
Steady Cu+ species via magnesium and boron co-modification for enhanced CO2 electroreduction to C2+ products: an in situ Raman spectroscopic study†
Hua
Yang
a,
Xuefan
Mu
a,
Jiexin
Guan
a,
Bo
Ouyang
b,
Huaming
Li
 a and
Yilin
Deng
*a
a and
Yilin
Deng
*a
aInstitute for Energy Research, Jiangsu University, Zhenjiang 212013, China. E-mail: yldeng@ujs.edu.cn
bDepartment of Applied Physics and Institution of Energy and Microstructure, Nanjing University of Science and Technology, Nanjing 210094, China
First published on 17th May 2024
Abstract
The electrochemical carbon dioxide reduction reaction (CO2RR) to produce high-value multi-carbon (C2+) compounds holds significant practical importance in realizing carbon neutrality. Copper-based electrocatalysts are promising for CO2-to-C2+ conversion. However, the labile Cu valence at high current densities impedes C2+ product generation. Here, we present an electrocatalyst derived from CuO, featuring a heterostructure of Cu/Cu2O/CuO/Mg(OH)2via the co-addition of Mg and B during the preparation (referred to as Cu5(B0.02 M)Mg1). The Cu5(B0.02 M)Mg1 shows an impressive C2+ yield, with a Faraday efficiency (FEC2+) of 79.59% at −1.57 V vs. RHE (reversible hydrogen electrode). Additionally, the partial current density of C2+ on the Cu5(B0.02 M)Mg1 catalyst is −317.03 mA cm−2, 2.7 and 3.5 times higher than those catalysts lacking B (Cu5Mg1 catalyst) or Mg (Cu(B0.02 M) catalyst), respectively. Over a wide potential range of 600 mV between −1.17 and −1.77 V vs. RHE, the overall FEC2+ surpasses 60% on the Cu5(B0.02 M)Mg1 catalyst. X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and high-resolution transmission electron microscopy (HRTEM) characterization studies indicate the NaBH4 reactant could promote the formation of crystalline Mg(OH)2 in the catalyst structure, which is found to better stabilize Cu+ at negative potentials compared to the amorphous phase. Further in situ Raman spectroscopy reveals that at increasingly negative potentials, the higher copper species (Cu2+) is inevitably reduced to the lower copper species (Cu0/Cu+). However, the synergy of Mg and B prolongs the presence of Cu+ on the catalyst surface across a broad potential range, and *CO and *CO2− could still be recorded at quite negative reduction potentials. This suggests enhanced binding strength of *CO intermediates on the catalyst surface, promoting the C–C coupling process.
1. Introduction
The electrocatalytic carbon dioxide reduction reaction (CO2RR) represents a pathway for producing high-value-added chemicals and fuels, offering a means to reduce global carbon dioxide (CO2) emissions and alleviate the shortage of non-renewable carbon resources.1–3 The CO2RR yields primarily hydrocarbons and oxygen-containing compounds. Among them, multi-carbon (C2+) products such as ethylene (C2H4) and ethanol (C2H5OH) have broader industrial applications and higher energy densities compared with C1 products like carbon monoxide (CO) and methane (CH4), thus commanding a higher economic value per unit mass.4–6 However, challenges persist due to the requirement of a significant amount of energy to break the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with the high dissociation energy of CO2 (about 750 kJ mol−1). Furthermore, the multi-step proton-coupling process contributes to the poor selectivity and yield of target products during the CO2RR.7–9 Addressing these issues requires the exploration and design of electrocatalysts with enhanced efficiency to facilitate CO2 conversion while minimizing product distribution variability during the CO2RR process.
O bond with the high dissociation energy of CO2 (about 750 kJ mol−1). Furthermore, the multi-step proton-coupling process contributes to the poor selectivity and yield of target products during the CO2RR.7–9 Addressing these issues requires the exploration and design of electrocatalysts with enhanced efficiency to facilitate CO2 conversion while minimizing product distribution variability during the CO2RR process.
To date, copper-based materials, which have been found crucial for C–C coupling, remain the major electrocatalysts for efficiently converting CO2 to long-chain hydrocarbons and multi-carbon oxygenates.10–12 Previous studies suggest that Cu+ species on Cu-based materials could enhance the selectivity of C2+ products.13,14 However, under high-current electrolysis, particularly at the negative potentials required for C2+ product formation, the catalyst structure becomes unstable, and Cu+ is readily reduced to Cu0 during the CO2RR. Consequently, prolonging the lifetime of Cu+ at reduction potentials is a current research focus. To preserve Cu+ during the CO2RR so as to enhance C2+ formation, essential steps involve generating Cu+ sites and optimizing intermediate adsorption. Researchers have developed several strategies for this purpose, including doping modification,15,16 addition of different metallic elements,17,18 heterogeneous interface engineering,19,20 oxidation state regulation,21–23 defect control,21,24–26etc. Until now, nonmetallic elements such as B, C, F, and S have been reported to effectively regulate the electronic structure and stabilize Cu+ in Cu lattices, facilitating the reduction of CO2 to C2+ products at large current densities.27–29 For example, Yao et al. devised a B-Cu2O catalyst which showed high selectivity for ethanol product. Their oxygen programmed desorption and density functional theory (DFT) calculations confirmed that B prevented lattice oxygen from separating out from Cu2O, thereby stabilizing Cu+ species on the B-Cu2O surface. This was believed to strengthen Cu–O bonds, significantly enhancing C–C coupling and facilitating the formation of C2+ products.30 Alkaline-earth metals such as Mg, Ba and Ca were found to not only maintain Cu+ on catalyst surfaces, but also enhance the adsorption of intermediates at active centers, thereby influencing the reaction kinetics of the CO2RR process.31 For instance, Peng et al. synthesized a nano-Cu2Mg intermetallic catalyst predominantly oriented along the (111) crystal face on a carbon black substrate (denoted as Cu2Mg(111)). In situ Raman spectroscopy and DFT calculations confirmed that the Cu3δ−–Mgδ+ active sites in Cu2Mg(111) enhanced the coverage of *CO on the catalyst surface, lowered the energy barrier for *CO–CO coupling, and stabilized the *CHCHOH intermediates to promote ethanol production.32 In addition, Xie et al. engineered a Mg-modified CuxO nanoparticle catalyst targeting the C2H4 product. In situ XRD, in situ Raman spectroscopy and DFT calculations demonstrated that the amorphous Mg(OH)2 species stabilized Cu+ species, promoting the presence of vital high *CO intermediates for C2+ products.33 However, challenges still exist regarding these Cu-based materials specially designed for C2+ products. Under current modification measures, Cu+ has a relatively short lifetime and could only exist within a narrow potential window, significantly limiting its effectiveness during the CO2RR. Furthermore, there is a lack of literature on the combined effects of alkaline-earth metals and non-metals on copper catalysts. Additionally, the influence of the crystalline or amorphous state of alkaline-earth metal oxides on the stability of Cu+ has not been thoroughly studied.
Based on the above considerations, we have designed a B-Cu/Cu2O/CuO/Mg(OH)2 catalyst through a simple two-step protocol involving calcination and wet chemical reduction, aiming to enhance C2+ products at high current densities across wide potential ranges. The target catalyst is referred to as Cu5(B0.02 M)Mg1, named according to the content of the elements during its preparation. X-ray diffraction (XRD), high-resolution transmission electron microscopy (HRTEM) and ex situ X-ray photoelectron spectroscopy (XPS) reveal that the addition of NaBH4 during catalyst synthesis could promote the formation of crystalline Mg(OH)2 in the catalyst structure, which we suggest would better stabilize Cu+ compared to the amorphous Mg(OH)2 phase. Further in situ Raman investigation confirms that Cu+ on the Cu5(B0.02 M)Mg1 catalyst could persist across a much broader potential window compared to those catalysts lacking crystalline Mg(OH)2 or B. Additionally, the *CO intermediate, a significant precursor of C2+ products, is recorded on the Cu5(B0.02 M)Mg1 surface even at rather negative potentials during the CO2RR. Benefiting from the stabilized Cu+, Cu5(B0.02 M)Mg1 shows high selectivity towards C2+ within −1.17 to −1.77 V vs. RHE (unless specified, all potentials in this work are referenced against RHE). Overall, our work reveals the significance of morphologies of Mg(OH)2 in Cu+ stabilization and provides a comprehensive understanding of the role of Cu+ in the efficient CO2RR towards C2+ products.
