
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh entropy hydroxide with a hollow nanocage structure promotes efficient and stable water/seawater electro-oxidation†
Rui Changab,
Yu Pangab,
Qin Yangab,
Ruotong Liuab,
Yu Yangab,
Yunmei Du a,
Kang Liuab,
Zexing Wuab,
Jianping Laiab,
Hongdong Li*ab and
Lei Wang*ab
a,
Kang Liuab,
Zexing Wuab,
Jianping Laiab,
Hongdong Li*ab and
Lei Wang*ab
aKey Laboratory of Eco-Chemical Engineering, Ministry of Education, International Science and Technology Cooperation Base of Eco-Chemical Engineering and Green Manufacturing, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: hdli@qust.edu.cn; inorchemwl@126.com
bCollege of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
First published on 27th May 2025
Abstract
High-entropy hydroxides (HEHs) exhibit excellent performance in electrocatalytic reactions, but controlling the synthesis of HEHs with a high specific surface area and rapid mass transfer kinetics is challenging. Herein, a series of hollow nanocage HEH catalysts decorated with nanosheet arrays were successfully synthesized by the “coordinated etching and precipitation” method. The hollow rich edge structure not only promotes the electrochemical mass transfer, but also has a high specific surface area and exposed active sites, which combined with the high entropy effect enhances the electrocatalytic performance. Theoretical calculations further confirm that the FeCoNiCuCrMn–OH HEH catalyst has a stronger adsorption capacity for OH− than Cl−, and can jointly improve the catalytic activity and corrosion resistance of the catalyst by effectively reducing the oxygen evolution reaction energy barrier and enhancing the resistance to Cl−. In alkaline seawater electrolytes, only 275 and 292 mV overpotentials are needed to reach 100 and 200 mA cm−2. In addition, it has excellent stability and corrosion resistance, and can work stably for more than 400 hours when operating at 100 mA cm−2. This study provides new ideas for the morphology control and composition control of high-efficiency electrocatalysts.
Introduction
Hydrogen, as an environmentally friendly and renewable energy carrier, has great potential for solving energy shortages.1,2 In order to achieve high-purity hydrogen production, electrochemical water splitting has attracted much attention as one of the most suitable and effective ways to produce hydrogen on a large scale.3,4 However, due to its slower kinetic rate and higher potential, the oxygen evolution reaction (OER) severely limits the overall water splitting efficiency.5–8 In addition, most of the current research on catalysts focuses on freshwater resources, which as a scarce energy source may exacerbate water scarcity in many regions. In this context, seawater, as an unlimited resource (96.5% of the Earth's total water content), has great potential for hydrogen production.9–15 However, due to the presence of Cl−, the chlorine electro-oxidation reaction (CER) usually competes with the OER to release toxic and corrosive chlorine substances (e.g. Cl2, ClO−), which reduces the efficiency of the electrolysis, further limiting the implementation of seawater electrolysis.16–20 Therefore, a key task is to explore electrocatalysts with high selectivity and durability to seawater oxidation.In recent years, transition metal based hydroxides with high OER activity have attracted great interest in seawater oxidation.21–23 Among them, CoFe-based materials, especially CoFe layered double hydroxides, exhibit excellent OER activity in seawater oxidation, but their low electrical conductivity hinders their application in practical energy devices.24–26 The lack of electrical conductivity can be improved by introducing metallic elements (e.g. Ni and Mn), which in turn effectively modulate the electronic structure and charge density of the metal atoms, providing favorable binding energy for intermediates, thus improving the catalytic activity and electrical conductivity.27,28 In particular, high entropy material catalysts (HEMs) with more than five components have excellent adsorption behavior, electron transfer and durability properties due to their unique high entropy structure, adjustable electronic structure and multi active sites.29–33 Ting et al. prepared a high-entropy FeNiCoMnCr-LDH catalyst by a simple hydrothermal method and demonstrated that the synergistic effect among the polymetallic components promoted the formation of highly active γ-NiOOH species, significantly enhancing the catalytic activity.29 Yang and others reported a high entropy Ni0.15Cr0.15Co0.4Zn0.1Fe0.2-LDH catalyst with integrated multiple metals and oxygen vacancies, which demonstrated remarkable and steady OER performance under industrial conditions.34 Emmerling et al. reported the synthesis of a high-entropy metal hydroxide organic framework under mild solvothermal conditions, which exhibited excellent electrocatalytic performance for the OER, and showed significant durability.35 In order to further achieve HEMs with high catalytic activity, combining hollow structures with high-entropy multi-component systems will provide a most promising modulation strategy for high entropy hydroxide catalysts (HEH) for overall water/seawater electrolysis. In addition, hollow nanocatalysts can synergistically improve catalytic activity by exposing a high-density outer surface to the reactants, increasing the contact interface between the electrolyte and the electrocatalyst, shortening the distance for mass transfer and electron transfer, and increasing the collision frequency by confining the reactants to a limited space.36–39 Therefore, compared with solid particles, the prepared hollow nanocatalysts showed significantly enhanced catalytic activity in the catalytic reaction. However, due to the complex nucleation/growth kinetics variations between different elements, it is extremely challenging to obtain high entropy hollow materials by precisely adjusting material size and morphology at the atomic level.
In this study, we overcome the large differences in the hydrolysis of different ions and synthesize a series of high-entropy nanocage catalysts by the coordinated etching and precipitation (CEP) method to achieve efficient and stable alkaline water/seawater oxidation. The prepared high-entropy HEH has a hollow structure with a dense array of nanosheets on its surface, thus having a larger electrochemically active surface area that enables exposure of more active sites. Therefore, the prepared FeCoNiCuCrMn–OH catalyst showed excellent OER catalytic activity in alkaline water/seawater due to its significant advantages in structure and composition. Overpotentials of only 249 and 268 mV were required to reach 100 and 200 mA cm−2 at 1 M KOH. The interaction between the polymetallic components and the phase stability induced by the high-entropy effect of the FeCoNiCuCrMn–OH catalyst enhances the durability, making it work well under alkaline seawater conditions. Importantly, the high entropy electrocatalyst has a strong adsorption capacity for OH−, and the adsorption capacity for OH− is stronger than that of Cl−, so it has a good OER selectivity. In alkaline seawater, it only requires overpotentials of 275 and 292 mV to drive current densities of 100 and 200 mA cm−2, respectively, and can operate stably for more than 400 hours at 100 mA cm−2. The design strategy provides a novel perspective for fine-regulating the development of high-entropy materials with special forms and expanding high-performance OER catalysts.
