
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The aromatic dianion metalloles
Junnian
Wei
 *,
Wen-Xiong
Zhang
and
Zhenfeng
Xi
*
*,
Wen-Xiong
Zhang
and
Zhenfeng
Xi
*
Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: jnwei@foxmail.com; zfxi@pku.edu.cn
First published on 4th December 2017
Abstract
Metalloaromatic species are unique and important both experimentally and theoretically. Significant progress has been made during the past few decades. New aromatic systems have challenged and extended the concept of aromaticity remarkably. In this perspective, recent results on the study of the dianion aromatic metalloles and their corresponding analogues are reviewed. These include the dilithio group 14 metalloles, group 13 metalloles and transition metal metalloles. X-ray crystallography has made a key contribution to the understanding of the structures. Various theoretical tools, such as NICS and AdNDP, make it possible to measure the aromaticity beyond Hückel’s rule. The dianion butadiene skeletons play a key role in these metalloles and can be regarded as non-innocent ligands, which accept the electrons from the metal center and thus form the aromatic rings. By simply changing the central metals to different metals, the metallole analogues such as dicupra[10]annulenes and spiroaromatic palladoles can also be generated, which opens a door to synthesize other metalla-macrocyclic aromatics. Key challenges and envisioned opportunities for the future, such as applying these dianion metalloles as novel ligands of transition metals and generating new types of organometallic aromatic system, are also discussed.
1. Introduction
Since the isolation of benzene by Faraday in 1825,1 aromaticity has become a fundamental and fascinating concept in chemistry. In most organic aromatic compounds, only the π-electrons participate in delocalization. However, by introducing metals into aromatic systems, σ-, π-, δ-, and φ-electron delocalizations can all be possible,2 which introduces various properties to these compounds and brings a revolution to aromatic chemistry. Significant progress has been made during the past few decades in metalla-aromatics, and these new aromatic systems have challenged and extended the concept of aromaticity remarkably.3Some milestones in aromatic organometallic chemistry should be noted first. In 1979, Thorn and Hoffmann predicted the existence of metallabenzenes.4 In 1982, the first metallabenzene, osmabenzene, was synthesized and characterized by Roper et al., which opened the door of metalla-aromatic chemistry.5 In 2001, Jia and co-workers reported the first isolation of metallabenzyne, osmabenzyne, which is also the smallest metal carbyne.6a,b Later, Paneque et al. reported the first isolated metallanaphthalene, iridanaphthalene.6c A metallaanthracene and derived metallaanthraquinone was recently synthesized by Wright and Frogley.6d In 2013, Xia reported the elegant synthesis of the first metallapentalyne, osmapentalyne,7 which involves the d orbitals of the transition metal center in the conjugation and thus switching of the Hückel anti-aromaticity of pentalyne into the Möbius aromaticity of metallapentalyne.
Unique and useful chemical and physical properties can always be expected based on novel metalla-aromatics. Although numerous metalla-aromatics and their analogues have been synthesized, the synthesis of new types of aromatic system with different metals still remains a great challenge.
Cyclopentadiene (Cp) anions are among the most studied classic aromatic systems. By introducing metals into Cp rings, various aromatic dianion metalloles have been synthesized and characterized recently, which has opened a new page in this field. Their novel bonding models and electronic structures have attracted much attention. In this perspective, aromatic dianion metalloles and their corresponding analogues will be summarized. The remaining challenges and future opportunities in this field are also discussed at the end.
2. Dianion metalloles
Before discussing the aromatic dianion metalloles, several widely used measurements of aromaticity will be introduced first, as new aromatics are usually significantly different from the typical organic aromatic systems documented in textbooks. Although some aromatic systems can be judged via the 4n + 2 Hückel’s rule or the 4n Baird’s rule, nowadays we can use more reliable criteria which are based on quantitative measurements and calculations instead of semi-empirical and even ambiguous descriptors. This makes comprehensive analysis of aromaticity possible.Among them, the nucleus-independent chemical shift (NICS)8 and its variants, the canonical molecular orbital contributions to NICS (CMO-NICS),9 are the most widely used theoretical indicators to judge aromaticity. Dilithio metalloles will be discussed in this perspective, and 7Li NMR can also be a direct experimental measurement to detect the shielding effect of the diatropic ring current.10 If a significant negative Li shift is observed, it means a strong shielding effect to Li atoms is present, indicating the extent of aromaticity. Moreover, energetic-based indicators such as resonance energies (REs) or isomerization stabilization energy (ISE)11 are also used in some aromatic systems.
Some qualitative and visual indications are also highly reliable and important, such as the adaptive natural density partitioning (AdNDP)12a analysis, and the anisotropy of the induced current density (ACID)12b analysis. They are useful and reliable tools to get theoretical insight into the nature of the delocalized bonding. With these theoretical tools in hand, the recently developed dilithio metalloles will be discussed.
2.1 Dilithio main group metalloles
A straightforward way to synthesize the metallole dianion is summarized in Scheme 1. The dianion compounds can be achieved via a common redox reaction using the added metal lithium as the reductant to react with the corresponding metalloles.
 | ||
| Scheme 1 A general method to prepare the dilithio metalloles. | ||
Joo and co-workers in 1987 reported the first generation of the silole dianion C4Ph4Si2−.15 In 1995, West and co-workers reported the single crystal structure of aromatic dilithiosilole 2 (dilithio-Si).14a These dilithio metalloles are synthesized via the reduction of dichloride metalloles 1 with excess lithium (Scheme 2). The structure contains two different lithium atoms. One Li atom is bonded with the silole ring with an η5 fashion, while the other one is η1-bonded to the Si atom. The C–C bond lengths in the butadiene skeleton are averaged. The reported 13C and 29Si NMR data also supported its aromatic character.16
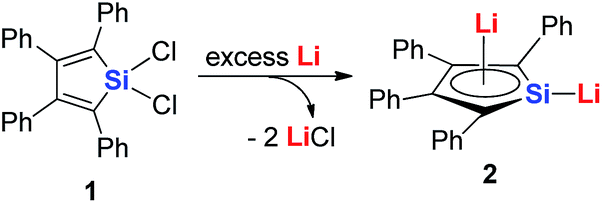 | ||
| Scheme 2 Preparation of dilithio-Si 2. | ||
Later, West and co-workers reported the first characterized delocalized germole dianion with the same butadiene skeleton as dilithio-Si, the dilithio tetraphenylgermole (dilithio-Ge).14b Interestingly, based on their X-ray structures, the dilithio-Ge 4 generated at −20 °C has a reverse-sandwich structure, with both lithium atoms lying above and below the center of the C4Ge ring in an η5 fashion (Scheme 3), while at 25 °C, 5 was obtained instead of 4 and the two lithium atoms were bonded in an η1/η5 fashion, similar to the dilithio-Si 2. In both cases, the C–C bond lengths in the germole ring are averaged, also pointing to delocalized π systems.
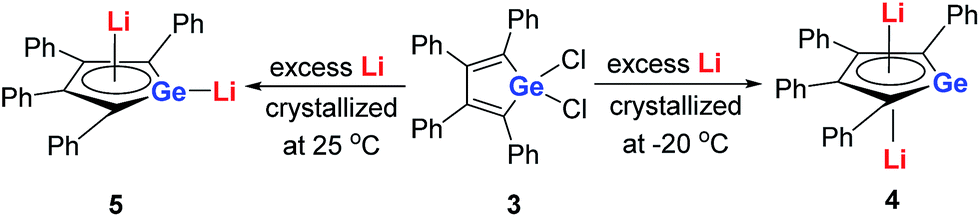 | ||
| Scheme 3 Preparation of dilithio-Ge 4 and 5. | ||
In 2005, Saito and co-workers reported group 14 aromatic dianion metalloles: dilithiostannole 7 (dilithio-Sn). As shown in Scheme 4, the dilithio-Sn 7 was synthesized via the reduction of hexaphenylstannole 6 with excess lithium in diethyl ether.17 The structure was confirmed by 1H, 13C, and 119Sn NMR spectroscopy, and X-ray analysis. Both lithium atoms are coordinated to the stannole ring in an η5 fashion.