2. Results and discussion
2.1 Morphological and structural characterization of the catalysts
The typical fabrication process of Cu5(B0.02 M)Mg1 is depicted in Fig. 1, with detailed synthesis described in the ESI.† The XRD pattern indicates diffraction peaks of Cu, Cu2O, CuO and Mg(OH)2 in the Cu5(B0.02 M)Mg1 catalyst (Fig. 2a). The characteristic peaks at 42.3°, 50.4°, and 74.1° correspond to the (111), (200) and (220) planes of Cu (PDF#04-0836), respectively. Additionally, four peaks at 29.5°, 36.4°, 42.3° and 61.3° correspond to the (110), (111), (200) and (220) planes of Cu2O (PDF#05-0667), respectively. Peaks at 35.4°, 39.7°, 48.7°, 53.5° and 66.4° are attributed to the (002), (111), (202), (020) and (310) planes of CuO (PDF#48-1548), respectively. Furthermore, peaks at 18.5°, 32.9°, 58.6° and 68.2° align well with the (001), (100), (110) and (103) planes of Mg(OH)2 (PDF#44-1482), respectively. The XRD pattern demonstrates that the Cu5(B0.02 M)Mg1 catalyst mainly comprises Cu, Cu2O, CuO and Mg(OH)2 crystal phases. On the other hand, XRD patterns of the corresponding reference Cu5Mg1, Cu5(B0.1 M)Mg1, Cu(B0.02 M), Cu10(B0.02 M)Mg1 and Cu1(B0.02 M)Mg1 catalysts are provided in Fig. S1.† The Cu5(B0.1 M)Mg1, Cu10(B0.02 M)Mg1 and Cu1(B0.02 M)Mg1 catalysts also consist of Cu, Cu2O, CuO and Mg(OH)2. The Cu(B0.02 M) catalyst comprises three phases: Cu, Cu2O and CuO. Notably, for the Cu5Mg1 catalyst (lacking the NaBH4 reactant during its preparation), XRD only reveals the existence of CuO (PDF#48-1548), while the Mg(OH)2 (PDF#44-1482) crystalline phases are absent. We propose that this results from the generation of amorphous Mg(OH)2 rather than crystalline Mg(OH)2 when the reducing agent NaBH4 is absent (Fig. S1a†).34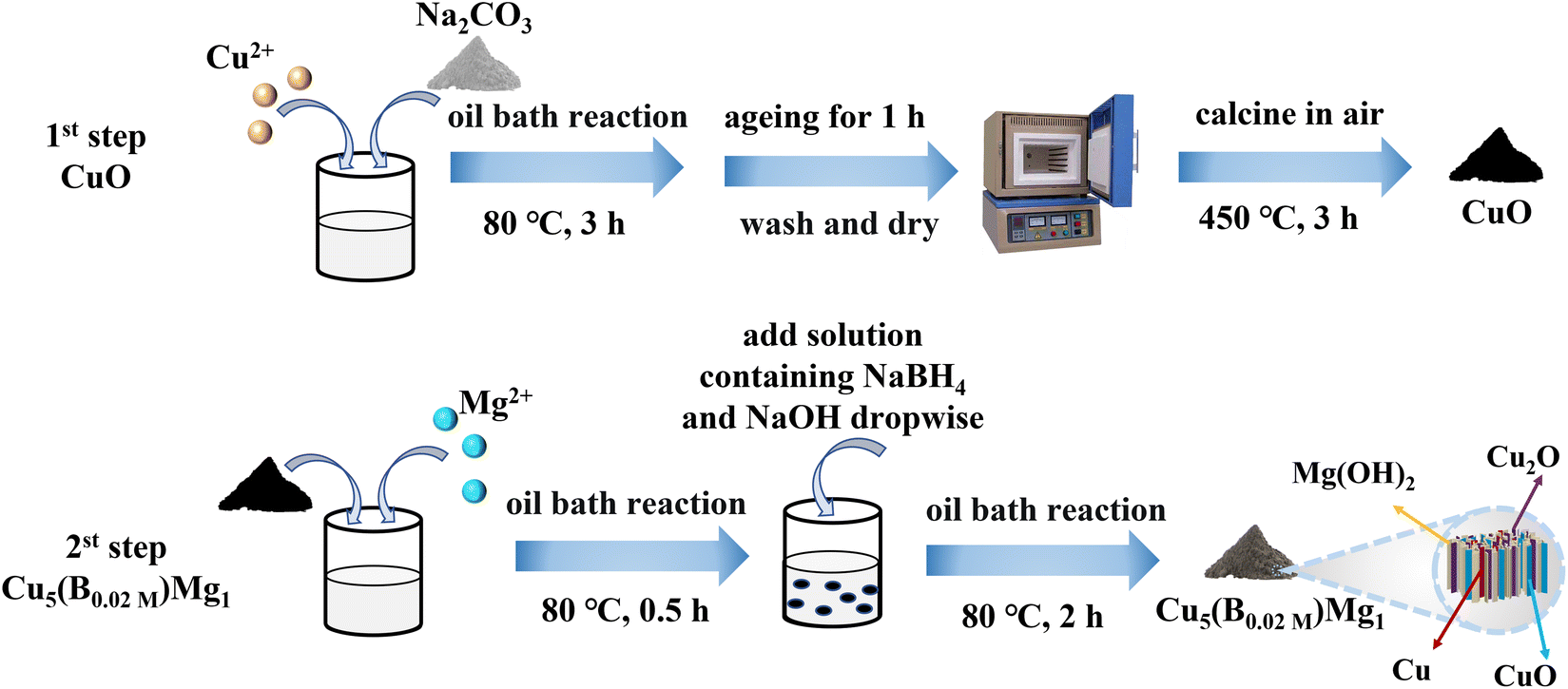 | ||
| Fig. 1 Graphical scheme for the synthesis of the Cu5(B0.02 M)Mg1 catalyst. | ||
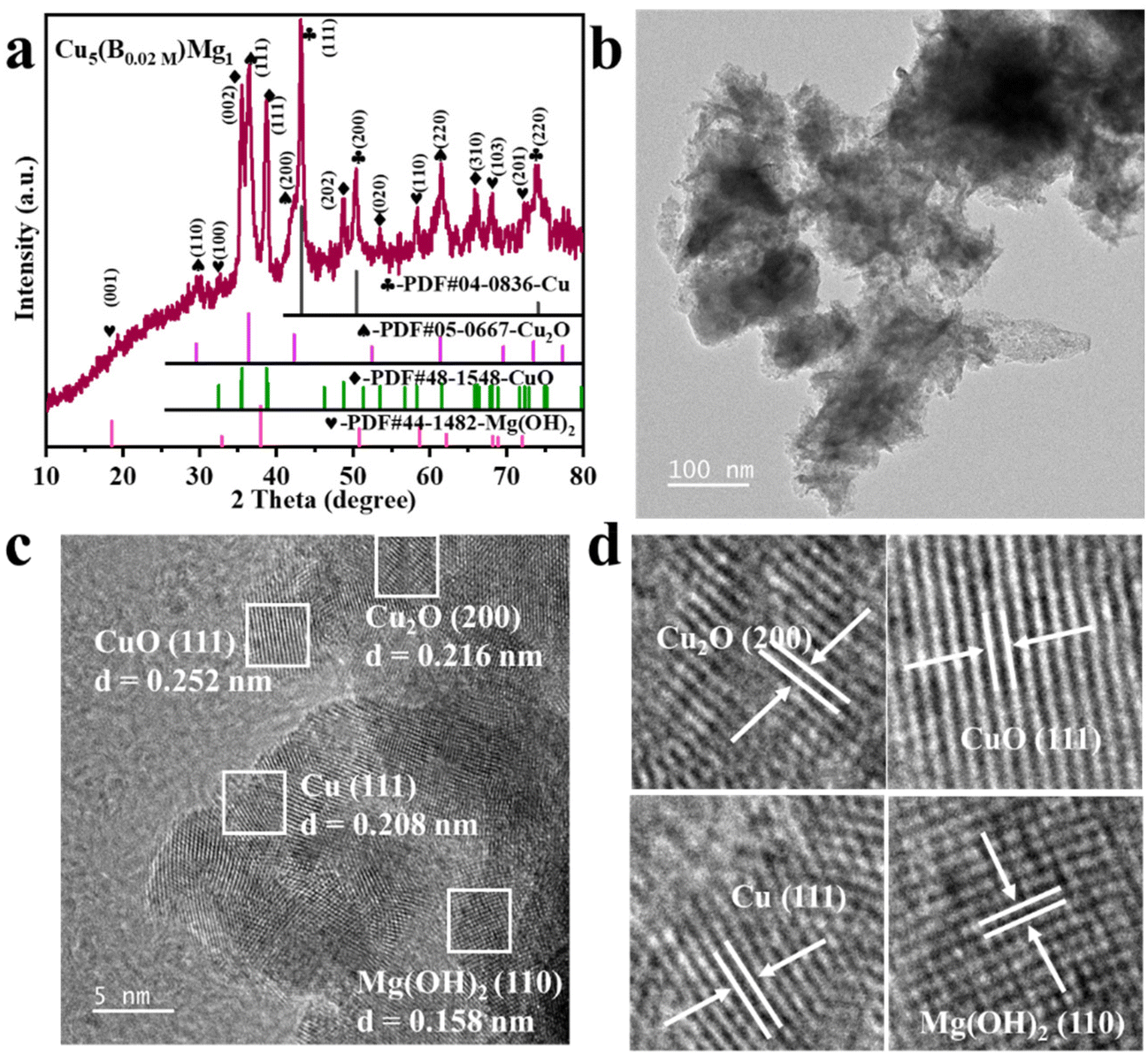 | ||
| Fig. 2 (a) XRD, (b) TEM and (c) HRTEM characterization of the Cu5(B0.02 M)Mg1 catalyst. (d) is the enlarged view of (c). | ||
Scanning electron microscopy (SEM) and TEM reveal that the as-prepared Cu5(B0.02 M)Mg1 catalyst is constructed of nanoclusters (Fig. S2† and Fig. 2b). Energy-dispersive X-ray spectroscopy (EDS) elemental mappings demonstrate a uniform distribution of Cu, Mg, B and O elements on the catalyst surface (Fig. S2c and d†). SEM and EDS characterization studies were performed on the Cu5Mg1, Cu5(B0.1 M)Mg1, Cu(B0.02 M), Cu10(B0.02 M)Mg1 and Cu1(B0.02 M)Mg1 catalysts as well, with the corresponding results shown in Fig. S3–S7.† Further HRTEM analysis reveals interplanar spacings of 0.208 (Cu (111)), 0.216 (Cu2O (200)), 0.252 (CuO (111)) and 0.158 nm (Mg(OH)2 (110)) in the Cu5(B0.02 M)Mg1 catalyst, consistent with its XRD result (Fig. 2c and d). TEM and HRTEM were also conducted on Cu(B0.02 M) and Cu5Mg1 to explore the effect of Mg and B on the structure of the catalyst (Fig. S8 and S9†). For the Cu(B0.02 M) catalyst, the HRTEM image reveals crystal lattices with interfacial spacings of 0.209, 0.247, and 0.253 nm, coinciding with the Cu (111), Cu2O (111), and CuO (111) lattices (Fig. S8†). Conversely, the Cu5Mg1 catalyst primarily exhibits the CuO (111) crystalline phase, along with some amorphous species (Fig. S9†). Considering TEM and XRD results collectively, we may speculate that the observed amorphous substance in Cu5Mg1 arises from some Mg-based amorphous species formed due to the absence of the NaBH4 reagent (Fig. S9b†).
