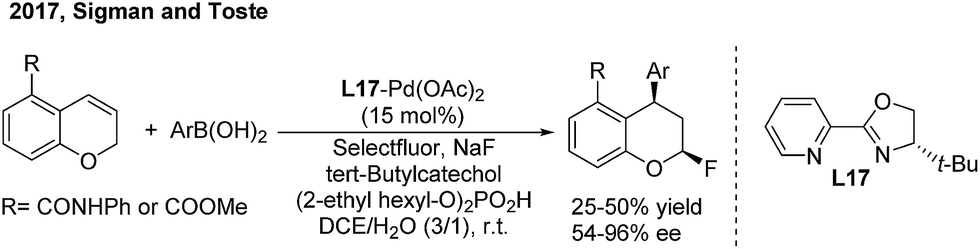
Recently, Sigman and Toste disclosed a palladium-catalyzed 1,3-regio and syn-diastereoselective arylfluorination of chromenes with arylboronic acids and selectfluor.27 With (S)-4-tert-butyl-2-(2-pyridyl)oxazoline (L17) as the chiral ligand, a wide spectrum of enantioenriched 2-fluoro-4-phenylchromanes were produced with up to 96% ee, albeit in moderate yields (Scheme 13). Meanwhile, an oxidative Heck-like mechanism was proposed based on the experimental studies in combination with computational and statistical analysis tools.
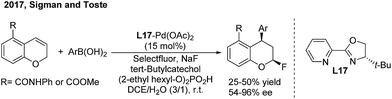 |
| | Scheme 13 Pd-catalyzed enantioselective 1,3-arylfluorination of chromenes. | |
In contrast to nucleophilic arylation, Gaunt recently reported a novel copper/bisoxazoline (L18)-catalyzed electrophilic arylation of allylic amides28 (Scheme 14). The protocol enables the asymmetric transfer of the electron-poor aryl group of diaryliodonium salts to the γ position of cinnamyl amides and provides chiral β,β-diaryl enamides with a high level of optical purity.
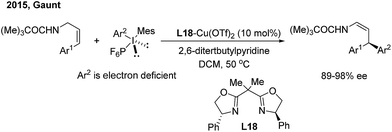 |
| | Scheme 14 Cu/BOX-catalyzed enantioselective electrophilic arylation of allylic amides. | |
2.4 1,2-Addition to arylketone and arylketimine derivatives
For ketone arylations, Fu reported the first enantioselective 1,2-addition of Ph2Zn to unactivated ketones catalyzed by 3-exo-(dimethylamino)isoborneol (L19)29 (Scheme 15). Although both aryl-alkyl and dialkyl ketones are reactive in the presence of MeOH, aryl-alkyl ketones gave better enantioselectivities (72–91%). Later on, Walsh and Yus/Ramón independently demonstrated that the easily accessible chiral isoborneolsulfonamide and camphorsulfonamide are good ligands.30 The catalytic system consisting of a combination of chiral diol ligands and Ti(OiPr)4 also promoted the enantioselective addition of Ph3Al, ArTi(OiPr)3 and ArMgBr to ketones, producing chiral diaryl alkyl carbinols.31
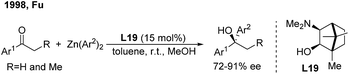 |
| | Scheme 15 The first catalytic asymmetric addition of organometallic reagents to ketones. | |
While arylboronic acids or derivatives are stable and frequently used in transition-metal catalyzed arylation reactions, their enantioselective additions to unactivated ketones are limited,32 probably due to the lack of effective chiral ligands. In 2011, Sakai/Korenaga32a discovered that electron-poor 2,6-bis(trifluoromethyl)-4-pyridyl (BFPy) phosphanes enable the acceleration of the Rh-catalyzed 1,2-addition of arylboronic acids to ketones. Accordingly, the enantioselective variant was obtained using BFPy derived biphep (L20) as the chiral ligand, albeit with only 39% ee (Scheme 16, left). Later on, a chiral diene ligand (L21) was demonstrated to promote the addition of arylborons to cyclic or acyclic arylketones with up to 68% ee32b (Scheme 16, middle). Recently, Deng and Tang32c reported a highly enantioselective addition of arylboroxines to simple aryl ketones catalyzed by the Rh/L22 complex, which produced a range of chiral diaryl alkyl carbinols with excellent ee (95–99%) (Scheme 16, right). The utility of this method was illustrated by the concise synthesis of the antidepressant drug escitalopram as well as the (+)-clemastine intermediate.
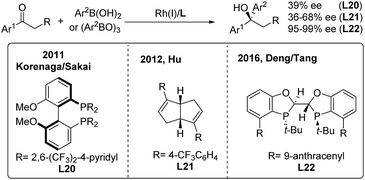 |
| | Scheme 16 Rh-catalyzed enantioselective 1,2-addition of α-aryl ketones. | |
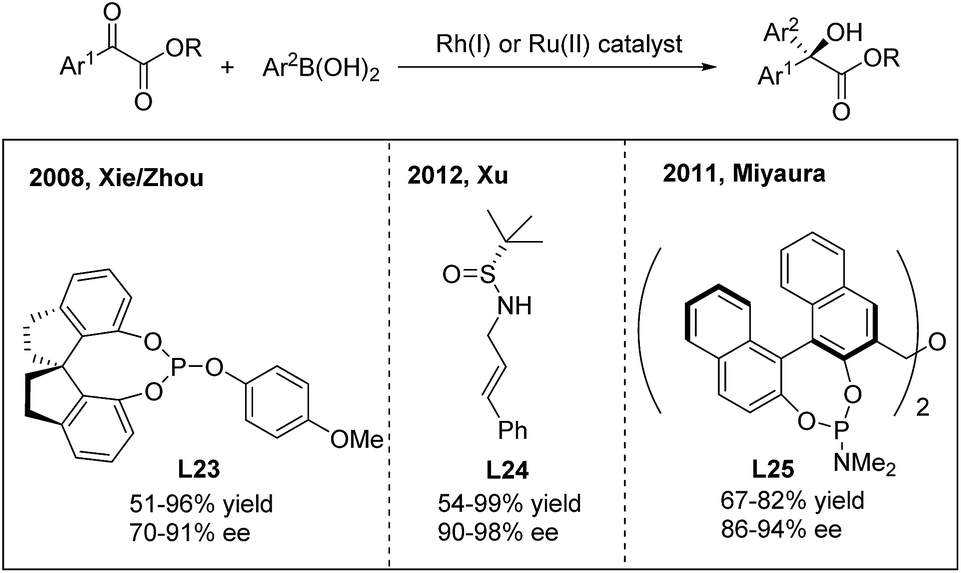
For the 1,2-arylation of activated ketones, Xie and Zhou developed the first highly enantioselective addition of arylboronic acids to α-ketoesters using a chiral Rh(I)-spirophosphite (L23) catalyst33a (Scheme 17). The method allows the synthesis of α-hydroxy-α-diaryl acetates with moderate to high ee (70–91%). A few years later, Xu and coworkers employed a simple N-(sulfinyl)cinnamylamine ligand (L24) in the arylation of α-ketoesters and α-diketones.33b Highly enantiopure α-hydroxy-α-diaryl acetates were afforded. In addition to rhodium catalysis, a ruthenium complex generated from [RuCl2(p-cymene)]2 and (R,R)-Me-BIPAM (L25) could also promote the asymmetric addition of arylboronic acids to α-ketoesters with high enantioselectivities.33c