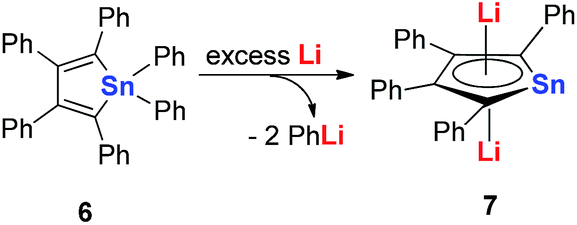 | ||
| Scheme 4 Preparation of dilithio-Sn 7. | ||
Based on DFT calculations, with Sn as the metal center, if one of the Li atoms was not bonded in an η5 fashion to the ring (7b), the energy would be 20 kcal mol−1 higher than that of the dilithio-Sn (7a) structure (Scheme 5). The bond lengths of the butadiene part in 7a are nearly equal, while the corresponding bonds of 7b alternate in length. These results indicate that both the central metal and Li atoms play an important role in forming the aromatic systems.
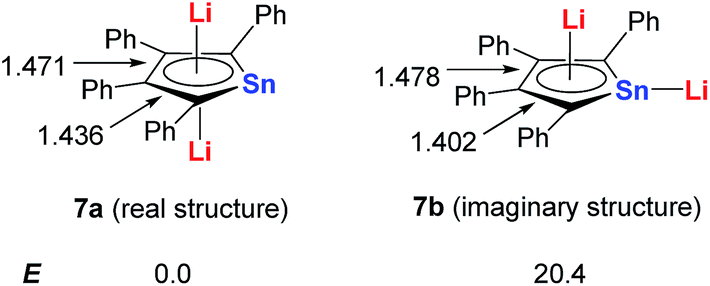 | ||
| Scheme 5 DFT calculations of dilithio-Sn 7. | ||
Later in 2014, Saito and co-workers reported more detailed studies on their dilithio-Sn.18 Based on theoretical studies, the aromaticity originates from the delocalization of the occupied p orbitals of the metal center toward the LUMO of the butadiene part as shown in Fig. 1. Thus, the decrease of the energy gap between the two orbitals would enhance the aromaticity. In fact, the silyl-substituted stannole dianions have more stannylene character and stronger aromaticity than their corresponding alkyl- and aryl-substituted compounds, as the silyl groups attached to the butadiene moiety can stabilize (lower) the LUMO through the σ–π* conjugation. The valences of the Sn atom in dilithio-Sn are all closer to Sn(II) than Sn(0) or Sn(IV), and were confirmed by 119Sn Mössbauer spectroscopy.
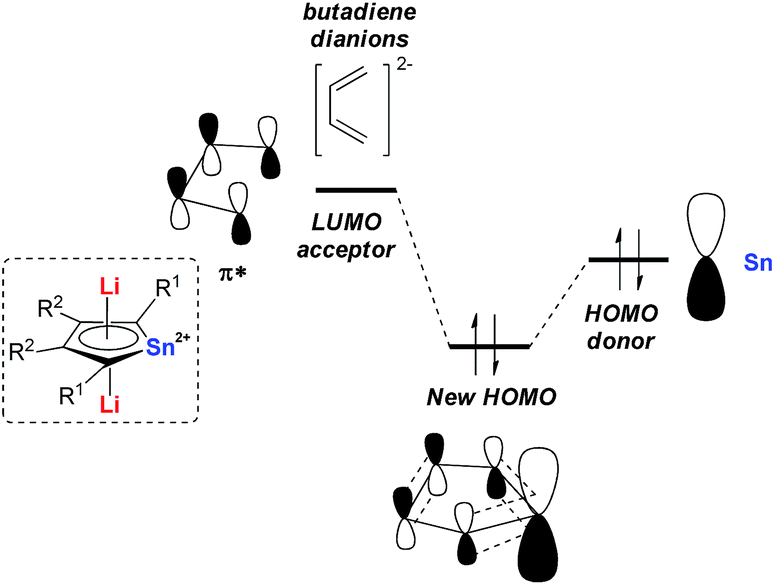 | ||
| Fig. 1 Origin of the aromatic nature of dilithio-Sn 7. | ||
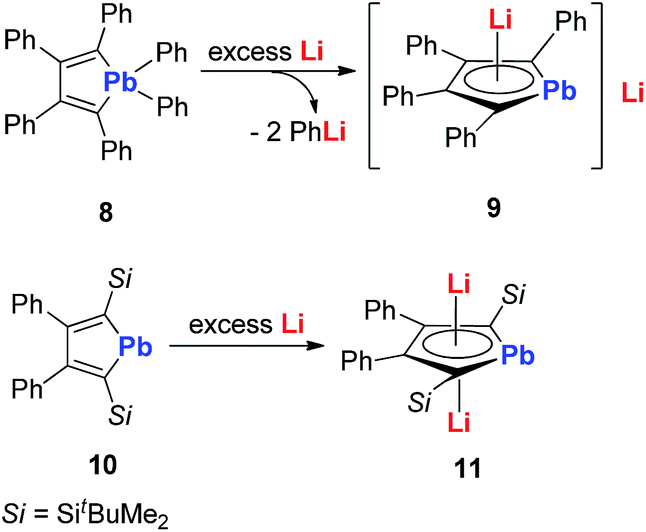
In 2010, Saito and co-workers reported the heaviest group 14 congener of the cyclopentadienyl (Cp) analog, dilithioplumbole 9 (dilithio-Pb) (Scheme 6).19a The dilithio-Pb 9 was synthesized by the reduction of hexaphenylplumbole with lithium, a similar synthetic method to that used for dilithio-Sn. Based on the single crystal X-ray structure, only one Li atom is coordinated with the plumbole ring in an η5 fashion, while the other Li atom is solvated and far away from the Pb center (more than 10 Å). Thus only one Li has interaction with the plumbole ring. The 7Li NMR spectrum has only one peak at −1.11 ppm, indicating a rapid exchange in solution between both Li atoms. The bond lengths are averaged inside the plumbole ring, suggesting that the dilithio-Pb 9 has considerable aromatic character. The remarkable negative NICS(1) value (−6.28 ppm) of free plumbole dianions also supports this conclusion.
 | ||
| Scheme 6 Preparation of dilithio-Pb 9 and 11. | ||
Interestingly, in 2015 the same group reported dilithio-Pb 11 from the reduction of plumbacyclopentadienylidene 10via adding an excess amount of Li in toluene.19b Compound 11 has both Li atoms coordinated with the plumbole ring in the η5 fashion. 11 has a similar structure to its analogue 7, thus it is also aromatic. And the 7Li NMR signal (−3.5 ppm) in C6D6 supported this conclusion. However, the 207Pb NMR signal for 11 (2573 ppm) shifted to a low field, compared with that for the tetraphenyl derivative 9 (1713 ppm), which indicates that its plumbylene character is enhanced by silyl groups.