Results and discussion
First, we synthesized a sequence of high-entropy hollow nanocages using Cu2O nanocubes (Fig. S1†) as sacrificial templates through the coordinated etching and co-precipitation method (Fig. 1). In the first reaction stage, CoCl2·6H2O was mixed with the Cu2O template, and the Cu2O@Co(OH)2 core–shell precursor was prepared through corrosion (Fig. S2†). Specifically, there is a soft interaction between Cu+ and S2O32− and the hydrolysis of S2O32− occurs in solution, resulting in the release of OH− to the surface of the Cu2O template (1) and (2). And then, surface OH− and Co2+ combine to form hydroxides with layered structures (3). With this imperative step, the structural collapse caused by the direct adding of five metals is avoided (Fig. S3†), and the constructed Co(OH)2 shell stably supports the framework during the subsequent reaction, making subsequent CEP easier to carry out.| Cu2O + xS2O32− + H2O → [Cu2(S2O32−)x]2−2x + 2OH− | (1) |
| S2O32− + H2O → HS2O3− + OH− | (2) |
| Co2+ + 2OH− → Co(OH)2 | (3) |
| Mx+ + xOH− → M(OH)x | (4) |
 | ||
| Fig. 1 Schematic diagram of the synthesis process of hollow nanocages. | ||
As shown in the second stage, Cu2O in the Cu2O@Co(OH)2 core–shell precursor is further corroded by adding sodium thiosulfate, and OH− is released, which reacts with Fe3+, Ni2+, Cr3+, Mn2+, and Zn2+ (4). With the continuous etching of the internal Cu2O template, these metal ions increasingly accumulate on the surface of the nanocage, and the high-entropy hollow nanocage structure is gradually formed. Importantly, when hydroxides proliferate on the nanoboxes, many interfaces are formed due to the uneven composition, thus providing more porous structures and interfacial heterostructures, thus expanding the active surface area.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses revealed that the obtained FeCoNiCuCrMn–OH nanocage had a cubic hollow structure with a uniform distribution and a size of about 700–800 nm (Fig. 2a, b and S4†). The results confirmed the complete removal of the Cu2O template and the formation of a hollow nanocage after the etching reaction. Moreover, TEM images further showed that a certain number of nanosheets were distributed around the hollow nanocages, and the wall thickness of the nanocage is approximately 32.3 nm. The two-dimensional layered and three-dimensional cubic structure of the nanocages is conducive to exposing more active sites, thus accelerating the electrocatalytic reaction.40–42 Furthermore, the N2 adsorption–desorption isothermal curve (Fig. S5†) of the catalyst indicates that the specific surface area of the FeCoNiCuCrMn–OH is as high as 98.2 m2 g−1. The pore size distribution obtained from N2 desorption reveals that there are narrow pores produced by layered structures in the nanocages, and the pore structure is irregular, with pore sizes ranging from 2 to 20 nm. The large amount of mesopores and the high BET surface area promote mass transfer, which makes the catalyst have better catalytic performance.43–45 The high-resolution TEM (HRTEM) image (Fig. 2c) shows that FeCoNiCuCrMn–OH has lattice spacings of 0.258 nm and 0.264, corresponding to the (012) and (101) planes. X-ray diffraction (XRD) further revealed the crystal structure of FeCoNiCuCrMn–OH (Fig. S6†), which correspond to the hydrated nickel hydroxide phase. In addition, further analysis by TEM-mapping (Fig. 2d) showed that elements (Cu, Co, Ni, Fe, Cr, Mn, O) in FeCoNiCuCrMn–OH were uniformly distributed, which was consistent with the results of SEM-mapping (Fig. S7a†), which confirmed the uniformity of the elemental composition and mixing of FeCoNiCuCrMn–OH. Meanwhile, the energy dispersive spectrometry (EDS) results showed (Fig. S7b and c†) that the metal atoms Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni:Fe:Cu:Cr:Mn of FeCoNiCuCrMn–OH synthesized were approximately 1:3:1:1:1:1. The success of the preparation of FeCoNiCuCrMn–OH was proved by the above-mentioned characterization.
:Ni:Fe:Cu:Cr:Mn of FeCoNiCuCrMn–OH synthesized were approximately 1:3:1:1:1:1. The success of the preparation of FeCoNiCuCrMn–OH was proved by the above-mentioned characterization.
 | ||
| Fig. 2 (a) SEM image, (b) TEM image, (c) HRTEM image, and (d) TEM image and the corresponding elemental maps of FeCoNiCuCrMn–OH. | ||
To further explore the valence states of the elements in FeCoNiCuCrMn–OH nanocages, X-ray photoelectron spectroscopy (XPS) tests were performed (Fig. 3). The survey spectrum confirmed the presence of major elements (Fe, Co, Ni, Cu, Cr, Mn, O) in the catalyst (Fig. 3a), which was consistent with the results of EDS. Fig. 3b–d show the XPS spectra of Fe 2p, Co 2p and Ni 2p. It can be seen that the introduction of the other elements can effectively adjust the electronic structure of the active elements (Co, Ni and Fe), thus enhancing the OER activity.46–48 Fig. 3e shows that the peaks of Cu are Cu2+ 2p3/2 (932.4 eV) and Cu2+ 2p1/2 (952.5 eV), respectively. For the Cr 2p spectrum (Fig. 3f), the peaks can be divided into Cr3+ (Cr3+ 2p3/2 and Cr3+ 2p1/2 at 577.5 eV and 587.8 eV, respectively). In the Mn spectrum (Fig. 3g), it is shown that Mn3+ 2p3/2 (641.9 eV), Mn3+ 2p1/2 (652.6 eV), Mn4+ 2p3/2 (644.3 eV) and Mn4+ 2p1/2 (654.5 eV) coexist. Among them, Mn has a high oxidation state for Mn4+, which is favorable for improving the catalyst activity. The O 1s spectrum (Fig. 3h) is fitted to metal–oxygen (M–O), metal–hydroxyl group (M–OH), and surface adsorbent water (H2Oads) at 530.3, 531.6, and 533.2 eV, respectively. Obviously, after the introduction of metal into FeCoNi, the O 1s spectrum moves towards the side with higher binding energy, which indicates that the addition of metal ions improves metal–oxygen covalency, consistent with the XPS shift results.