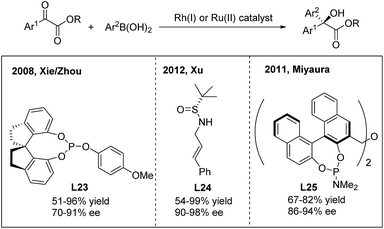 |
| | Scheme 17 Catalytic asymmetric 1,2-addition of arylboronic acids to α-ketoesters. | |
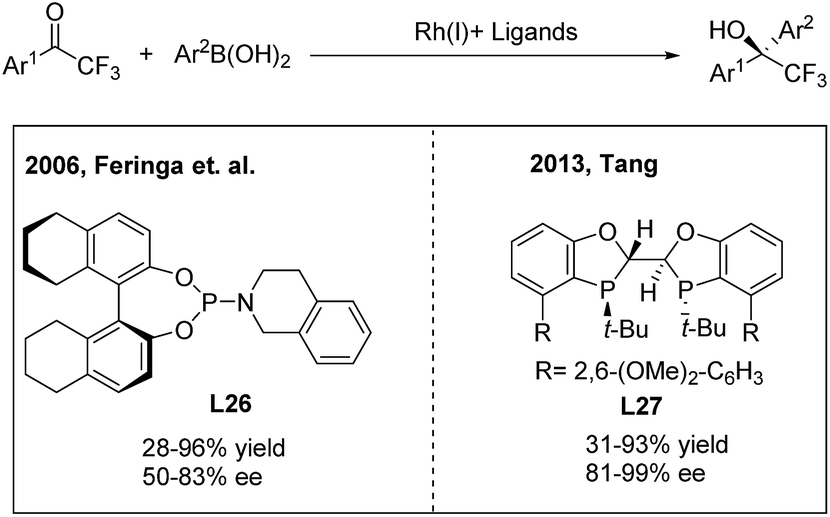
Due to the unique biological activities of fluorinated compounds, many scientists focused on the development of catalytic asymmetric methods for the synthesis of α-chiral CF3-containing compounds. However, enantioselective synthesis of diaryl trifluoroethanes through TMCAAr has rarely been reported.34 In 2006, Vries, Feringa and Minnaard34a reported the first asymmetric approach towards 2-hydroxy-2,2-diaryl trifluoroethanes through the rhodium(I)/phosphoramidite (L26) catalysed 1,2-addition of arylboronic acids to 2,2,2-trifluoroacetophenones (Scheme 18, left). In 2010, Iuliano and coworkers34b found that optically active 2-hydroxy-2,2-diaryl trifluoroethanes could also be produced using a deoxycholic acid derived monophosphite as the chiral ligand, albeit with moderate enantioselectivities. Recently, Tang34c demonstrated that a new C2-symmetrical chiral bisphosphorus ligand (L27) was highly effective in the Rh-catalyzed arylation of trifluoroacetophenones (Scheme 18, right).
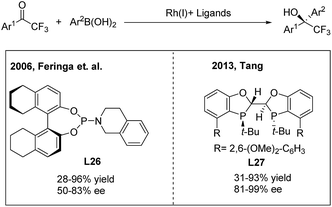 |
| | Scheme 18 Rh-catalyzed asymmetric 1,2-addition of arylboronic acids to 2,2,2-trifluoroacetophenones. | |
3-Hydroxy-3-aryl-2-oxindoles are important biologically active candidates in recent pharmaceutical studies. The Rh/Ir(I),35 Pd(II),36 Cu(I)37 and Ru(II)38-catalyzed enantioselective additions of arylboronic acids or esters to isatins and derivatives provide efficient methods for the synthesis of these compounds. In 2006, Shintani and Hayashi35a reported the first rhodium-catalyzed asymmetric addition of arylboronic acids to isatins using (R)-MeO-mop as a chiral ligand. A variety of optically active 3-hydroxy-3-aryl-2-oxindoles were afforded in good to excellent yields (49–98%) with high enantioselectivities (72–91%) (Scheme 19, left). Meanwhile, Vries, Feringa and Minnaard35b examined a chiral phosphoramidite in the Rh(I)-catalyzed arylation of NH isatin but obtained a poor enantioselectivity (55%). Liao and co-workers35c demonstrated that the chiral sulfoxide–phosphine (SOP) ligand L28 is also compatible with the NH isatin arylation process and gives an improved efficiency (Scheme 19, right).
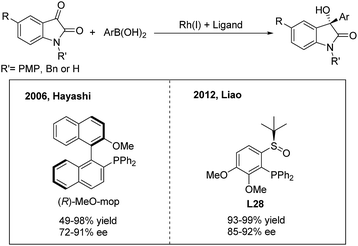 |
| | Scheme 19 Catalytic asymmetric 1,2-addition of arylboronic acids to isatins. | |
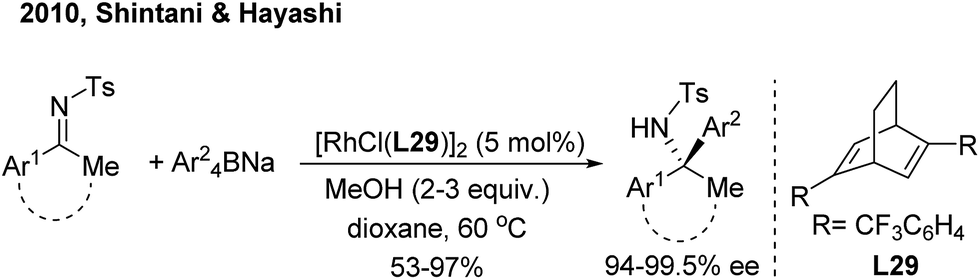
For ketimine arylation, Hayashi/Shintani39 pioneered the Rh-catalyzed asymmetric arylation of N-tosyl ketimines with sodium tetraarylborates by employing a chiral diene ligand (L29) (Scheme 20). The method is practically useful for the synthesis of chiral arylethanone-, indanone- and tetralone-derived amines.
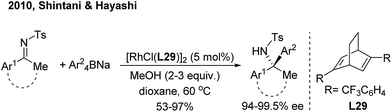 |
| | Scheme 20 Rh/diene-catalyzed enantioselective 1,2-arylation of ketimines. | |
Benzosultams containing a chiral α-amino acid unit and benzosulfamidates containing a CF3 group are attractive to organic and medicinal chemists. In 2013, Xu and coworkers developed a rhodium-catalyzed asymmetric addition of arylboronic acids to CF3- or alkoxycarbonyl-substituted cyclic ketimines.40a In this reaction, they utilized a chiral sulfur–olefin ligand (L30) which they developed themselves to provide such molecules in high yields with excellent enantioselectivities (Scheme 21). The analogous alkyl-substituted cyclic N-sulfonyl ketimines can also produce enantioenriched α-arylalkyl-substituted benzosulfamidates and benzosultams with excellent ee.40b,c These adducts allow for further transformation to versatile chiral α-diaryl alkylamines and some bioactive analogues.