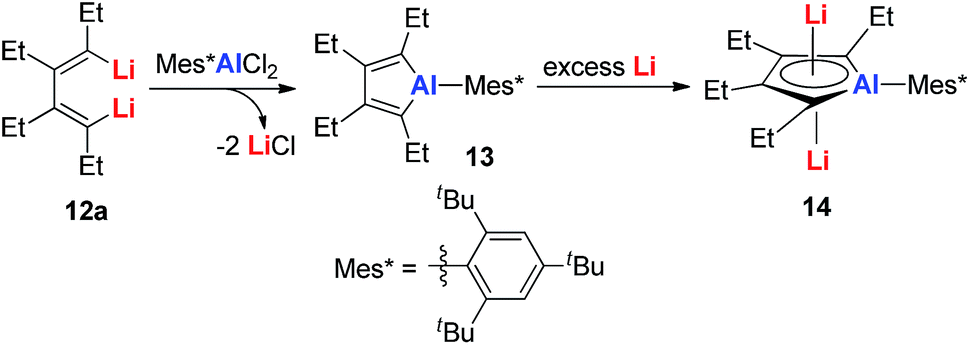 | ||
| Scheme 7 Preparation of dilithio-Al 14. | ||
Dilithio-Al has a similar structure to dilithio-Sn, with averaged bond lengths and two η5 coordinated Li atoms. As the Li atoms lie above and below the alumole core both in an η5 fashion, if the five-membered ring core is aromatic, significant negative shifts should be observed, because of the strong shielding effect of the diatropic ring current. The 7Li NMR chemical shift of dilithio-Al (−6.0 ppm) corresponds well with the calculated value (−5.5 ppm), indicating that the contact ion-pair structure is retained in solution and that the AlC42− skeleton is aromatic. The significant negative NICS(0) value (−15.01 ppm) also supports this conclusion.
With a similar strategy, the dilithio-Ga 16 compounds were also synthesized by the same group in 2015 (Scheme 8).21a The dilithio-Ga showed almost the same structures as dilithio-Al and also showed remarkable aromaticity based on the 7Li NMR (−6.0 to −6.8 ppm) and NICS(1) (−15.0 to −15.5 ppm) values.
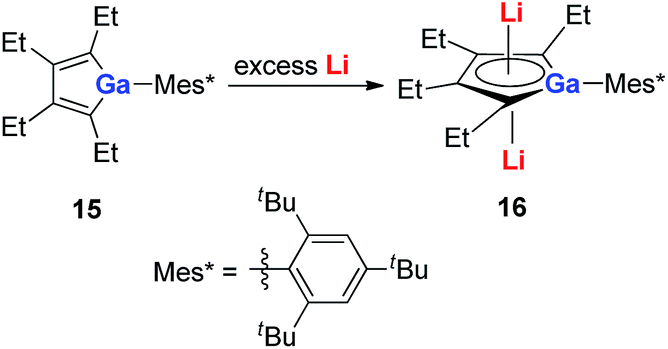 | ||
| Scheme 8 Preparation of dilithio-Ga 16. | ||
Very recently, our group reported the synthesis of aromatic tetralithiodigalloles 18 with a Ga–Ga bond via a similar strategy (Scheme 9).21b The two dilithio-Ga parts are connected with a Ga–Ga bond. Both of the dilithio-Ga units are aromatic, similar to 16, based on the X-ray structure and 7Li NMR (−6.68 ppm) and NICS(1) (−11.1 ppm) values.
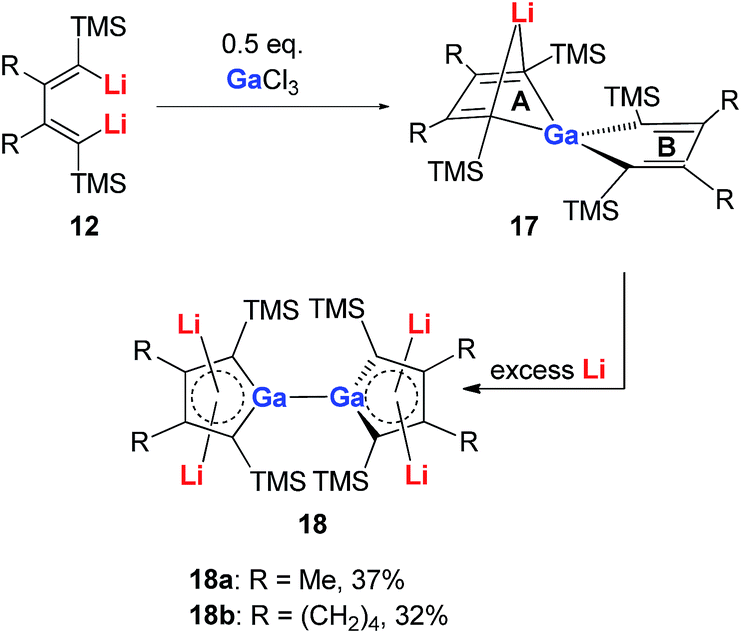 | ||
| Scheme 9 Preparation of tetralithiodigalloles 18. | ||
2.2 Dilithio transition metal metalloles
All the recently reported aromatic compounds contain two Li atoms and a butadiene skeleton. Their synthetic strategies were normally based on the redox reaction between an excess amount of the lithium metal and their corresponding metalloles. However, the synthesis of metalloles via transmetalation is in many cases impossible or extremely difficult, especially with transition metals whose corresponding salts are moderate or strong oxidizing reagents. In these cases, the corresponding aromatic dilithio metalloles could not be prepared, which limits the scope of this methodology.To hunt for synthetic methods to create novel aromatic dilithio complexes, it is necessary to go back and re-examine the structures of the dilithio metalloles. The dilithio metalloles consist of a dilithio butadiene compound and a metal center. In fact, the LUMO of dilithio butadienes 12 is significantly lower than that of their corresponding butadienes based on our calculations. Thus, it is possible for the dilithio reagents 12 to react with appropriate low-valent metal complexes directly, providing a pair of electrons to the vacant LUMO of the dilithio skeletons.
Our group has been working on dilithio reagents 12 for a long time22 and we envision that the final product could also be regarded as the combination of the conjugated dilithio butadiene part and the metal center part (Fig. 2). From this viewpoint, it is possible to synthesize various interesting metalla-aromatics readily via the reactions of the dilithio reagents with appropriate metal complexes.