The electronic structures of the catalysts with different ratios of Cu, Mg and B additions were characterized by XPS (Fig. 3). The Cu 2p XPS spectra demonstrate two peaks corresponding to Cu 2p3/2 and Cu 2p1/2 (Fig. 3a and b).35,36 The peaks at 933.98 and 953.78 eV are attributed to the Cu2+ of CuO in Cu5Mg1 (Fig. 3a and Table S1†),37–39 indicating that in the absence of NaBH4 reducing agent, the Cu species in the catalyst mainly exists in the form of Cu2+. Notably, upon NaBH4 addition, the Cu2+ bands of Cu5(B0.02 M)Mg1 and Cu5(B0.1 M)Mg1 catalysts shift to higher electron binding energies by 0.55 and 0.64 eV, respectively, and at the same time Cu0/Cu+ is produced. Additionally, with increasing NaBH4 content, the Cu0/Cu+ electron binding energies decrease from 932.43/952.23 eV (Cu5(B0.02 M)Mg1) to 932.17/951.97 eV (Cu5(B0.1 M)Mg1) (Fig. 3a and Table S1†).40–42 These results demonstrate that the NaBH4 content is a key factor affecting the electronic structure of Cu centers. To further differentiate between Cu0 and Cu+, Cu LMM Auger electron spectroscopy analysis was performed on Cu5(B0.02 M)Mg1 and Cu5(B0.1 M)Mg1 catalysts (Fig. S10†). The two peaks at around 568.00 and 569.75 eV correspond to Cu0 and Cu+,43,44 respectively, confirming the co-existence of Cu0 and Cu+ species on these two catalysts. According to previous studies, Cu0 and Cu+ played different roles during the CO2RR. Cu0 contributed to activating CO2 molecules.45 At the initial stage of the reaction, CO2 tended to be above the Cu0 zone. After CO2 activation, Cuδ+ (0 < δ ≤ 1) was proposed to stabilize CO2 molecules, which can promote the dimerization process between *CO intermediates and enhance the subsequent C–C coupling.46,47 In addition, we have investigated the effects of Mg on the valence state of Cu compounds, as provided in Fig. 3b and Table S2.† It can be seen that Mg plays a similar role to B, since with the increasing Mg content, the characteristic XPS band of Cu2+ shifts to a higher electron binding energy, while the Cu0/Cu+ peak shifts in the opposite direction. These results suggest that the introduced B and Mg elements interact with the surrounding atoms, altering the electron density at the Cu sites. Fig. 3c shows that regardless of the presence of NaBH4, the Mg(OH)2 species persists on the catalysts, as indicated by its characteristic peak at ∼1303.60 eV.48 Taking the XPS and the above XRD/TEM results into comprehensive consideration, it is suggested that the NaBH4 reactant plays a significant role in tuning the crystalline state of the Mg(OH)2 species. In the absence of NaBH4, Mg(OH)2 exists in an amorphous phase, while with NaBH4 addition, Mg(OH)2 exists as crystals. We propose that the existing form of Mg(OH)2 could be a critical influencing factor on the CO2RR performance of the catalyst. Nonetheless, the high-resolution B 1s spectra of Cu5(B0.02 M)Mg1 and Cu5(B0.1 M)Mg1 show two peaks at 191.92 and 191.81 eV, belonging to B–O (Fig. 3d).49,50 Additionally, the contents of Cu, Mg and B elements on the Cu5(B0.02 M)Mg1 catalyst surface were determined via XPS analysis, as provided in Table S3.†
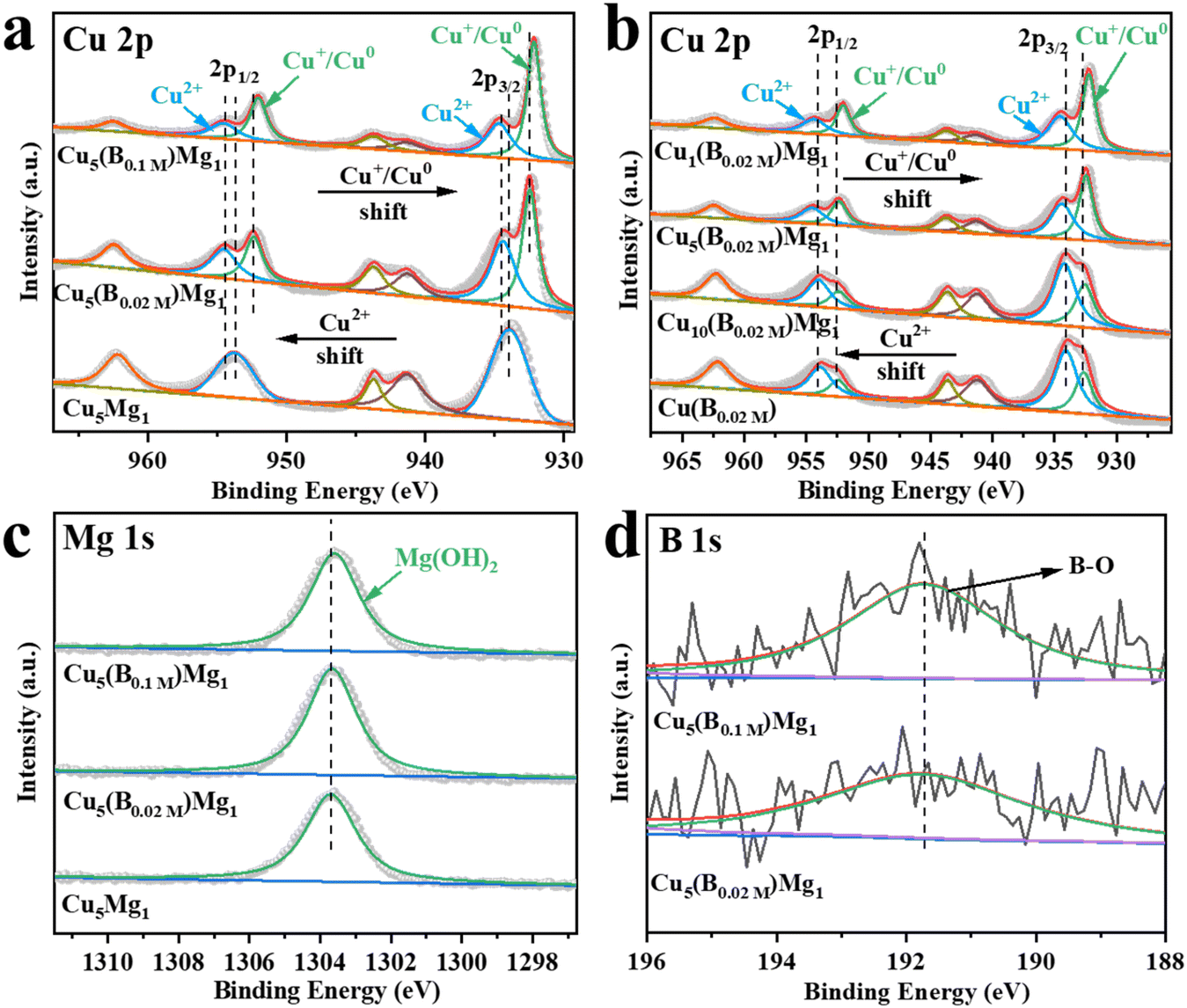 | ||
Fig. 3 XPS spectra of Cu 2p in (a) Cu5Mg1, Cu5(B0.02 M)Mg1, Cu5(B0.1 M)Mg1 (varying the NaBH4 feeding amount while keeping the Cu/Mg ratio at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and (b) Cu(B0.02 M), Cu10(B0.02 M)Mg1, Cu5(B0.02 M)Mg1 and Cu1(B0.02 M)Mg1 (varying the ratio of Cu/Mg while keeping the NaBH4 feeding amount the same at 0.02 M). (c) XPS spectra of Mg 1s in Cu5Mg1, Cu5(B0.02 M)Mg1, and Cu5(B0.1 M)Mg1. (d) The corresponding B 1s XPS spectra of Cu5(B0.02 M)Mg1 and Cu5(B0.1 M)Mg1 catalysts. :1) and (b) Cu(B0.02 M), Cu10(B0.02 M)Mg1, Cu5(B0.02 M)Mg1 and Cu1(B0.02 M)Mg1 (varying the ratio of Cu/Mg while keeping the NaBH4 feeding amount the same at 0.02 M). (c) XPS spectra of Mg 1s in Cu5Mg1, Cu5(B0.02 M)Mg1, and Cu5(B0.1 M)Mg1. (d) The corresponding B 1s XPS spectra of Cu5(B0.02 M)Mg1 and Cu5(B0.1 M)Mg1 catalysts. | ||
2.2 Electrochemical CO2RR performance in a flow cell
The CO2RR performances of Cu5(B0.02 M)Mg1, Cu5(B0.1 M)Mg1, Cu5Mg1, Cu10(B0.02 M)Mg1, Cu1(B0.02 M)Mg1 and Cu(B0.02 M) catalysts were evaluated in 1.0 M KOH electrolyte saturated with CO2 at potentials ranging from −0.77 to −1.97 V. Typical gaseous products (C2H4, CH4, CO, and H2) and liquid products (CH3CH2OH, CH3COOH, CH3OH, and HCOOH) were analyzed (Fig. S11 and S12†). Among these different materials, Cu5(B0.02 M)Mg1 (Cu:Mg = 5:1, NaBH4 = 0.02 M) shows excellent selectivity towards C2+ products (Fig. 4 and S13†).