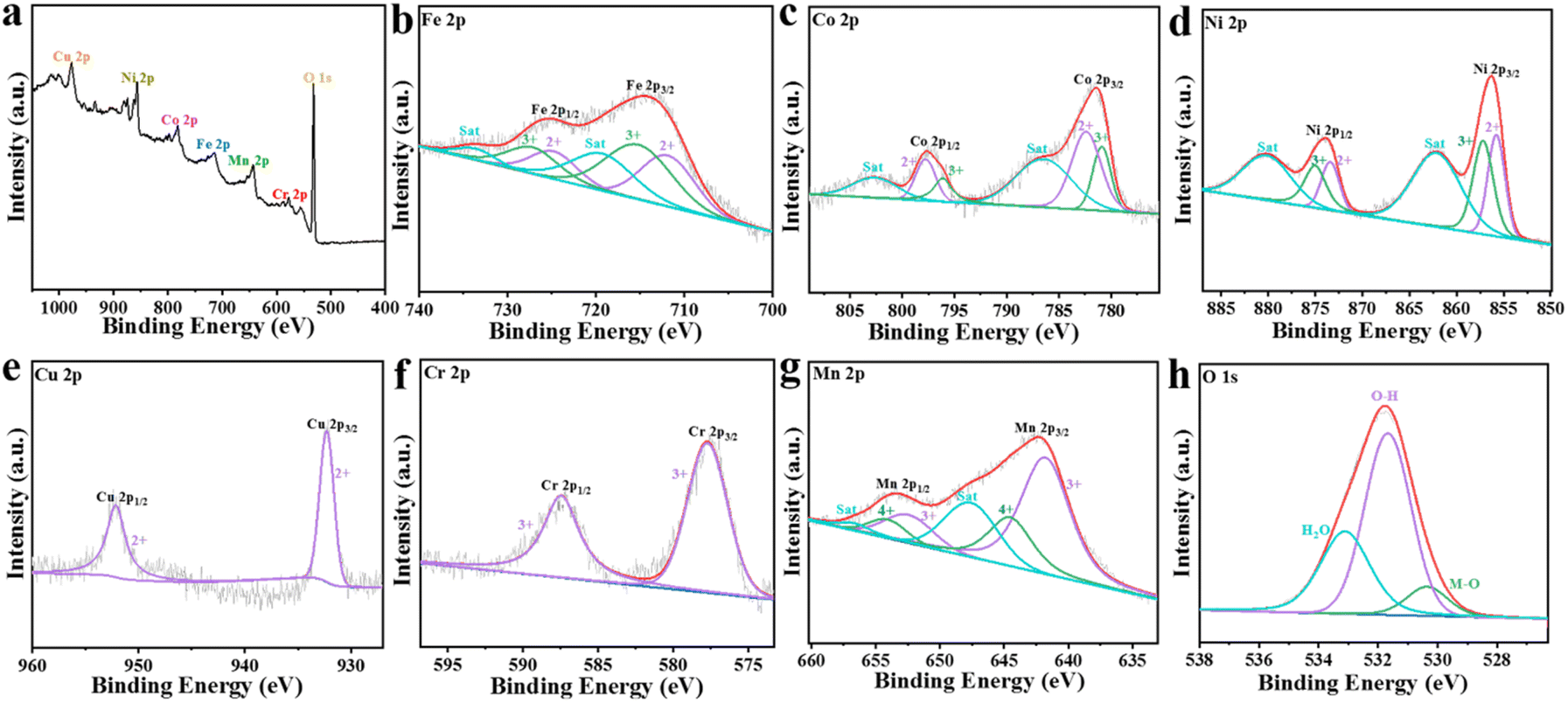 | ||
| Fig. 3 (a) XPS survey spectrum of the FeCoNiCuCrMn–OH catalyst. (b–h) XPS spectra of the elemental Fe, Co, Ni, Cu, Cr, Mn, and O in the catalyst: (b) Fe 2p, (c) Co 2p, (d) Ni 2p, (e) Cu 2p, (f) Cr 2p, (g) Mn 2p, (h) O 1s. | ||
In addition to FeCoNiCuCrMn–OH, FeCoNiCuCrZn–OH, FeCoNiCuCr–OH and FeCoNiCu–OH were also prepared by altering the metal salts, and the catalysts were characterized by SEM, SEM-EDS and XRD. The SEM images showed that the prepared HEHs had a hollow structure, and a certain number of nanosheets were distributed around it, and the elemental mapping further showed that the element distribution was uniform (Fig. S8 and S9†). At the same time, XRD comparison of all samples (Fig. S10†) confirmed that the prepared samples were high-entropy hydroxides, and no significant phase transition was caused by cation substitution. EDS showed (Table S1†) that the stoichiometric ratios of elements in the catalyst were the same as those of FeCoNiCuCrMn–OH. Through the above characterization, the results demonstrated that the samples FeCoNiCuCrZn–OH, FeCoNiCuCr–OH and FeCoNiCu–OH were also successfully prepared.