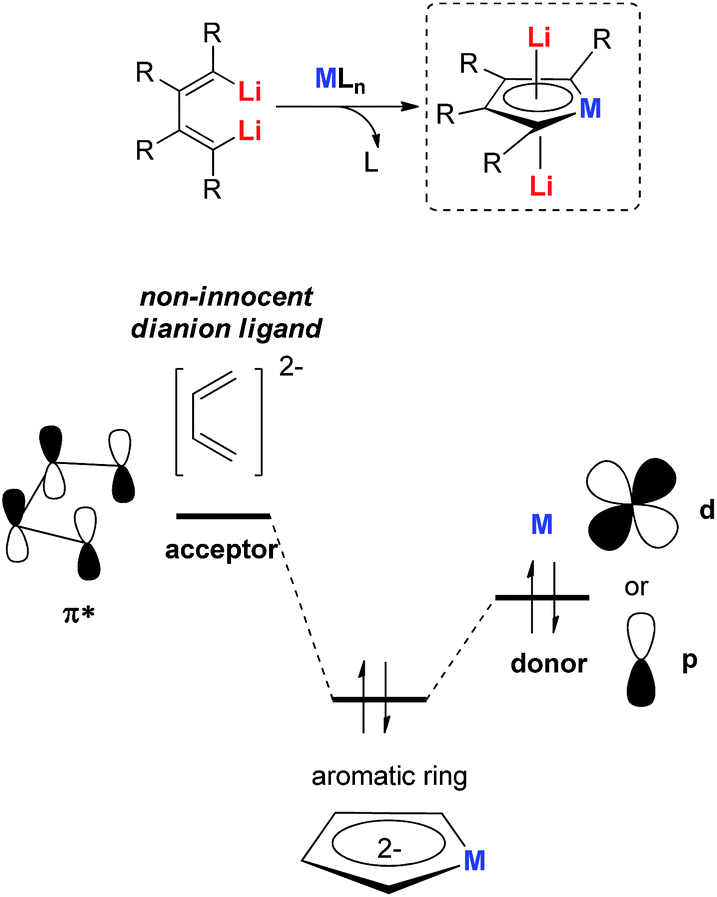 | ||
| Fig. 2 Novel method to prepare dilithio metalloles. | ||
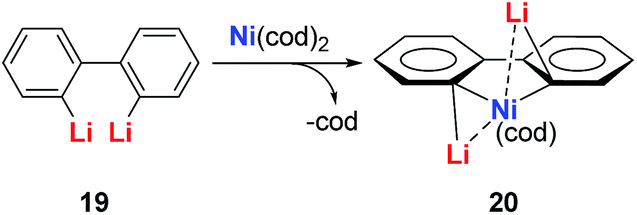 | ||
| Scheme 10 Reaction of diphenyl dilithio reagent with Ni(cod)2. | ||
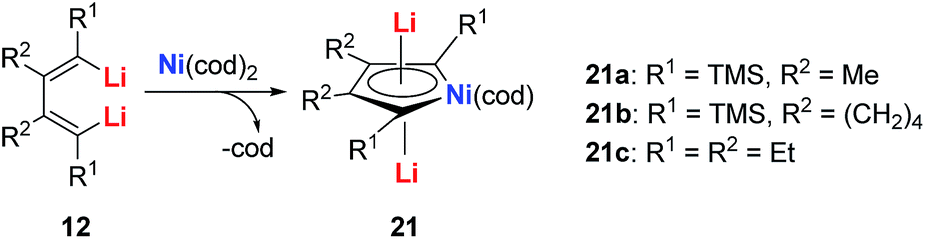 | ||
| Scheme 11 Preparation of dilithio-Ni 21. | ||
The dilithio-Ni 21 has similar skeletons to its main group analogues (e.g., dilithio-Sn). With these results, we could envision that the Ni in dilithio-Ni 21 should be Ni(II). To confirm this hypothesis, X-ray photoelectron spectroscopy (XPS) measurements of compounds 20 and 21 were carried out. XPS of 20 detected the Ni 2p3/2 binding energy at 852.5 eV, falling within the range of Ni(0). To compare, XPS of 21 detected the Ni 2p3/2 binding energy at 855.5 eV, falling within the range of Ni(II). This result strongly indicates that the chemical environments of the Ni atoms in 20 and 21 are clearly different, supporting the hypothesis that in compound 21, one of the occupied orbitals of the metal center delocalizes its electron pairs toward the LUMO of the butadiene part.
Direct evidence of delocalization is that the C–C bond lengths are averaged in the skeleton of compound 21, based on the single crystal X-ray structures, while the corresponding C–C bonds in 20 alternate in length. As expected, the 7Li NMR shifts in 21 are all around −6 ppm, which also strongly suggest aromaticity. To compare, the 7Li NMR shift in compound 20 is only −1.7 ppm. Considerable negative NICS(0) (−8.6 ppm) and NICS(1) (−10.3 ppm) values were also obtained in compound 21, indicating aromaticity. This is also the first example of dilithio-TM (TM = transition metal) aromatic systems.
In 2015, our group reported the synthesis and characterization of aromatic dilithiorhodacycles 22 (dilithio-Rh).25 The reaction of [RhCl(cod)]2 with dilithio reagent 12 could offer dilithio-Rh 22 as dark red crystalline compounds (Scheme 12). In fact, this was a two-step reaction; the Rh(I) starting material first reacted with 12 to generate the rhodium ate complexes 23, and 23 then reacted with another equivalent of dilithio reagent 12 to provide the final product 22.
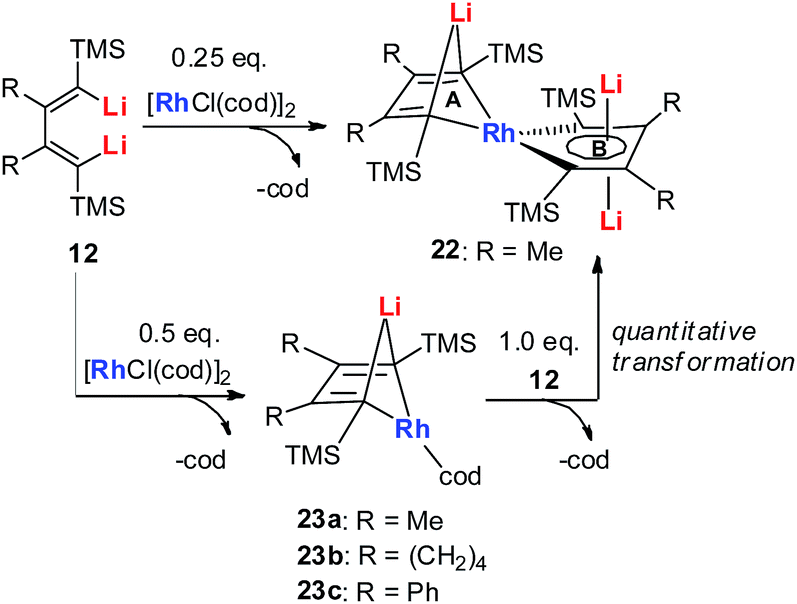 | ||
| Scheme 12 Preparation of dilithio-Rh 22. | ||
Dilithio-Rh compound 22 is a good example to show the roles of the butadiene parts. We label the two rings in 22 as Ring A (the Rh/Li double bridge part) and Ring B (five-membered dilithio rhodium ring). Ring A is, in fact, a normal Rh ate complex part, while Ring B has the metalla-aromatic structure, similar to the dilithio-M system mentioned above.
The Rh–C(sp2) bond lengths in Ring B are shorter than those in Ring A, implying a higher Rh–C bond order (Fig. 3). In fact, based on the Wiberg bond order, the Rh–C bond order in Ring A is only 0.65, while the Rh–C bond order in Ring B is around 0.90. Another difference is that the C–C bond lengths in Ring A show a clear 1,3-diene character with bond alternation, while in Ring B the bond lengths are averaged, suggesting a considerable delocalization effect. Thus, the formation of Ring B could also be explained by the cyclometallation mechanism as shown in Scheme 13.
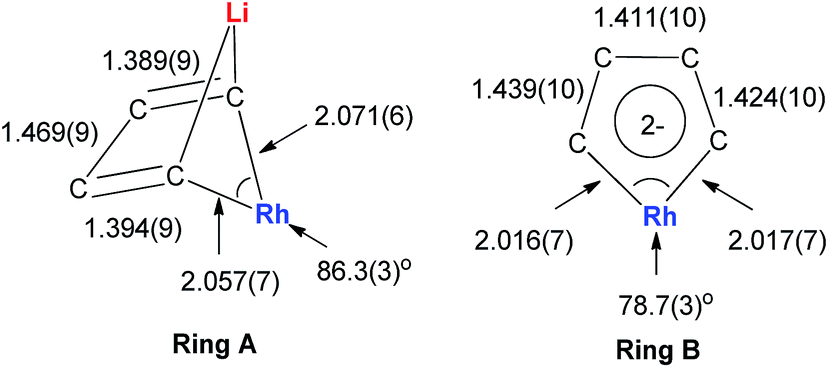 | ||
| Fig. 3 Molecular structure and selected bond lengths (Å) of 22. | ||
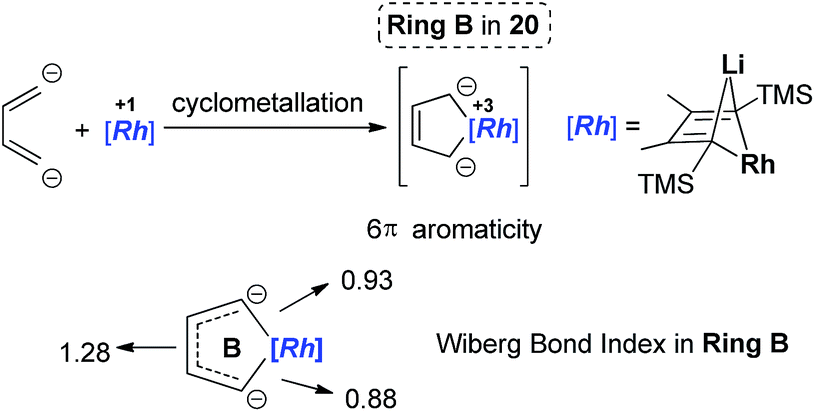 | ||
| Scheme 13 An alternative explanation of the formation of Ring B. | ||
The 7Li NMR spectrum also clearly shows the difference between Ring A and Ring B. The chemical shift of Li in Ring A is much less negative (−4.1 ppm) than that in Ring B (−6.3 ppm). These results also correspond well with NICS calculations (−4.6 ppm for Ring A and −7.8 ppm for Ring B).