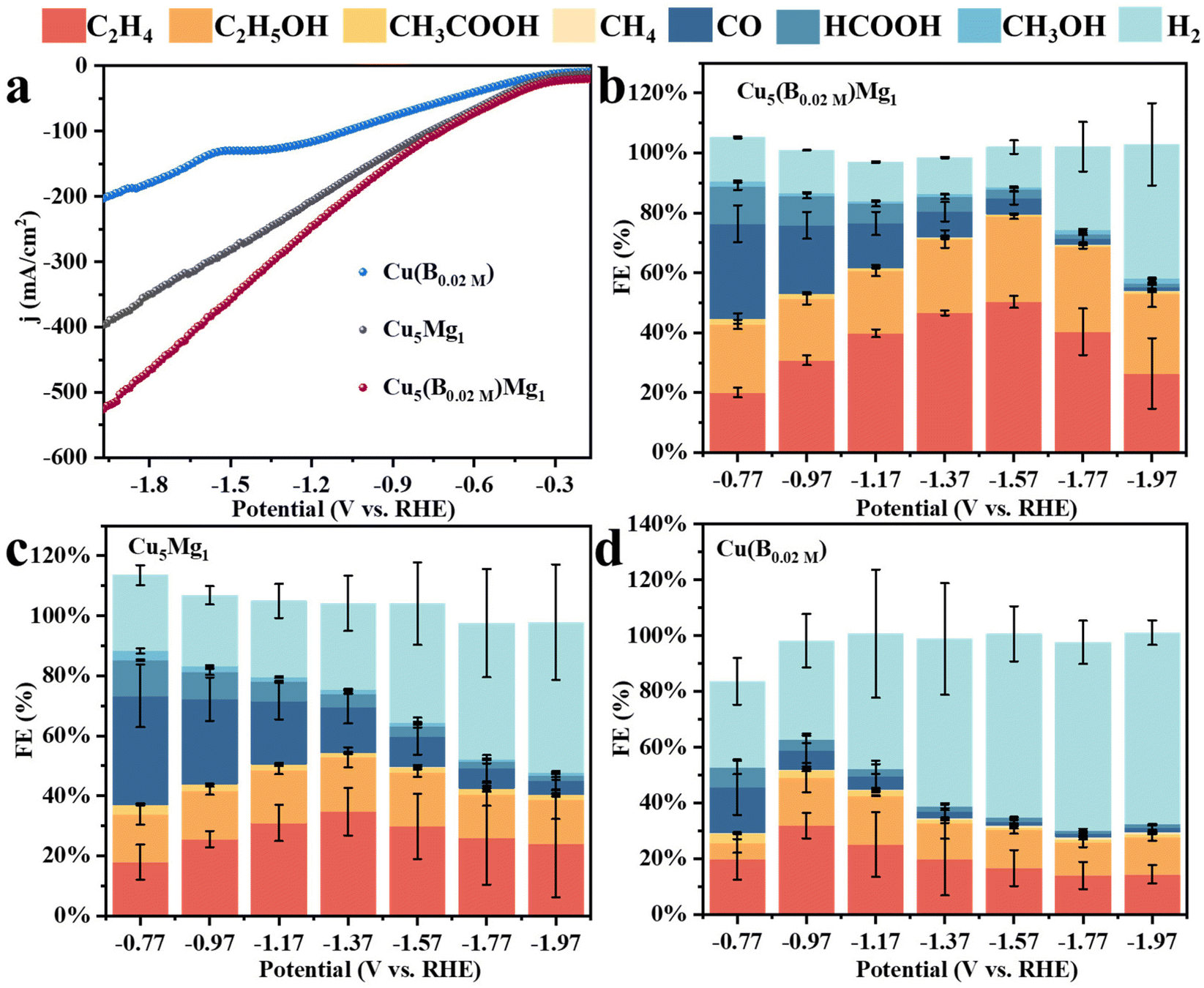 | ||
| Fig. 4 (a) LSVs of Cu(B0.02 M), Cu5Mg1 and Cu5(B0.02 M)Mg1. The Faraday efficiencies (FEs) of all products for different catalysts: (b) Cu5(B0.02 M)Mg1, (c) Cu5Mg1, and (d) Cu(B0.02 M). | ||
Linear sweep voltammetry (LSV) was first employed to determine the current densities of these Cu catalysts modified by Mg and/or B under CO2RR conditions (Fig. 4a). It is found that the Cu5(B0.02 M)Mg1 catalyst exhibits a significantly higher current density as compared to the Cu5Mg1 and Cu(B0.02 M) catalysts, suggesting a potentially better CO2RR activity. The electrocatalytic products of the three catalysts were then collected during chronoamperometry measurements, and their Faraday efficiencies (FEs) are presented in Fig. 4b–d. Among all the catalysts, Cu5(B0.02 M)Mg1 shows the highest C2+ product selectivity (Fig. 4b). Notably, the FEC2+ for the Cu5(B0.02 M)Mg1 catalyst remains >60% across a wide potential range of 600 mV (from −1.17 to −1.77 V), significantly outperforming Cu5Mg1 and Cu(B0.02 M) (Fig. 4b–d). It should be noted that the Cu5(B0.02 M)Mg1 catalyst also shows a low FE for H2 (FEH2) of <15% within the potential range of −0.77 to −1.57 V, indicating that the hydrogen evolution reaction is effectively inhibited on the catalyst. In particular, a maximal FEC2+ of 79.59% at −1.57 V is recorded for Cu5(B0.02 M)Mg1 (FEC2H4 = 50.31%, FEC2H5OH = 28.58%, with a small amount of CH3COOH), significantly higher than those of Cu5Mg1 (FEC2+ = 49.53%) and Cu(B0.02 M) catalysts (FEC2+ = 31.21%) (Fig. 5a and Table S4†). Furthermore, Cu5(B0.02 M)Mg1 shows prominently enhanced partial current densities for C2+ products (jC2+). As depicted in Fig. 5b, jC2+ of Cu5(B0.02 M)Mg1 (−317.03 mA cm−2) is 2.7 and 3.5 times higher than those of Cu5Mg1 (−116.74 mA cm−2) and Cu(B0.02 M) (−91.09 mA cm−2) at the same working potential of −1.57 V, suggesting the synergistic promotion of C2+ formation on Cu surfaces by Mg and B. In addition, jC2+ of Cu5(B0.02 M)Mg1, Cu5Mg1 and Cu(B0.02 M) at more different voltages are shown in Table S5.† Additionally, we have investigated the influence of different ratios of B and Mg during preparation on the electrocatalytic CO2RR activities of the catalysts (Fig. S13†). The results reveal that appropriate amounts of NaBH4 and Mg reactants during synthesis are conducive to enhanced C2+ selectivity, while excessive usage will reduce C2+ products. This could be because the selective promoting and blocking effects of these elements in Cu5(B0.02 M)Mg1 reach a relatively balanced state for efficient CO2RR catalysis (Table S4†).33 Furthermore, we have compared the CO2RR activities of Cu5(B0.02 M)Mg1 with other different catalysts in previous literature, which shows that the synergy of Mg and B contributes to a competitive selectivity towards C2+ products (Table S6†).