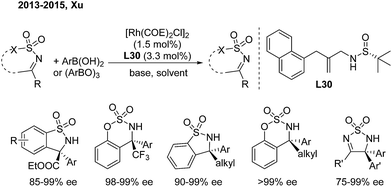 |
| | Scheme 21 Rh/sulfur–olefin-catalyzed enantioselective 1,2-addition of ketimines. | |
Pd-catalyzed enantioselective additions of arylboronic acids to cyclic N-sulfonyl ketimines were disclosed by Zhang41a and Lu/Hayashi42 using chiral pyridine-oxazoline (L32) and phosphine-oxazoline (L31) ligands, respectively (Scheme 22). Analogously, the enantioselective 1,2-addition of arylboronic acids to 3-ketimino oxindoles, by the Zhang group,41b was catalysed by a Pd(II)/L33 complex and enables the synthesis of enantioenriched 3-amino-3-aryl-2-oxindoles with high ee. Zhang also demonstrated the first Ni(II)-catalyzed asymmetric addition of arylboronic acids to cyclic imines using a tropos phosphine-oxazoline biphenyl ligand.41c
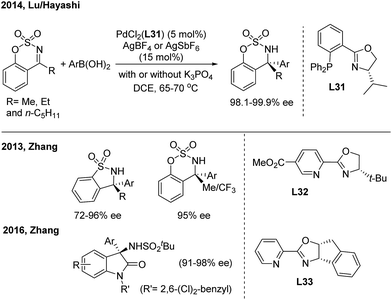 |
| | Scheme 22 Pd-catalyzed 1,2-addition of arylboronic acids to cyclic ketimines. | |
3 Asymmetric allylic arylation (AAAr) reactions
Asymmetric allylic arylation (AAAr) reactions of cinnamyl electrophiles are one of the most important strategies to access chiral 1,1-diarylpropene molecules. Although the transfer of aryl groups to γ-aryl substituted substrates resulted mainly in the achiral α product with palladium catalysis, the γ-regioselectivity is facile for iridium and copper catalysis. In 2007, Alexakis43 reported the first AAAr of arylzinc reagents to cinnamyl carbonates catalyzed by chiral Ir(I)/L34 complexes, which afforded the γ product with a high enantioselectivity but moderate γ-regioselectivity (Scheme 23, top). Recently, Fu44 realized the Ir/L35-catalyzed enantioselective arylation of racemic secondary allylic alcohols with aniline derivatives using BF3·Et2O (30 mol%) as the promoter. The formal SN2-substituted products, gem-diarylpropenes, were obtained with excellent ee (Scheme 23, bottom).
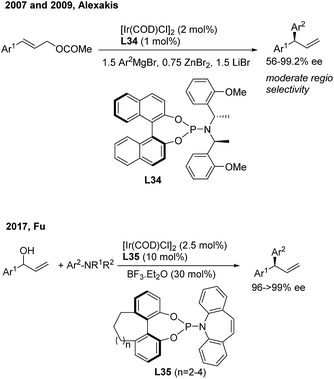 |
| | Scheme 23 Ir(I)-catalyzed AAAr using chiral phosphoramidite ligands. | |
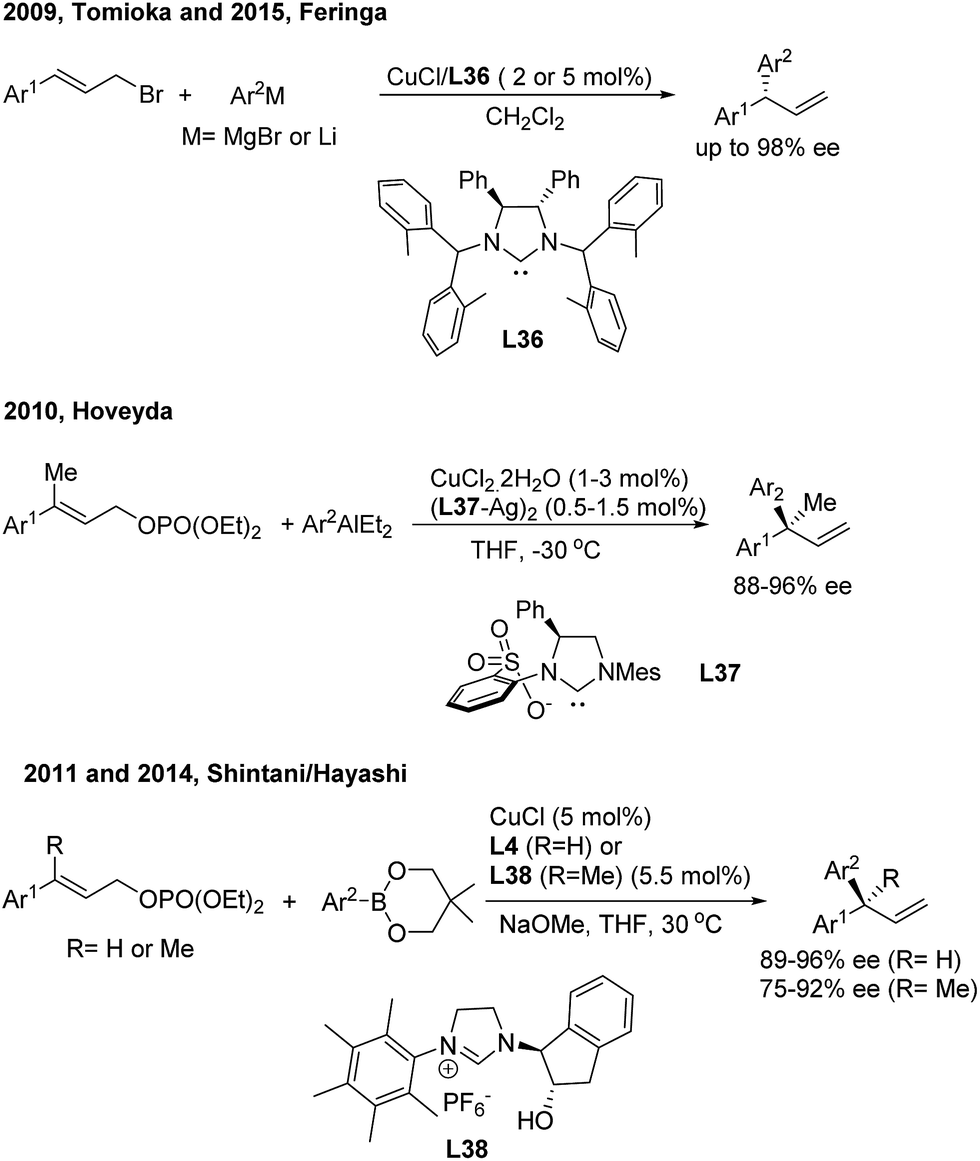
In the field of Cu(I)-catalyzed AAAr,45 chiral N-heterocyclic carbenes (L36, L37, L4 and L38) displayed remarkably high γ-regioselectivity as well as excellent enantioselectivity (Scheme 24). In these transformations, an array of arylmetallic (i.e. Mg, Li, Al and B) reagents can couple with cinnamyl bromides or carbonates to construct tertiary and quaternary gem-diarylmethine stereogenic centres.
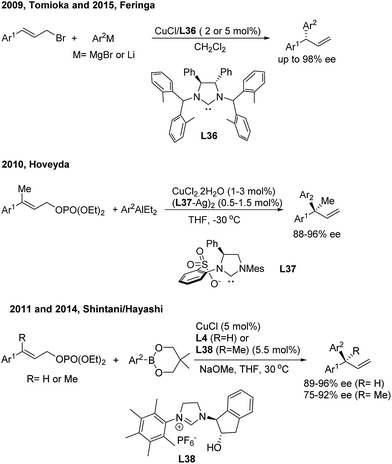 |
| | Scheme 24 Cu-catalyzed AAAr using chiral NHC–carbene ligands. | |
4 Asymmetric aryl cross-coupling to benzyl C–X bonds
4.1 Enantioconvergent cross-coupling reactions of racemic benzylic substrates
Transition metal-catalyzed stereospecific aryl cross-couplings allow for the transformation of secondary enantioenriched benzylic electrophiles or nucleophiles to 1,1-diarylalkane compounds. However, the catalytic enantioselective transformations of racemic benzylic compounds to enantiomerically enriched products still remain limited46 (Scheme 25).
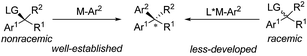 |
| | Scheme 25 The conceptual strategies for asymmetric cross-coupling of benzyl electrophiles and arylmetallic reagents. | |
In 2013, Fu46a developed the first successful enantioconvergent Negishi reactions of racemic benzylic mesylates with arylzinc reagents (Scheme 26). A broad range of 1,1-diarylalkanes with a high level of optical purity (81–95% ee) were produced when using a chiral nickel(II)/L39 catalyst. The method was applied to a gram-scale synthesis of (S)-sertraline tetralone from the available racemic 4-hydroxy-4-phenylbutanoate. Other efforts to attempt the enantioconvergent arylation of racemic benzylic chloride or trifluoroborate, also by Ni(II)/bis(oxazoline) catalysis, revealed the moderate stereoselectivity. In 2017, Reisman47 disclosed an elegant enantioselective Ni-catalyzed reductive cross-coupling between racemic secondary benzylic chlorides and (hetero)aryl iodides with 4-heptyl substituted bioxazoline (L40) as the chiral ligand (Scheme 27, top). In particular, 5-iodo-2-substituted pyridines were quite reactive under standard conditions and a wide range of 1,1-diarylalkanes were prepared with a generally high enantiopurity.