In addition, we further validated the properties of HEHs by analyzing the XPS of different materials to understand their electronic structures (Fig. S11–S14†). First, Fe, Co and Ni have been shown to have high OER activity in the literature. Therefore, the triple FeNiCo was chosen as the benchmark catalyst. As shown in Fig. S13,† the introduction of low-electronegative Cr into FeCoNiCu–OH makes the Fe, Co, Ni and Cu move towards the direction of low binding energy. Similarly, for FeCoNiCuCrZn–OH, the electronegativity order is Zn (1.65) < Cr (1.66) < Fe (1.83) < Co (1.88) < Ni (1.91), and differences in electronegativity between different metals result in strong local electron transfer (Fig. S14†). The strong local electron interaction caused by the electronegativity difference reduces the adsorption energy of the reactant intermediates and products, thus effectively enhancing the OER. In FeCoNiCuCrMn–OH, the electronegativity order is Mn (1.55) < Cr (1.66) < Fe (1.83) < Co (1.88) < Cu (1.90) < Ni (1.91). When these metals form HEH, the difference in electronegativity leads to the redistribution of the electron cloud density. Among them, Cu and Ni move towards lower binding energy, while Mn, Cr, Fe and Co move towards higher binding energy, increasing the local electron density at Ni and Cu sites and decreasing the local electron density at Mn, Co and Fe sites. Furthermore, the presence of Cr has been proven to increase the OER activity of the material. The introduction of Cr has a significant impact on the oxidation states of the active sites of Fe, Ni and Co, and enhances the OER activity by promoting the high oxidation states of the active elements (Co, Ni and Fe).46–48
Then, we mixed the synthesized HEHs with carbon black (Fig. S15†) for testing and study in 1 M KOH solution. Fig. 4a and S16† show the LSV curves of four kinds of prepared HEH catalysts and commercial RuO2. It can be seen that the FeCoNiCuCrMn–OH catalyst exhibited the best OER activity. In Fig. 4b, FeCoNiCuCrMn–OH showed the smallest overpotentials of 249 mV at 100 mA cm−2 and 268 mV at 200 mA cm−2. It is superior to FeCoNiCuCrZn–OH, FeCoNiCuCr–OH, and FeCoNiCu–OH, which further indicates that FeCoNiCuCrMn–OH has the best electrocatalytic OER activity. And, the Tafel slope of FeCoNiCuCrMn–OH is only 62.0 mV dec−1 (Fig. 4c). It was much lower than that of FeCoNiCuCrZn–OH (63.1 mV dec−1), FeCoNiCuCr–OH (86.7 mV dec−1), FeCoNiCu–OH (101.2 mV dec−1) and RuO2 (103.7 mV dec−1). Next, the OER reaction kinetics was evaluated using electrochemical impedance spectroscopy (EIS). As expected, the Nyquist diagram shows that the charge transfer resistance (Rct) at the FeCoNiCuCrMn–OH interface (Fig. 4d) is much lower than at the other reference electrodes. The Tafel slope and EIS diagram prove that FeCoNiCuCrMn–OH has excellent OER kinetics and fast electron transfer rate. The double-layer capacitance (Cdl) was measured to assess the electrochemical surface area (ECSA) of the sample, and the CV curves of the sample were recorded at different scanning rates (Fig. 4e, S17–S19 and Table S2†). The Cdl values of FeCoNiCuCrMn–OH, FeCoNiCuCrZn–OH, FeCoNiCuCr–OH and FeCoNiCu–OH in 1 M KOH solution were 22.42 mF cm−2, 14.62 mF cm−2, 12.34 mF cm−2 and 9.70 mF cm−2, respectively, indicating that FeCoNiCuCrMn–OH has more active sites than the other HEHs. Turnover frequency (TOF) curves and mass activities were calculated using all metals as active sites (Fig. S20 and S21†). When the overpotential is 300 mV, the TOF value of FeCoNiCuCrMn–OH is 0.383 s−1, which is much larger than that of FeCoNiCuCrZn–OH (0.095 s−1), FeCoNiCuCr–OH (0.056 s−1) and FeCoNiCu–OH (0.036 s−1), and the mass activity of FeCoNiCuCrMn–OH is 2.57 A mg−1, higher than that of FeCoNiCuCrZn–OH (0.62 A mg−1), FeCoNiCuCr–OH (0.39 A mg−1) and FeCoNiCu–OH (0.24 A mg−1). In summary, FeCoNiCuCrMn–OH exhibits the best OER catalytic performance in the prepared HEHs samples. In addition, FeCoNiCuCrMn–OH nanocages have excellent electrochemical properties compared to recently reported catalysts (Fig. 4f and Table S3†). At the same time, FeCoNiCuCrMn–OH has excellent stability, being maintained for over 400 h at 100 mA cm−2 (Fig. 4f).
 | ||
| Fig. 4 (a) OER LSV curves, (b) overpotential comparison, (c) Tafel slope values, (d) Nyquist plots, (e) double-layer capacitance (Cdl) measurements, (f) comparison of overpotentials (at 100 mA cm−2) of FeCoNiCuCrMn–OH with other electrocatalysts, and (g) the long-term stability test of FeCoNiCuCrMn–OH at 100 mA cm−2 in 1.0 M KOH. | ||
Inspired by the excellent OER activity in alkaline KOH solutions, we further investigated the OER performance of the catalysts in alkaline simulated seawater and alkaline natural seawater. Impressively, the catalytic activity of FeCoNiCuCrMn–OH did not decrease significantly in alkaline simulated seawater, which was very close to that in 1 M KOH (Fig. 5a, S22 and S23†). In the alkaline natural seawater, FeCoNiCuCrMn–OH still exhibits excellent catalytic activity, requiring only overpotentials as low as 275 mV and 292 mV at 100 mA cm−2 and 200 mA cm−2 (Fig. 5b, S24 and S25†), superior to most of the reported catalysts (Table S4†). The Nyquist impedance diagram and Tafel diagram can reveal the corrosion resistance of catalysts in different electrolyte systems. The Nyquist diagram (Fig. 5c) shows that the Rct at the interface of FeCoNiCuCrMn–OH in seawater is much lower than that of the other HEHs. The Tafel diagram of the catalyst in alkaline natural seawater is shown in Fig. 5d, from which the corrosion current density (Icorr) can be calculated. Generally, the lower Icorr indicates a better corrosion resistance performance.49 The results show that the corrosion current density of FeCoNiCuCrMn–OH is 1.69 × 10−5, which is much lower than that of FeCoNiCuCrZn–OH (2.12 × 10−5), FeCoNiCuCr–OH (2.38 × 10−5) and FeCoNiCu–OH (6.39 × 10−5). The stability of the electrocatalyst is very important in practical application. The durability of the FeCoNiCuCrMn–OH catalyst in alkaline natural seawater was evaluated by a continuous time–current test. Fig. 5e and S26† show that compared with the FeCoNiCuCr–OH and FeCoNiCu–OH catalysts, the FeCoNiCuCrMn–OH catalyst can run continuously for 400 h at 100 mA cm−2 without significant attenuation, indicating that it has high corrosion resistance and stability in alkaline natural seawater, and this excellent OER stability can be attributed to the phase stability induced by the high entropy effect. Therefore, OER activity and electrochemical durability tests have confirmed its great potential in the overall electrolysis of seawater. A series of structural characterization analyses were performed to investigate the structural evolution of FeCoNiCuCrMn–OH after stabilization tests. SEM and TEM images (Fig. S27–S29†) show that FeCoNiCuCrMn–OH still maintains the original shape of the hollow nanocage, and the element mapping image shows that all elements are evenly distributed. The XRD pattern shows that the diffraction peak corresponding to FeCoNiCuCrMn–OH has no obvious displacement, and no other diffraction peaks appear. The results showed that the morphology and crystal structure of HEHs remained basically unchanged after the reaction, which proved its outstanding stability. After the long-term seawater oxidation reaction, we also measured hypochlorite (ClO−) in the electrolyte solution by the o-tolidine method (Fig. S30†) and colorimetry (Fig. S31†) in order to verify that the inhibition of chlorine chemistry led to an increase in catalyst lifetime. However, hypochlorite was not detected, indicating that FeCoNiCuCrMn–OH has excellent corrosion resistance and high selectivity to seawater oxidation in alkaline seawater.50–52 In addition, in order to evaluate the overall water splitting stability of the FeCoNiCuCrMn–OH catalyst, we conducted a comprehensive seawater electrolysis test by combining FeCoNiCuCrMn–OH with the HER catalyst (Pt/C). As shown in Fig. S32,† under the room temperature condition of 1.0 M KOH + seawater, the assembled double-electrode electrolytic cell can maintain excellent performance during the 200 hours electrolysis process at a current density of 100 mA cm−2 without obvious degradation, demonstrating good stability in seawater electrolysis.