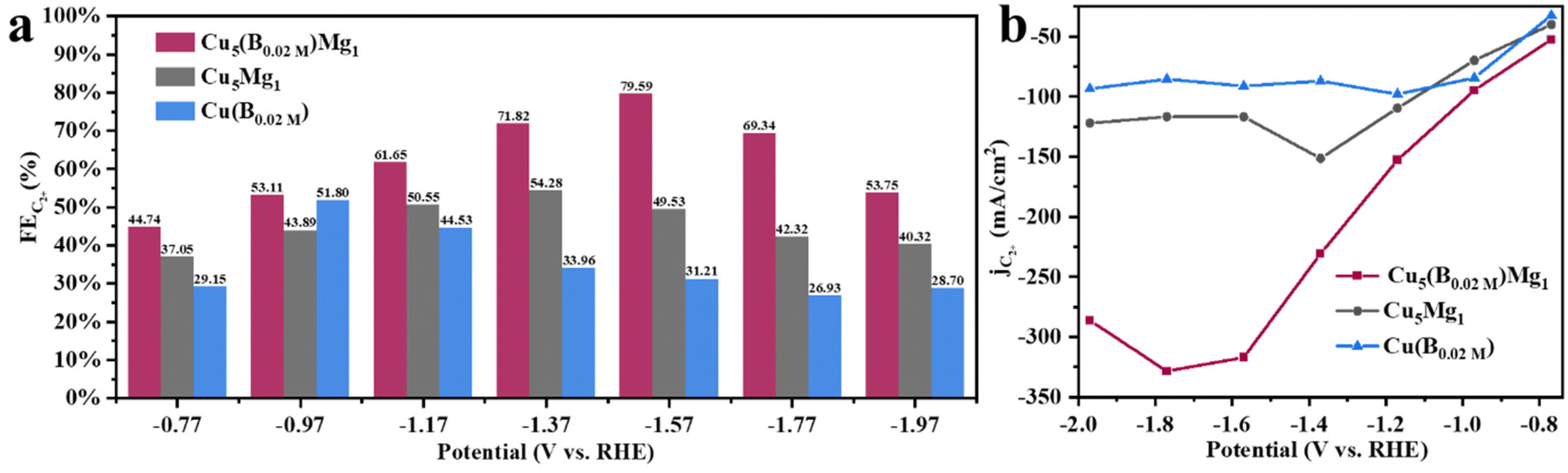 | ||
| Fig. 5 Electrochemical CO2RR performance of the Cu5(B0.02 M)Mg1 catalyst: (a) FEs of C2+ (C2H4, C2H5OH and CH3COOH) at different potentials and (b) partial current densities of C2+. | ||
The long-term catalytic performance of Cu5(B0.02 M)Mg1 for the CO2RR was measured through chronoamperometry at −1.57 V (Fig. S14†). Within a 10 h period, the current density shows negligible attenuation, but the yield of C2H4 gradually decreases over time. After the long-term CO2RR measurement, Cu5(B0.02 M)Mg1 was subjected to XRD characterization. The results indicate that the bulk of the catalyst post CO2RR is primarily composed of Cu (PDF#04-0836), Cu2O (PDF#05-0667) and Mg(OH)2 (PDF#44-1482), with CuO completely reduced. Notably, the XRD peaks of Mg(OH)2 diminish post CO2RR, which we believe leads to the decline in the stability of Cu5(B0.02 M)Mg1 (Fig. S15†). Besides, previous literature studies have suggested that the accumulation of impurities like NOx, SOx, and carbon on the catalyst surface can also affect the active sites for the CO2RR, thereby resulting in a gradual decrease in performance.51–53 In addition to XRD, XPS characterization was employed to analyze the elemental distribution on the Cu5(B0.02 M)Mg1 catalyst surface (Fig. S16†). As depicted in Fig. S16a,† copper species, in the form of Cu0 and Cu+, predominantly exist on the catalyst surface. The crystalline Mg(OH)2 species nearly vanishes after the long-term CO2RR, therefore contributing to the decline in the performance of the Cu5(B0.02 M)Mg1 catalyst (Fig. S16b†). Additionally, XPS results show that the B element still exists on the catalyst (Fig. S16c†).
To study the enhanced CO2RR performance of the Cu5(B0.02 M)Mg1 catalyst, the electrochemically active surface areas of all the electrocatalysts were estimated by calculating their double layer capacitances (Cdl) (Fig. S17 and S18†). Obviously, compared with Cu5Mg1 (2.83 mF cm−2), Cu5(B0.1 M)Mg1 (1.61 mF cm−2), Cu(B0.02 M) (0.99 mF cm−2), Cu10(B0.02 M)Mg1 (2.20 mF cm−2) and Cu1(B0.02 M)Mg1 (1.19 mF cm−2), Cu5(B0.02 M)Mg1 (3.28 mF cm−2) has the largest Cdl value, revealing that the Cu5(B0.02 M)Mg1 catalyst can provide more active sites for the CO2RR. Meanwhile, it is worth noting that the active surface area of Cu5Mg1 is lower than that of Cu5(B0.02 M)Mg1, which may be due to the formation of amorphous Mg(OH)2 on the catalyst surface, causing part of the active sites to be covered due to the “stacking” effect, thus reducing the overall catalytic activity of Cu5Mg1.54 Electrochemical impedance spectroscopy measurements have also been performed to investigate the charge transfer properties of the catalysts (Fig. S19†). It is obvious that an appropriate co-modification of Mg and B could accelerate the charge transfer process during the CO2RR.
In situ Raman spectroscopy was further conducted to examine the effects of B and/or Mg on the valence of Cu (Cu0, Cu+ or Cu2+) as well as to probe probable intermediates during the CO2RR (Fig. 6). Fig. 6a presents the in situ Raman spectra of the Cu5(B0.02 M)Mg1 catalyst in 1.0 M KOH electrolyte (saturated with CO2) during the CO2RR. At the open circuit voltage, three characteristic peaks are observed at around 147, 522 and 626 cm−1, attributed to Cu2O.55–57 After applying a cathodic potential of −0.37 V, a new Raman band appears at 703 cm−1 on the Cu5(B0.02 M)Mg1 catalyst, corresponding to surface δCO2−, which represents the umbrella motion of oxygen atoms of the *CO2− intermediate. Previous studies recognized *CO2− as the initial intermediate just after CO2 activation.58 At the same time, Raman peaks at 290 and 374 cm−1 are recorded, assigned to the frustrated rotation and tensile vibrations of Cu-CO, respectively.59–61 Besides, the band at 1071 cm−1 corresponds to the symmetric C–O stretching band of CO32− from the electrolyte.62,63 It is worth noting that over the wide potential range of −0.37 to −1.37 V, Cu2O peaks at 147, 522 and 626 cm−1 consistently appear on the Cu5(B0.02 M)Mg1 catalyst, indicating that the CuO species on the catalyst surface has been completely reduced, while Cu+ persists, probably being the reactive sites during the electrochemical CO2RR. We propose that the robust stability of Cu2O species over a wide potential window is attributed to the synergy of B and crystalline Mg(OH)2. According to previous literature, the Cu+ on the catalyst surface is crucial for the formation of multi-carbon products.64–67 The Raman spectra over −1.37 V cannot be collected because of the accumulation of gaseous products on the catalyst surface that block signals. Nevertheless, Raman signals for Cu5Mg1 and Cu(B0.02 M) are also recorded under the same conditions as Cu5(B0.02 M)Mg1 to explore the influences of Mg and B on the Cu catalysts, as shown in Fig. 6b and c. In contrast to the Cu5(B0.02 M)Mg1 catalyst, Cu2O peaks vanish at only −0.87 V and −0.97 V for Cu5Mg1 and Cu(B0.02 M) catalysts, respectively. In addition, unlike the Cu5(B0.02 M)Mg1 catalyst which displays characteristic bands of *CO and *CO2− at −0.37 V, neither Cu5Mg1 nor the Cu(B0.02 M) catalyst shows these bands until −0.47 V. Hence, it is evident that compared to the Cu5Mg1 and Cu(B0.02 M) catalysts, the collaborative effect of Mg and B, which induces the formation of crystalline Mg(OH)2 species in the catalyst structure, facilitates the early formation of *CO2− and *CO intermediates during the CO2RR.