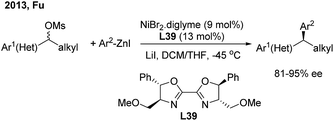 |
| | Scheme 26 Ni/BOX-catalyzed enantioconvergent Negishi reactions of racemic benzylic electrophiles. | |
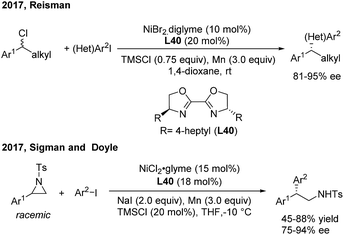 |
| | Scheme 27 Ni/BiOx-catalyzed stereoconvergent reductive cross-coupling of racemic styrenyl aziridines and aryl iodides. | |
The catalytic asymmetric α-arylation of styrenyl aziridines is one of the most important methods to access nonracemic 2,2-diarylethylamine derivatives. However, successful cross-coupling reactions rely on the stereospecific transformation of enantiomerically enriched aziridines. Recently, Sigman and Doyle developed an elegant Ni-catalyzed stereoconvergent reductive cross-coupling of racemic N-Ts aziridines and aryl iodides with Mn(0) as the reductant (Scheme 27, bottom). Intrigued by the discovery that enantiopure aziridine produces the corresponding amine as the racemate, they examined chiral amine- and phosphine-based ligands and found that 4-heptyl substituted bioxazoline (L40, BiOx) was the best ligand for the asymmetric transformation.48 An array of 2,2-diarylethylamines were afforded with high enantioselectivities and moderate to good yields.
4.2 Enantioselective arylation of benzyl C–H bonds
Transition metal-catalyzed asymmetric functionalizations of unreactive C–H bonds have been extensively investigated in recently years.49 The enantioselective arylation of benzylic C–H bonds enables direct access to optically active gem-diarylalkanes, wherein the precoordination of the metal catalyst with prochiral substrates in a bidentate or monodentate manner is usually demanded. In 2015, Duan50 firstly introduced a chiral phosphoric amide (L41) into the Pd(II)-catalyzed direct β-arylation of aminoquinoline derived aliphatic amides with aryl iodides. An array of β,β-diaryl carboxylic acid derivatives were produced in moderate to good enantiomeric ratios (Scheme 28, top). One year later, He and Chen51 investigated the enantioselective Γ-arylation of N-picolinic protected alkylamines with a combination of chiral phosphoric acid and Pd(II) catalysts. In the end, both high yields and enantioselectivities were obtained using a substoichiometric amount of chiral phosphoric acid (L42) under solvent-free conditions (Scheme 28, bottom).
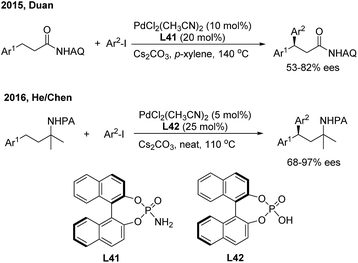 |
| | Scheme 28 Pd-catalyzed and phosphate-mediated enantioselective benzylic C–H arylation reactions. | |
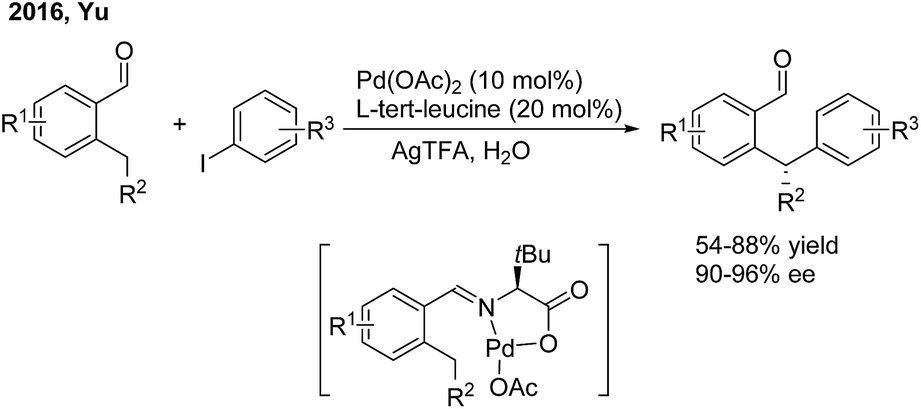
In 2016, Yu employed chiral α-amino acids as transient directing groups in the enantioselective benzylic C(sp3)–H arylation of benzaldehydes via the precoordination of Pd(II) with the in situ generated imine intermediate52 (Scheme 29). In the presence of 20 mol% L-tert-leucine, 10 mol% Pd(OAc)2 and 3 equiv. H2O, o-alkyl benzaldehydes reacted with a wide range of aryl iodides to produce 1,1-diaryl alkanes in moderate yields with high enantiomeric ratios.
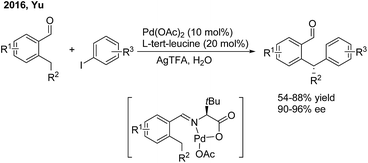 |
| | Scheme 29 Pd-catalyzed and amino acid-mediated enantioselective benzylic C–H arylation of benzaldehydes. | |
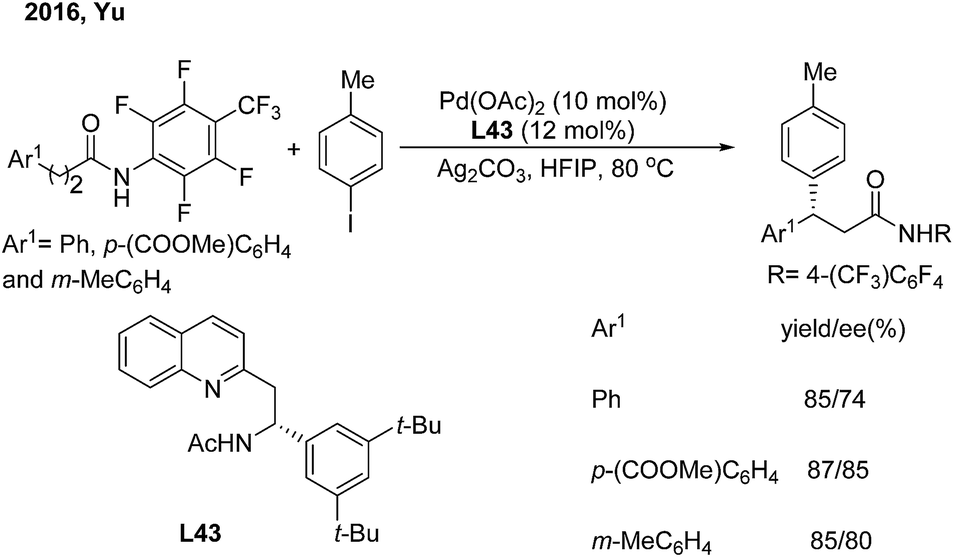
Soon afterwards, the same group employed chiral acetyl-protected aminoethyl quinoline (L43) ligands in the Pd(II)-catalyzed monodentate auxiliary directed C(sp3)–H arylation of aliphatic amides.53 This strategy enables the enantioselective construction of chiral 3,3-diaryl amides on subjecting 3-aryl propenamides to the catalytic system (Scheme 30).