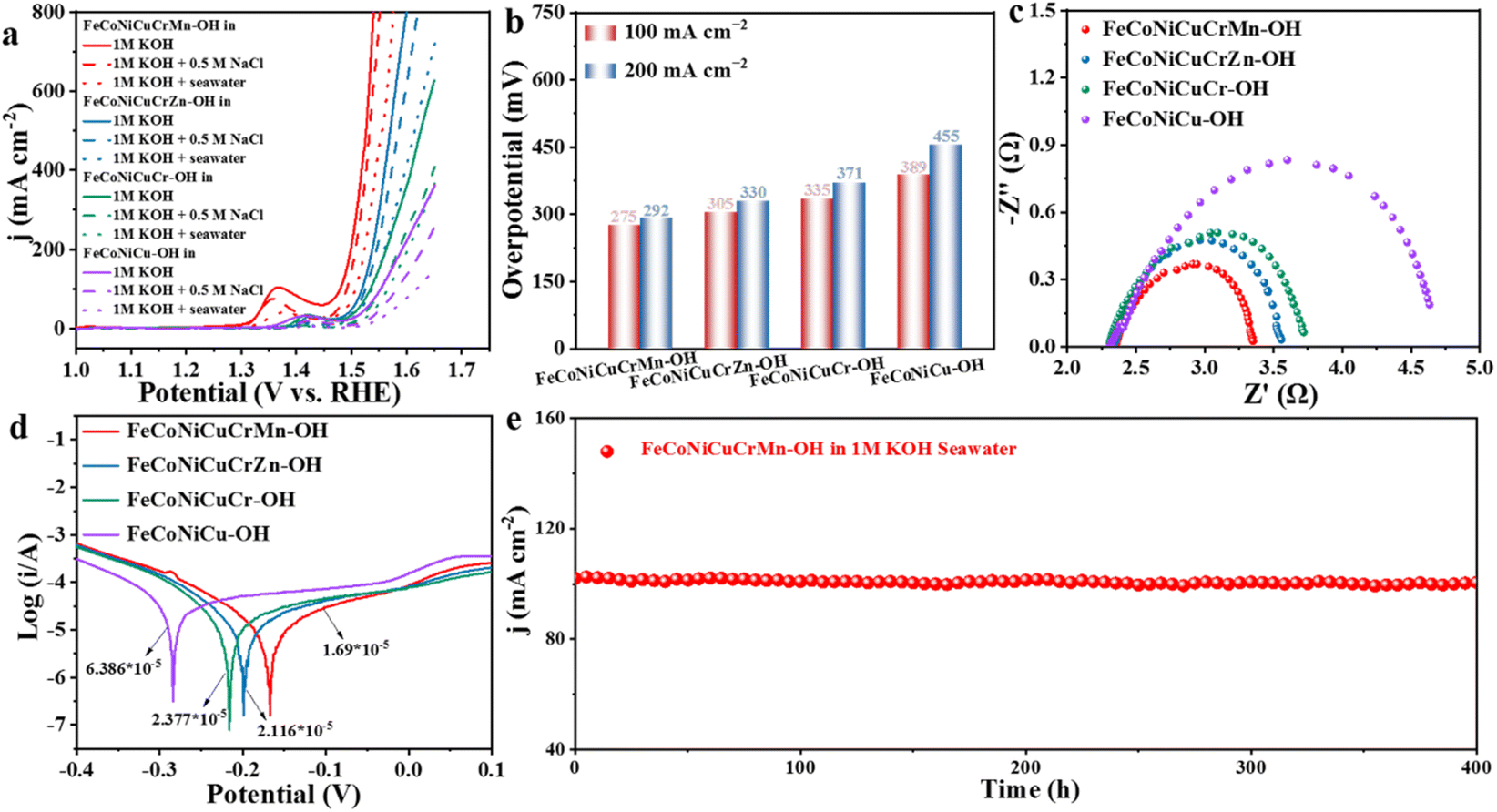 | ||
| Fig. 5 (a) LSV curves in different electrolytes, (b) Overpotential comparison, (c) and (d) Nyquist impedance plots and Tafel plots in 1.0 M KOH seawater, and (e) the long-term OER stability test of FeCoNiCuCrMn–OH at 100 mA cm−2 in 1.0 M KOH seawater. | ||
In order to further understand the catalytic reaction, the chemical state of the hollow FeCoNiCuCrMn–OH catalyst was analyzed. A slight shift towards high binding energy was observed (Fig. S33†), indicating that the oxidation state increased after the electrochemical reaction.29,53 And, the O 1s spectrum indicates that the M–O bond strength increases after the OER. According to the catalytic mechanism of the OER under alkaline conditions, the catalyst reacts with OH− to release O2, and a hydroxyl group is then adsorbed to form a hydroperoxy compound. In this process, hydrogen–oxygen transition metals provide an abundance of high-valence metals (Co3+, Ni3+, and Fe3+) as catalytically active sites for direct catalytic OER processes. Based on the above results, hydroxides with high valence states are defined as the catalytic active sites of FeCoNiCuCrMn–OH nanocages.