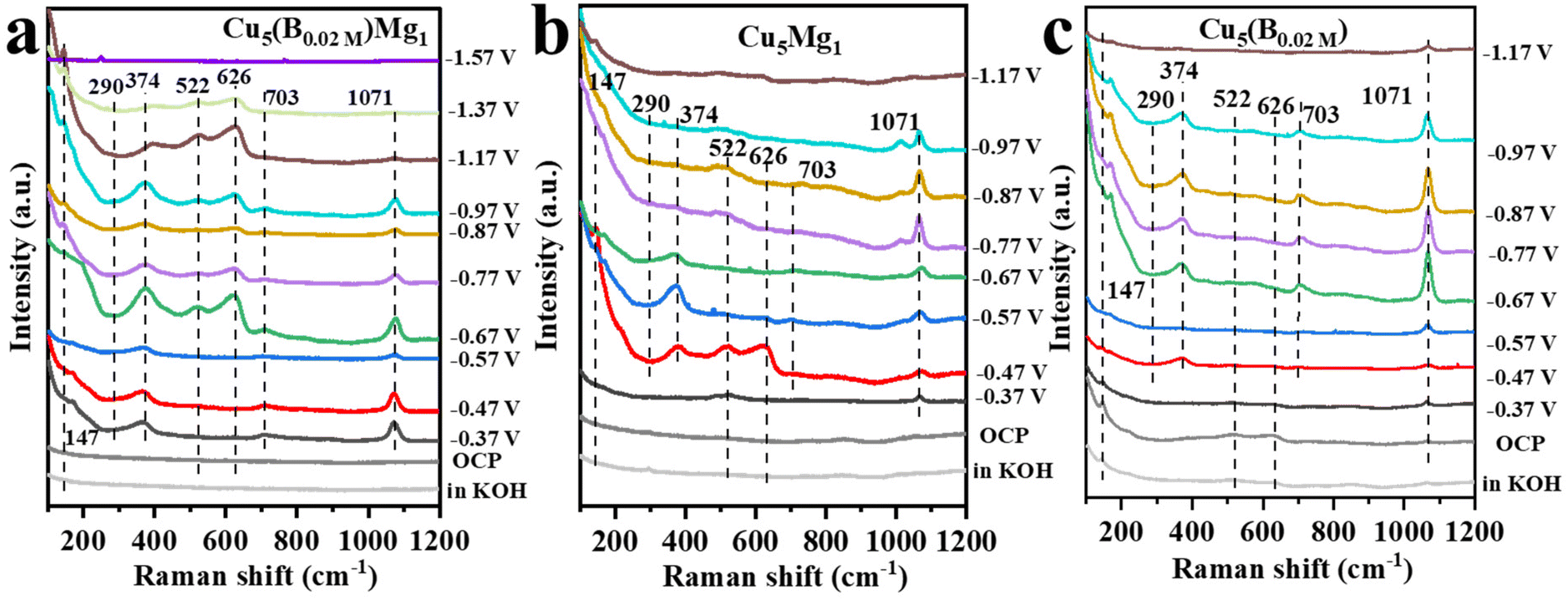 | ||
| Fig. 6 The in situ Raman spectra of (a) Cu5(B0.02 M)Mg1, (b) Cu5Mg1 and (c) Cu(B0.02 M) catalysts at various potentials in 1.0 M KOH electrolyte during CO2RR catalysis. | ||
A previous study by Yang et al. utilized cerium as a self-sacrificing agent to stabilize Cu+ in their catalyst, taking advantage of the easy redox properties of Ce3+/Ce4+. Their in situ Raman and Fourier transform infrared spectroscopy results demonstrated that the stabilized Cu+ components promoted C–C coupling under the CO2RR. DFT calculations further revealed that strong *CO adsorption and low C–C coupling energy were conducive to the formation of C2H5OH.61 Wang et al. prepared an ultra-thin 2D Cu2O nanosheet (Cu2O-NS) catalyst with abundant oxygen vacancies. In situ Raman spectroscopy and DFT calculations supported that Cu2O-NS prevented the reduction of surface Cu oxides, thereby stabilizing Cu+ and enhancing the conversion of CO2 to C2+.68 Similarly, in our work, Cu+ stabilization is achieved through the introduction of appropriate Mg and B into Cu materials. We found that the addition of the NaBH4 reactant can tune the amorphous or crystalline phases of Mg(OH)2. Crystallized Mg(OH)2 is crucial for prolonging the lifetime of Cu+ during the CO2RR and facilitating the activation of CO2 on the Cu surface to generate key *CO intermediates for C–C coupling. Consequently, this enhances the selectivity towards C2+ products of our catalyst.
3 Conclusion
Overall, a simple two-step method involving calcination and subsequent wet chemical reduction is developed to construct a Cu-based electrocatalyst modified by crystalline Mg(OH)2 and B. XRD, TEM, and XPS analyses indicate that the addition of the NaBH4 reactant can induce the formation of crystalline Mg(OH)2, which is proposed to enhance the conversion of CO2 to C2+ during the CO2RR compared to amorphous Mg(OH)2. In support of this, in situ Raman spectroscopy reveals that the synergy of crystalline Mg(OH)2 and B can stabilize Cu+ at rather cathodic potentials during the CO2RR process, effectively producing and preserving the *CO intermediates, therefore greatly enhancing the *CO–CO coupling. As a result, the FEC2+ surpasses 60% on the Cu5(B0.02 M)Mg1 electrocatalyst over a wide potential window of 600 mV (−1.17 to −1.77 V). In particular, at −1.57 V, the catalyst exhibits a FEC2+ as high as 79.59%, demonstrating enormous potential for its scalable applications. This work not only provides a low-cost, simple and effective synthesis method for efficient electrocatalysts but also offers an in-depth understanding of the active sites and reaction intermediates during CO2-to-C2+ conversion. We believe that this will help inspire the rational design of future CO2RR electrocatalysts.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21905118 and 12304020), the Natural Science Foundation of Jiangsu Province (BK20230909), and the Funding for Scientific Research Startup of Jiangsu University (18JDG033).References
- H. H. Wang, N. Wen, Y. Q. Wang, X. L. Jiao, Y. G. Xia and D. R. Chen, Boosting Electrochemical Reduction of CO2 to Formate over Oxygen Vacancy Stabilized Copper–Tin Dual Single Atoms Catalysts, Adv. Funct. Mater., 2023, 33, 2303473 CrossRef CAS.
- J. Y. Liu, P. S. Li, J. H. Bi, S. Q. Jia, Y. Wang, X. C. Kang, X. F. Sun, Q. G. Zhu and B. X. Han, Switching between C2+ Products and CH4 in CO2 Electrolysis by Tuning the Composition and Structure of Rare-Earth/Copper Catalysts, J. Am. Chem. Soc., 2023, 145, 23037–23047 CrossRef CAS PubMed.
- J. Yin, J. Jin, Z. Y. Yin, L. Zhu, X. Du, Y. Peng, P. Xi, C. H. Yan and S. H. Sun, The built-in electric field across FeN/Fe3N interface for efficient electrochemical reduction of CO2 to CO, Nat. Commun., 2023, 14, 1724 CrossRef CAS PubMed.
- P. S. Li, J. H. Bi, J. Y. Liu, Y. Wang, X. C. Kang, X. F. Sun, J. L. Zhang, Z. M. Liu, Q. G. Zhu and B. X. Han, p–d Orbital Hybridization Induced by p-Block Metal-Doped Cu Promotes the Formation of C2+ Products in Ampere-Level CO2 Electroreduction, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
- Z. Y. Zhang, H. Tian, L. Bian, S. Z. Liu, Y. Liu and Z. L. Wang, Cu-Zn-based alloy/oxide interfaces for enhanced electroreduction of CO2 to C2+ products, J. Energy Chem., 2023, 83, 90–97 CrossRef CAS.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- X. T. Wang, Z. Z. Wang, Y. Li, J. T. Wang and G. K. Zhang, Efficient photocatalytic CO2 conversion over 2D/2D Ni-doped CsPbBr3/Bi3O4Br Z-scheme heterojunction: Critical role of Ni doping, boosted charge separation and mechanism study, Appl. Catal., B, 2022, 319, 121895 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, CO2 Reduction: From the Electrochemical to Photochemical Approach, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- Z. Li, R. Wu, L. Zhao, P. Li, X. Wei, J. Wang, J. S. Chen and T. Zhang, Metal-support interactions in designing noble metal-based catalysts for electrochemical CO2 reduction: Recent advances and future perspectives, Nano Res., 2021, 14, 3795–3809 CrossRef CAS.
- Y. Yang, S. Louisia, S. M. Yu, J. B. Jin, I. Roh, C. B. Chen, M. V. F. Guzman, J. Feijóo, P. C. Chen, H. S. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. D. Yang, Operando studies reveal active Cu nanograins for CO2 electroreduction, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- S. Y. Lee, J. M. Kim, G. Bak, E. C. Lee, D. Kim, S. Yoo, J. Kim, H. Yun and Y. J. Hwang, Probing Cation Effects on *CO Intermediates from Electroreduction of CO2 through Operando Raman Spectroscopy, J. Am. Chem. Soc., 2023, 145, 23068–23075 CrossRef CAS PubMed.
- T. Ahmad, S. Liu, M. Sajid, K. Li, M. Ali, L. Liu and W. Chen, Electrochemical CO2 reduction to C2+ products using Cu-based electrocatalysts: A review, Nano Res. Energy, 2022, 1, e9120021 CrossRef.