With dilithio-Rh in hand, we could further reduce this complex by reacting it with an excess amount of the Li metal in THF at room temperature.26 As shown in Scheme 14, the pentalithio spiroaromatic rhodacycle 24 could be obtained in 65% isolated yield and ready for X-ray single crystal analysis. One extra Li atom is located between the two Rh rings, and the corresponding Li signal in 7Li NMR was found at 6.37 ppm. Each of the five membered rings in the spiro rhodacycle has a similar structure to Ring B in dilithio-Rh.
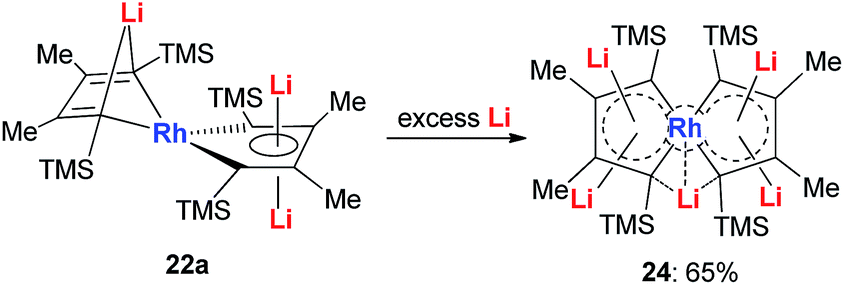 | ||
| Scheme 14 Preparation of pentalithio spiroaromatic rhodacycle 24. | ||
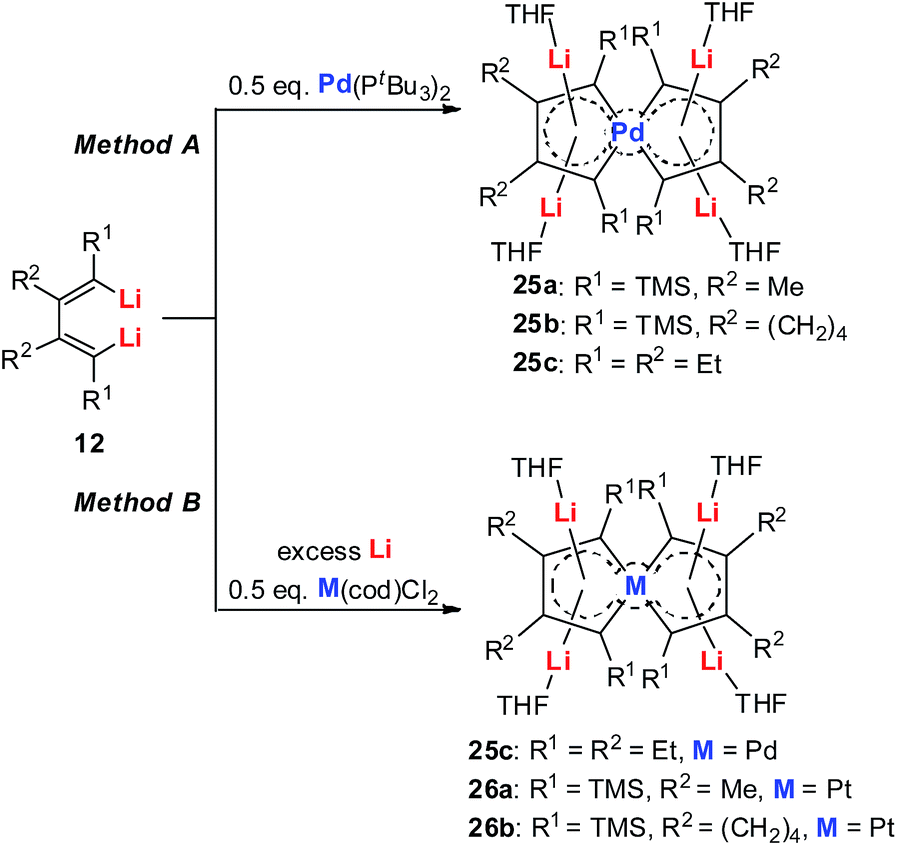 | ||
| Scheme 15 Preparation of tetralithio spiroaromatic palladoles 25 and platinacycles 26. | ||
Tetralithio spiroaromatic palladoles 25 could be prepared by the reaction between dilithio reagents 12 with 0.5 equivalents of Pd(PtBu3)2 in a mixed solvent, while the tetralithio spiroaromatic platinacycles could be isolated in high yields by reacting dilithio reagents 12 with M(cod)Cl2 (M = Pt or Pd) in the presence of an excess amount of lithium. X-ray structural characterization shows that these spiro aromatics contain two identical metalloles that share the central metal. Each metallole is planar and has averaged C–C bond lengths. Similarly, the four Li atoms are located above and below the metallole and are bonded in the η5 fashion. The low frequency resonance 7Li NMR peaks (−4.1 to −5.2 ppm) and the corresponding NICS values (about −15.7 ppm) also support the aromaticity of these spiro metalla-aromatics.
Based on the AdNDP study, we proposed that these spiro-aromatics are 10 π-systems with two delocalized 7c-2e π-bonds on each metallole and one 13c-2e delocalized bond. The aromaticity was further confirmed by the anisotropy of the induced current density (AICD) analysis. The clockwise current density vectors indicate a diatropic ring current along the periphery of the spiropalladole ring.26
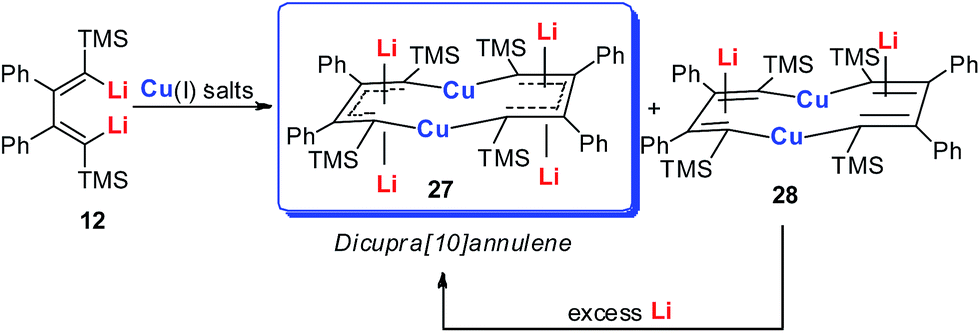 | ||
| Scheme 16 Preparation of dicupra[10]annulenes 27. | ||
Annulenes with a general formula C2nH2n are classic cyclic conjugated systems and have been well studied. The well-known annulenes including cyclobutadiene and benzene correspond well with the Hückel rule. However, the [10]annulene that has 10 π-electrons is non-aromatic, because of the steric hindrance of the two internal hydrogens. In dicupra[10]annulenes 27, the steric hindrance was avoided; each of the two metals could offer one electron to form delocalized π-bonds, thus it turned out to be aromatic. 27 could also be generated via the reduction of 28 by adding excess Li. The C–C bond lengths in 27 are averaged, whilst they alternate in the corresponding cuprate compound 28 (Fig. 4).