Further, in order to reveal the reaction mechanism of the catalyst, in situ Raman analysis of the FeCoNiCuCrMn–OH catalyst was conducted under 532 nm laser excitation, and the oxidation evolution of active species under different working voltages (1.2–1.6 V vs. RHE) was further monitored. In Fig. S34,† without applying any bias pressure, the catalyst shows two major Raman peaks at 450 and 526 cm−1, which is attributed to the vibrations of M–O and M–OH in the metal hydroxide. When the voltage increases to 1.2 V, the Raman shift remains unchanged. Until 1.3 V and above, the truly active species (MIII–OOH) appeared at 473 cm−1 (δMIII–O) and 557 cm−1 (νMIII–O). With the increase of the potential, the Raman signal of MIII–O vibration shows a continuous enhancement, the Raman peak widens and shifts, indicating an increase in the bonding strength of the M–O bond, representing the formation and accumulation of the M–OOH material on the surface, which is a key active site required for the subsequent conversion to O2 in the OER.28,29,44,54 In addition, the peak at 1038 cm−1 corresponds to the adsorbed OH* at the metal site.55 Meanwhile, as the potential increases, the peak intensity of OH* gradually strengthens, and more OH* is adsorbed on the surface of the catalyst, which is conducive to the progress of the OER. Moreover, the M–Cl absorption peak of the catalyst was not observed near 190 cm−1, indicating that the interaction between the FeCoNiCuCrMn–OH catalyst and Cl− is relatively weak.56 This is mainly because during the reaction process, a stable hydroxide passivation layer forms on the surface of HEH, which hinders the contact between Cl− and the active site. Finally, the high entropy stability suppressed the erosion of Cl− and improved the corrosion resistance of the catalyst. Traditional catalysts are prone to erosion by Cl−, resulting in the dissolution of active sites. However, the high configurational entropy of HEH can suppress element segregation and maintain structural integrity.
To further reveal the relationship between the OER performance and the electronic structure of the catalyst, we have carried out theoretical calculations. The HEH structure model was constructed based on XRD results. In nickel-based hydroxide electrocatalysts, high-valence nickel cations are considered as active species, and Ni sites are high in content, so Ni sites are selected as the main reactive sites (Fig. S34 and S35†).29,57,58 To better understand the localized electronic environment, we calculated the d-orbital bias density of states (d-PDOS). As shown in Fig. S36,† the d orbitals of Ni in the four catalysts all have high state density distribution near the Fermi level, which means that these catalysts have rich electron density and fast electron transport speed.57 The catalytic performance is closely related to the adsorption energy of the reaction intermediates (i.e. *OH, *O and *OOH), so the OER free energy is calculated (Fig. 6a). Fig. 6b shows the energy barrier at each step, it can be inferred that the rate-determining step (RDS) of FeCoNiCu–OH and FeCoNiCuCr–OH is the step from M–*OH to M–*O. However, the step from M–*O to M–*OOH is the RDS of FeCoNiCuCrMn–OH. The corresponding overpotential (η) derived from the RDS is 0.59 V (FeCoNiCu–OH), 0.52 V (FeCoNiCuCr–OH), and 0.22 V (FeCoNiCuCrMn–OH). Therefore, the addition of Cr and Mn elements on the basis of the four-element alloy (FeCoNiCu–OH) reduces the overpotential of the RDS during the OER.59–61 The experimental results of overpotential are in agreement with the conclusion of theoretical calculation. We further calculated the OER and CER free energy barriers of the FeCoNiCu–OH, FeCoNiCuCr–OH and FeCoNiCuCrMn–OH catalysts at U = 1.23 V. As shown in Fig. 6c and d, the energy barrier required for the reaction intermediate to change from *O → *OCl is significantly larger than that from *O → *OOH, indicating that the interaction between the catalyst and Cl− is relatively weak.62,63 Furthermore, the energy barrier value of *O → *OCl of FeCoNiCuCrMn–OH is significantly greater than that of FeCoNiCu–OH and FeCoNiCuCr–OH, indicating that FeCoNiCuCrMn–OH has a strong ability to inhibit the adsorption of Cl− while selectively adsorbing OH−. In addition, FeCoNiCuCrMn–OH adsorbed OH− stronger than Cl−, proving that the samples have excellent OER selectivity in seawater,64,65 which is favourable to prevent Cl− corrosion and undesirable CER (Fig. 6e and f). Therefore, the experimental results described above show that the synthesized catalyst has excellent selectivity and stability.
 | ||
| Fig. 6 (a) Proposed reaction mechanism of the OER on FeCoNiCuCrMn–OH. Reaction free energy diagrams for the (b) OER at U = 0 V, (c) OER and (d) CER at U = 1.23 V on different catalyst surfaces. Adsorption energy of (e) *Cl and (f) *OH on different catalyst surfaces. | ||
Conclusions
In conclusion, a series of high entropy hollow nanocages with high OER catalytic activity and stability were synthesized through a simple CEP method. Among them, FeCoNiCuCrMn–OH can provide 100 mA cm−2 with an overpotential of only 275 mV in seawater, and can remain stable at 100 mA cm−2 for at least 400 hours. This excellent catalytic performance stems from the multiple synergistic mechanisms between the high-entropy effect and the hollow structure. Firstly, the three-dimensional open structure of the hollow nanocage provides abundant mass transfer channels and a relatively high specific surface area, fully exposing the active sites. Secondly, the high entropy effect of the multi-metallic components induces lattice distortion and electronic structure reconstruction, optimizing the D-band electronic structure of the active centers of Fe, Ni, and Co, and significantly enhancing the adsorption capacity of *OH. Theoretical calculations further confirm that the selective adsorption characteristics of the high-entropy material effectively suppress the competitive adsorption of Cl−, thereby ensuring high selectivity for the catalyst in seawater. This study not only provides a simple method for the preparation of high-entropy catalysts, but also offers important guidance for the design of a new generation of high-performance electrocatalysts by revealing the synergistic mechanism of “component entropy nanostructure”.Data availability
The data that support the findings of this study are available on request from the corresponding author.Author contributions
L. W. and H. L. conceived and supervised the project. R. C., Y. P., Q. Y., R. L., and H. L. conducted the experiments. R. C., Y. P., Y. Y., H. L., Y. D., K. L., Z. W., J. L. analysed the data and contributed to writing the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52472217, 52272222 and 22302108), Taishan Scholar Young Talent Program (tsqn201909114), Youth Innovation and Technology Foundation of Shandong Higher Education Institutions, China (2023KJ313), Natural Science Foundation of Qingdao (23-2-1-12-zyyd-jch), Qingdao Postdoctoral Researcher Applied Research Project (QDBSH20220202043), and Qingdao University of Science and Technology 2024 innovative training program for college students (202410426063).References
- L. Zhuang, J. Li, K. Wang, Z. Li, M. Zhu and Z. Xu, Adv. Funct. Mater., 2022, 32, 2201127 CrossRef CAS.