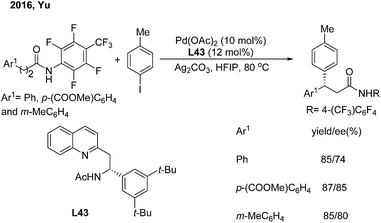 |
| | Scheme 30 Pd-catalyzed enantioselective β-arylation of aliphatic amides induced by chiral acetyl-protected aminoethyl quinoline ligands. | |
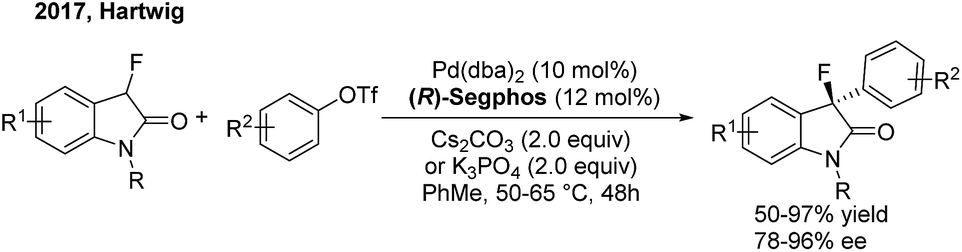
Although transition-metal catalysed asymmetric α-arylation of carbonyl compounds has been widely reported, the use of this method for the construction of gem-diarylalkanes has rarely been studied. In 2009, Buchwald54 disclosed a highly enantioselective Pd-catalyzed intermolecular C–C coupling of oxindoles and arylbromides using an axially chiral P-stereogenic ligand. Enantioenriched oxindoles containing a gem-diaryl quaternary center were afforded with 95–99% ee. Recently, Hartwig55 reported a palladium-catalyzed enantioselective α-arylation of α-fluorooxindoles with aryl triflates, using (R)-segphos as a chiral ligand. Enantioenriched 3-aryl-3-fluorooxindoles including a chiral quaternary center were obtained in high yields with excellent enantioselectivities (Scheme 31).
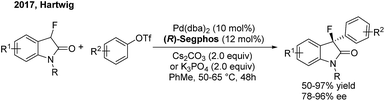 |
| | Scheme 31 Pd-catalyzed enantioselective α-arylation of α-fluorooxindoles. | |
4.3 Enantioselective arylation of benzyl carbene precursors
In 2015, Zhu and Zhou56 reported an enantioselective arylation of α-aryl-α-diazoacetates with anilines catalysed by dirhodium(II) trifluoroacetate and a chiral spiro phosphoric acid (SPA) (Scheme 32). Chiral α-diaryl acetates were produced in good yields (up to 95%) and high enantioselectivities (up to 97% ee). A step-wise reaction mechanism was proposed based on deuterium-labeling experiments. The Rh2(TFA)4 catalyst is responsible for the generation of the zwitterion (I). The 1,2-proton shift occurs via a proton shuttle model, which is mediated and stereochemically controlled by the chiral SPA (L44).
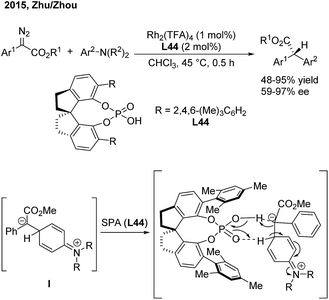 |
| | Scheme 32 Catalytic asymmetric arylation of α-aryl-α-diazoacetates with aniline derivatives. | |
5 Asymmetric aryl cross-coupling across CC bonds
Inspired by the efficiency of direct aryl-benzyl coupling, transition metal-catalyzed three-component cross-coupling reactions of olefins have been developed as an important and complementary method in the construction of gem-diaryl moieties. The conceptual strategy of this method involves the enantioselective formation from the styrene and stereospecific coupling of metal bound benzyl intermediates. These species are either nucleophilic or electrophilic depending on the nature of the initiator (M1-R′) (Scheme 33). In this regard, initiators include in situ generated Pd–H, Cu–H and Cu-Bpin and some electrophilic radicals (i.e. CF3 or amino radicals).
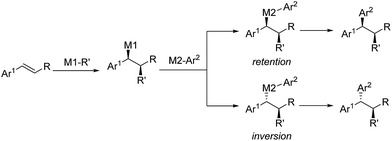 |
| | Scheme 33 The conceptual strategy for the three-component cross-couplings. | |
5.1 The net hydroarylation of styrene derivatives
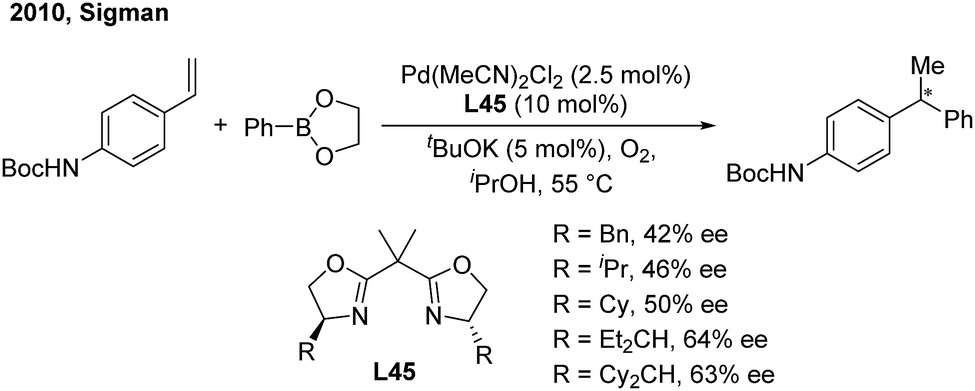
In 2010, the Sigman group initially studied the palladium-catalyzed asymmetric hydroarylation of styrenes with arylboron esters in the presence of an i-PrOH solvent and in an O2 atmosphere57 (Scheme 34). Through investigating chiral NHC and bisoxazoline ligands, they found that bisoxazoline ligands (L45) could give the best enantioselective induction (up to 64%).
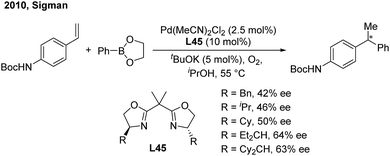 |
| | Scheme 34 Pd-catalyzed hydroarylation of styrenes using bisoxazoline ligands. | |
In 2016, Sigman and Toste developed an elegant enantioselective 1,1-diarylation method via double aryl cross-coupling to acrylates.58 They introduced the chiral anion phase transfer strategy into this diarylation transformation. Catalyzed by chiral phosphoric acid L46 and Pd2(dba)3, optically active 3,3-diaryl esters with a high enantioselectivity were produced (Scheme 35). The process possibly involves a stereospecific hydroarylation of a chiral benzyl cinnamate-associated Pd(II)–H complex intermediate.
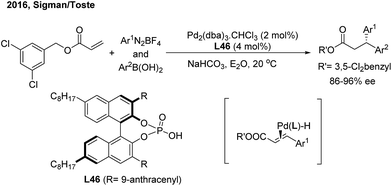 |
| | Scheme 35 Pd-catalyzed enantioselective three-component cross-coupling of benzyl acrylates, aryldiazonium salts and arylboronic acids. | |
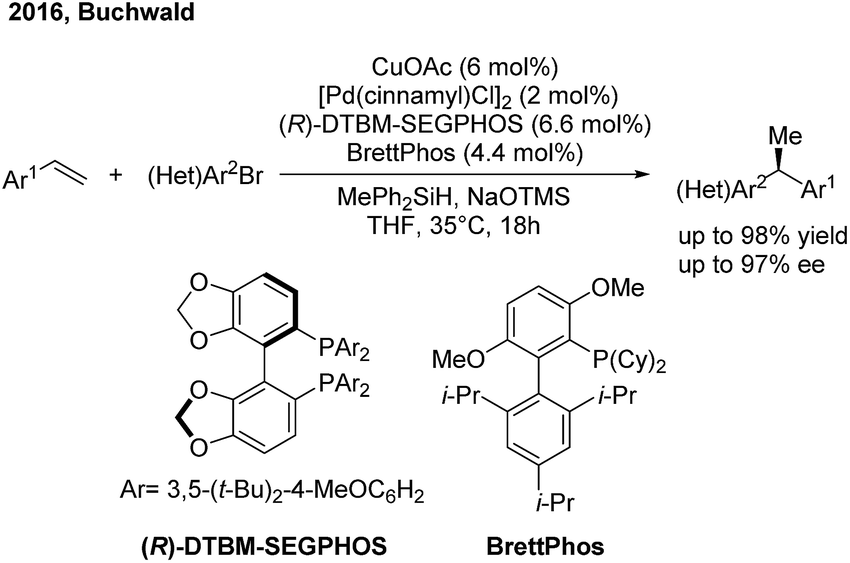
Recently the Buchwald group developed an alternative strategy to realize highly enantioselective hydroarylation of styrenes through CuH/Pd(0) cooperative catalysis59 (Scheme 36). In the presence of a chiral copper and achiral palladium catalyst, the three-component cross-coupling of styrenes, arylbromides and MePh2SiH proceeded smoothly to produce enantioenriched 1,1-diarylethanes in good yields with good to excellent enantioselectivities.
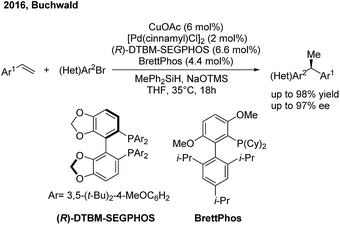 |
| | Scheme 36 The cooperative Cu/Pd-catalyzed enantioselective hydroarylation of styrenes. | |
5.2 Borylarylation of styrene derivatives
The Cu/Pd cooperatively catalysed enantioselective 1,2-arylboration of styrenes was also demonstrated by the Brown60 and Liao61 groups independently. Brown found that the chiral NHC–carbene ligand (L47) was compatible with a variety of 1,2-bisubstituted alkyenylarene substrates with excellent diastereo- and enantioselectivities. The syn/trans selectivity of the arylboration addition of 1,2-dihydronaphthalene was facilely switched by changing the achiral ligands on the Pd(II)-complex (Scheme 37).
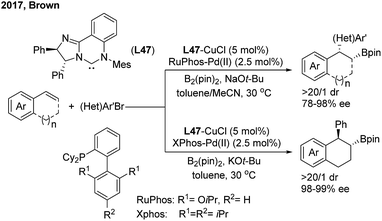 |
| | Scheme 37 The cooperative Cu/Pd-catalyzed enantioselective 1,2-arylboration of vinylarenes using chiral NHC–carbene ligands. | |
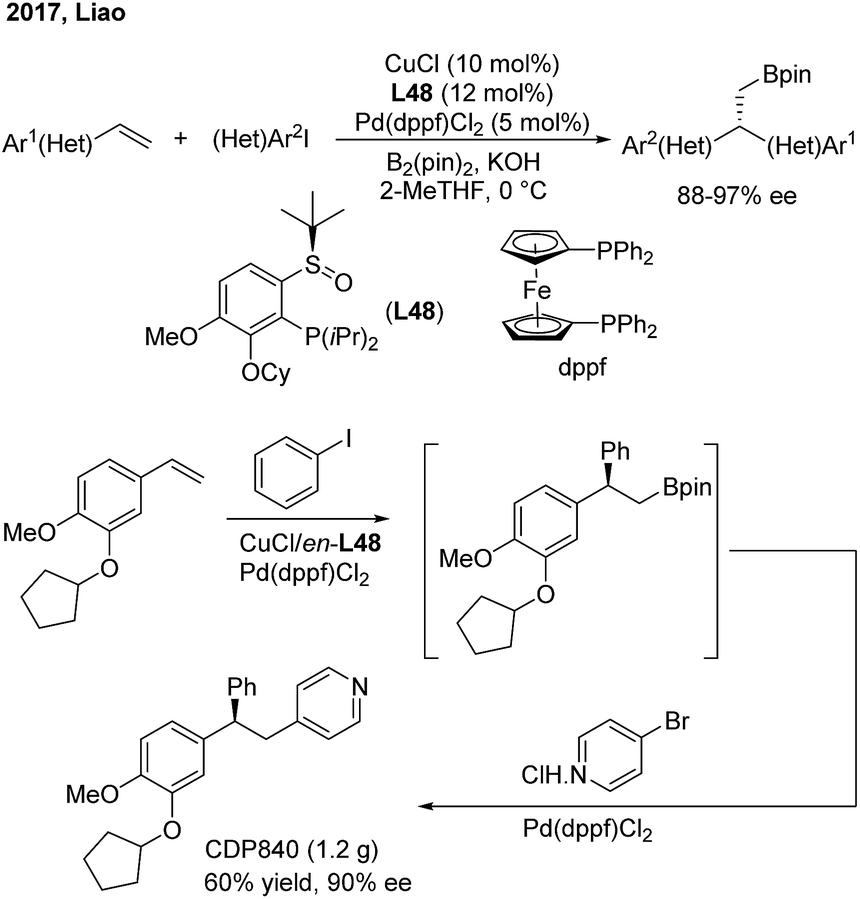
Liao and co-workers utilized a chiral sulfoxide–phosphine ligand (L48) to promote the Cu/Pd-catalyzed enantioselective arylboration of terminal vinylarenes with aryl iodides under mild conditions (Scheme 38). The method was particularly effective for the synthesis of chiral 2,2′-heteroaryl-aryl-ethylborates from either heteroaryl alkenes or heteroaryl iodides. Furthermore, the author merged this transformation and Suzuki–Miyaura coupling into a streamlined procedure for the modular synthesis of a series of important 1,1,2-triarylethane molecules, including CDP840.
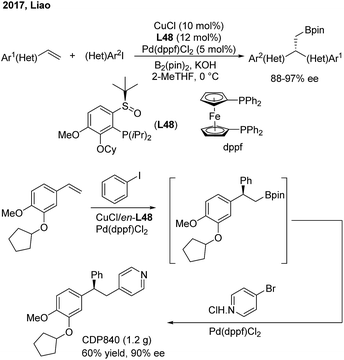 |
| | Scheme 38 Chiral triarylethane synthesis through the enantioselective arylboration of styrenes. | |
5.3 Trifluoromethyl and aminoarylation of styrene derivatives
Recently, Liu and coworkers developed a novel copper catalysis strategy to construct a gem-diarylmethine stereogenic center via enantioselective arylation of a secondary benzyl radical intermediate.62 In the presence of a Cu(I)/L49 catalyst, the enantioselective trifluoromethyl and aminoarylation of styrenes proceeded smoothly and afforded gem-diarylethane derivatives in moderate to high yields and with good ees (Scheme 39).
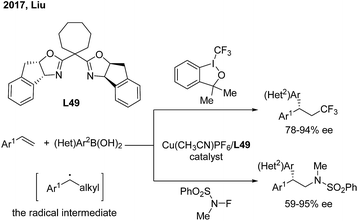 |
| | Scheme 39 Cu/BOX-catalyzed enantioselective trifluoromethyl and aminoarylation of styrenes. | |
6 Conclusion and perspective
In this review, a large number of TMCAAr reactions, which target the construction of chiral gem-diaryl tertiary or quaternary stereogenic centers, have been described. These reactions are versatile methods to site-selectively and stereochemically couple prochiral or racemic starting materials with various aryl reagents (almost always aryl metals or halides) to provide nonracemic gem-diarylalkane compounds. Due to distinguishing features including the wide range of substrate scope, good functional group tolerance and the use of easily accessible substrates, the related methodologies have received increasing interest from synthetic and pharmaceutical chemists, aiding the latter in synthesising medicinal molecules in a highly efficient manner.