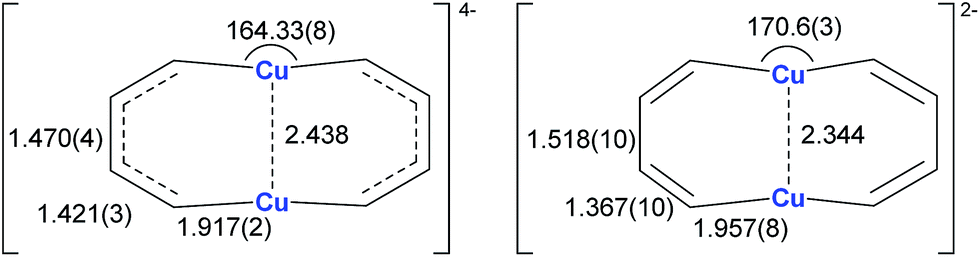 | ||
| Fig. 4 Selected bond lengths (Å) of 27 (left) and 28 (right). | ||
In fact, based on the single crystal structures, 7Li NMR (−5.1 to −6.2 ppm), and the NICS values (−9.8 to −12.7 ppm), there is no doubt that the dicupra[10]annulene 27 is aromatic. AdNDP suggests that there are two delocalized 8c-2e π-bonds for both half rings, together with one 14c-2e delocalized bond. Grande-Aztatzi et al. made a theoretical analysis of this dicupra[10]annulene system and suggested that this system could also be regarded as metalla-naphthalene.27b Another DFT calculation work by Zhu et al. suggested that the dicupra[10]annulene 27 might be 16e Craig-type Möbius aromatic, whilst compound 28 is antiaromatic.27c
3 Conclusions and outlook
In this perspective, a series of recently reported aromatic dilithio metalloles are discussed, which extend the concept of aromaticity in organometallic chemistry. However, there are still some questions and challenges remaining in this field.The first question is whether the Li atoms are essential or not. Based on the results above, the lithium atoms play an important role on the aromaticity and should not be regarded only as the counter ion.27c If other alkali metals, transition metals, or even some organic cations could be introduced as the counter ion parts, then the aromatic dianion metalloles would be used as ligands, which should have a wide range of applications.
The other question is whether other types of aromatic system can be obtained by utilizing different types of the central metal. As shown in this perspective, although most of the dianion systems have similar dilithio metallole cores, a novel dicupra[10]annulene could be generated by simply changing the metals to Cu. By changing the metal center from Ni to Pd and Pt, spiro metalla-aromatics could be observed instead of the dilithio metalloles. Until now, only metals from limited groups have been introduced into this system, yet other types of metalla-aromatics should also be possible and need to be studied.
In regards to the first question, as the p orbitals of the Li atoms make significant contributions to the HOMO of these aromatic systems based on theoretical calculations, we expect that other alkali metals (Na, K and Cs) will have weaker interactions with the butadiene part than Li and decrease the aromaticity. In fact, there are some limited examples for the synthesis of these aromatics systems with other alkali metals. Tilley and co-workers reported the aromatic dianion silole 29 with [K([18]crown-6)+]2 as the cation parts 20 years ago (Scheme 17).13a Unfortunately, there is no further work reported on this system.
 | ||
| Scheme 17 The comparison of the synthesis of the di-Li and di-K silole dianion complexes. | ||
Other main group metals are relatively hard to introduce into this system. However, the two negative charges of the metallole might enable the coordination to transition metals. The coordination chemistry of some typical metallole anions has been investigated by Tilley’s group.28 Thus, it is possible to regard all the above mentioned new aromatic systems as dianion ligands of transition metals.
Recently Saito and co-workers successfully replaced the Li atoms with Ru via transmetalation using CpRuCl (Scheme 18).29 The di-Ru products are still aromatic and the Ru atoms coordinate with the metallole both in an η5 fashion. Based on the orbital analysis, the HOMO − 2 shows that the stannole dianion moiety coordinates the ruthenium atoms as an allyl anion, while the p(Sn) overlaps with d(Ru) via the HOMO − 7. The silyl groups on the dilithio part might also play an important role to form these μ–η5:η5-fashions, as only the dilithio-Sn with silyl-substituted alpha-carbons can form this triple-decker structure.
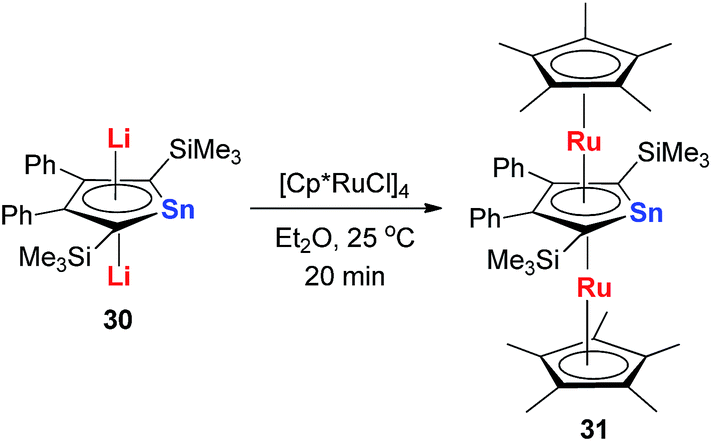 | ||
| Scheme 18 Synthesis of μ–η5:η5-stannole dianion complexes 31. | ||
However, interestingly, when CpHfCl2 is used in the same transmetalation reaction, an η1 coordination fashion could be observed instead of the triple-decker structure (Scheme 19). As expected, remarkable C–C bond alternation was found in the butadiene part in 33, indicating the loss of aromaticity.30 These examples suggest that the electronic nature of these metallole dianions is highly dependent on the coordination mode and the counter ions.
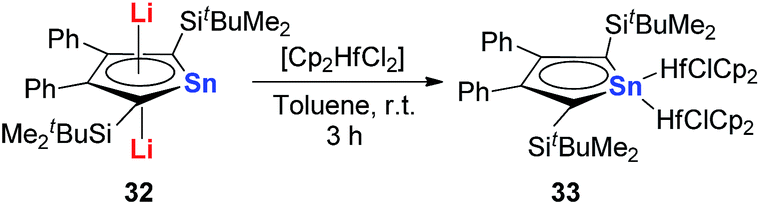 | ||
| Scheme 19 Reaction of dilithiostannole 32 with 2 equivalents of Cp2HfCl2 in toluene. | ||
In regards to the second question, although studies of the transmetalation of the dilithio-M have been limited until now, the products of the transmetalation, especially the triple-decker aromatic complexes, could be generated via other strategies. These results may lead to further syntheses of other kinds of metalla-aromatics.
Very recently, Loginov and co-workers reported the synthesis and characterizations of two impressive triple-decker structures 36 and 37 (Scheme 20).31 In particular for compound 37, although the authors did not realize that it was an aromatic rhodacycle dianion, it should have similar electronic structures to the dianion metalloles mentioned in this perspective based on the average C–C bond lengths in the C4H4Rh ring measured by single X-ray analysis.