- C. Liu, M. M. Wang, J. Y. Ye, L. B. Liu, L. G. Li, Y. H. Li and X. Q. Huang, Highly Selective CO2 Electroreduction to C2+ Products over Cu2O-Decorated 2D Metal–Organic Frameworks with Rich Heterogeneous Interfaces, Nano Lett., 2023, 23, 1474–1480 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C. T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. D. Yang and E. H. Sargent, Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- L. C. Ding, N. N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. C. Wu and Y. J. Xiong, Over 70% Faradaic Efficiency for CO2 Electroreduction to Ethanol Enabled by Potassium Dopant–Tuned Interaction between Copper Sites and Intermediates, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed.
- C. Peng, G. Luo, J. B. Zhang, M. H. Chen, Z. Q. Wang, T. K. Sham, L. J. Zhang, Y. F. Li and G. F. Zheng, Double sulfur vacancies by lithium tuning enhance CO2 electroreduction to n-propanol, Nat. Catal., 2021, 12, 1580 CAS.
- Y. R. Lin, D. U. Lee, S. Q. Tan, D. M. Koshy, T. Y. Lin, L. Wang, D. Corral, J. E. Avilés Acosta, J. A. Z. Zeledon, V. A. Beck, S. E. Baker, E. B. Duoss, C. Hahn and T. F. Jaramillo, Vapor–Fed Electrolyzers for Carbon Dioxide Reduction Using Tandem Electrocatalysts: Cuprous Oxide Coupled with Nickel–Coordinated Nitrogen–Doped Carbon, Adv. Funct. Mater., 2022, 32, 2113252 CrossRef CAS.
- X. P. Qin, S. Q. Zhu, F. Xiao, L. L. Zhang and M. H. Shao, Active Sites on Heterogeneous Single-Iron-Atom Electrocatalysts in CO2 Reduction Reaction, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef CAS.
- Y. J. Shi, Y. J. Wang, J. Y. Yu, Y. K. Chen, C. Q. Fang, D. Jiang, Q. H. Zhang, L. Gu, X. W. Yu, X. Li, H. Liu and W. J. Zhou, Superscalar Phase Boundaries Derived Multiple Active Sites in SnO2/Cu6Sn5/CuO for Tandem Electroreduction of CO2 to Formic Acid, Adv. Energy Mater., 2023, 13, 2203506 CrossRef CAS.
- P. T. Wang, M. Qiao, Q. Shao, Y. C. Pi, X. Zhu, Y. F. Li and X. Q. Huang, Phase and structure engineering of copper tin heterostructures for efficient electrochemical carbon dioxide reduction, Nat. Catal., 2018, 9, 4933 Search PubMed.
- D. G. Park, J. W. Choi, H. J. Chun, H. S. Jang, H. B. Lee, W. H. Choi, B. C. Moon, K. H. Kim, M. G. Kim, K. M. Choi, B. C. Han and J. K. Kang, Increasing CO Binding Energy and Defects by Preserving Cu Oxidation State via O2-Plasma-Assisted N Doping on CuO Enables High C2+ Selectivity and Long-Term Stability in Electrochemical CO2 Reduction, ACS Catal., 2023, 13, 9222–9233 CrossRef CAS.
- G. Park, H. Kim, G. H. Han, J. Ha, J. Y. Seo, M. J. Kang, M. G. Seo, Y. H. Choi, S. Y. Kim and S. H. Ahn, The effect of Cu oxidation states on C2H4 production from electrochemical CO2 conversion, J. CO2 Util., 2023, 75, 102569 CrossRef CAS.
- R. B. Sun, C. Wei, Z. X. Huang, S. W. Niu, X. Han, C. Chen, H. R. Wang, J. Song, J. D. Yi, G. Wu, D. W. Rao, X. S. Zheng, Y. Wu, G. M. Wang and X. Hong, Cu2+1O/CuOx heterostructures promote the electrosynthesis of C2+ products from CO2, Nano Res., 2022, 16, 4698–4705 CrossRef.
- H. H. Yang, S. Y. Li and Q. Xu, Efficient strategies for promoting the electrochemical reduction of CO2 to C2+ products over Cu-based catalysts, Chin. J. Catal., 2023, 48, 32–65 CrossRef CAS.
- J. Wang, J. J. Liu, Y. Q. Song, S. B. Geng, Z. H. Peng, J. L. Yu, F. Liu, Y. H. Wang, S. B. Xi, Z. J. Zhang and Z. X. Fan, Simultaneous Defect and Size Control of Metal–Organic Framework Nanostructures for Highly Efficient Carbon Dioxide Electroreduction to Multicarbon Products, ACS Mater. Lett., 2023, 5, 2121–2130 CrossRef CAS.
- J. W. Su, D. H. Pan, Y. Dong, Y. Y. Zhang, Y. L. Tang, J. Sun, L. J. Zhang, Z. Q. Tian and L. Chen, Ultrafine Fe2C Iron Carbide Nanoclusters Trapped in Topological Carbon Defects for Efficient Electroreduction of Carbon Dioxide, Adv. Energy Mater., 2023, 13, 2204391 CrossRef CAS.
- R. G. Cai, M. Z. Sun, F. Yang, M. Ju, Y. P. Chen, M. D. Gu, B. L. Huang and S. H. Yang, Engineering Cu(I)/Cu(0) interfaces for efficient ethanol production from CO2 electroreduction, Chem, 2023, 10, 1–23 Search PubMed.
- Y. P. Zang, T. F. Liu, P. F. Wei, H. F. Li, Q. Wang, G. X. Wang and X. H. Bao, Selective CO2 Electroreduction to Ethanol over a Carbon–Coated CuOx Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed.
- C. F. Wen, M. Zhou, P. F. Liu, Y. W. Liu, X. F. Wu, F. X. Mao, S. Dai, B. B. Xu, X. L. Wang, Z. Jiang, P. Hu, S. Yang, H. F. Wang and H. G. Yang, Highly Ethylene–Selective Electrocatalytic CO2 Reduction Enabled by Isolated Cu−S Motifs in Metal–Organic Framework Based Precatalysts, Angew. Chem., Int. Ed., 2021, 61, e202111700 CrossRef PubMed.
- Y. Yao, Y. Zhou, X. Liu, Y. Li, D. Wang, X. Chi, X. Wang, R. Zhao, H. Zhang, Y. Sun, Z.-Y. Yang, Y. Wei and Y.-M. Yan, Restraining lattice oxygen of Cu2O by enhanced Cu–O hybridization for selective and stable production of ethylene with CO2 electroreduction, J. Mater. Chem. A, 2022, 10, 20914–20923 RSC.
- S. Sinha and J. J. Jiang, Main group elements in electrochemical hydrogen evolution and carbon dioxide reduction, Chem. Commun., 2023, 59, 11767–11779 RSC.
- C. Peng, J. Ma, G. Luo, S. Yan, J. Zhang, Y. Chen, N. Chen, Z. Wang, W. Wei, T. K. Sham, Y. Zheng, M. Kuang and G. Zheng, (111) Facet-oriented Cu2Mg Intermetallic Compound with Cu3-Mg Sites for CO2 Electroreduction to Ethanol with Industrial Current Density, Angew. Chem., Int. Ed. Engl., 2024, e202316907 CAS.
- M. C. Xie, Y. Shen, W. C. Ma, D. Y. Wei, B. Zhang, Z. H. Wang, Y. H. Wang, Q. H. Zhang, S. J. Xie, C. Wang and Y. Wang, Fast Screening for Copper–Based Bimetallic Electrocatalysts: Efficient Electrocatalytic Reduction of CO2 to C2+ Products on Magnesium–Modified Copper, Angew. Chem., Int. Ed., 2022, 61, e202213423 CrossRef CAS PubMed.
- J. Fan, X. Zhang, M. Han, X. Xiang, C. Guo, Y. Lin, N. Shi, D. Xu, Y. Lai and J. Bao, Amorphous Ni–Fe–Mo Oxides Coupled with Crystalline Metallic Domains for Enhanced Electrocatalytic Oxygen Evolution by Promoted Lattice–Oxygen Participation, Small, 2023, 20, 2303927 CrossRef PubMed.
- Y. Z. Li, Z. B. Wang, C. Li, F. Qi, P. W. Yan, Y. P. Wang, M. F. He, Z. L. Chen, Q. Wang, Y. Wang, H. Zheng, A. Ikhlaq, J. Kumirska, E. Maria Siedlecka and O. Ismailova, Reducing agents enhanced prometon degradation by CuBi2O4/peroxymonosulfate: Development of interfacial electron transport and circulation of Cu+/Cu2+, Chem. Eng. J., 2023, 470, 144387 CrossRef CAS.
- Z. P. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y. H. Chou, X. Y. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. J. Han and R. Amal, Atomically Dispersed Cu Catalysts on Sulfide-Derived Defective Ag Nanowires for Electrochemical CO2 Reduction, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- Z. Q. Zhang, J. L. Liang, W. Zhang, M. Zhou, X. L. Zhu, Z. Y. Liu, Y. Li, Z. Q. Guan, C. S. Lee, P. K. Wong, H. M. Li and Z. F. Jiang, Modified-pollen confined hybrid system: A promising union for visible-light-driven photocatalytic antibiotic degradation, Appl. Catal., B, 2023, 330, 122621 CrossRef CAS.