- X. Xing, R. Wang, N. Bauer, P. Ciais, J. Cao, J. Chen, X. Tang, L. Wang, X. Yang, O. Boucher, D. Goll, J. Peñuelas, I. A. Janssens, Y. Balkanski, J. Clark, J. Ma, B. Pan, S. Zhang, X. Ye, Y. Wang, Q. Li, G. Luo, G. Shen, W. Li, Y. Yang and S. Xu, Nat. Commun., 2021, 12, 3159 CrossRef CAS PubMed.
- Y. N. Zhou, W. L. Yu, H. J. Liu, R. Y. Fan, G. Q. Han, B. Dong and Y. M. Chai, EcoEnergy, 2023, 1, 425–436 CrossRef.
- Y. Gu, N. Nie, J. Liu, Y. Yang, L. Zhao, Z. Lv, Q. Zhang and J. Lai, EcoEnergy, 2023, 1, 405–413 CrossRef.
- Q. Zhou, L. Liao, H. Zhou, D. Li, D. Tang and F. Yu, Mater. Today Phys., 2022, 26, 100727 CrossRef CAS.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- J. Huang, B. Hu, J. Meng, T. Meng, W. Liu, Y. Guan, L. Jin and X. Zhang, Energy Environ. Sci., 2024, 17, 1007–1045 RSC.
- G. Gao, Z. Sun, X. Chen, G. Zhu, B. Sun, X. Long Huang, H. K. Liu and S. X. Dou, Coord. Chem. Rev., 2024, 509, 215777 CrossRef CAS.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202210753 CrossRef CAS PubMed.
- S. Zhang, W. Xu, H. Chen, Q. Yang, H. Liu, S. Bao, Z. Tian, E. Slavcheva and Z. Lu, Adv. Mater., 2024, 36, e2311322 CrossRef PubMed.
- X. Xiao, L. Yang, W. Sun, Y. Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, 2105830 CrossRef CAS PubMed.
- C. Wang, H. Shang, L. Jin, H. Xu and Y. Du, Nanoscale, 2021, 13, 7897–7912 RSC.
- Z. Chen, N. Han, W. Wei, D. Chu and B. J. Ni, EcoEnergy, 2024, 2, 114–140 CrossRef.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, eScience, 2023, 3, 100111 CrossRef.
- H.-M. Zhang, L. Zuo, Y. Gao, J. Guo, C. Zhu, J. Xu and J. Sun, J. Mater. Sci. Technol., 2024, 173, 1–10 CrossRef CAS.
- F. Zhang, L. Yu, L. Wu, D. Luo and Z. Ren, Trends Chem., 2021, 3, 485–498 CrossRef CAS.
- X. H. Wang, Y. Ling, B. Wu, B. L. Li, X. L. Li, J. L. Lei, N. B. Li and H. Q. Luo, Nano Energy, 2021, 87, 106160 CrossRef CAS.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- C. Huang, Z. Wang, S. Cheng, Y. Liu, B. Deng, S. Xu, L. Yu and Y. Yu, Sci. China: Chem., 2024, 67, 3198–3208 CrossRef CAS.
- Y. S. Park, J.-Y. Jeong, M. J. Jang, C.-Y. Kwon, G. H. Kim, J. Jeong, J.-H. Lee, J. Lee and S. M. Choi, J. Energy Chem., 2022, 75, 127–134 CrossRef CAS.
- L. Wu, L. Yu, Q. Zhu, B. McElhenny, F. Zhang, C. Wu, X. Xing, J. Bao, S. Chen and Z. Ren, Nano Energy, 2021, 83, 105838 CrossRef CAS.
- W. Liu, K. Jiang, Y. Hu, Q. Li, Y. Deng, J. Bao and Y. Lei, J. Colloid Interface Sci., 2021, 604, 767–775 CrossRef CAS PubMed.
- Y. Yao, C. Yang, S. Sun, H. Zhang, M. Geng, X. He, K. Dong, Y. Luo, D. Zheng, W. Zhuang, S. Alfaifi, A. Farouk, M. S. Hamdy, B. Tang, S. Zhu, X. Sun and W. Hu, Small, 2023, 20, 2307294 CrossRef PubMed.
- R. A. Marquez, E. Kalokowski, M. Espinosa, J. T. Bender, Y. J. Son, K. Kawashima, C. E. Chukwuneke, L. A. Smith, H. Celio, A. Dolocan, X. Zhan, N. Miller, D. J. Milliron, J. Resasco and C. B. Mullins, Energy Environ. Sci., 2024, 17, 2028–2045 RSC.
- S. Chen, Y. Zhuo, X. Wang, S. Li, J. Lu, D. Liu, H. Pan and Z. Wang, Coord. Chem. Rev., 2024, 510, 215832 CrossRef CAS.
- L.-F. Li, Y.-F. Li and Z.-P. Liu, ACS Catal., 2020, 10, 2581–2590 CrossRef CAS.
- P. W. Menezes, S. Yao, R. Beltran-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- T. X. Nguyen, C.-C. Tsai, V. T. Nguyen, Y.-J. Huang, Y.-H. Su, S.-Y. Li, R.-K. Xie, Y.-J. Lin, J.-F. Lee and J.-M. Ting, Chem. Eng. J., 2023, 466, 143352 CrossRef CAS.
- Y. Xin, S. Li, Y. Qian, W. Zhu, H. Yuan, P. Jiang, R. Guo and L. Wang, ACS Catal., 2020, 10, 11280–11306 CrossRef CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- H. Li, M. Sun, Y. Pan, J. Xiong, H. Du, Y. Yu, S. Feng, Z. Li, J. Lai, B. Huang and L. Wang, Appl. Catal., B, 2022, 312, 121431 CrossRef CAS.
- X. Tian, H. Li, R. Chang, Y. Yang, Z. Wang, T. Dong, J. Lai, S. Feng and L. Wang, J. Mater. Chem. A, 2024, 12, 3276–3282 RSC.
- Y. Zhai, X. Ren, T. Gan, L. She, Q. Guo, N. Yang, B. Wang, Y. Yao and S. Liu, Adv. Energy Mater., 2025, 2502065 CrossRef.
- A. Roy, S. Kumar, A. Guilherme Buzanich, C. Prinz, E. Gotz, A. Retzmann, T. Hickel, B. Bhattacharya and F. Emmerling, Adv. Mater., 2024, 36, e2408114 CrossRef PubMed.
- K. Huang, Y. Sun, Y. Zhang, X. Wang, W. Zhang and S. Feng, Adv. Mater., 2018, 31, 1801430 CrossRef PubMed.
- Z. Li, M. Song, W. Zhu, W. Zhuang, X. Du and L. Tian, Coord. Chem. Rev., 2021, 439, 213946 CrossRef CAS.
- L. Tian, Y. Liu, C. He, S. Tang, J. Li and Z. Li, Chem. Rec., 2022, 23, e202200213 CrossRef PubMed.
- Y. Pan, H. Li, J. Xiong, Y. Yu, H. Du, S. Li, Z. Wu, S. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 306, 121111 CrossRef CAS.
- J. Huang, S. Wang, J. Nie, C. Huang, X. Zhang, B. Wang, J. Tang, C. Du, Z. Liu and J. Chen, Chem. Eng. J., 2021, 417, 128055 CrossRef CAS.
- L. Zheng, Y. Zhong, J. Cao, M. Liu, Y. Liao, H. Xu, S. Chen, F. Xiong, Y. Qing and Y. Wu, Small, 2023, 20, 2308928 CrossRef PubMed.
- H. Wang, G. Zhan, C. Tang, D. Yang, W. Liu, D. Wang, Y. Wu, H. Wang, K. Liu, J. Li, M. Huang and K. Chen, ACS Nano, 2023, 17, 4790–4799 CrossRef CAS PubMed.
- X. Luo, N. Xu, Y. Zhou, X. Yang, W. Yang, G. Liu, J. K. Lee and J. Qiao, eScience, 2024, 4, 100290 CrossRef.
- L. Liu, T. Liu, C. Xu, W. Zhao, J. Fan, J. Liu, X. Ma and W. Fu, Nano Lett., 2024, 24, 2831–2838 CrossRef CAS PubMed.
- W. Cao, J. Wu, C. Zhou, X. Gao, E. Hu, J. Zhang and Z. Chen, Small, 2024, 20, e2309769 CrossRef PubMed.
- A. Abdelhafiz, B. Wang, A. R. Harutyunyan and J. Li, Adv. Energy Mater., 2022, 12, 2200742 CrossRef CAS.
- Z. J. Chen, T. Zhang, X. Y. Gao, Y. J. Huang, X. H. Qin, Y. F. Wang, K. Zhao, X. Peng, C. Zhang, L. Liu, M. H. Zeng and H. B. Yu, Adv. Mater., 2021, 33, 2101845 CrossRef CAS PubMed.
- P. Ma, S. Zhang, M. Zhang, J. Gu, L. Zhang, Y. Sun, W. Ji and Z. Fu, Sci. China Mater., 2020, 63, 2613–2619 CrossRef.
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang, Z. Tian, Z. Lu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 22740–22744 CrossRef CAS PubMed.
- X. Duan, Q. Sha, P. Li, T. Li, G. Yang, W. Liu, E. Yu, D. Zhou, J. Fang, W. Chen, Y. Chen, L. Zheng, J. Liao, Z. Wang, Y. Li, H. Yang, G. Zhang, Z. Zhuang, S. F. Hung, C. Jing, J. Luo, L. Bai, J. Dong, H. Xiao, W. Liu, Y. Kuang, B. Liu and X. Sun, Nat. Commun., 2024, 15, 1973 CrossRef CAS PubMed.
- Y. Zhang, W. Zhang, X. Zhang, X. Wang, J. Wang, Q. Zhao, Y. Sun, J. Li, G. Liu and X. Han, Energy Environ. Sci., 2024, 17, 3347–3357 RSC.
- C. Yang, Z. Cai, J. Liang, K. Dong, Z. Li, H. Sun, S. Sun, D. Zheng, H. Zhang, Y. Luo, Y. Yao, Y. Wang, Y. Ren, Q. Liu, L. Li, W. Chu, X. Sun and B. Tang, Nano Res., 2024, 17, 5786–5794 CrossRef CAS.
- Y. Yang, L. Dang, M. J. Shearer, H. Sheng, W. Li, J. Chen, P. Xiao, Y. Zhang, R. J. Hamers and S. Jin, Adv. Energy Mater., 2018, 8, 1703189 CrossRef.
- Y. Yuan, Z. Li, X. He, H. Wang, H. Tang, X. Tang, Q. Dai, D. Zheng, S. Sun, Y. Luo, M. S. Hamdy, F. A. Ibrahim, T. Li, B. Tang and X. Sun, Mater. Today Phys., 2024, 49, 101592 CrossRef CAS.
- Z. Zhao, J. Sun, X. Li, S. Qin, C. Li, Z. Zhang, Z. Li and X. Meng, Nat. Commun., 2024, 15, 7475 CrossRef CAS PubMed.
- C. Zhang, P. Shan, Y. Zou, T. Bao, X. Zhang, Z. Li, Y. Wang, G. Wei, C. Liu and C. Yu, Nat. Sustain., 2025, 8, 542–552 CrossRef.
- C. Feng, Y. Zhou, M. Chen, L. Zou, X. Li, X. An, Q. Zhao, P. Xiaokaiti, A. Abudula, K. Yan and G. Guan, Appl. Catal., B, 2024, 349, 123875 CrossRef CAS.
- Z. Qiu, C.-W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581 RSC.
- P. Li, B. Wu, K. Du, Z. Liu, E. Gao, H. Yin and D. Wang, ACS Sustainable Chem. Eng., 2023, 11, 14246–14254 CrossRef CAS.
- F. Wang, P. Zou, Y. Zhang, W. Pan, Y. Li, L. Liang, C. Chen, H. Liu and S. Zheng, Nat. Commun., 2023, 14, 6019 CrossRef CAS PubMed.
- D. Liu, X. Yan, P. Guo, Y. Yang, Y. He, J. Liu, J. Chen, H. Pan and R. Wu, ACS Catal., 2023, 13, 7698–7706 CrossRef CAS.
- J. Wang, Y. Liu, G. Yang, Y. Jiao, Y. Dong, C. Tian, H. Yan and H. Fu, Nat. Commun., 2025, 16, 1319 CrossRef CAS PubMed.
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Nat. Sustainability, 2024, 7, 158–167 CrossRef.
- Y. Yang, J. Li, W. Qiao, H. Yang, Y. Huang, F. Li, Y. Yu, J. Fang and P. Li, ACS Catal., 2025, 15, 6954–6968 CrossRef CAS.
- N. Wang, P. Ou, S. F. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao, Y. Yan, J. Zhang, Z. Wang, R. Dorakhan, A. Badreldin, A. Abdel-Wahab, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Adv. Mater., 2023, 35, e2210057 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01961c |
| This journal is © The Royal Society of Chemistry 2025 |