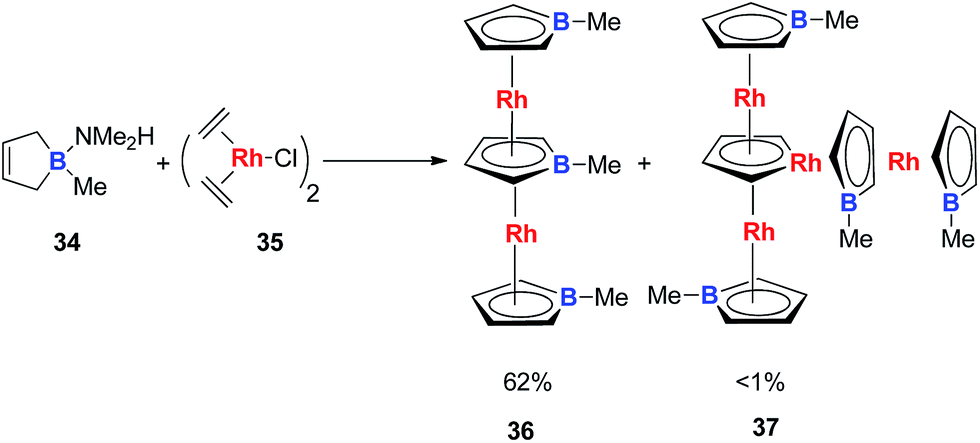 | ||
| Scheme 20 Reaction of C4H6BMe·NMe2H with [(C2H4)2RhCl]2. | ||
Although the dilithio-Fe has not been synthesized yet, its triple-decker analogue has been reported and is well-studied.32 As shown in Scheme 21, the final product 40 has almost equal C–C bond lengths in the ferracycle. Thus it might also be regarded as an analogue of dianion metalloles. This result indicates that other kinds of dianion ferracycle are also possible.
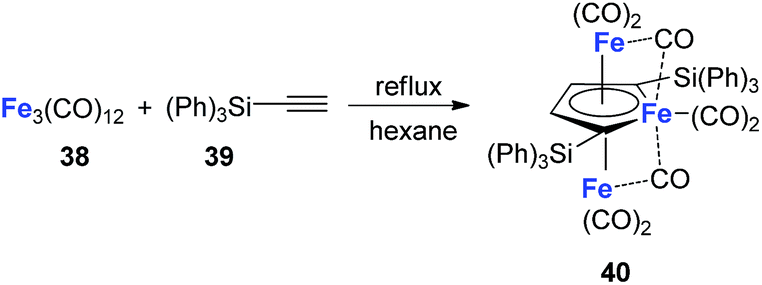 | ||
Scheme 21 Reaction of Fe3(CO)12 with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSi(Ph)3. CSi(Ph)3. | ||
Another example is with Ru. Following a similar strategy to that with Fe, the triple-decker ruthenacycle 43 could be prepared (Scheme 22).33
 | ||
| Scheme 22 Reaction of Ru3(CO)12 with MeCCSi(Me)3. | ||
It should also be noted that all the examples provided in this perspective focus on the main group and late transition metals; the appropriate earlier transition metals might also be able to form dianion metalloles or other different aromatic structures. As there are various transition metals with variable valency and atomic radius, new types of aromatic system could be expected. In 2011, Murugesu and co-workers reported a novel chromium complex 46 (Scheme 23).34 The starting material 44 is a typical Cr(II) lithium ate complex. Interestingly, based on their DFT calculations, all the three Cr atoms in the final product 46 are remaining Cr(II). And the center Cr five membered ring could be regarded as a [(C4H4)Cr(L)]2− species. Additionally, the C–C bond lengths in the (C4H4)Cr(L) ring are also averaged. These results are similar to the dianion metalloles mentioned in this perspective, which indicates that it is possible to generate more kinds of aromatic metallole with early transition metals.
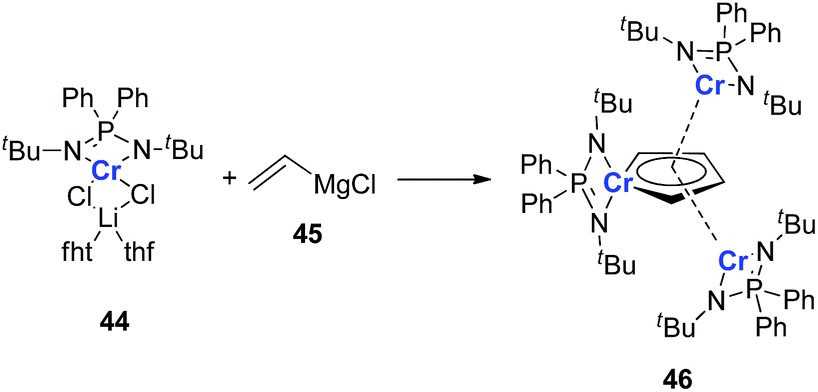 | ||
| Scheme 23 Generation of chromium complex 46. | ||
From the works shown in this perspective, it becomes apparent that aromaticity is an attractive topic in chemistry even after decades of studies. Nowadays, the aromaticity of metalloaromatic species can be judged by reliable theoretical measurements such as NICS and AdNDP. However, a few things still remain unclear in many situations. For instance, it could be argued that the dilithio metalloles should be regarded as spherical aromatics instead of MC42− planar aromatics. In this case, future works need to be done to better understand aromaticity and help in the design of new compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21372012 and 21690061).References
- M. Faraday, Philos. Trans. R. Soc. London, 1825, 115, 440–446 CrossRef.
- (a) A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed; (b) C. A. Tsipis, Coord. Chem. Rev., 2005, 249, 2740–2762 CrossRef CAS; (c) D. Yu. Zubarev, B. B. Averkiev, H.-J. Zhai, L. S. Wang and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 257–267 RSC; (d) F. Feixas, E. Matito, J. Poater and M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 105–122 CrossRef CAS.
- (a) J. R. Bleeke, Chem. Rev., 2001, 101, 1205–1228 CrossRef CAS PubMed; (b) G. Jia, Acc. Chem. Res., 2004, 37, 479–486 CrossRef CAS PubMed; (c) L. J. Wright, Dalton Trans., 2006, 1821–1827 RSC; (d) C. W. Landorf and M. M. Haley, Angew. Chem., Int. Ed., 2006, 45, 3914–3936 CrossRef CAS PubMed; (e) J. Chen and G. Jia, Coord. Chem. Rev., 2013, 257, 2491–2521 CrossRef CAS; (f) B. J. Frogley and L. J. Wright, Coord. Chem. Rev., 2014, 270–271, 151–166 CrossRef CAS; (g) X.-Y. Cao, Q. Zhao, Z. Lin and H. Xia, Acc. Chem. Res., 2014, 47, 341–354 CrossRef CAS PubMed; (h) S. Roy, U. Rosenthal and E. D. Jemmis, Acc. Chem. Res., 2014, 47, 2917–2930 CrossRef CAS PubMed; (i) B. J. Frogley and L. J. Wright, Chem.–Eur. J., DOI:10.1002/chem.201704888.
- D. L. Thorn and R. Hoffmann, Nouv. J. Chim., 1979, 3, 39–45 CAS.
- G. P. Elliott, W. R. Roper and J. M. Waters, J. Chem. Soc., Chem. Commun., 1982, 811–813 RSC.
- (a) T. B. Wen, Z. Y. Zhou and G. Jia, Angew. Chem., Int. Ed., 2001, 40, 1951–1954 CrossRef CAS; (b) W. Y. Huang, B. Liu, W. Shou, T. B. Wen, C. Shi, H. H.-Y. Sung, I. D. Williams, Z. Lin and G. Jia, J. Am. Chem. Soc., 2011, 133, 18350–18360 CrossRef PubMed; (c) M. Paneque, C. M. Posadas, M. L. Poveda, N. Rendón, V. Salazar, E. Oñate and K. Mereiter, J. Am. Chem. Soc., 2003, 125, 9898–9899 CrossRef CAS PubMed; (d) B. J. Frogley and L. J. Wright, Angew. Chem., Int. Ed., 2017, 56, 143–147 CrossRef CAS PubMed.