Predictably, the development of strategies that transform commercially available feedstocks to highly valuable gem-diaryl molecules has recently been highlighted and will be the focus of continuous research. The present methods, including hydro- or borylarylation, direct benzyl C–H bond arylation and so on, need improvement of the efficiency (i.e. enantioselectivities and catalyst loadings) and broadening of the substrate scope, and their use in the construction of quaternary carbon stereogenic centres remains challenging.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the NSFC (No. 21472184, 21272226, 21572218 and 21402186) for financial support.
Notes and references
- D. Ameen and T. J. Snape, Med. Chem. Commun., 2013, 4, 893 RSC.
-
(a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed;
(b) F. Schmidt, R. T. Stemmler, J. Rudolph and C. Bolm, Chem. Soc. Rev., 2006, 35, 454 CAS;
(c) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093 RSC;
(d) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95 CrossRef CAS;
(e) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed.
-
(a) J.-F. Paquin, C. Defieber, C. R. J. Stephenson and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 10850 CrossRef CAS PubMed;
(b) J.-F. Paquin, C. R. J. Stephenson, C. Defieber and E. M. Carreira, Org. Lett., 2005, 7, 3821 CrossRef CAS PubMed;
(c) C. Defieber, J.-F. Paquin, S. Serna and E. M. Carreira, Org. Lett., 2004, 6, 3873 CrossRef CAS PubMed.
- T. Itoh, T. Mase, T. Nishikata, T. Iyama, H. Tachikawa, Y. Kobayashi, Y. Yamamoto and N. Miyaura, Tetrahedron, 2006, 62, 9610 CrossRef CAS.
-
(a) T. Nishikata, Y. Yamamoto, I. D. Gridnev and N. Miyaura, Organometallics, 2005, 24, 5025 CrossRef CAS;
(b) T. Nishikata, Y. Yamamoto and N. Miyaura, Adv. Synth. Catal., 2007, 349, 1759 CrossRef CAS;
(c) K. Kobayashi, T. Nishikata, Y. Yamamoto and N. Miyaura, Bull. Chem. Soc. Jpn., 2008, 81, 1019 CrossRef CAS.
- G. Chen, N. Tokunaga and T. Hayashi, Org. Lett., 2005, 7, 2285 CrossRef CAS PubMed.
- G. Chen, J. Xing, P. Cao and J. Liao, Tetrahedron, 2012, 68, 5908 CrossRef CAS.
- J. Ming and T. Hayashi, Org. Lett., 2016, 18, 6452 CrossRef CAS PubMed.
-
(a) H. F. Duan, J. H. Xie, X. C. Qiao, L. X. Wang and Q. L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4351 CrossRef CAS PubMed;
(b) X. Feng, Y. Nie, J. Yang and H. Du, Org. Lett., 2012, 14, 624 CrossRef CAS PubMed.
-
(a) J. Wang, M. Wang, P. Cao, L. Jiang, G. Chen and J. Liao, Angew. Chem., Int. Ed., 2014, 53, 6673 CrossRef CAS PubMed;
(b) J. Wang, B. Wang, P. Cao and J. Liao, Tetrahedron Lett., 2014, 55, 3450 CrossRef CAS.
- A. Lee and H. Kim, J. Am. Chem. Soc., 2015, 137, 11250 CrossRef CAS PubMed.
- K. Takatsu, R. Shintani and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 5548 CrossRef CAS PubMed.
- C. Wu, G. Yue, C. D.-T. Nielsen, K. Xu, H. Hirao and J. Zhou, J. Am. Chem. Soc., 2016, 138, 742 CrossRef CAS PubMed.
-
(a) A. Duursma, R. Hoen, J. Schuppan, R. Hulst, A. J. Minnaard and B. L. Feringa, Org. Lett., 2003, 5, 3111 CrossRef CAS PubMed;
(b) A. Duursma, D. Peña, A. J. Minnaard and B. L. Feringa, Tetrahedron: Asymmetry, 2005, 16, 1901 CrossRef CAS.
- V. R. Jumde and A. Iuliano, Adv. Synth. Catal., 2013, 355, 3475 CrossRef CAS.
- Z. Q. Wang, C. G. Feng, S. S. Zhang, M. H. Xu and G. Q. Lin, Angew. Chem., Int. Ed., 2010, 49, 5780 CrossRef CAS PubMed.
- K. C. Huang, B. Gopula, T. S. Kuo, C. W. Chiang, P. Y. Wu, J. P. Henschke and H. L. Wu, Org. Lett., 2013, 15, 5730 CrossRef CAS PubMed.
- R. Li, Z. Wen and N. Wu, Org. Biomol. Chem., 2016, 14, 11080 CAS.
-
(a) F. Lang, G. Chen, L. Li, J. Xing, F. Han, L. Cun and J. Liao, Chem.–Eur. J., 2011, 17, 5242 CrossRef CAS PubMed;
(b) J. Xing, G. Chen, P. Cao and J. Liao, Eur. J. Org. Chem., 2012, 2012, 1230 CrossRef CAS.
- X. Bao, Y.-X. Cao, W.-D. Chu, H. Qu, J.-Y. Du, X.-H. Zhao, X.-Y. Ma, C.-T. Wang and C.-A. Fan, Angew. Chem., Int. Ed., 2013, 52, 14167 CrossRef CAS PubMed.
- F. Xue, D. Wang, X. Li and B. Wan, J. Org. Chem., 2012, 77, 3071 CrossRef CAS PubMed.
- J. D. Sieber, D. Rivalti, M. A. Herbage, J. T. Masters, K. R. Fandrick, D. R. Fandrick, N. Haddad, H. Lee, N. K. Yee, B. F. Gupton and C. H. Senanayakea, Org. Chem. Front., 2016, 3, 1149 RSC.
- Q. He, F. Xie, G. Fu, M. Quan, C. Shen, G. Yang, I. D. Gridnev and W. Zhang, Org. Lett., 2015, 17, 2250 CrossRef CAS PubMed.
-
(a) P. Mauleoón and J. C. Carretero, Org. Lett., 2004, 6, 3195 CrossRef PubMed;
(b) P. Mauleoón and J. C. Carretero, Chem. Commun., 2005, 4961 RSC;
(c) P. Mauleoón, I. Alonso, M. R. Rivero and J. C. Carretero, J. Org. Chem., 2007, 72, 9924 CrossRef PubMed.
- T. Nishimura, Y. Takiguchi and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 9086 CrossRef CAS PubMed.
- Y.-N. Yu and M.-H. Xu, Acta Chim. Sin., 2014, 72, 815 CrossRef CAS.
- R. T. Thornbury, V. Saini, T. de A. Fernandes, C. B. Santiago, E. P. A. Talbot, M. S. Sigman, J. M. McKenna and F. D. Toste, Chem. Sci., 2017, 8, 2890 RSC.
- E. Cahard, H. P. J. Male, M. Tissot and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 7986 CrossRef CAS PubMed.
- P. I. Dosa and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 445 CrossRef CAS.
-
(a) C. García and P. J. Walsh, Org. Lett., 2003, 5, 3641 CrossRef PubMed;
(b) V. J. Forrat, O. Prieto, D. J. Ramón and M. Yus, Chem.–Eur. J., 2006, 12, 4431 CrossRef CAS PubMed;
(c) V. J. Forrat, D. J. Ramón and M. Yus, Tetrahedron: Asymmetry, 2009, 20, 65 CrossRef CAS.
-
(a) C.-A. Chen, K.-H. Wu and H.-M. Gau, Angew. Chem., Int.Ed., 2007, 46, 5373 CrossRef CAS PubMed;
(b) C.-A. Chen, K.-H. Wu and H.-M. Gaua, Adv. Synth. Catal., 2008, 350, 1626 CrossRef CAS;
(c) K.-H. Wu, Y.-Y. Kuo, C.-A. Chen, Y.-L. Huang and H.-M. Gaua, Adv. Synth. Catal., 2013, 355, 1001 CrossRef CAS;
(d) E. F. Mateos, B. Maciá and M. Yus, Eur. J. Org. Chem., 2014, 6519 CrossRef.
-
(a) T. Korenaga, A. Ko, K. Uotani, Y. Tanaka and T. Sakai, Angew. Chem., Int. Ed., 2011, 50, 10703 CrossRef CAS PubMed;
(b) Y.-X. Liao, C.-H. Xing and Q.-S. Hu, Org. Lett., 2012, 14, 1544 CrossRef CAS PubMed;
(c) L. Huang, J. Zhu, G. Jiao, Z. Wang, X. Yu, W.-P. Deng and W. Tang, Angew. Chem., Int. Ed., 2016, 55, 4527 CrossRef CAS PubMed.
-
(a) H. F. Duan, J. H. Xie, X. C. Qiao, L. X. Wang and Q. L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4351 CrossRef CAS PubMed;
(b) T. S. Zhu, S. S. Jin and M. H. Xu, Angew. Chem., Int. Ed., 2012, 51, 780 CrossRef CAS PubMed;
(c) Y. Yamamoto, T. Shirai, M. Watanabe, K. Kurihara and N. Miyaura, Molecules, 2011, 16, 5020 CrossRef CAS PubMed.
-
(a) S. L. Martina, R. B. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2006, 4093 RSC;
(b) V. R. Jumde, S. Facchetti and A. Iuliano, Tetrahedron: Asymmetry, 2010, 21, 2775 CrossRef CAS;
(c) R. Luo, K. Li, Y. Hu and W. Tang, Adv. Synth. Catal., 2013, 355, 1297 CrossRef CAS;
(d) V. Valdivia, I. Fernández and N. Khiar, Org. Biomol. Chem., 2014, 12, 1211 RSC.
-
(a) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS PubMed;
(b) P. Y. Toullec, R. B. C. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Org. Lett., 2006, 8, 2715 CrossRef CAS PubMed;
(c) J. Gui, G. Chen, P. Cao and J. Liao, Tetrahedron: Asymmetry, 2012, 23, 554 CrossRef CAS;
(d) X. Feng, Y. Nie, L. Zhang, J. Yang and H. Du, Tetrahedron Lett., 2014, 55, 4581 CrossRef CAS;
(e) Y. Zhuang, Y. He, Z. Zhou, W. Xia, C. Cheng, M. Wang, B. Chen, Z. Zhou, J. Pang and L. Qiu, J. Org. Chem., 2015, 80, 6968 CrossRef CAS PubMed;
(f) C. S. Marques and A. J. Burke, ChemCatChem, 2016, 8, 3518 CrossRef.
-
(a) H. Lai, Z. Huang, Q. Wu and Y. Qin, J. Org. Chem., 2008, 74, 283 CrossRef PubMed;
(b) Z. Liu, P. Gu, M. Shi, P. McDowell and G. Li, Org. Lett., 2011, 13, 2314 CrossRef CAS PubMed;
(c) Q. Li, P. Wan, S. Wang, Y. Zhuang, L. Li, Y. Zhou, Y. He, R. Cao, L. Qiu and Z. Zhou, Appl. Catal., A, 2013, 458, 201 CrossRef CAS.
-
(a) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946 CrossRef CAS PubMed;
(b) R. Shintani, K. Takatsu and T. Hayashi, Chem. Commun., 2010, 46, 6822 RSC.
- Y. Yamamoto, M. Yohda, T. Shirai, H. Ito and N. Miyaura, Chem.–Asian J., 2012, 7, 2446 CrossRef CAS PubMed.
- R. Shintani, M. Takeda, T. Tsuji and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 13168 CrossRef CAS PubMed.
-
(a) H. Wang, T. Jiang and M. H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed;
(b) H. Wang, Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 3962 CrossRef CAS PubMed;
(c) T. Jiang, Z. Wang and M.-H. Xu, Org. Lett., 2015, 17, 528 CrossRef CAS PubMed.
-
(a) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540 CrossRef CAS PubMed;
(b) Q. He, L. Wu, X. Kou, N. Butt, G. Yang and W. Zhang, Org. Lett., 2016, 18, 288 CrossRef CAS PubMed;
(c) M. Quan, L. Tang, J. Shen, G. Yang and W. Zhang, Chem. Commun., 2017, 53, 609 RSC.
- C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936 CrossRef CAS PubMed.
-
(a) A. Alexakis, S. E. Hajjaji, D. Polet and X. Rathgeb, Org. Lett., 2007, 9, 3393 CrossRef CAS PubMed;
(b) D. Polet, X. Rathgeb, C. A. Falciola, J.-B. Langlois, S. E. Hajjaji and A. Alexakis, Chem.–Eur. J., 2009, 15, 1205 CrossRef CAS PubMed.
- H. Tian, P. Zhang, F. Peng, H. Yang and H. Fu, Org. Lett., 2017, 19, 3775 CrossRef CAS PubMed.
-
(a) K. B. Selim, K. Yamada and K. Tomioka, Chem. Commun., 2008, 5140 RSC;
(b) K. B. Selim, Y. Matsumoto, K. Yamada and K. Tomioka, Angew. Chem., Int. Ed., 2009, 48, 8733 CrossRef CAS PubMed;
(c) F. Gao, Y. Lee, K. Mandai and A. H. Hoveyda, Angew. Chem., Int. Ed., 2010, 49, 8370 CrossRef CAS PubMed;
(d) R. Shintani, K. Takatsu, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 8656 CrossRef CAS PubMed;
(e) S. Guduguntla, V. Hornillos, R. Tessier, M. Fanñanaás-Mastral and B. L. Feringa, Org. Lett., 2016, 18, 252 CrossRef CAS PubMed.
-
(a) H.-Q. Do, E. R. R. Chandrashekar and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 16288 CrossRef CAS PubMed;
(b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed;
(c) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC.
- K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684 CrossRef CAS PubMed.
- B. P. Woods, M. Orlandi, C. Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688 CrossRef CAS PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed.
- S.-B. Yan, S. Zhang and W.-L. Duan, Org. Lett., 2015, 17, 2458 CrossRef CAS PubMed.
- H. Wang, H.-R. Tong, G. He and G. Chen, Angew. Chem., Int. Ed., 2016, 55, 15387 CrossRef CAS PubMed.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed.
- G. Chen, W. Gong, Z. Zhuang, M. S. Andrä, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023 CrossRef CAS PubMed.
- A. M. Taylor, R. A. Altman and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 9900 CrossRef CAS PubMed.
- Y.-S. Jin, M. Chen, S.-Z. Ge and J. F. Hartwig, Org. Lett., 2017, 19, 1390 CrossRef CAS PubMed.
- B. Xu, M. L. Li, X. D. Zuo, S. F. Zhu and Q. L. Zhou, J. Am. Chem. Soc., 2015, 137, 8700 CrossRef CAS PubMed.
- S. M. Podhajsky, Y. Iwai, A. Cook-Sneathen and M. S. Sigman, Tetrahedron, 2011, 67, 4435 CrossRef CAS PubMed.
- E. Yamamoto, M. J. Hilton, M. Orlandi, V. Saini, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15877 CrossRef CAS PubMed.
- S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372 CrossRef CAS PubMed.
- K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 851 CrossRef CAS PubMed.
- B. Chen, P. Cao, X. Yin, Y. Liao, L. Jiang, J. Ye, M. Wang and J. Liao, ACS Catal., 2017, 7, 2425 CrossRef CAS.
-
(a) L. Wu, F. Wang, X. Wan, D. W. Chen, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed;
(b) D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac
*ac