- X. L. Zhou, J. Q. Shan, L. Chen, B. Y. Xia, T. Ling, J. J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, Stabilizing Cu2+ Ions by Solid Solutions to Promote CO2 Electroreduction to Methane, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- D. X. Tan, J. L. Zhang, L. Yao, X. N. Tan, X. Y. Cheng, Q. Wan, B. X. Han, L. R. Zheng and J. Zhang, Multi-shelled CuO microboxes for carbon dioxide reduction to ethylene, Nano Res., 2020, 13, 768–774 CrossRef CAS.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, Alloying as a Strategy to Boost the Stability of Copper Nanocatalysts during the Electrochemical CO2 Reduction Reaction, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- P. T. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Q. Huang and S. Z. Qiao, Boosting electrocatalytic CO2−to–ethanol production via asymmetric C–C coupling, Nat. Catal., 2022, 13, 3754 CAS.
- W. Lai, L. H. Ge, H. Yang, Y. L. Deng, H. M. Li, B. Ouyang, L. Xu and J. Bao, Reprogramming the redox states of nickel via interface engineering and heteroatom doping to boost overall water splitting, J. Mater. Chem. A, 2022, 10, 10525–10539 RSC.
- J. H. Jang, S. Q. Zhu, E. P. Delmo, T. H. Li, Q. L. Zhao, Y. N. Wang, L. L. Zhang, H. W. Huang, J. J. Ge and M. H. Shao, Facile design of oxide–derived Cu nanosheet electrocatalyst for CO2 reduction reaction, EcoMat, 2023, 5, e12334 CrossRef CAS.
- Y. Zhang, Z. B. Si, H. H. Du, Y. L. Deng, Q. K. Zhang, Z. L. Wang, Q. Yu and H. Xu, Selective CO2 Reduction to Ethylene Over a Wide Potential Window by Copper Nanowires with High Density of Defects, Inorg. Chem., 2022, 61, 20666–20673 CrossRef CAS PubMed.
- X.-J. Cui, Y.-Q. Qiu, H.-Q. Wang and C.-G. Liu, Direct coupling of two inert CO2 molecules to form a C–C bond on the Cu0 atomic interfaces of the nitrogen-doped graphene-supported Cu4 cluster, Inorg. Chem. Front., 2024, 11, 85–97 RSC.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Potential Link between Cu Surface and Selective CO2 Electroreduction: Perspective on Future Electrocatalyst Designs, Adv. Mater., 2020, 32, e1908398 CrossRef PubMed.
- Z. Li, Z. Liu, S. Li, Y. Pei, D. Li, J. Mao, R. Zhou, C. Qiu, Y. Lu and B. Zhang, Modulating the localized electronic distribution of Cu species during reconstruction for enhanced electrochemical CO2 reduction to C2+ products, J. Mater. Chem. A, 2024 10.1039/d4ta01184h.
- S. J. Han, L. L. Mao, T. Wu and H. Z. Wang, Homogeneous polyethersulfone hybrid membranes prepared with in-suit synthesized magnesium hydroxide nanoparticles by phase inversion method, J. Membr. Sci., 2016, 516, 47–55 CrossRef CAS.
- H. Zhang, D. Y. Zhang, M. Y. Guo, Z. Huang, X. Wang, C. Q. Gao, F. Gao, M. Terrones and Y. Q. Wang, Combustion Activation Induced Solid–State Synthesis for N, B Co–Doped Carbon/Zinc Borate Anode with a Boosting of Sodium Storage Performance, Adv. Sci., 2023, 10, 2207751 CrossRef CAS PubMed.
- H. Q. Peng, X. Ming, K. Pang, Y. R. Chen, J. Zhou, Z. Xu, Y. J. Liu and C. Gao, Highly electrically conductive graphene papers via catalytic graphitization, Nano Res., 2022, 15, 4902–4908 CrossRef CAS.
- J. Leverett, J. A. Yuwono, P. Kumar, T. Tran-Phu, J. Qu, J. Cairney, X. Wang, A. N. Simonov, R. K. Hocking, B. Johannessen, L. Dai, R. Daiyan and R. Amal, Impurity Tolerance of Unsaturated Ni-N-C Active Sites for Practical Electrochemical CO2 Reduction, ACS Energy Lett., 2022, 7, 920–928 CrossRef CAS.
- S. Van Daele, L. Hintjens, S. Hoekx, B. Bohlen, S. Neukermans, N. Daems, J. Hereijgers and T. Breugelmans, How flue gas impurities affect the electrochemical reduction of CO2 to CO and formate, Appl. Catal., B, 2024, 341, 123345 CrossRef CAS.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Stability Issues in Electrochemical CO2 Reduction: Recent Advances in Fundamental Understanding and Design Strategies, Adv. Mater., 2023, 35, e2306288 CrossRef PubMed.
- G. X. Zhang, X. L. Zheng, X. M. Cui, J. Wang, J. H. Liu, J. F. Chen and Q. Xu, Doping of Vanadium into Bismuth Oxide Nanoparticles for Electrocatalytic CO2 Reduction, ACS Appl. Nano Mater., 2022, 5, 15465–15472 CrossRef CAS.
- D. Ren, Y. L. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- S. J. Mu, H. L. Lu, Q. B. Wu, L. Li, R. I. Zhao, C. Long and C. H. Cui, Hydroxyl radicals dominate reoxidation of oxide-derived Cu in electrochemical CO2 reduction, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- H. T. Du, L. X. Liu, P. Li, Q. H. Min, S. J. Guo and W. L. Zhu, Enriching Reaction Intermediates in Multishell Structured Copper Catalysts for Boosted Propanol Electrosynthesis from Carbon Monoxide, ACS Nano, 2023, 17, 8663–8670 CrossRef CAS PubMed.
- Y. Zhao, X. G. Zhang, N. Bodappa, W. M. Yang, Q. Liang, P. M. Radjenovica, Y. H. Wang, Y. J. Zhang, J. C. Dong, Z. Q. Tian and J. F. Li, Elucidating electrochemical CO2 reduction reaction processes on Cu(hkl) single-crystal surfaces by in situ Raman spectroscopy, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- Y. L. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, On the Role of Sulfur for the Selective Electrochemical Reduction of CO2 to Formate on CuSx Catalysts, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 CrossRef CAS PubMed.
- D. Ren, J. H. Fong and B. S. Yeo, The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction, Nat. Commun., 2018, 9, 925 CrossRef PubMed.
- Z. Yang, D. G. Ji, Z. Li, Z. D. He, Y. Hu, J. Yin, Y. H. Hou, P. X. Xi and C. H. Yan, Ceo2/Cus Nanoplates Electroreduce Co2 to Ethanol with Stabilized Cu+ Species, Small, 2023, 19, 2303099 CrossRef CAS PubMed.
- X. P. Yan, M. L. Zhang, Y. Z. Chen, Y. H. Wu, R. Z. Wu, Q. Wan, C. X. Liu, T. T. Zheng, R. J. Feng, J. Zhang, C. J. Chen, C. Xia, Q. G. Zhu, X. F. Sun, Q. L. Qian and B. X. Han, Synergy of Cu/C3N4 Interface and Cu Nanoparticles Dual Catalytic Regions in Electrolysis of CO to Acetic Acid, Angew. Chem., Int. Ed., 2023, 62, e202301507 CrossRef CAS PubMed.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. R. Qiu, A. Bergmann, B. R. Cuenya and O. M. Magnussen, Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- H. Zhang, Y. Qiao, Y. Wang, Y. Zheng and H. Huang, In situ oxidative etching-enabled synthesis of hollow Cu2O nanocrystals for efficient CO2RR into C2+ products, Sustainable Energy Fuels, 2022, 6, 4860–4865 RSC.
- M. Fang, M. Wang, Z. Wang, Z. Zhang, H. Zhou, L. Dai, Y. Zhu and L. Jiang, Hydrophobic, Ultrastable Cuδ+ for Robust CO2 Electroreduction to C2 Products at Ampere-Current Levels, J. Am. Chem. Soc., 2023, 145, 11323–11332 CrossRef CAS PubMed.
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y. M. Yan, Stabilization of Cu+ via Strong Electronic Interaction for Selective and Stable CO2 Electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202205832 CrossRef CAS PubMed.
- H. Cheng, S. Jia, J. Jiao, X. Chen, T. Deng, C. Xue, M. Dong, J. Zeng, C. Chen, H. Wu, M. He and B. Han, Stabilization of Cu+ sites by amorphous Al2O3 to enhance electrochemical CO2 reduction to C2+ products, Green Chem., 2024, 26, 2599–2604 RSC.
- P. Wang, S. Meng, B. Zhang, M. He, P. Li, C. Yang, G. Li and Z. Li, Sub-1 nm Cu2O Nanosheets for the Electrochemical CO2 Reduction and Valence State-Activity Relationship, J. Am. Chem. Soc., 2023, 145, 26133–26143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi00842a |
| This journal is © the Partner Organisations 2024 |