- (a) C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lin, T. Wen, Z. Xie, P. von R. Schleyer and H. Xia, Nat. Chem., 2013, 5, 698–703 CrossRef CAS PubMed; (b) T. Wang, H. Zhang, F. Han, L. Long, Z. Lin and H. Xia, Angew. Chem., Int. Ed., 2013, 52, 9251–9255 CrossRef CAS PubMed; (c) C. Zhu, M. Luo, Q. Zhu, J. Zhu, P. von R. Schleyer, J. I.-C. Wu, X. Lu and H. Xia, Nat. Commun., 2014, 5, 3265 Search PubMed; (d) C. Zhu, Y. Yang, J. Wu, M. Luo, J. Fan, J. Zhu and H. Xia, Angew. Chem., Int. Ed., 2015, 54, 7189–7192 CrossRef CAS PubMed; (e) M. Luo, L. Long, H. Zhang, Y. Yang, Y. Hua, G. Liu, Z. Lin and H. Xia, J. Am. Chem. Soc., 2017, 139, 1822–1825 CrossRef CAS PubMed.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- T. Heine, P. von R. Schleyer, C. Corminboeuf, G. Seifert, R. Reviakine and J. Weber, J. Phys. Chem. A, 2003, 107, 6470–6475 CrossRef CAS.
- (a) R. H. Cox, H. W. Terry Jr and L. W. Harrison, J. Am. Chem. Soc., 1971, 93, 3297–3298 CrossRef CAS; (b) R. H. Cox and H. W. Terry Jr, J. Magn. Reson., 1974, 14, 317–322 CAS; (c) L. A. Paquette, W. Bauer, M. R. Sivik, M. Bühl, M. Feigel and P. v. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 8776–8789 CrossRef CAS.
- M. K. Cyrański, Chem. Rev., 2005, 105, 3773–3811 CrossRef PubMed.
- (a) D. Yu. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC; (b) D. Geuenich, K. Hess, F. Kohler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- (a) W. P. Freeman, T. D. Tilley, G. P. A. Yap and A. L. Rheingold, Angew. Chem., Int. Ed. Engl., 1996, 35, 882–884 CrossRef CAS; (b) P. Dufour, J. Dubac, M. Dartiguenave and Y. Dartiguenave, Organometallics, 1990, 9, 3001–3003 CrossRef CAS; (c) W. P. Freeman, T. D. Tilley, F. P. Arnold, A. L. Rheingold and P. K. Gantzel, Angew. Chem., Int. Ed. Engl., 1995, 34, 1887–1890 CrossRef CAS.
- (a) R. West, H. Sohn, U. Bankwitz, J. Calabrese, Y. Apeloig and T. Müller, J. Am. Chem. Soc., 1995, 117, 11608–11609 CrossRef CAS; (b) R. West, H. Sohn, D. R. Powell, T. Müller and Y. Apeloig, Angew. Chem., Int. Ed. Engl., 1996, 35, 1002–1004 CrossRef CAS; (c) S.-B. Choi, P. Boudjouk and J.-H. Hong, Organometallics, 1999, 18, 2919–2921 CrossRef CAS; (d) B. Goldfuss, P. von R. Schleyer and F. Hampel, Organometallics, 1996, 15, 1755–1757 CrossRef CAS; (e) B. Goldfuss and P. von R. Schleyer, Organometallics, 1997, 16, 1543–1552 CrossRef CAS.
- W.-C. Joo, Y. C. Park, S. K. Kang, J. H. Hong and Y.-K. Kong, Bull. Korean Chem. Soc., 1987, 8, 270–272 CAS.
- J.-H. Hong, P. Boudjouk and S. Castellino, Organometallics, 1994, 13, 3387–3389 CrossRef CAS.
- M. Saito, R. Haga, M. Yoshioka, K. Ishimura and S. Nagase, Angew. Chem., Int. Ed., 2005, 44, 6553–6556 CrossRef CAS PubMed.
- T. Kuwabara, J.-D. Guo, S. Nagase, M. Minoura, R. H. Herber and M. Saito, Organometallics, 2014, 33, 2910–2913 CrossRef CAS.
- (a) M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura, S. Nagase and M. Hada, Science, 2010, 328, 339–342 CrossRef CAS PubMed; (b) M. Saito, M. Nakada, T. Kuwabara and M. Minoura, Chem. Commun., 2015, 51, 4674–4676 RSC.
- T. Agou, T. Wasano, P. Jin, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10031–10034 CrossRef CAS PubMed.
- (a) T. Agou, T. Wasano, T. Sasamori and N. Tokitoh, J. Phys. Org. Chem., 2015, 28, 104–107 CrossRef CAS; (b) Y. Zhang, Y. Chi, J. Wei, Q. Yang, Z. Yang, H. Chen, R. Yang, W.-X. Zhang and Z. Xi, Organometallics, 2017, 36, 2982–2986 CrossRef CAS.
- Z. Xi, Acc. Chem. Res., 2010, 43, 1342–1351 CrossRef CAS PubMed.
- (a) I. Mitteilung, K. R. Pörschke, K. Jonas, G. Wilke, R. Benn, R. Mynott, R. Goddard and C. Krüger, Chem. Ber., 1985, 118, 275–297 CrossRef; (b) K.-R. Pörschke, K. Jonas and G. Wilke, Chem. Ber., 1988, 121, 1913–1919 CrossRef.
- J. Wei, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2015, 54, 5999–6002 CrossRef CAS PubMed.
- J. Wei, Y. Zhang, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2015, 54, 9986–9990 CrossRef CAS PubMed.
- Y. Zhang, J. Wei, Y. Chi, X. Zhang, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2017, 139, 5039–5042 CrossRef CAS PubMed.
- (a) J. Wei, Y. Zhang, Y. Chi, L. Liu, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2016, 138, 60–63 CrossRef CAS PubMed; (b) R. Grande-Aztatzi, J. M. Mercero, E. Matito, G. Frenking and J. M. Ugalde, Phys. Chem. Chem. Phys., 2017, 19, 9669–9675 RSC; (c) K. An, T. Shen and J. Zhu, Organometallics, 2017, 36, 3199–3204 CrossRef CAS.
- (a) W. P. Freeman, T. D. Tilley, A. L. Rheingold and R. L. Ostrander, Angew. Chem., Int. Ed. Engl., 1993, 32, 1744–1745 CrossRef; (b) W. P. Freeman, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1994, 116, 8428–8429 CrossRef CAS; (c) J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 1998, 120, 8245–8246 CrossRef CAS; (d) J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 3097–3105 CrossRef CAS; (e) J. M. Dysard and T. D. Tilley, Organometallics, 2000, 19, 2671–2675 CrossRef CAS; (f) W. P. Freeman, J. M. Dysard, T. D. Tilley and A. L. Rheingold, Organometallics, 2002, 21, 1734–1738 CrossRef CAS.
- T. Kuwabara, J.-D. Guo, S. Nagase, T. Sasamori, N. Tokitoh and M. Saito, J. Am. Chem. Soc., 2014, 136, 13059–13064 CrossRef CAS PubMed.
- T. Kuwabara and M. Saito, Organometallics, 2015, 34, 4202–4204 CrossRef CAS.
- D. A. Loginov, D. V. Muratov, Y. V. Nelyubina, J. Laskova and A. R. Kudinov, J. Mol. Catal. A: Chem., 2017, 426, 393–397 CrossRef CAS.
- (a) D. Lentz, H. Michael-Schulz and M. Reuter, Organometallics, 1992, 8, 2916–2922 CrossRef; (b) A. S. Estrada-Montaño, M. A. Leyva, R. Grande-Aztatzi, A. Vela and M. J. Rosales-Hoz, J. Organomet. Chem., 2014, 751, 420–429 CrossRef.
- V. González-López, M. A. Leyva and M. J. Rosales-Hoz, Dalton Trans., 2013, 42, 5401–5411 RSC.
- K. Albahily, V. Fomitcheva, S. Gambarotta, I. Korobkov, M. Murugesu and S. I. Gorelsky, J. Am. Chem. Soc., 2011, 133, 6380–6387 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |